

ABSTRACT
The detailed pathogenesis of endometriosis remains largely unclear despite decades of research. Recent studies have demonstrated that miRNAs plays an important role in endometriosis. The expression of miR-142-3p was decreased in ectopic endometrial tissues, while KLF9 and VEGFA expression levels were increased. Overexpression of miR-142-3p or knockdown of KLF9 significantly suppressed CRL-7566 cell proliferation and metastasis, induced cell apoptosis, and decreased both cell autophagy and vascularization. Additionally, KLF9 was confirmed to be a direct target of miR-142-3p and to directly bind to the promoter of the VEGFA gene, regulating its expression. Finally, intraperitoneal injection of miR-142-3p lentivirus significantly attenuated ectopic endometriotic lesions in vivo.miR-142-3p directly targeted KLF9, regulated VEGFA expression, and was protective against the growth of ectopic endometriotic lesions. Therefore, the miR-142-3p/KLF9/VEGFA signalling pathway may be a potential target in endometriosis treatment.
KEYWORDS: Endometriosis, miR-142-3p, KLF9, autophagy, VEGFA
Introduction
Endometriosis is a benign gynaecological disease and is characterized by ectopic growth of functional endometrial glands [1]. It is an estrogen-dependent chronic condition and results in infertility over 25% of women; about 30%-50% of women with endometriosis are born with pain and infertility [2–4]. The global incidence rate of endometriosis is approximately 2.5 ‰ per year [5]. Histopathological diagnosis and laparoscopic visual examination are the gold standard methods for the diagnosis of endometriosis [6], which may be developed into endometrioid and ovarian cancer [7,8]. Genetic predisposition, hormonal aberrations, environmental factors, abnormal immune responses and individual anatomy may be risk factors for the development of endometriosis [9–11]. Although a number of studies have been performed to understand the aetiology of endometriosis, the details and underlying mechanisms in the pathogenesis of endometriosis have not yet been fully elucidated.
MicroRNAs (miRNAs) are a group of non-coding single-stranded RNAs with 18 to 25 nucleotides, which play important roles in regulating post-transcriptional gene expression [12]. miRNAs usually exhibits imperfect binding to the 3ʹ-untranslated regions (3ʹUTR) of their target mRNA, resulting in degradation or suppressed translation of mRNA [13,14]. Many studies have detailed the critical role of miRNAs in a wide variety of cellular processes, including cell differentiation, cell proliferation, apoptosis, autophagy and vascularization [15–18]. Dysregulation of miRNAs contributes to cancer, acute respiratory distress syndrome (ARDS), stroke, depression, endometriosis, and other diseases [15,19–21]. Wang et al. found that miR-7 is downregulated in endometriosis and IL-4 receptor and IL-6 receptor are target genes of miR-7, and suggesting miR-7 and IL-4 may be potential biomarkers for the detection of endometriosis [22]. Yang et al. found that miR-543 is significantly decreased in endometrial samples with endometriosis and is associated with endometrial receptivity; downregulation of miR-543 may contribute to endometriosis-related infertility [23]. miR-142-3p is located at chromosome 17q22. Recently studies have reported that miR-142-3p regulates cell proliferation, apoptosis, and migration in different human cancers [24,25] and increases the population of cancer stem cells (CSCs) through the bone marrow-derived mesenchymal stem (BM-MSCs) derived exosomal miR-142-3p [26]. Additional, miR-142-3p also increases chemosensitivity by inhibition of high mobility group box 1 (HMGB1)-mediated autophagy in human non-small-cell lung cancer (NSCLC) and acute myelogenous leukaemia [27,28]. Microarray analyses show downregulation of miR-142-3p in human ectopic endometrial tissues compared with eutopic endometrial tissues, while functional analysis suggested the involvement of CREB-binding protein, c-Jun, Akt, and cyclin D1signaling pathway in human endometriosis [29].
Although miR-142-3p is decreased in human ectopic endometrial tissues, the contribution of miR-142-3p to human endometriosis is not known, and the underlying molecular mechanisms of miR-142-3p in the regulation of endometriosis remain unclear. Our study describes novel protective roles for miR-142-3p against the development and progression of endometriosis. In the present study, we confirmed that miR-142-3p is decreased in human ectopic endometrial tissues, and overexpression suppresses CRL-7566 cell growth and migration. Furthermore, we found Krüppel-like factor 9 (KLF9) is a target gene of miR-142-3p, and miR-142-3p induces apoptosis by promoting KLF9-mediated autophagy. Moreover, KLF9 also promotes angiogenesis by regulating vascular endothelial growth factor A (VEGFA) expression directly. According to these findings, themiR-142-3p/KLF9/VEGFA signalling pathway may be a potential target for endometriosis treatment.
Results
Mir-142-3p is decreased in human ectopic endometrial tissues and negatively correlated with KLF9 and VEGFA expression
To investigate the function of miR-142-3p in human endometriosis, we analysed the expression level of miR-142-3p in ectopic endometrial tissues. Firstly, H&E staining revealed small interstitial cells, glandular epithelial vacuolar degeneration, and reduction of ectopic endometrium (Figure 1(a)). Furthermore, qRT-PCR indicated that the expression of miR-142-3p was decreased in human ectopic endometrial tissues compared with eutopic endometrial tissues, and FISH demonstrated cytoplasmic localization of miR-142-3p (Figure 1(b,c). In addition, qPCR, Western blot and IHC showed that the mRNA and protein expression of KLF9 and VEGFA were increased in ectopic endometrial tissues compared with eutopic endometrial tissues (Figure 1(d-h). Moreover, KLF9 expression levels were negatively associated with miR-miR-142-3p expression levels in 20 ectopic endometrial tissue samples (p = 0.001, R = −0.667, Figure 1(i)), while VEGFA expression levels were positively correlated with KLF9 expression levels (P = 0.019, R = 0.520, Figure 1(j)). Altogether, these findings suggest that downregulation of miR-142-3p and upregulation of KLF9 may contribute to the progression of human endometriosis.
Figure 1.
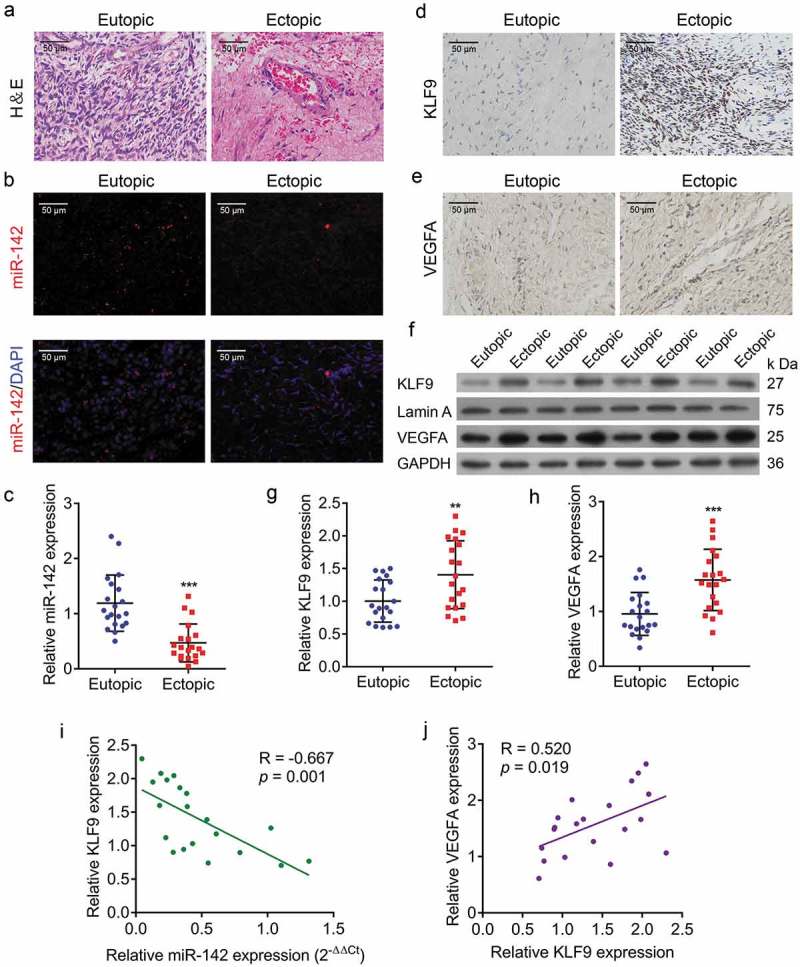
Decreased miR-142-3p level and increased KLF9 and VEGFA levels in human ectopic endometrial tissues. (a) H&E-stained sections of human ectopic and eutopic endometrial tissues. (b) FISH analysis of the expression of miR-142-3p in human ectopic and eutopic endometrial tissues. (c) miR-142-3p expression in human ectopic endometrial tissues is lower than in eutopic tissue, *** P< 0.001. (d) IHC for KLF9 in human ectopic and eutopic endometrial tissues. (e) IHC examination for VEGFA in human ectopic and eutopic endometrial tissues. (f) Western blot analysis of KLF9 and VEGFA protein expression in human ectopic and eutopic endometrial tissues. (g, h) qRT-PCR analysis of KLF9 and VEGFA protein expression in human ectopic and eutopic endometrial tissues, respectively (n = 20), ** P< 0.01, *** P< 0.001. (i) KLF9 protein expression levels were negatively correlated with miR-142-3p expression levels (P= 0.001, R = −0.667). (j) VEGFA protein expression levels were positively correlated with KLF9 protein expression levels (P = 0.019, R = 0.520).
Overexpression of mir-142-3p inhibits endometrial cell proliferation
To investigate the role of miR-142-3p in endometrial cell proliferation, the growth ability of endometrial cell was analysed after manipulation of miR-142-3p. Firstly, the expression of miR-142-3p was lower in ECSCs and CRL-7566 cells than in NESCs and hEM15A, respectively (Figure 2(a)). Analysis of qRT-PCR and Western blot assays showed increased levels of KLF9 and VEGFA in ECSCs and CRL-7566 cells (Figure 2(b–d). Furthermore, the expression of miR-142-3p was modified by miR-142-3p mimics and transfection inhibitor transfection (Figure 2(e)). EdU staining and CCK-8 assay indicated that cell proliferation viability was inhibited after miR-142-3p mimics transfection (Figure 2(f,g). Moreover, flow cytometry revealed that upregulation of miR-142-3p in CRL-7566 cells decrease the proportion of cells in S phase and increase the proportion in G1 phase, while downregulation of miR-142-3p decrease the proportion in G phase and increase the proportion in S phase (Figure 2(h,i). These findings demonstrated miR-142-3p alteration of the cell cycle, supporting its role in endometrial cell proliferation.
Figure 2.
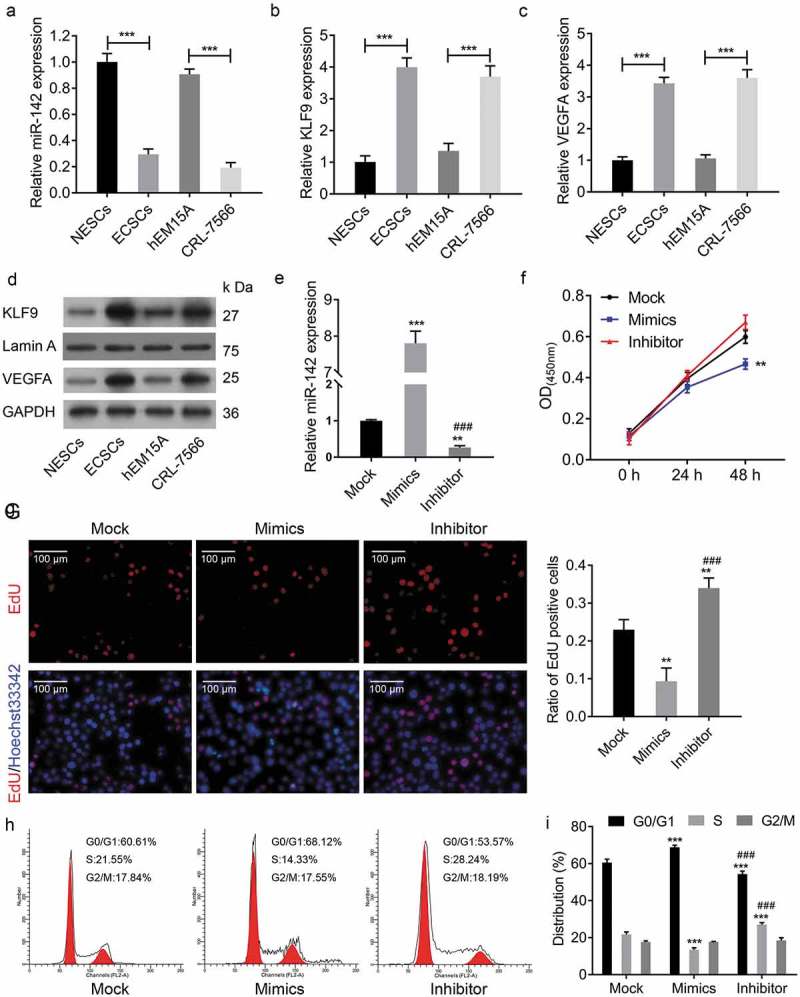
Overexpression of miR-142-3p inhibited endometrial cell proliferation. (a) qRT-PCR analysis of the expression of miR-142-3p in ECSCs, NESCs, CRL-7566 and hEM15A cells, *** P < 0.001. (b-d) qRT-PCR and Western blot analysis of the mRNA and protein of KLF9 and VEGFA expression levels in ECSCs, NESCs, CRL-7566 and hEM15A cells, *** P < 0.001. (e) The expression of miR-142-3p was altered by miR-142-3p mimics and transfection inhibitor, ** P < 0.01, *** P < 0.001. (f) CCK-8 assay to analyse the effect of miR-142-3p on CRL-7566 cell proliferation, ** P < 0.01. (g) EdU assay to evaluate the proliferation of CRL-7566 cells treated with miR-142-3p mimics and inhibitor, ** P < 0.01. (h, i) Flow cytometry to measure the contribution of miR-142-3p to the cell cycle, *** P < 0.001.
Overexpression of mir-142-3p inhibits endometrial cell metastasis and vascular endothelial cell tube formation
Transwell migration and invasion assay and scratch wound healing assay were performed to determine the effects of miR-142-3p on cell migration and invasion. The transwell migration and invasion assay showed that miR-142-3p mimics transfection inhibited the migratory and invasive viability of CRL-7566 cells, while miR-142-3p inhibitor transfection promoted the migratory and invasive viability of CRL-7566 cells (Figure 3a–c). Following 48hs of culture, cell wound scratch assay results showed that CRL-7566 cells travelled longer in miR-142-3p mimics-transfected group, whereas travelled shorter in miR-142-3p inhibitor-transfected group compared with the control group. (Figure 3(d) and e). Furthermore, the expression of VEGFA was decreased in CRL-7566 cells transfected with miR-142-3p mimics and increased in those transfected with miR-142-3p inhibitor (Figure 3(f–h)), Similar alteration of VEGFA protein in culture medium was observed (Figure 3(i)). The CRL-7566 cell culture medium was collected and used for HUVEC culture. The tube formation assay showed that the medium from miR-142-3p mimics-transfected CRL-7566 cells inhibited vascularization, and the medium from miR-142-3p inhibitor-transfected CRL-7566 cells enhanced vascularization (Figure 3(j) and k). Thus, miR-142-3p regulates endometrial cell migration and invasion and reduces VEGFA expression, resulting in the inhibition of vascularization.
Figure 3.
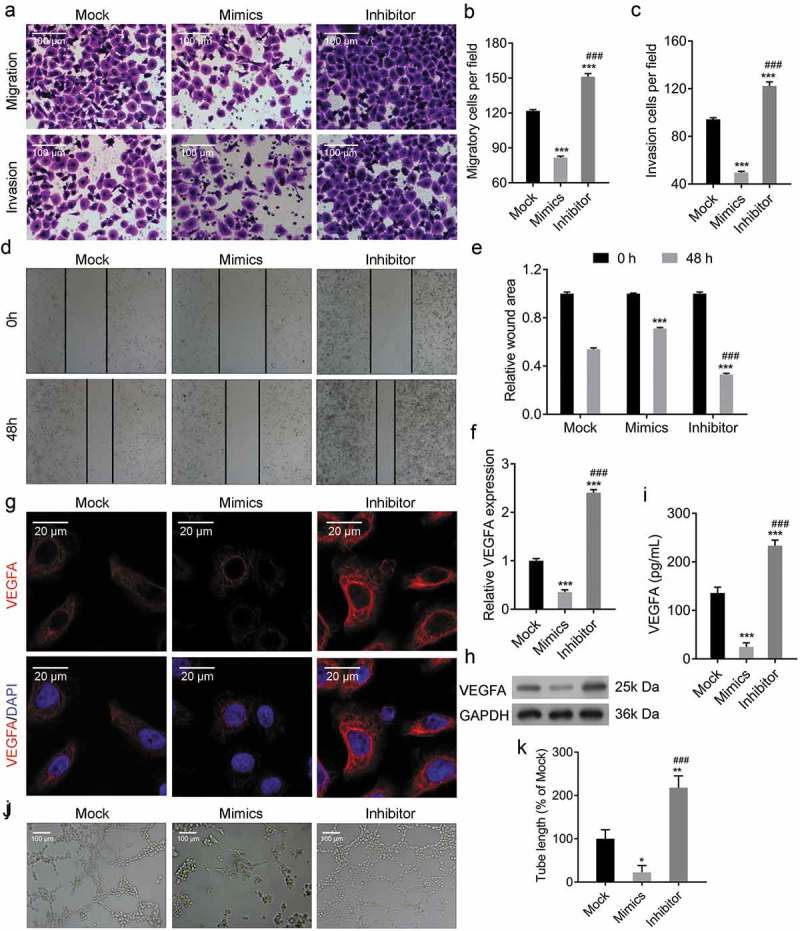
Overexpression of miR-142-3p inhibited endometrial cell metastasis and vascular endothelial cell tube formation. (a-c) Transwell migration and invasion assays to evaluate the metastasis of CRL-7566 cells treated with miR-142-3p mimics and inhibitor, *** P < 0.001. (d, e) Cell wound scratch assay to evaluate the migration of CRL-7566 cells transfected with miR-142-3p mimics and inhibitor, *** P < 0.001. (f-h) qRT-PCR, IF and Western blot analysis of VEGFA expression in CRL-7566 cells transfected with miR-142-3p mimics and inhibitor, *** P < 0.001. (I) ELISA analysis of VEGFA concentration in the culture medium of treated CRL-7566 cells, *** P < 0.001. (j, k) Tube formation assay to evaluate the culture medium gathered from treated CRL-7566 cell on the vascularization viability of HUVEC, * P < 0.05, ** P < 0.01.
Overexpression of mir-142-3p inhibits autophagy and induces cell apoptosis
To investigate the role of miR-142-3p in cell apoptosis, miR-142-3p mimics or inhibitor was transfected into CRL-7566 cells. Flow cytometry showed a significant increase in the apoptosis rate in CRL-7566 cells transfected with miR-142-3p mimics and a decrease in those transfected with miR-142-3p inhibitor (Figure 4(a)). Similarly, Hoechst 33,258 staining showed the number of cells with condensed and fragmented nuclei indicative of apoptosis was significantly increased in miR-142-3p mimics-transfected CRL-7566 cells (Figure 4(b)). Transmission electron microscopy showed that the number of autophagosomes was decreased in miR-142-3p mimics-transfected cells (Figure 4(f)). As expected, KLF9 expression was suppressed in miR-142-3p overexpressing cells, according to qPCR, Western blot and IF analysis (Figure 4(c-e)). In addition, Western blot analysis showed that upregulation of miR-142-3p in CRL-7566 cells induces the expression of Bax and p62 and inhibit the expression of Bcl-2 and LC3B (Figure 4(g)). The findings suggested that miR-142-3p might regulate cell apoptosis by targeting KLF9-mediated autophagy.
Figure 4.
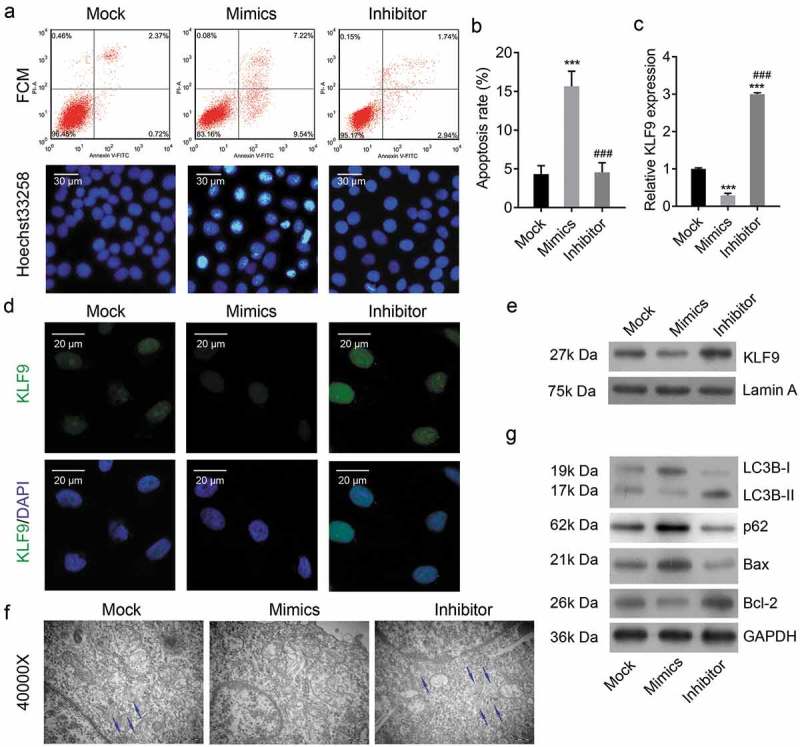
Overexpression of miR-142-3p inhibited autophagy and induced cell apoptosis. (a, b) Flow cytometry and Hoechst 33,258 staining were performed to analysis apoptosis of CRL-7566 cells transfected with of miR-142-3p mimics or inhibitor, *** P < 0.001. (c-e) qRT-PCR, IF and Western blot analysis of the expression of KLF9 in miR-142-3p mimics or inhibitor-transfected CRL-7566 cells, *** P < 0.001. (f) Transmission electron microscopy to evaluate the autophagy of CRL-7566 cells treated with miR-142-3p mimics and inhibitor. The autophagosome was indicated with an arrowhead. (g) Western blot analysis of Bcl-2, Bax, LC3B and p62 expression in the CRL-7566 cells transfected with miR-142-3p mimics or inhibitor.
KLF9 is a target gene of mir-142-3p
Our above results showed a negative correlation between KLF9 expression and miR-142-3p expression. To demonstrate the direct interaction between KLF9 and miR-142-3p, the first step was miRNA-target prediction using the TargetScan database (www.targetscan.org). As expected, miR-142-3p has one predicted target site in the human KLF9-3ʹ-UTR (Figure 5(a)). Dual-luciferase reporter assay showed that the luciferase activity of the reported was downregulated by miR-142-3p mimics, but the luciferase activity of mutant KLF9-3ʹ-UTR was not regulated by miR-142-3p (Figure 5(b)). These findings indicated that KLF9 is a direct target gene of miR-142-3p.
Figure 5.
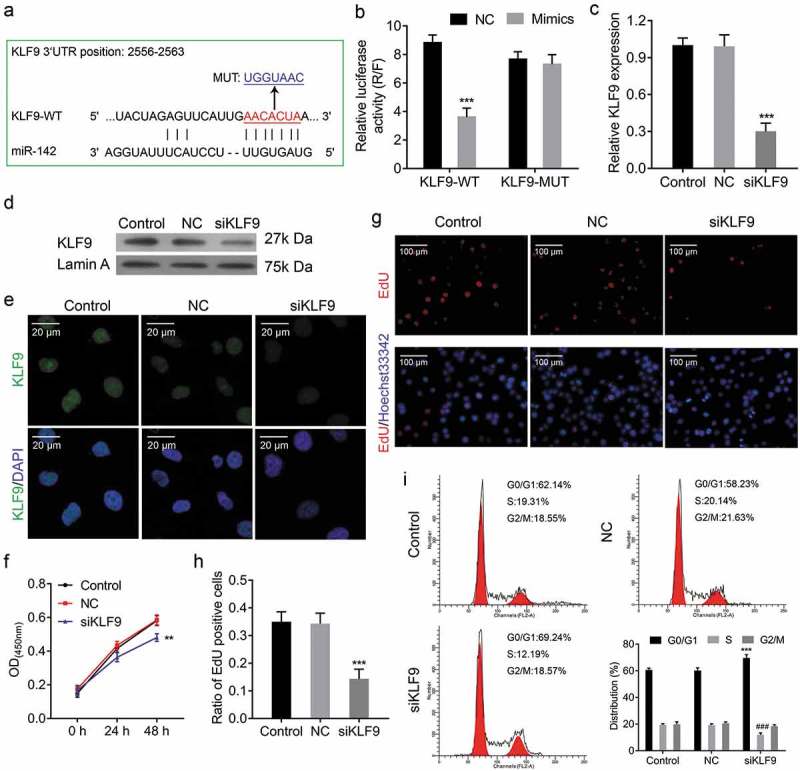
KLF9 is a target gene of miR-142-3p, and knockdown of KLF9 suppresses cell proliferation. (a) Schematic representation of wild type and mutant psi-CHECK2-KLF90-3ʹUTR miRNA expression vectors used in luciferase reporter assays. (b) Relative luciferase activities in 293T cells corresponding to the binding site, *** P < 0.001. (c-e) qRT-PCR, Western blot and IF analysis of the expression of KLF9 in CRL-7566 cells treated with KLF9-siRNA. (f) CCK-8 assay to evaluate the effect of KLF9-siRNA on CRL-7566 cell proliferation, ** P < 0.01. (g, h) EdU assay to assess the proliferation of CRL-7566 cells treated with KLF9-siRNA, *** P < 0.001. (i) Flow cytometry to measure the contribution of KLF9-siRNA to the cell cycle, *** P < 0.001.
Knockdown of KLF9 suppresses cell proliferation
To investigate the role of KLF9 in endometrial cell proliferation, the growth ability of endometrial cells was analysed after knockdown of KLF9. Firstly, the mRNA and protein expression levels of KLF9 were decreased by KLF9-siRNA transfection (Figure 5(c-e)). CCK-8 assay and EdU staining indicated that cell proliferation viability was reduced after KLF9-siRNA transfection (Figure 5(f-h)). Moreover, flow cytometry indicated that knockdown of KLF9 increases G1 phase and decreases S phase (Figure 5(i)). Thus, our findings suggested that KLF9 alters the cell cycle to regulate endometrial cell proliferation.
Knockdown of KLF9 inhibits endometrial cell metastasis and vascular endothelial cell tube formation
The transwell migration and invasion assay and scratch wound healing assay were performed to determine the effects of KLF9 on cell migration and invasion. The transwell migration and invasion assay showed that knockdown of KLF9 inhibited the migratory and invasive viability of CRL-7566 cells(Figure 6(a-c)). Following 48hs of culturing, the distance travelled by KLF9-siRNA transfected CRL-7566 cells were significantly longer than the control group (Figure 6(d) and e). Furthermore, the expression of VEGFA was decreased in CRL-7566 cells after KLF9-siRNA transfection (Figure 6(g-i)), and VEGFA protein released into the culture medium was also decreased (Figure 6(f)). The CRL-7566 cell culture medium was collected and used for culturing HUVEC. The tube formation assay showed that the medium from KLF9-siRNA transfected CRL-7566 cells inhibited vascularization (Figure 6(j) and k). These findings demonstrated that KLF9 regulates endometrial cell migration and invasion and reduce VEGFA expression, resulting vascularization inhibition.
Figure 6.
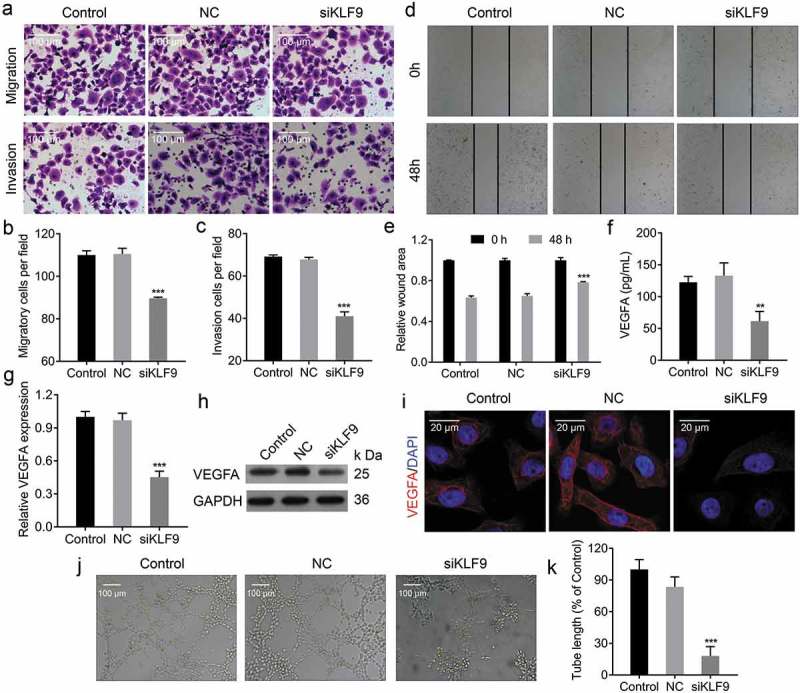
Knockdown of KLF9 inhibited endometrial cell metastasis and vascular endothelial cell tube formation. (a-c) Transwell migration and invasion assays to evaluate the metastasis of CRL-7566 cells treated with KLF9-siRNA, *** P < 0.001. (d, e) Cell wound scratch assay to evaluate the migration of CRL-7566 cells transfected with KLF9-siRNA, *** P < 0.001. (f) ELISA analysis of VEGFA concentration in the culture medium of treated CRL-7566 cells, ** P < 0.01. (g-i) qRT-PCR, IF and Western blot analysis of the VEGFA expression in CRL-7566 cells transfected with KLF9-siRNA, *** P < 0.001. (j, k) Tube formation assay to evaluate the effects of culture medium gathered from treated CRL-7566 cells on vascularization in HUVEC, *** P < 0.001.
Knockdown of KLF9 inhibits autophagy and induces cell apoptosis
Flow cytometry and Hoechst 33,258 staining were performed to analyse the effect of KLF9 on cell autophagy and apoptosis. Flow cytometry showed that the apoptosis rate was significantly induced in CRL-7566 cells transfected with KLF9-siRNA (Figure 7(a) and b). Furthermore, the number of cells with condensed and fragmented nuclei was significantly increased in KLF9-siRNA-transfected CRL-7566 cells (Figure 7(a)). Transmission electron microscopy showed that the number of autophagosome was decreased in CRL-7566 cells after knockdown of KLF9 (Figure 7(c)). In addition, Western blot and IF showed that knockdown of KLF9 induced the expression of Bax and p62 and decreased the expression of Bcl-2 and LC3B in CRL-7566 cells ()). Finally, the online tool (www.genomatix.de) was used to identify putative KLF9 binding sites in the genomic sequence adjacent to the transcription start site(TSS) of the VEGFA gene, regulating its transcription. We found a potential KLF9 binding site in the −1140 to −1127 genomic region relative to the 5ʹ initiation site of VEGFA, which was designated as +1 (Figure 8(a)). Data from the dual-luciferase reporter assay, ChIP PCR and EMSA-supershift assay confirmed that KLF9 directly bound to the VEGFA promoter in the −1140 to −1127 genomic region (Figure 8(b)-d). Altogether, these findings suggested that KLF9 might regulate cell apoptosis through autophagy and directly regulate the expression VEGFA at the transcriptional level.
Figure 7.
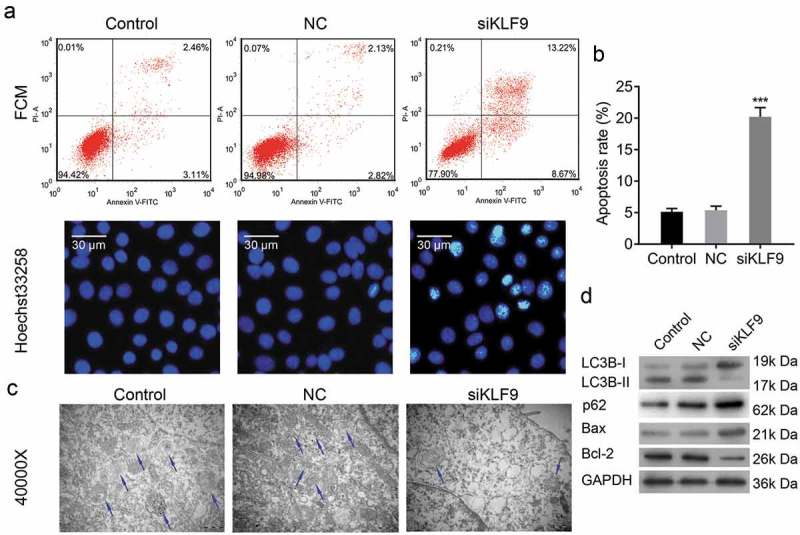
Knockdown of KLF9 inhibited autophagy and induced cell apoptosis. (a, b) Flow cytometry and Hoechst 33,258 staining methods to analyse apoptosis of KLF9-siRNA transfected CRL-7566 cells, *** P< 0.001. (c) Transmission electron microscopy to evaluate autophagy in CRL-7566 cells treated with LKF9-siRNA. The autophagosome was indicated with arrowhead. (d) Western blot analysis of Bcl-2, Bax, LC3B and p62 expression after transfection of CRL-7566 cell with KLF9-siRNA.
Figure 8.
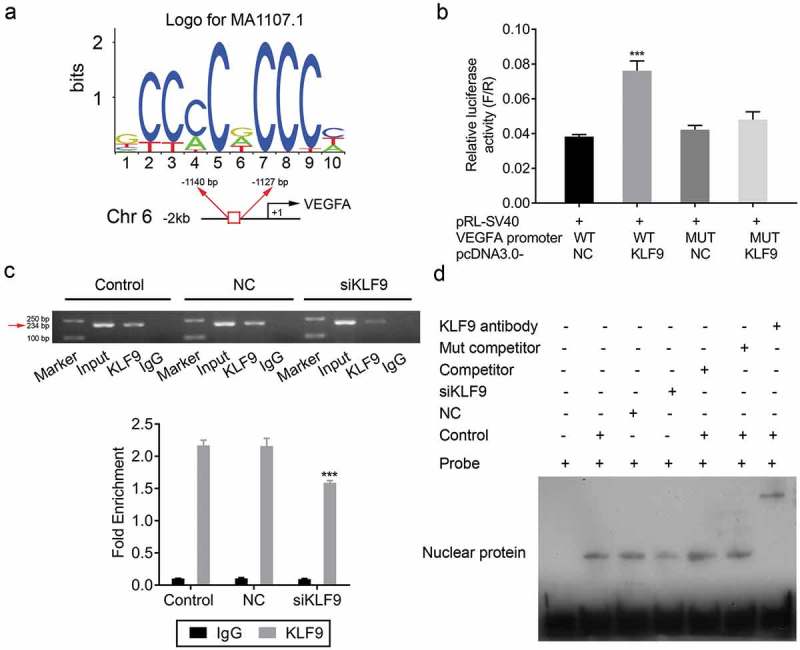
KLF9 directly binds to the VEGFA promoter. (a) Consensus sequence of the KLF9 binding site. (b) Relative luciferase activities in 293T cell corresponding to binding site, *** P < 0.001. (c) ChIP analysis of the binding of KLF9 to the endogenous VEGFA promoter. (d) EMSA analysis of the binding of KLF9 to the endogenous VEGFA promoter.
Knockdown of KLF9 ameliorated the mir-142-3p inhibitor-mediated cell proliferation and metastasis promotion, and apoptosis inhibition
KLF9 expressions were significantly decreased in si-KLF9-transfected CRL-7566 cells, as shown by qRT-PCR, Western Blot and IF (Figure 9a-c), indicating that si-KLF9 was successfully transfected and resulted in KLF9 knockdown. Furthermore, knockdown of KLF9 in miR-142-3p inhibitor-transfected CRL-7566 cells significantly decreased the ability of miR-142-3p inhibitor to promote CRL-7566 cell proliferation and metastasis (Figure 9(d)-g, Figure 10(a-c). Knockdown of KLF9 in miR-142-3p inhibitor-transfected CRL-7566 cells significantly decreased the inhibitory effect of miR-142-3p inhibitor on CRL-7566 cell apoptosis (Figure 9(h-j)). In addition, the expression of VEGFA was significantly decreased in miR-142-3p inhibitor-transfected CRL-7566 cells after KLF9-siRNA transfection (Figure 10(d-f)). The tube formation viability was decreased in HUVEC that were cultured in the medium gathered from CRL-7566 cells co-transfected with KLF9-siRNA and miR-142-3p inhibitor (Figure 10(g)).
Figure 9.
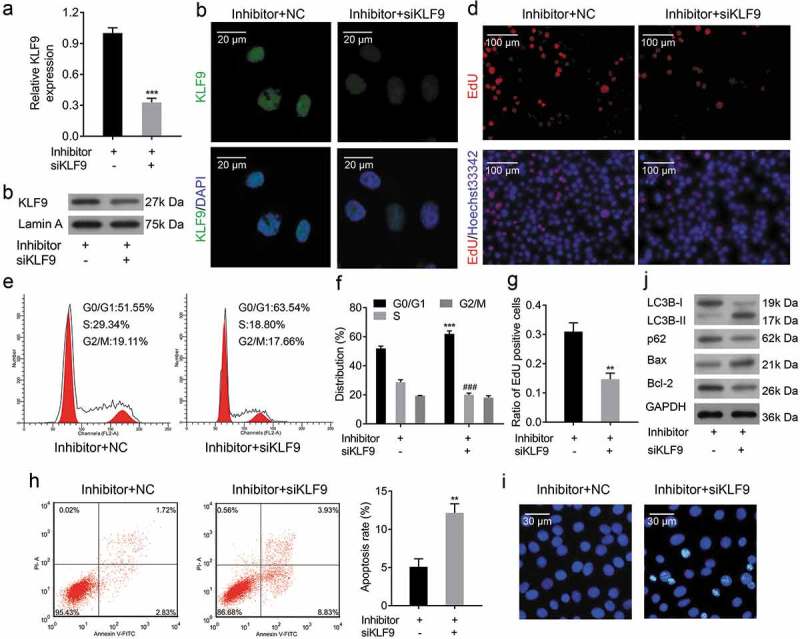
Knockdown of KLF9 ameliorated the miR-142-3p inhibitor-mediated cell proliferation promotion and apoptosis inhibition. (a-c) qRT-PCR, Western blot and IF analysis of the expression of KLF9 in miR-142-3p inhibitor transfected CRL-7566 cells after KLF9-siRNA transfection, *** P < 0.001. (d, g) EdU staining analyse the cell proliferation viability of miR-142-3p inhibitor transfected CRL-7566 cell transfected with KLF9-siRNA, *** P < 0.001. (e, f) Flow cytometry to measure the effect of KLF9-siRNA on cell cycle in miR-142-3p inhibitor transfected CRL-7566 cells, *** P< 0.001. (h,i) Flow cytometry and Hoechst 33,258 staining methods were performed to analyse the effect of KLF9-siRNA on cell apoptosis in miR-142-3p inhibitor transfected CRL-7566 cells, ** P < 0.01. (j) Western blot analysis of VEGFA, Bcl-2, Bax, LC3B and p62 expression in miR-142-3p inhibitor transfected CRL-7566 cells after KLF9-siRNA transfection.
Figure 10.
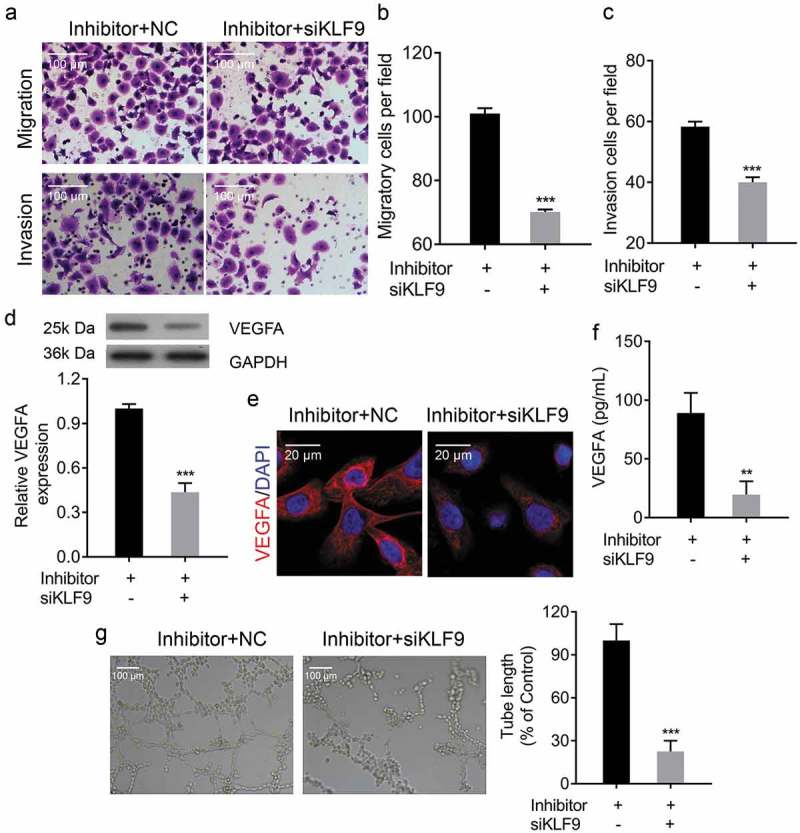
Knockdown of KLF9 ameliorated miR-142-3p inhibitor-mediated increase in metastasis. (a-c) Transwell migration and invasion assays to evaluate the metastasis of miR-142-3p transfected CRL-7566 cells treated with KLF9-siRNA, *** P < 0.001. (d, e) qRT-PCR, IF and Western blot analysis of the VEGFA expression in miR-142-3p transfected CRL-7566 cells treated with KLF9-siRNA *** P < 0.001. (f) ELISA analysis of VEGFA concentration in the culture medium of treated CRL-7566 cells, ** P < 0.01. (g) Tube formation assay to evaluate effects of the culture medium gathered from treated CRL-7566 cells on vascularization viability of HUVEC, *** P < 0.001.
Mir-142-3p suppresses of the growth of endometriotic lesions in vivo
To investigate whether miR-142-3p is a critical for the progression of endometriosis and determine the effect of miR-142-3p in vivo, a rat model of endometriosis was established. The body weight in the model group was significantly decreased compared with the control group and was significantly increased in the miR-142-3p treatment group at the end of the experiment (Figure 11(a)). Additionally, the concentrations of VEGFA and MDA in rat serum were increased in the model group compared with the control group, and miR-142-3p inhibits the expression of VEGFA and MDA Figure 11(b and d)). The concentrations of SOD and CAT in rat serum were decreased in the model group compared with the control group, and miR-142-3p induced the expression of SOD and CAT (Figure 11(c and e)). Furthermore, the expression of miR-142-3p was significantly decreased in the model group and was increased in the miR-142-3p treatment group (Figure 11(f)). We also found that the expression levels of KLF9 and VEGFA were negatively correlated with miR-142-3p expression (Figure 11(g and h)). Pathology analysis revealed small interstitial cells, degeneration of glandular epithelial vacuolar and reduction of ectopic endometrium and increasing apoptosis in the model group; overexpression of miR-142-3p ameliorates endometriosis (Figure 11(i)). Finally, Western blot analysis in the model group showed increased expression of VEGFA, LCB3, KLF9 and Bcl-2 and decreased expression of p62 and Bax; overexpression of miR-142-3p can ameliorate this endometriosis-mediated protein expression pattern (Figure 11(j)). Therefore, miR-142-3p exerts a therapeutic effect in a rat endometriosis model.
Figure 11.
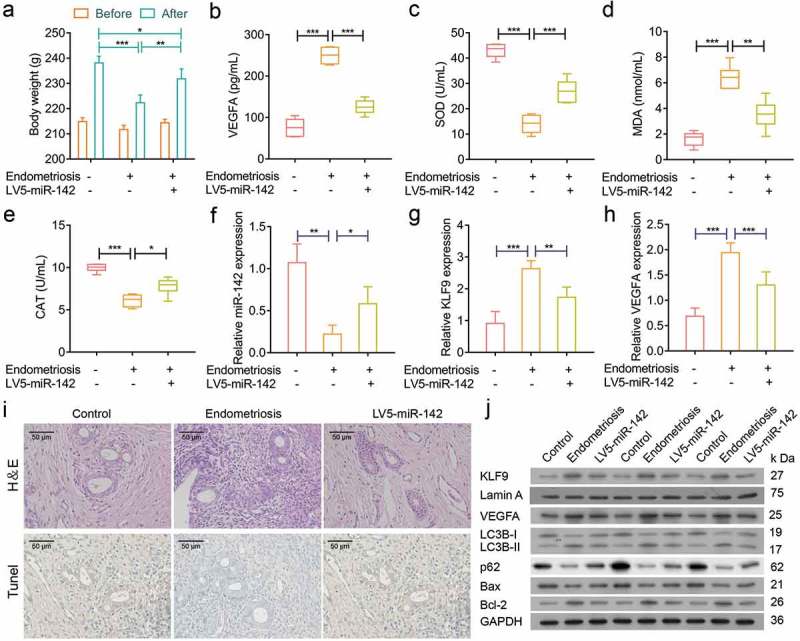
miR-142-3p suppressed endometriotic lesions in vivo. (a) The bodyweight of rats was assessed in control, model and miR-142-3p treated groups, * P < 0.05, ** P < 0.01, *** P < 0.001. (b-e) ELISA analysis of VEGFA, SOD, MDA and CAT concentrations in rat sera from different treatment groups, * P < 0.05, ** P < 0.01, *** P < 0.001. (f-h) qRT-PCR analysis of miR-142-3p, KLF9 and VEGFA expression in endometrial tissues from control, model and miR-142-3p treated groups, * P < 0.05, ** P< 0.01, *** P < 0.001. (I) H&E-stained and Tunel-stained sections of the endometrial tissues from control, model and miR-142-3p treated groups. (J) Western blot analysis of VEGFA, Bcl-2, Bax, LC3B, p62 and KLF9 expression in endometrial tissues from control, model and miR-142-3p treated groups.
Discussion
A growing number of studies indicate that miRNAs play critical roles in a wide range of cellular processes [30] and dysregulation of miRNAs contributes to the development and progression of human diseases [31]. In the present study, we found that miR-142-3p was significantly decreased in human ectopic endometrial tissues compared with eutopic endometrial tissues. In addition, overexpression of miR-142-3p suppressed CRL-7566 cell proliferation, migration and invasion and induced cell apoptosis. Furthermore, overexpression of miR-142-3p inhibited cell autophagy and vascularization of HUVEC. Treatment with miR-142-3p led to an increase in body weight and ameliorated endometriosis in our rat model of endometriosis.
The KLF family is composed of 17 members with diverse regulatory functions [32]. The Krüppel-like factor 9 (KLF9) is a member of the KLFs family and is also known as BTE-B1 (basic transcription element-binding protein 1). KLF9 has been played a major role in aberrant estrogen receptor (ESR) and progesterone receptor (PGR) mediated reproductive dysfunctional states, including infertility, endometrial cancer, endometriosis and uterine hypoplasia [33–35]. KLF9 may be a coregulator of PRGand a regulator of ESR1 signalling, contributing to reproductive dysfunction [36,37]. Previous studies showed that knockdown of KLF9 and PRG in human endometrial stromal cells resulted in blocking of Wnt, cytokine, and IGF signalling in vitro, all of which are PGR- and ESR1-regulated pathways [33]. In the current study, KLF9 expression was increased in human ectopic endometrial tissues compared with eutopic endometrial tissues, and was negatively correlated with miR-142-3p expression. The luciferase reporter assay confirmed that KLF9 is a target gene of miR-142-3p. The attenuated expression of KLF9 inhibited CRL-7566 cell proliferation, migration and invasion, and promoted cell apoptosis in vitro. Furthermore, knockdown of KLF9 induced cell autophagy and inhibited vascularization of HUVEC.
Autophagy is an evolutionarily conserved pathway of non-apoptotic programmed cell death, contributing to the degradation of cytoplasmic components in eukaryotic cells, including cytoplasmic organelles, long-lived proteins and other macromolecules [38,39]. Cellular components can be degraded in lysosomes, and bioenergetic metabolites can be recycled through the autophagy pathway [40,41]. The microtubule-associated protein light chain 3 beta (LC3) is a mammalian homolog of yeast autophagy-related gene 8 (Atg8), which is present on autophagosomes, and the conversion of LC3 I to LC3 II is widely used as an indicator of autophagy [42]. Extensive activation of autophagy contributes to cellular homeostasis [39,43] and plays an important role in inhibiting cell apoptosis in some normal cells and cancer cells [44,45] Interestingly, autophagy was observed in ectopic endometrium tissues from ovarian endometriosis patients [46]. Here, we found that overexpression of miR-142-3p or knockdown of KLF9 inhibited CRL-7566 cell autophagy and promoted cell apoptosis in vitro. Our rat endometriosis model showed increased expression rate of LC3 II/LC3 I and decreased p62 expression levels in transplanted ectopic endometrial tissues, indicating that autophagy was increased in the model group. When treated with miR-142-3p, the expression of LC3 II/LC3 I and p62 were altered. These findings, suggest that autophagy may play an important role in endometriosis and possibly contribute to the decreased death of endometriotic cells.
The dense web of blood vessels is one of the distinct features of endometriotic lesions, and angiogenesis is essential for eutopic endometrium. However, the expression of angiogenic growth factors, particularly vascular endothelial growth factor (VEGF) was increased in ectopic endometrium tissues compared with normal tissues. Previous studies had demonstrated that inhibition of angiogenesis by blocking VEGF decreased the growth of endometriotic lesions in vivo. In the present study, we found that the expression of VEGFA was increased in human ectopic endometrium tissues compared with normal tissues. Overexpression of miR-142-3p or knockdown of KLF9 suppressed the expression VEGFA and inhibited vascular endothelial cell tube formation. In addition, we confirmed that KLF9 directly targeted the promoter of VEGFA. Therefore, miR-142-3p may regulate KLF9-mediated VEGFA expression, which affects the development of eutopic endometrium.
In conclusion, our results indicated that in ectopic endometrial tissues, miR-142-3p expression level was decreased while KLF9 and VEGFA expression levels were increased. The current study also confirmed that KLF9 is a direct target gene of miR-142-3p and that KLF9 protein can bind to the promoter of the VEGFA gene. Overexpression of miR-142-3p or knockdown of KLF9 suppressed cell proliferation, migration and invasion, inhibited cell autophagy, and promoted cell apoptosis. Overexpression of miR-142-3p or knockdown of KLF9 also promoted the vascularization viability of HUVEC. Therefore, the miR-142-3p-KLF9-VEGFA signalling pathway may be a potential target for endometriosis treatment.
Materials and methods
Tissue collection
Twenty ectopic endometrial tissue samples were collected from women with AFS stage II–IV endometriosis during laparoscopic surgical procedures. For the control group, 20 eutopic endometrial tissues were obtained from women with infertility related to fallopian tube pathology. All of the women were not taking any medication and had regular menstrual cycles. All the procedures were approved by the human ethics committee of The Seventh Affiliated Hospital, Sun Yat-sen University. Written informed consent was obtained from all of the volunteers. All tissues were collected and fixed with 4% paraformaldehyde or stored at −80°C.
Cell culture
The endometrial cell line CRL-7566 and hEM15A were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). Fresh human ectopic endometrial tissues (ECSCs) and eutopic endometrial tissues (NESCs) were processed for cell isolation, as previously described [47,48]. The cells were cultured in high-glucose complete Dulbecco’s modified Eagle medium (DMEM; Invitrogen, San Diego, USA) containing 10% fetal bovine serum (FBS; Invitrogen, San Diego, USA) and 1% penicillin–streptomycin (100 U/mL penicillin and 100 μg/mL streptomycin). The cells were maintained in a humidified incubator with an atmosphere comprising 5% CO2 at 37°C.
Quantitative real-time PCR (qrt-PCR)
Total RNA was extracted from tissues or cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), followed by quantification using the Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). A total of 2 μg of total RNA was used for cDNA synthesis using a Bestar qPCR RT Kit (DBI® Bioscience, Shanghai, China). DBI Bestar® SybrGreen qPCR Master Mix (DBI® Bioscience, Shanghai, China) was used for real-time PCR according to the manufacturer’s protocol. Amplification and detection were performed using a Stratagene Real-time PCR Detection System (Mx3000P, Agilent, Santa Clara, CA, USA) for 40 cycles, each cycle consisting of denaturation at 94°C for 2 min, heating at 94°C for 20 s, annealing at 58°C for 20 s, and extension at 72°C for 20 s. The value of relative mRNA or miRNA expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or U6, respectively. The 2−△△Ct method was used to quantify resulting data. Primers were constructed for VEGFA: 5ʹ-ATCTTCAAGCCATCCTGTGTG-3ʹ (forward), 5ʹ- ACCAACGTACACGCTCCA-3ʹ (reverse); for KLF9: 5ʹ-AGAGTGCATACAGGTGAACGG-3ʹ (forward), 5ʹ-AGTGTGGGTCCGGTAGTGG-3ʹ (reverse); for GAPDH: 5ʹ-TGTTCGTCATGGGTGTGAAC −3ʹ (forward), 5ʹ-ATGGCATGGACTGTGGTCAT-3ʹ (reverse); for miR-142-3p: 5ʹ-ACACTCCAGCTGGGTGTAGTGTTTCCTACTTT-3ʹ (forward), 5ʹ-CTCAACTGGTGTCGTGGA-3ʹ (reverse); and for U6: 5ʹ-CTCGCTTCGGCAGCACA-3ʹ (forward), 5ʹ- AACGCTTCACGAATTTGCGT-3ʹ (reverse).
Western blot assay
Total proteins were extracted from tissues and cells using a sodium dodecyl sulfate (SDS) lysis buffer (Beyotime, Shanghai, China), and the concentrations of total protein were determined usingNanodrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). 20 μg of total protein were separated in 10% sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Roche, Mannheim, Germany). The membranes were blocked in Tris-buffered saline containing 5% bull serum albumin (BAS) for 2 h at room temperature and then incubated with primary antibody (anti-KLF9 (1: 2000), anti-VEGFA (1: 1000), anti-LC3B(1: 1000), anti-p62 (1: 1000), anti-Bax (1: 1000) and anti-Bcl-2 (1: 1000)) at 37°C for 1 h. The membranes were subsequently incubated with goat anti-rabbit IgG conjugated to horse radish peroxidase (HRP) secondary antibody (1: 5000) and visualized by chemiluminescence. All antibodies were purchased from Abcam (abcam, Cambridge, UK).
Plasmid construction
The 3ʹ-untranslated region (3ʹ-UTR) of KLF9 gene containing putative binding sites for miR-142-5p was amplified and cloned into psi-CHECK2 vectors (Promega, Madison, WI, USA). The resulting plasmid was named KLF9-3ʹUTR-WT. KLF9 gene mutant 3ʹ-UTR recombinant plasmid, expressed as KLF9-3ʹUTR-MUT, was generated using the TaKaRa MutanBEST Kit (TaKaTa, Beijing, China) with the 8-bp mutation, AACACUAA to UGGUAACG, in the predicted miR-142-5p binding site.
The KLF9 gene overexpression vector was obtained by cloning the amplified cDNA into pcDNA3.1 vectors (V79020, Invitrogen, San Diego, USA) and was verified by DNA sequencing (BGI, Shenzhen, China).
The promoter of VEGFA gene containing putative binding sites for KLF9 protein was amplified and cloned into pGL3 vectors (Promega, Madison, WI, USA). The resulting plasmid was named as VEGFA-promoter-WT. VEGFA gene promoter mutant recombinant plasmid was generated using the TaKaRa MutanBEST Kit (TaKaTa, Beijing, China), containing the 13-bp mutation, CCCCCCACCCCCT to GATGAGTGAGGTC, in the predicted KLF9 binding site. The resulting plasmid was named VEGFA-promoter-WT.
Cell transfection
The miR-142-3p mimics (chemically modified double-stranded small RNAs), inhibitor (chemically modified single-stranded small RNA molecules), scrambled sequence RNA (NC) and small interfering RNA (siRNA) targeting the KLF9 gene were purchased from GenePharma (Suzhou, China). About 100 μL (1 × 105 cells/ml) cells were seeded into each well of 96-well plates. The cells were incubated for 24 h and then transfected with RNAs (100 nM) or plasmids (20 μM) using Lipofectamine 2000 (11,668,019, Invitrogen, San Diego, USA) in serum-free medium in accordance with the manufacturer’s instructions.
Dual-luciferase reporter assay
For miRNA-target gene dual-luciferase reporter assay, 1 × 105 293T cells were seeded into 24-well plates and cultured for 24 h, and then co-transfected with 100 ng KLF9-3ʹUTR-WT or -MUT vectors and 100 nM miR-142-3p mimics or NC using Lipofectamine 2000. After 48 h of transfection, luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacturer’s instructions. Renilla luciferase activity was normalized to Firefly luciferase activity.
For transcriptional factors (TF) promoter dual-luciferase reporter assay, 1 × 105 293T cells were seeded into 24-well plates and cultured for 24 h, and then co-transfected with 100 ng VEGFA-promoter-WT or -MUT vectors and 100 ng pcDNA3.1-KLF9 or pcDNA3.1 As the internal control, empty vectors were co-transfected using Lipofectamine 2000, also pRL-TK vectors (Promega, USA) in each cell group. After 48 h of transfection, luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega, USA) according to manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity.
Chromatin immunoprecipitation (chip)
The ChIP assay was performed using a Magna ChIP™ A Kit (Millpore, Burlington, MA, USA) according to the manufacturer’s protocol.Briefly, 1 × 106 cells were crosslinked with 1% formaldehyde at room temperature for 10 min. After washing with cold PBS, 5 μL of Protease Inhibitor Cocktail II was added. Subsequently, 2 mL of 10× Glycine was added at room temperature for 5 min to quench unreacted formaldehyde. The cells were washed with cold PBS, collected into a separate microfuge tube, and pelleted at 800 × g at 4°C for 5 min. The lysate was resuspended in 0.5 ml of cell Lysis Buffer containing 1× Protease Inhibitor Cocktail II and incubated on ice for 15 min. The cell lysate was centrifuged at 800 × g at 4°C for 5 min, and the cell pellet was resuspended in 0.5 ml of Nuclear Lysis Buffer. Chromatin was sheared to ~150–900 base pairs in length using a ultrasonic cell disruptor (JY99-IIDN, scientz, Zhejiang, China) with 20% power, 20 cycles, 15 s on and 10 s off on ice (Figure S1). Then, the chromatin solution was incubated with the indicated antibody overnight at 4°C with rotation. Beads were with ChIP wash buffer. Immunocomplexes were eluted from beads using ChIP elution buffer and then incubated overnight at 62°C to reverse cross-linking. DNA was purified and analysed by PCR. The primer sequences used for ChIP PCR analysis were 5ʹ-GAAGCAACTCCAGTCCCAAAT-3ʹ (forward), 5ʹ-GCACACACTCACTCACCCAC-3ʹ (reverse).
Electrophoretic mobility shift assay (EMSA)
The EMSA was performed using a LightShift® Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, 1 μg/μL Poly (dI•dC) and probes were incubated with 3 μL nuclear extract in 20 μL binding buffer for 20 min at 25°C. Anti-KLF9 antibody or specific mutant competitors were pre-incubated with the nuclear extract. Samples (20 μL) were separated by polyacrylamide gel electrophoresis (PAGE) on a 6% gel for 3 h at 100 V. Then the probes were transferred to a soak nylon membrane at 380 mA for 30 min and crosslinked at a distance of approximately 0.5 cm from the membrane for 5–10 min with a hand-held UV lamp equipped with 254 nm bulbs. The biotin-labelled DNA was detected by chemiluminescence. The probes used for were purchased from GenePharma (Suzhou, China).
Cell proliferation assay
The cell proliferation ability was performed using Cell Counting Kit 8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) assay and EdU stain assay. For CCK-8 assay, the cells were treated in different conditions for 24 and 48 h. Then, cell proliferation was determined in accordance with the manufacturer’s instructions. For EdU staining, the cells were treated in different conditions for 48 h. The cells were then fixed with formaldehyde and stained as previously described [49]. The ratio of EdU-positive cells was calculated as EdU-stained cells/Hoechst 33,342-stained cells. All experiments were performed in triplicate.
Cell migration and invasion assays
The 24-well transwell chambers were used for the cell migration and invasion assay in accordance with the manufacturer’s protocol. The details have been described previously [50]. All experiments were performed in triplicate.
Cell wound scratch assay
After transfection of RNAs or plasmids for 48 h, the wound healing assay was performed to analyse cell migration ability, as described previously [51]. The cells were counted manually at 0 h and after 48 h post-scratch from at least six images.
Flow cytometry
Cell apoptosis and cell cycle were determined using an Annexin-V fluorescein isothiocyanate/propidium iodid (FITC/PI) apoptosis detection kit (Life Technologies, Waltham, MA, USA) and cell cycle staining kit (MultiSciences, Shanghai, China) with flow cytometry in accordance with the manufacturer’s instruction. For apoptosis detection, cell samples were analysed using a FACScan (BD, Biosciences, UK). Annexin-V(+)/PI(−) represented the cells in early apoptosis and Annexin-V(+)/PI(+) represented the cells in late apoptosis. For cell cycle, the cells were stained with propidium iodide solution for 30 min and then analysed by flow cytometry on a FFACScan (BD, Biosciences, UK).
Hoechst 33,258 stain
The treated cells were stained using Hoechst 33,258 (Sigma-Aldrich, Burlington, MA, USA) as recommended by the manufacturer. Briefly, the cells were treated for 48 h and harvested. Then, the cells were fixed and stained with 100 μL Hoechst33258 solution for 5 min at room temperature. The morphology of the cell nuclei was observed using a fluorescence microscope (Olympus Corporation, Japan).
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of VEGFA in the culture medium were measured using a human VEGFA ELISA kit (Elabscience, Wuhan, China) following the manufacturer’s instructions. Concentrations of VEGFA, superoxide dismutase (SOD), malondialdehyde (MDA) and catalase (CAT) in rat serum were measured using a rat ELISA kit for the respective proteins (Elabscience, Wuhan, China), following the manufacturer’s instructions. The ELISA plates were read with a microplate reader (Thermo Scientific, Shanghai, China) and VEGFA, SOD, MDA and CAT concentrations were derived from standard curve.
Tube formation assay
Tube formation assay was performed as previously described [52]. The parent or transfected CRL-7566 cells were cultured for 48 h, and then the medium was collected and centrifuged at 1000 × g for 5 min at 4°C. The collected media were used for human umbilical vein endothelial cells (HUVEC) culture. After culturing for 4 h, images were taken using an Olympus microscope (Olympus Corporation, Japan) with the 10× objective lens. Total tube length of each group was quantified using Image J software.
Hematoxylin-eosin (H&E) staining
Tissues were stored in 10% formalin buffer and embedded in paraffin. Sections (4 μm) were cut and stained with H&E. These sections were examined under a light microscope (Olympus, Tokyo, Japan) at 40x magnification.
Transmission electron microscopy
The treated CRL-7566 cells were harvested and then fixed with 3% glutaraldehyde and 2% paraformaldehyde in 0.1 M PBS buffer for 30 min at room temperature. The cells were post-fixed with 1% osmium tetroxide for 2 h and then stained with 3% aqueous uranyl acetate for 1 h at 37°C. The cells were dehydrated and then cut into ultrathin sections using an ultramicrotome. Cells were observed with a Zeiss transmission electron microscope (Zeiss Inc., Thornwood, NY, USA).
Immunohistochemistry (IHC)
IHC was used to examine protein expression in tissues. Tissues were fixed with 4% paraformaldehyde solution and embedded in paraffin, and 4-μm sections were cut and immunostained. Slides were immersed in xylene and ethanol for dehydration, incubated with 3% H2O2 in the dark for 25 min, and then blocked with 3% bovine serum albumin (BSA) for 30 min at 25°C. The slides were incubated with primary antibody KLF9 (1:100) and VEGFE (1:150) for 1 h at 37°C. The slides were washed and incubated with HRP-conjugated secondary antibodies for 60 min at room temperature. Followed, nuclear counterstained with hematoxylin for 5 min. Images were collected under a microscope (Olympus, Tokyo, Japan) at a magnification of 40× .
Terminal-deoxynucleoitidyl transferase-mediated nick end labelling (TUNEL) staining
The sections prepared as mentioned above were used for TUNEL staining. In brief, the sections were incubated with proteinase K (2 mg/mL) and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS). Cell apoptosis was determined by TUNEL assay kit (Roche, IN, USA) following the manufacturer’s instructions, and the nuclei were counterstained with hematoxylin. The images were captured with a light microscope (Olympus) at a magnification of 40 × .
Immunofluorescence (IF)
Treated cells were fixed and permeabilized. Cells were incubated with IgG control (Rabbit IgG – Isotype Control (Alexa Fluor® 647) at 1/500 dilution (ab199093) or Rabbit IgG Isotype Control (FITC) at 1/100 dilution (ab223339)), Anti-KLF9 and anti-VEGFA primary antibodies overnight at 4°C, followed by incubation with FITC-tagged/Alexa Fluor® 647-tagged secondary antibodies for 1 h at room temperature in the dark. Afterwards, cells were washed and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were analysed with a fluorescence microscope (Olympus Corporation, Japan). In the IgG control group, only the cell nucleus was stained with DAPI (Figure S2).
Fluorescence in situ hybridization (FISH)
Cy5-labelled locked nucleic acid miR-142-3p probes were obtained from RiboBio (Guangzhou, China), and the FISH assay was performed as previously described [11]. All data were captured using a fluorescence microscope (Olympus Corporation, Japan).
Establishment of the endometriosis rat model
The rat endometriosis model was established by autotransplantation of endometrial tissues, as previously reported [12]. Thirty six-week-old female Sprague–Dawley (SD) rats (200–210 g) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Briefly, 30 rats were anaesthetized with 10% chloral hydrate (m/v) at a dose of 0.4 ml/100 g body weight, and then the abdominal skin was disinfected opened. The uterine vessel was ligated, and the right uterine horn was removed to prepare two squares of 5 × 5 mm open uterus. Each endometrium segment was placed in the peritoneal side of the bilateral abdominal wall. Subsequently, the abdominal muscle and skin was closed. The sham group underwent surgery without autotransplantation of endometrial tissues. The rats were intraperitoneally injected with cephalosporin for 3 day. For miR-142-3p treatment, 10 endometriosis model rats were intraperitoneally injected with miR-142-3p lentivirus for 4 weeks. After a period of 28 days, the bodyweight of the rat was measured and autografts were collected for further study.
Statistical analysis
All experiments were performed at least three times and the data are presented as the mean ± standard deviation (SD). All statistical analyses were performed using SPSS 20.0 (IBM Corp, Armonk, NY, USA). Unpaired Student’s t-test or one-way analysis of variance (ANOVA) was used to analyse the statistical significance differences between groups. P values <0.05 were considered statistically significant.
Funding Statement
This work was supported by the Science and Technology Planning Projects of Guangzhou City (China, 2016201604030009).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Leyland N, Casper R, Laberge P, et al. Endometriosis: diagnosis and management. J Obstetrics Gynaecol Can JOGC. 2010;32(7 Suppl 2):S1–32. [PubMed] [Google Scholar]
- [2].Sanchez AM, Quattrone F, Pannese M, et al. The cannabinoid receptor CB1 contributes to the development of ectopic lesions in a mouse model of endometriosis. Hum Reprod. 2017;32(1):175–184. [DOI] [PubMed] [Google Scholar]
- [3].Ilangavan K, Kalu E.. High prevalence of endometriosis in infertile women with normal ovulation and normospermic partners. Fertil Steril. 2010;93(3): e10 author reply e12. [DOI] [PubMed] [Google Scholar]
- [4].de Ziegler D, Borghese B, Chapron C. Endometriosis and infertility: pathophysiology and management. Lancet. 2010;376(9742):730–738. [DOI] [PubMed] [Google Scholar]
- [5].Hummelshoj L, Prentice A, Groothuis P. Update on endometriosis. Women’s Health. 2006;2(1):53–56. [DOI] [PubMed] [Google Scholar]
- [6].Fassbender A, Burney RO, D’Hooghe ODF, et al. Update on biomarkers for the detection of endometriosis. Biomed Res Int. 2015;2015:130854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Prowse AH, Manek S, Varma R, et al. Molecular genetic evidence that endometriosis is a precursor of ovarian cancer. Int J Cancer J Inter Du Cancer. 2006;119(3):556–562. [DOI] [PubMed] [Google Scholar]
- [8].Vigano P, Somigliana E, Chiodo I, et al. Molecular mechanisms and biological plausibility underlying the malignant transformation of endometriosis: a critical analysis. Hum Reprod Update. 2006;12(1):77–89. [DOI] [PubMed] [Google Scholar]
- [9].Guo SW. Epigenetics of endometriosis. Mol Hum Reprod. 2009;15(10):587–607. [DOI] [PubMed] [Google Scholar]
- [10].Lebovic DI, Mueller MD, Taylor RN. Immunobiology of endometriosis. Fertil Steril. 2001;75(1):1–10. [DOI] [PubMed] [Google Scholar]
- [11].Simpson JL, Bischoff FZ. Heritability and molecular genetic studies of endometriosis. Ann NY Acad Sci. 2002;955: 239–251. discussion 293-235, 396-406. [DOI] [PubMed] [Google Scholar]
- [12].Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18(5):504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. [DOI] [PubMed] [Google Scholar]
- [14].Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–114. [DOI] [PubMed] [Google Scholar]
- [15].Ma YS, Lv ZW, Yu F, et al. MicroRNA-302a/d inhibits the self-renewal capability and cell cycle entry of liver cancer stem cells by targeting the E2F7/AKT axis. J Exp Clin Cancer Res. 2018;37(1):252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu H, Liu Y, Bian Z, et al. Circular RNA YAP1 inhibits the proliferation and invasion of gastric cancer cells by regulating the miR-367-5p/p27 (Kip1) axis. Mol Cancer. 2018;17(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen S, Wu J, Jiao K, et al. MicroRNA-495-3p inhibits multidrug resistance by modulating autophagy through GRP78/mTOR axis in gastric cancer. Cell Death Dis. 2018;9(11):1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Miscianinov V, Martello A, Rose L, et al. MicroRNA-148b targets the TGF-beta pathway to regulate angiogenesis and endothelial-to-mesenchymal transition during skin wound healing. Mol Ther. 2018;26(8):1996–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ling Y, Li ZZ, Zhang JF, et al. MicroRNA-494 inhibition alleviates acute lung injury through Nrf2 signaling pathway via NQO1 in sepsis-associated acute respiratory distress syndrome. Life Sci. 2018;210:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [20].Zhao H, Wang J, Gao L, et al MiRNA-424 protects against permanent focal cerebral ischemia injury in mice involving suppressing microglia activation. Stroke. 2013;44(6):1706–1713. [DOI] [PubMed] [Google Scholar]
- [21].Cai H, Zhu XX, Li ZF, et al. MicroRNA dysregulation and steroid hormone receptor expression in uterine tissues of rats with endometriosis during the implantation window. Chin Med J. 2018;131(18):2193–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang F, Wang H, Jin D, et al. Serum miR-17, IL-4, and IL-6 levels for diagnosis of endometriosis. Medicine. 2018;97(24):e10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang P, Wu Z, Ma C, et al. Endometrial miR-543 is downregulated during the implantation window in women with endometriosis-related infertility. Reprod Sci. 2019,26(7):900–908. [DOI] [PubMed] [Google Scholar]
- [24].Schwickert A, Weghake E, Brüggemann K, et al microRNA miR-142-3p inhibits breast cancer cell invasiveness by synchronous targeting of WASL, integrin alpha V, and additional cytoskeletal elements. PLoS One. 2015;10(12):e0143993–e0143993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pan D, Du Y, Ren Z, et al. Radiation induces premature chromatid separation via the miR-142-3p/Bod1 pathway in carcinoma cells. Oncotarget. 2016;7(37):60432–60445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li H, Li F. Exosomes from BM-MSCs increase the population of CSCs via transfer of miR-142-3p. Br J Cancer. 2018;119(6):744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Y, Liu Y, Xu X. Upregulation of miR-142-3p improves drug sensitivity of acute myelogenous leukemia through reducing P-Glycoprotein and repressing autophagy by targeting HMGB1. Transl Oncol. 2017;10(3):410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen Y, Zhou X, Qiao J, et al. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem. 2017;41(4):1370–1382. [DOI] [PubMed] [Google Scholar]
- [29].Ohlsson Teague EM, Van der Hoek KH, Van der Hoek MB, et al. MicroRNA-regulated pathways associated with endometriosis. Mol Endocrinol. 2009;23(2):265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lages E, Ipas H, Guttin A, et al. MicroRNAs: molecular features and role in cancer. Front Biosci. 2012;17:2508–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. [DOI] [PubMed] [Google Scholar]
- [32].Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4(2):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pabona JM, Simmen FA, Nikiforov MA, et al. Kruppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2012;97(3):E376–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Simmen RC, Eason RR, McQuown JR, et al. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Kruppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem. 2004;279(28):29286–29294. [DOI] [PubMed] [Google Scholar]
- [35].Panda H, Pelakh L, Chuang TD, et al. Endometrial miR-200c is altered during transformation into cancerous states and targets the expression of ZEBs, VEGFA, FLT1, IKKbeta, KLF9, and FBLN5. Reprod Sci. 2012;19(8):786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Velarde MC, Iruthayanathan M, Eason RR, et al. Progesterone receptor transactivation of the secretory leukocyte protease inhibitor gene in Ishikawa endometrial epithelial cells involves recruitment of Kruppel-like factor 9/basic transcription element binding protein-1. Endocrinology. 2006;147(4):1969–1978. [DOI] [PubMed] [Google Scholar]
- [37].Zhang D, Zhang XL, Michel FJ, et al. Direct interaction of the Kruppel-like family (KLF) member, BTEB1, and PR mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology. 2002;143(1):62–73. [DOI] [PubMed] [Google Scholar]
- [38].Hale AN, Ledbetter DJ, Gawriluk TR, et al. Autophagy: regulation and role in development. Autophagy. 2013;9(7):951–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang YP, Liang ZQ, Gu ZL, et al. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin. 2005;26(12):1421–1434. [DOI] [PubMed] [Google Scholar]
- [40].Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kretowski R, Borzym-Kluczyk M, Stypulkowska A, et al. Low glucose dependent decrease of apoptosis and induction of autophagy in breast cancer MCF-7 cells. Mol Cell Biochem. 2016;417(1–2):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Allavena G, Carrarelli P, Del Bello B, et al. Autophagy is upregulated in ovarian endometriosis: a possible interplay with p53 and heme oxygenase-1. Fertil Steril. 2015;103(5):1244–1251.e1241. [DOI] [PubMed] [Google Scholar]
- [47].Nishida M, Nasu K, Fukuda J, et al. Down-regulation of interleukin-1 receptor type 1 expression causes the dysregulated expression of CXC chemokines in endometriotic stromal cells: a possible mechanism for the altered immunological functions in endometriosis. J Clin Endocrinol Metab. 2004;89(10):5094–5100. [DOI] [PubMed] [Google Scholar]
- [48].Nishida M, Nasu K, Ueda T, et al. Endometriotic cells are resistant to interferon-gamma-induced cell growth inhibition and apoptosis: a possible mechanism involved in the pathogenesis of endometriosis. Mol Hum Reprod. 2005;11(1):29–34. [DOI] [PubMed] [Google Scholar]
- [49].Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA. 2008;105(7):2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ruan W-D, Wang P, Feng S, et al. MicroRNA-497 inhibits cell proliferation, migration, and invasion by targeting AMOT in human osteosarcoma cells. Onco Targets Ther. 2016;9:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Joseph R, Srivastava OP, Pfister RR. Downregulation of β-actin and its regulatory gene HuR affect cell migration of human corneal fibroblasts. Mol Vis. 2014;20:593–605. [PMC free article] [PubMed] [Google Scholar]
- [52].Wei Y, Gong J, Xu Z, et al. Nrf2 in ischemic neurons promotes retinal vascular regeneration through regulation of semaphorin 6A. Proc Natl Acad Sci USA. 2015;112(50):E6927–E6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.