

Abstract
Lung cancer is a malignant tumor responsible for the highest mortality rate in humans. The identification of novel functional genes is of great importance in the treatment of lung cancer. The reported roles of replication factor C subunit 3 (RFC3) in tumorigenesis are contradictory. The present study aimed to explore the role and mechanism of RFC3 in lung cancer cells. An immunohistochemical study of 165 lung cancer and adjacent tissues was conducted (123 lung adenocarcinoma tissues and 42 lung squamous cell carcinoma tissues). Kaplan-Meier analysis and Cox multivariate analysis were employed to explore the relationship between RFC3 and patient prognosis. In addition, the proliferation, cell cycle distribution and apoptosis of A549 and H1299 cells were determined by MTT assay and flow cytometry, respectively, following cell transfection to induce overexpression and knockdown of RFC3. A Boyden chamber assay and wound-healing assay were conducted to determine the invasive and migratory abilities of A549 and H1299 cells. Western blotting was used to analyze the effects of RFC3 overexpression and RFC3 small interfering RNA-induced knockdown, and to explore the potential mechanism and pathway underlying the effects of RFC3. Positive expression of RFC3 was detected in lung adenocarcinoma, and overexpression of RFC3 shortened the survival time of patients with lung adenocarcinoma. Furthermore, overexpression of RFC3 increased the invasion and migration of A549 cells, whereas knockdown of RFC3 significantly reduced the invasion and migration of H1299 cells. Ectopic expression of RFC3 induced epithelial-mesenchymal transition (EMT), as determined by downregulation of E-cadherin, and upregulation of N-cadherin, vimentin and Wnt signaling target genes, including c-MYC, Wnt1 and β-catenin, and the ratio of phosphorylated-glycogen synthase kinase 3 (GSK3)-β (Ser9)/GSK3-β. In conclusion, RFC3 may be considered a coactivator that promotes the Wnt/β-catenin signaling pathway, and induces EMT and metastasis in lung adenocarcinoma.
Keywords: lung adenocarcinoma, replication factor C subunit 3, cell invasion and migration, epithelial-mesenchymal transition, Wnt/β-catenin signaling pathway
Introduction
Lung cancer is a malignant tumor responsible for the highest mortality rate in humans worldwide (1). Non-small cell lung cancer (NSCLC) is responsible for 80-85% of cases of diagnosed lung cancer (2). The incidence of lung adenocarcinoma has been increasing in recent years, and despite the application of various methods for treating lung adenocarcinoma, the mortality rate remains high (3). Although targeted drugs can be used in some cases to treat patients with mutations in genes such as epidermal growth factor receptor and anaplastic lymphoma kinase, the proportion of such patients is limited and drug resistance limits the long-term efficacy of targeted therapy (4). Therefore, the identification of key genes associated with lung cancer is of great significance for the development of novel therapeutic strategies. The classical Wnt/β-catenin pathway serves a critical role in the multiplication and differentiation of progenitor cells in various adult epithelia (5). A strong link has been identified between overactivation of the Wnt/β-catenin pathway, and the occurrence and progression of several types of cancer, including colorectal cancer (6). It was been reported that overactivation of β-catenin in the pulmonary epithelium of genetically engineered mice could induce epithelial differentiation defects, promoting cell multiplication, basal cell amplification and lung tumorigenesis (7). This previous finding suggested that overactivation of the Wnt/β-catenin pathway may also lead to lung cancer. It has further been shown that the Wnt/β-catenin signaling pathway is extensively involved in NSCLC as a controller of cellular proliferation, apoptosis, differentiation, cell cycle progression, invasion and migration (8-10). Epithelial-mesenchymal transition (EMT) is a critical event, characterized by loss of cellular polarity and contact, leading to migration of cancer cells and tumor progression, which is induced by activation of the Wnt/β-catenin pathway (11-13). Mutations in β-catenin or adenomatous polyposis coli, which represent the most universal mechanisms underlying abnormal activation of the Wnt/β-catenin pathway, are infrequent in NSCLC (14). Therefore, the cause of overactivation of the Wnt/β-catenin pathway and its effect on EMT in NSCLC require further exploration.
Replication factor C (RFC) is a component of the eukaryotic DNA polymerase (15). In all eukaryotic cell cycles, it is essential for DNA duplication, DNA injury repair and checkpoint control (16-18). RFC consists of five subunits (RFC1-5). The interrelationships between RFC2, RFC3 and the c-MYC oncogene (transcription factor) can induce cell division and proliferation (19). With the exception of RFC1, the other four subunits are highly expressed in various malignant tumors (20-24). It has been reported that RFC3 can combine with proliferating cell nuclear antigen (PCNA) to form a complex, and decreased expression of RFC3 can restrict the multiplication of cancer cells (25). Previous studies have demonstrated that RFC3 is associated with liver, breast, esophageal and ovarian cancer, and serves a critical role in cellular proliferation, invasion and metastasis (25-28). Notably, 9-cis retinoic acid-activated retinoid X receptor α can inhibit the growth of retinoid-sensitive breast cancer and embryonic cells, and arrest S phase entry by disrupting the RFC3-PCNA complex (29). These previous findings suggest that RFC3 may be an important cancer antigen; however, the effect of RFC3 on the occurrence and progression of NSCLC remains unclear.
This study explored the expression of RFC3 in NSCLC and corresponding paracancerous tissues, and investigated the association between clinicopathological features and RFC3 expression. Furthermore, MTT, flow cytometry, Boyden chamber and wound-healing assays were performed to explore the effects of RFC3 on proliferation, invasion and migration of A549 and H1299 cells. Western blot analysis was finally applied to confirm that RFC3 induces EMT of lung adenocarcinoma H1299 and A549 cells via the Wnt/β-catenin pathway.
Materials and methods
Tissue specimens, cell lines and culture conditions
Tissue samples were obtained from patients with lung adenocarcinoma or squamous cell carcinoma who underwent surgery at The First Affiliated Hospital of China Medical University between March 2010 and June 2013. This study was approved by the Institutional Research Ethics Committee of China Medical University and written informed consent was obtained from all 165 patients. A total of 42 patients with lung squamous cell carcinoma and 123 patients with lung adenocarcinoma were included in this study. Each case comprised cancerous tissue and paracancerous tissue (≥5 cm from the neoplasm border) samples. All samples had complete follow-up data and were pathologically diagnosed with lung squamous cell carcinoma or adenocarcinoma. The NSCLC H460, A549, LK2, H1299, H3255, H1650, H1975 and H292 cell lines were used in this study, and normal human bronchial epithelial 135-E6E7 (HBE135-E6E7) cells were used as a control. The tissue sections were stored in liquid nitrogen. HBE cells were obtained from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences. The NSCLC H460, A549, LK2, H1299, H3255, H1650, H1975 and H292 cell lines were acquired from the American Type Culture Collection. HBE, A549 and LK2 cells were cultured in DMEM (cat. no. 10569069; Gibco; Thermo Fisher Scientific, Inc.) and the remaining cell lines were cultured in RPMI-1640 (cat. no. 61870044; Gibco; Thermo Fisher Scientific, Inc.), with all recommended supplements: 10% fetal bovine serum (cat. no. 10099141C), 100 U/ml penicillin and 100 µg/ml streptomycin (cat. no. 15140122) PBS (cat. no. 70013073) and trypsin (cat. no. R001100) (all from Gibco; Thermo Fisher Scientific, Inc.). All cells were maintained in a humidified container with 95% air and 5% CO2 at 37°C.
Immunohistochemistry
Immunohistochemistry was performed to investigate RFC3 expression in cancerous and paracancerous tissues. The tissues were fixed in 10% formalin for 4 h/mm at room temperature and embed in paraffin blocks. The 4-µm paraffin-embedded sections of lung cancer and paracancerous tissues were prepared and deparaffinized three times in xylene (5 min each time) and were then dehydrated in graded ethanol solutions. The sections were washed twice in 100% ethanol (15 min/wash), then twice in 90% ethanol (15 min/wash). After heat treatment at 95°C for 5 min with 0.01 mol/l citrate buffer (pH 6.0; cat. no. 005000; Thermo Fisher Scientific, Inc.) for antigen retrieval, immunohistochemical analysis of paraffin-embedded sections was performed by incubation with a mouse monoclonal antibody (anti-RFC3; 1:100; cat. no. sc-390293; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature, followed by incubation with a secondary antibody (1:1,000; cat. no. 7076; Cell Signaling Technology, Inc.) for 1 h at room temperature. Finally, the sections were counterstained with hematoxylin for 2 min at room temperature. Rabbit IgG (1:100; cat. no. 3900s; Cell Signaling Technology, Inc.) was employed as a negative control. Two pathologists who were ignorant of the clinical data independently analyzed the immunohistochemistry results. A semi-quantitative method was used to determine the expression levels of RFC3. The proportion and staining intensity of positive cells were scored under a light microscope. The proportion of positive cells was scored as follows: 0, 0; 1, 1-25; 2, 26-50; 3, 51-75; and 4, 76-100%; the positive staining intensity was scored as follows: 0, no staining; 1, light brown staining; 2, moderate brown staining; 3, dark brown staining. The final score was obtained by multiplying the two results for each sample; scores ranged between 0 and 12. Receiver operating characteristic curve analysis was performed to determine the cut-off scores for low or high RFC3 expression. All samples were separated into low expression (0-4) or high expression (5-12) groups.
Cell transfection for overexpression and knockdown of RFC3
A549 cells were treated with 0.25% trypsin and evenly inoculated in 6-well culture plates at a density of 1.5×105 cells/well. After incubation at constant temperature (37°C, 5% CO2), cells were transfected once they adhered to the plates. Cells were starved of serum for 1 h prior to transfection. The transfection mixture consisted of serum-free medium, 2.5 µg/ml RFC3 plasmid (cat. no. RC201655; OriGene Technologies, Inc.) or control plasmid (cat. no. PS100001; OriGene Technologies, Inc.), and Lipofectamine® 3000 (cat. no. L3000150; Invitrogen; Thermo Fisher Scientific, Inc.), and was added to the wells drop-wise. The culture plate was then gently shaken and incubated at constant temperature (37°C, 5% CO2) for 5 h. The transfection media were then discarded, and DMEM containing 10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc.) was added for further culture (37°C, 5% CO2, 24 h).
H1299 cells were digested with 0.25% trypsin and evenly inoculated into 6-well culture plates at a density of 1×105 cells/well. Following incubation at constant temperature (37°C, 5% CO2), cells were transfected once they adhered to the plates. Cells were starved of serum for 1 h prior to transfection. The transfection mixture consisted of serum-free medium, 100 nmol RFC3 small interfering (si)RNA (cat. no. sc-37635; Santa Cruz Biotechnology, Inc.) or control siRNA (cat. no. sc-37007; Santa Cruz Biotechnology, Inc.), and DHARMAFECT (cat. no. T-2001-02; GE Healthcare Dharmacon, Inc.), and was added to the wells dropwise. The culture plate was then gently shaken and incubated at constant temperature (37°C, 5% CO2) for 5 h. Transfection media were discarded and RPMI-1640 medium containing 10% fetal calf serum was added for further culture (37°C, 5% CO2, 48 h).
Western blot analysis
Cells were rinsed with PBS three times. After removing the buffer, cells were collected in a culture bottle. The prepared lysis buffer (cat. no. FNN0021; Thermo Fisher Scientific, Inc.) was added to the culture bottle, and cell lysis was allowed to occur on ice for 30 min. Protein concentration in the supernatants was determined using a BCA kit (Pierce; Thermo Fisher Scientific, Inc.). Proteins (20 µg/lane) were separated by SDS-PAGE using 10% gels and were transferred to PVDF membranes (EMD Millipore) for analysis. The PVDF membranes were then transferred to 0.05% Tween-TBS (TTBS) and rinsed for 5 min; this was repeated three times. PVDF membranes were blocked with 5% milk in TTBS for 1 h at room temperature, and were probed with primary antibodies against RFC3 (1:500; cat. no. sc-390293; Santa Cruz Biotechnology, Inc.), Vimentin (1:1,000, cat. no. 5741S; Cell Signaling Technology, Inc.), N-cadherin (1:1,000, cat. no. 13116S, Cell Signaling Technology, Inc.), E-cadherin (1:1,000, cat. no. 14472S; Cell Signaling Technology, Inc.), phosphorylated (p)-glycogen synthase kinase 3 (GSK3)-β (Ser9) (1:1,000, cat. no. 5558S; Cell Signaling Technology, Inc.), β-catenin (1:1,000, cat. no. 8480S, Cell Signaling Technology, Inc.), GSK3-β (1:1,000, cat. no. 12456S; Cell Signaling Technology, Inc.), c-MYC (1:1,000, cat. no. 13987S; Cell Signaling Technology, Inc.), Wnt1 (1:1,000, cat. no. 27935-1-AP; Proteintech Group, Inc.), and GAPDH (1:1,000, cat. no. 5174S; Cell Signaling Technology, Inc.) or actin (1:2,000, cat. no. 3700S; Cell Signaling Technology, Inc.) overnight at 4°C. Subsequently, the PVDF membranes were rinsed with TTBS three times prior to incubation with horse anti-mouse IgG-HRP (1:2,000; cat. no. 7076V; Cell Signaling Technology, Inc.) or goat anti-rabbit IgG-HRP (1:2,000; cat. no. 7074V; Cell Signaling Technology, Inc.) secondary antibodies at indoor temperature for 2 h. The PVDF membranes were finally rinsed three times and observed with an ECL kit (Thermo Fisher Scientific, Inc.). The intensity of each protein band was semi-quantified using image analysis software (ImageJ; National Institutes of Health).
Cell growth curve analysis
The MTT assay was used to observe the effects of RFC3 upregulation or downregulation on lung adenocarcinoma cell proliferation. After A549 and H1299 cells were transfected for 48 h, they were transferred to 96-well plates; cell density was maintained at ~2,000 cells per well. At scheduled times on days 1-5, 5 mg/ml MTT solution was added to each well, and the reaction was allowed to occur at 37°C for 4 h. Subsequently, DMSO was added to the wells. The absorbance of each well was calculated using a microplate reader at a wavelength of 490 nm.
Flow cytometry
To detect the proportion of cells in different stages of the cell cycle and apoptotic cells, flow cytometry (FACSCalibur; BD Biosciences) was conducted. A total of 3 days post-transfection, the A549 and H1299 cells were collected, rinsed with PBS and treated with 75% ethanol at 4°C for 2 h. After washing with PBS, the cells were incubated with 200 µg/ml RNAse (cat. no. 10109134001; Sigma-Aldrich; Merck KGaA) for 20 min at room temperature and stained with 1 µg/ml propidium iodide (PI; BD Biosciences) in 1 ml PBS at 4°C for 30 min. Subsequently, the cell cycle was measured by flow cytometry. Cell apoptosis was detected after washing with 500 µl PBS. Briefly, 10% Annexin V-FITC (BD Biosciences) and 50 µg/ml PI staining was performed for 15 min in the dark at room temperature. To promote apoptosis, A549 cells were treated with paclitaxel (Shanghai YuanYe Bio-Technology Co. Ltd.) at a final concentration of 50 nM, followed by incubation for 18 h at 37°C. In addition, H1299 cells were treated with erlotinib (MAYA Reagent) at a final concentration of 2 µM and were incubated for 18 h at 37°C. Apoptosis was measured by flow cytometry using a FACSCalibur analysis system (BD Biosciences). Data were analyzed with Modfit 2.0 (Verity Software House, Inc.).
Boyden chamber assay
Cell invasion and migration were analyzed using Transwell assays. For the migration analysis, 1×105 cells were added to the upper Boyden chamber. After 24 h, non-migrating cells were removed from the upper chamber with a soft cotton swab and the cells that had migrated to the lower chamber were stained with hematoxylin for 15 min at room temperature and counted under light microscopy. For the invasion analysis, the upper Boyden chamber was precoated with 50 mg/l Matrigel (BD Biosciences) prior to the addition of 1×105/ml cells to the upper chamber. The other steps remained almost the same as for the migration analysis. Finally, the cells that had invaded into the lower chamber through the membranes were stained with hematoxylin for 15 min at room temperature and counted under light microscopy.
Wound-healing assay
Cells were transferred to 6-well plates at 5×105 cells/well and allowed to reach 90% confluence. A single layer of cells was scratched in a straight line using a 10-µl pipette tip and was washed with PBS. Subsequently, the plates were cultured in serum-free medium at 37°C for 48 h. The width of the healing monolayer wound was recorded after 0 and 48 h. Quantitative analysis of the wound area from three independent experiments was performed using ImageJ software with the macro MRI Wound Healing Tool. Migration rate was presented as a percentage of the initial wound area: Wound area (%)=(wound area at 0 h-wound area at 48 h)/wound area at 0 hx100. 0 h is the time when the scratch was initially created; 48 is 48 h after scratch creation.
Statistical analysis
Each experiment was repeated at least three times. The immunohistochemistry results were assessed using χ2 test, whereas the other results were assessed using Student's t-test or one-way ANOVA, in order to examine the differences between groups, and data were expressed as the mean ± SD. Multiple comparisons between the groups were performed using the S-N-K method. Repeated-measures two-way ANOVA was used to determine whether there were statistical differences between the growth curves. Cox regression was employed for univariate and multivariate analyses. The overall survival rate was assessed by Kaplan-Meier survival analysis and compared with the log-rank test. SPSS 23.0 (SPSS, Inc.) was employed for statistical analysis, and P<0.05 was considered to indicate a statistically significant difference.
Results
RFC3 expression in lung cancer specimens
RFC3 exhibited high expression in lung adenocarcinoma and RFC3 expression was associated with clinicopathological characteristics; this was not observed in lung squamous cell carcinoma. RFC3 expression was measured in various tissues, as shown in Figs. 1 and 2; and Table I. RFC3 expression in 123 lung adenocarcinoma specimens was significantly higher than in the corresponding non-neoplastic lung tissues (P<0.0001). However, no significant differences were found in 42 cases of lung squamous cell carcinoma (P=0.533). The association between RFC3 expression and the clinicopathological characteristics of 123 patients with lung adenocarcinoma, including sex, age, smoking history, TNM classification (30), differentiation, tumor size and node status, is presented in Table II. RFC3 expression was markedly associated with TNM classification, differentiation and node status. Univariate analysis revealed that RFC3 expression, TNM classification, differentiation, tumor size and node status had significant effects on overall survival in patients with lung adenocarcinoma. The expression of RFC3, differentiation and node status were confirmed as critical independent risk factors by multivariate analysis (Table III). Kaplan-Meier analysis indicated that high RFC3 expression could lead to a poor prognosis in cases of lung adenocarcinoma; similar results were determined in analysis of disease-free survival (Fig. 3; P<0.05). In 42 cases of lung squamous cell carcinoma, univariate analysis revealed that RFC3 expression had no significant effect on prognosis (P=0.57; data not shown). The expression of RFC3 in three cases of lung adenocarcinoma and corresponding normal lung tissues was evaluated by western blotting. The protein expression levels of RFC3 in cancer tissues were higher than in normal lung tissues; these findings were consistent with the immunohisto-chemistry results (Fig. 4A). RFC expression in NSCLC cell lines and the HBE cell line was evaluated by western blotting (Fig. 4B). RFC3 expression in the lung adenocarcinoma A549 cell line was slightly lower than that in HBE, but no significant difference was found (P=0.1049). RFC3 expression in the lung squamous cell carcinoma LK2 cell line was slightly higher than that in HBE, but no significant difference was found (P=0.7724). RFC3 expression in the other NSCLC cell lines was significantly higher than that in HBE cells (P<0.05).
Figure 1.

Immunohistochemistry staining of RFC3 expression in different tissues. (A and B) RFC3 expression in lung adenocarcinoma tissues (high expression). (C and D) RFC3 expression in non-tumor lung tissues (low expression). (E and F) RFC3 expression in non-tumor lung tissues (negative expression). (A, C and E) Magnification, ×200; (B, D and F) magnification, ×400. RFC3, replication factor C subunit 3.
Figure 2.

Immunohistochemistry staining of RFC3 expression in different tissues. (A and B) RFC3 expression in lung squamous cell carcinoma (low expression). (C and D) RFC3 expression in non-tumor lung tissues (low expression). (A and C) Magnification, ×200; (B and D) magnification, ×400. RFC3, replication factor C subunit 3.
Table I.
Expression of RFC3 in different tissues.
| Tissue type | n | RFC3 expression
|
χ2 | P-value | |
|---|---|---|---|---|---|
| Low expression (%) | High expression (%) | ||||
| Lung adenocarcinoma | 123 | 66 (53.66) | 57 (46.34) | 18.732 | <0.0001a |
| Non-tumor lung tissue | 123 | 98 (79.67) | 25 (20.33) | ||
| Lung squamous carcinoma | 42 | 35 (83.33) | 7 (16.67) | 0.389 | 0.533 |
| Non-tumor lung tissue | 42 | 37 (88.10) | 5 (11.90) | ||
P<0.05, Pearson χ2 test. RFC3, replication factor C subunit 3.
Table II.
Association between RFC3 expression and clinicopathological characteristics of 123 patients with lung adenocarcinoma.
| Variables | RFC3 expression
|
Total | χ2 | P-value | |
|---|---|---|---|---|---|
| Low expression | High expression | ||||
| Total cases | 66 | 57 | |||
| Sex | 0.012 | 0.914 | |||
| Male | 26 | 23 | 49 | ||
| Female | 40 | 34 | 74 | ||
| Age | 0.879 | 0.349 | |||
| ≤60 years | 38 | 28 | 66 | ||
| >60 years | 28 | 29 | 57 | ||
| Smoking history | 2.897 | 0.089 | |||
| Yes | 27 | 15 | 42 | ||
| No | 39 | 42 | 81 | ||
| TNM stage | 6.409 | 0.011a | |||
| Stage I-II | 60 | 42 | 102 | ||
| Stage III-IV | 6 | 15 | 21 | ||
| Differentiation | 5.139 | 0.023a | |||
| Well | 39 | 22 | 61 | ||
| Moderate/poor | 27 | 35 | 62 | ||
| Size | 2.328 | 0.127 | |||
| ≤3 cm | 49 | 35 | 84 | ||
| >3 cm | 17 | 22 | 39 | ||
| Node status | 5.747 | 0.017a | |||
| Positive | 9 | 18 | 27 | ||
| Negative | 57 | 39 | 96 | ||
P<0.05, Pearson χ2 test. RFC3, replication factor C subunit 3.
Table III.
Univariate and multivariate analyses of the factors associated with overall survival of patients with lung adenocarcinoma.
| Variables | Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | P-value | Hazard ratio | 95% CI | P-value | |
| RFC3 expression | 3.46 | 1.660-7.209 | 0.001a | 2.339 | 1.103-4.959 | 0.027a |
| Sex | 1.812 | 0.933-3.517 | 0.079 | |||
| Age | 1.927 | 0.980-3.792 | 0.057 | |||
| Smoking history | 1.504 | 0.770-2.938 | 0.233 | |||
| TNM stage | 4.088 | 2.051-8.148 | <0.0001a | 0.588 | 0.211-1.636 | 0.309 |
| Differentiation | 4.907 | 2.141-11.249 | <0.0001a | 3.143 | 1.321-7.478 | 0.010a |
| Size | 2.78 | 1.431-5.401 | 0.003a | 1.558 | 0.747-3.248 | 0.237 |
| Node status | 5.578 | 2.859-10.882 | <0.0001a | 4.922 | 1.913-12.665 | 0.001a |
P<0.05. RFC3, replication factor C subunit 3.
Figure 3.

Kaplan-Meier analysis of the association of RFC3 expression with overall survival and disease-free survival in 123 lung adenocarcinoma cases. Patients with high RFC3 expression exhibited markedly shortened survival time compared with the low expression group (log-rank test, P<0.05). RFC3, replication factor C subunit 3.
Figure 4.
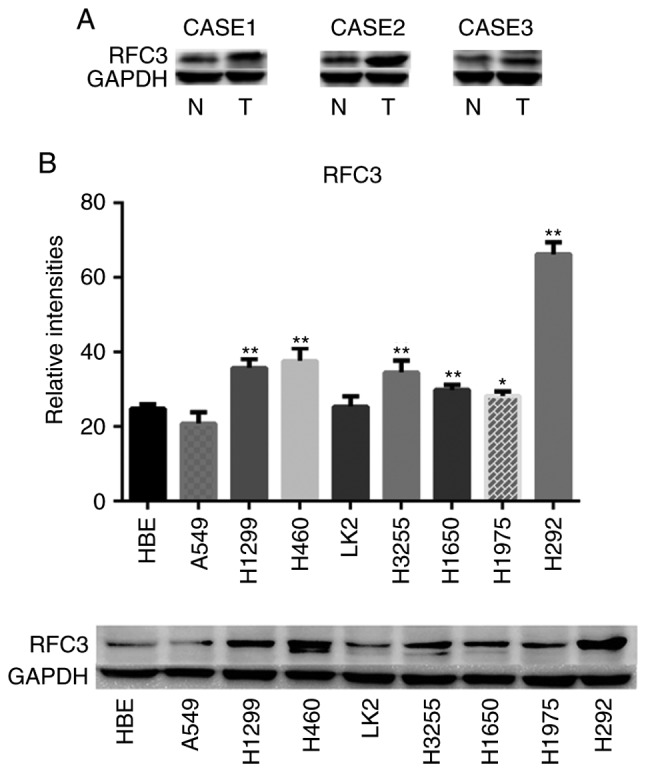
(A) Western blotting of RFC3 protein expression in three human primary lung adenocarcinoma (T) and paired adjacent non-tumor lung tissues (N); for each case, paired T and N tissues were analyzed. (B) Comparison of RFC3 protein expression in eight lung cancer cell lines (H460, A549, LK2, H1299, H3255, H1650, H1975 and H292) and one normal cell line (HBE). Semi-quantified data of three separate western blot analyses and representative images are presented. Relative intensities refer to the gray value of RFC3 protein divided by the gray value of the internal reference protein GAPDH. *P<0.05; **P<0.01 vs. HBE cells. HBE, human bronchial epithelial; RFC3, replication factor C subunit 3.
Role of RFC3 in cell proliferation, cell cycle progression and apoptosis
Based on the aforementioned results, a plasmid containing cloned RFC3 cDNA was transfected into the A549 cell line to induce overexpression of RFC3. In addition, a RFC3 siRNA was transfected into the H1299 cell line to knock down RFC3. Western blotting was employed to verify the effects (Fig. 5). As shown in Fig. S1, the proliferative ability of H1299 and A549 cells was not significantly affected by overexpression or knockdown of RFC3 compared with in the control groups (P>0.05). Flow cytometry revealed that the percentage of A549 cells in G0/G1 stage was decreased when RFC3 was overexpressed, whereas the percentage of S stage cells was increased, indicating that RFC3 overexpression induced more cells to enter S stage from G1 stage (P<0.05). The proportion of H1299 cells in G0/G1 stage was increased when RFC3 was knocked down, whereas the proportion of S stage cells was decreased, indicating that more cells were arrested at G0/G1 stage after RFC3 was knocked down (P<0.05; Fig. S2). Although fewer apoptotic cells were detected in the A549 cell line when RFC3 was overexpressed, the difference was not significant compared with in the control group (P=0.2666). In addition, more apoptotic cells were detected in the H1299 cell line when RFC3 was knocked down in comparison with the control group (P<0.01; Fig. S3). When paclitaxel was added to the A549 cell line to induce apoptosis, fewer apoptotic cells were detected in the A549 cell line when RFC3 was overexpressed in comparison with the control group (P<0.01). Conversely, when erlotinib was added to the H1299 cell line to induce apoptosis, more apoptotic cells were detected in the H1299 cell line when RFC3 was knocked down in comparison with the control group (P<0.05; Fig. S4).
Figure 5.
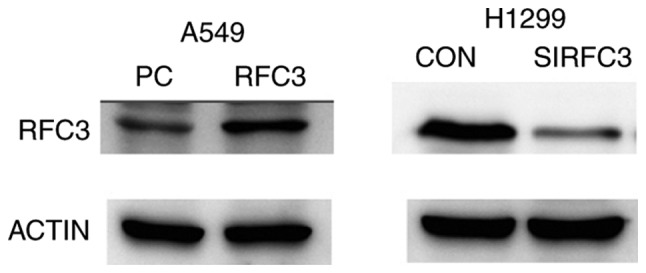
Representative data of immunoblotting of RFC3 in A549 cells transfected with the empty vector or RFC3 overexpression plasmid, and in H1299 cells transfected with the control siRNA or RFC3-specific siRNA. RFC3, replication factor C subunit 3; siRNA, small interfering RNA.
Effects of RFC3 on cell invasion and migration
The Boyden chamber assay indicated that the invasion and migration of A549 cells was significantly increased following overexpression of RFC3 (P<0.05), whereas the invasion and migration of H1299 cells was significantly decreased after RFC3 downregulation (P<0.05; Figs. 6 and 7). The wound-healing assay demonstrated that the migration rate of A549 cells following overexpression of RFC3 was markedly higher than that in the control group (P<0.05), indicating that the migratory ability of A549 cells was increased (Fig. 8). Conversely, after the knockdown of RFC3, the opposite result was observed in H1299 cells (P<0.05; Fig. 9).
Figure 6.

Effects of overexpression or knockdown of RFC3 on cell migration. Following transfection of RFC3 overexpression plasmid and empty vector, or RFC3-specific siRNA and non-specific control siRNA, cell migration was assessed by Transwell migration assay. (A) Representative image data of A549 cells transfected with RFC3 overexpression plasmid and empty vector that migrated through the membrane (magnification, ×200); quantitative representation of the cell number obtained from three separate experiments. (B) Representative image data of H1299 cells transfected with non-specific control siRNA and RFC3-specific siRNA that migrated through the membrane (magnification, ×200); quantitative representation of the cell number obtained from three separate experiments. *P<0.05. RFC3, replication factor C subunit 3; siRNA, small interfering RNA.
Figure 7.

Effects of overexpression or knockdown of RFC3 on cell invasion. Following transfection of RFC3 overexpression plasmid and empty vector, or RFC3-specific siRNA and non-specific control siRNA, cell invasion was assessed by Transwell invasion assay. (A) Representative image data of A549 cells transfected with RFC3 overexpression plasmid and empty vector that invaded through the membrane (magnification, ×200); quantitative representation of the cell number obtained from three separate experiments. (B) Representative image data of H1299 cells transfected with non-specific control siRNA and RFC3-specific siRNA that invaded through the membrane (magnification, ×200); quantitative representation of the cell number obtained from three separate experiments. *P<0.05. RFC3, replication factor C subunit 3; siRNA, small interfering RNA.
Figure 8.

Effects of RFC3 overexpression on wound healing. Following transfection of RFC3 overexpression plasmid and empty vector, cell migration was further assessed by wound-healing assay in A549 cells. Representative image data of cells migrated during the wound-healing assay (magnification, ×100); quantitative representation of migration rate from three separate experiments. *P<0.05. RFC3, replication factor C subunit 3.
Figure 9.

Effects of RFC3 knockdown on wound healing. Following transfection of RFC3-specific siRNA and control siRNA, cell migration was further assessed by wound-healing assay in H1299 cells. Representative image data of cells migrated during the wound-healing assay (magnification, ×100); quantitative representation of migration rate from three separate experiments. *P<0.05. RFC3, replication factor C subunit 3; siRNA, small interfering RNA.
RFC3 induces EMT in lung adenocarcinoma cells via the Wnt/β-catenin pathway
Western blotting was employed to detect alterations in the expression of proteins associated with the Wnt/β-catenin signaling pathway and EMT in A549 cells transfected with the RFC3 overexpression plasmid and H1299 cells transfected with the RFC3 siRNA. After the overexpression of RFC3 in A549 cells, the expression levels of Wnt1, β-catenin, c-MYC, N-cadherin, Vimentin and the ratio of p-GSK3-β (Ser9)/GSK3-β were increased, whereas E-cadherin expression was decreased. However, following RFC3 knockdown in H1299 cells, the expression levels of Wnt1, N-cadherin, β-catenin, Vimentin, c-MYC and the ratio of p-GSK3-β (Ser9)/GSK3-β were decreased, whereas E-cadherin expression was increased. These results suggested that RFC3 may trigger the Wnt/β-catenin signaling pathway in A549 and H1299 cells and promote lung adenocarcinoma migration and invasion through EMT (Fig. 10).
Figure 10.

Effects of RFC3 on the regulation of proteins associated with epithelial-mesenchymal transition and the Wnt/β-catenin signaling pathway. (A) RFC3 overexpression plasmid and empty vector were transfected into the lung cancer A549 cell line. (B) RFC3-specific siRNA and non-specific control siRNA were transfected into the lung cancer H1299 cell line. Total protein was extracted and subjected to immunoblotting of Wnt1, p-GSK3-β (Ser9), GSK3-β, β-catenin, c-MYC, E-cadherin, N-cadherin and Vimentin. Actin was used as a loading control. (A and B) Representative image data of immunoblotting, semi-quantitative representation of protein expression and ratios of p-GSK3-β/GSK3-β from three separate experiments. *P<0.05, **P<0.01. GSK3, glycogen synthase kinase 3; p-, phosphorylated; RFC3, replication factor C subunit 3; siRNA, small interfering RNA.
Discussion
NSCLC is the main cause of malignant tumor-associated mortality in humans and adenocarcinoma is the main subtype worldwide (1). Novel diagnostic and therapeutic technologies have been developed; however, overall survival and prognosis are still poor (2,3). Therefore, it is important to identify new biomarkers and pathways for the treatment of lung adenocarcinoma. In several types of malignant tumor, RFC3 is overexpressed and is associated with the regulation of tumor development through various mechanisms, as observed in triple-negative breast cancer, ovarian cancer, esophageal cancer and liver cancer (25-28). However, it has also been reported that RFC3 mutations in gastric and colorectal cancer result in downregulation of expression or loss of function (31). These findings indicated that RFC3 might serve different roles in different types of cancer. Therefore, the mechanism and function of RFC3 in NSCLC remains to be elucidated.
Through immunohistochemistry, this study revealed that RFC3 expression was markedly higher in lung adenocarcinoma tissue compared with in corresponding paracancerous lung tissue, and RFC3 expression was significantly associated with clinicopathological characteristics; these findings were not observed in lung squamous cell carcinoma. Notably, RFC3 expression was markedly associated with TNM classification, differentiation and node status in lung adenocarcinoma. Furthermore, RFC3 expression was determined to be a critical independent risk factor through multivariate analysis. Kaplan-Meier analysis indicated that RFC3 overexpression could lead to a poor prognosis in patients with lung adenocarcinoma, and similar results were obtained in analysis of disease-free survival. These results demonstrated that RFC3 may present prognostic value in human lung adenocarcinoma. The outcomes of this research were highly similar to findings obtained in triple-negative breast cancer, esophageal cancer, ovarian cancer and liver cancer (25-28). However, to the best of our knowledge, RFC3 has not been reported as an independent risk factor in lung adenocarcinoma. Unlike triple-negative breast cancer, RFC3 expression was not related to the size of lung adenocarcinoma (P=0.127) (25), which suggests that RFC3 may not be associated with the proliferation of lung adenocarcinoma cells.
In the present cell line experiments, the highest expression of RFC3 was detected in H292 cells and the second highest in H460 cells. However, H292 and H460 cells are a mucoepidermoid carcinoma of the bronchus cell line and a large-cell lung cancer cell line, respectively. The incidence of these cancers is much lower than that of lung adenocarcinoma; therefore, their value for further clinical research is limited. Finally, the lung adenocarcinoma H1299 cell line was selected for use in the present study for the RFC3 knockdown experiment; this cell line exhibited the third highest expression of RFC3. Previous studies have observed changes in cell biological behavior after knocking down RFC3 (25-28). In addition, RFC3 overexpression was induced in A549 cells with low basal expression, in order to observe changes in their biological behavior. The proliferative abilities of A549 and H1299 cells were not significantly affected by overexpression or knockdown of RFC3 compared with in the control groups. These findings confirmed that RFC3 may not be associated with the proliferation of lung adenocarcinoma cells. However, previous studies have reported that the proliferation curve is significantly decreased when RFC3 is knocked down (25-28) in the following cell lines: Liver cancer SMMC-7721, ovarian cancer OVCAR-3, breast cancer MDA-MB-231 and MDA-MB-468, and esophageal cancer, OE33 and OE19. The difference in these findings might suggest that RFC3 serves different roles in cancer cell proliferation in different types of cancer.
Flow cytometry suggested that upregulation of RFC3 resulted in a greater number of cells entering S phase from G1 phase in A549 cells, whereas downregulation of RFC3 resulted in a greater number of cells arrested at G0/G1 phase in H1299 cells; however, no significant change in the ratio of cells in G2/M stage was found. The proportion of G0/G1 and S phase cells was altered by only ~4%; therefore, it was hypothesized that RFC3 may exert a certain effect on the cell cycle progression of lung adenocarcinoma, but this effect is weaker than that on liver cancer and ovarian cancer. Notably, hepatocellular and ovarian cancer cell lines were arrested in S phase after RFC3 knockdown (26,27). Changes in some cell cycle regulatory proteins have been detected after RFC3 knockdown in hepatocellular carcinoma cell lines, which explains why the hepatocellular carcinoma cell cycle was arrested in S stage after RFC3 knockdown in this previous study (26). The present study demonstrated that upregulation of RFC3 or knockdown of RFC3 could result in corresponding changes in the downstream protein c-MYC in the Wnt/β-catenin pathway. Notably, c-MYC can promote cell cycle G1-S progression (32-34). Unlike the effects on hepatocellular carcinoma cells (26), in this study, the effects of RFC3 on the cell cycle progression of lung adenocarcinoma resulted in G1-S progression, not S-G2 progression. Why RFC3 has different effects on the cell cycle progression of hepatocellular carcinoma and lung adenocarcinoma cells requires further study.
Upregulation of RFC3 reduced apoptosis in the A549 cell line; however, the difference was not significant when compared with the control group. When paclitaxel was added to the A549 cell line to induce apoptosis, overexpression of RFC3 more obviously reduced apoptosis in the A549 cell line; this finding was statistically significant. Whether or not erlotinib was added to H1299 cells to induce apoptosis, knockdown of RFC3 resulted in a significant increase in apoptosis. However, the percentage of apoptotic cells changed very little, all <2%. Furthermore, after 72 h of incubation, it was observed that the apoptotic cells were almost all early apoptotic cells. Unlike these results, downregulation of RFC3 in ovarian cancer has been shown to induce a higher proportion of apoptotic cells, and after 24 h incubation, late apoptotic cells were revealed to account for the majority of apoptotic cells (27). In summary, RFC3 has a weaker effect on cell cycle progression and apoptosis in lung adenocarcinoma cells than in other cancer cells.
In this study, the results of Boyden chamber and wound-healing assays suggested that RFC3 may increase lung cancer cell invasion and migration. The expression levels of Wnt1 and the ratio of p-GSK3-β (Ser9)/GSK3-β were upregulated by overexpression of RFC3, which together may result in activation of the Wnt/β-catenin signaling pathway. The upregulation of p-GSK3-β (Ser9)/GSK3-β can lead to a decrease in the ability of GSK3-β to phosphorylate and degrade β-catenin (35). β-catenin is a prime downstream protein of the classical Wnt signaling pathway, which has a critical function in EMT. The accumulation of β-catenin leads to upregulation of the expression of the downstream protein c-MYC and affects EMT-related proteins (increases Vimentin and N-cadherin, and decreases E-cadherin) (36,37). When RFC3 was knocked down, the aforementioned effects of RFC3 overexpression were reversed. In a previous study, microarray analysis and Ingenuity Pathway Analysis software were employed to explore the biological pathways and gene networks of RFC3 in esophageal adenocarcinoma (28). Pathway analysis of genes related to RFC3 expression indicated that the Wnt/β-catenin signaling pathway was enriched, but this prediction has not been further confirmed (28). RFC3 has been reported to promote EMT in triple-negative breast cancer cell lines; however, there is no evidence that RFC3 promotes EMT in triple-negative breast cancer cells by affecting the Wnt/β-catenin signaling pathway (25). This study indicated that RFC3 may induce EMT in lung adenocarcinoma cells via the Wnt/β-catenin pathway.
This research had some limitations. A relatively small number of cases was included in the study, and the TNM classifications were mainly stages I and II. To address this problem, extensive identification of cases with different TNM classifications and complete case follow-up data are required. Secondly, in vivo experiments and improved exploration of the RFC3 mechanism are required in the future. STRING database (38) and WebGestalt database (39) were used for bioinformatics analysis, however, the target protein through which RFC3 can affect the Wnt pathway has not yet been identified (data not shown). When the target protein has been identified, we aim to study its association with RFC3 in vivo. Thirdly, the study is retrospective; therefore, prospective studies and double-blind control studies are required to further verify the current outcomes. Finally, RFC3 expression in "normal" lung tissue was compared and analyzed by immunohistochemistry. The 'normal' lung tissues came from the paracancerous tissues of the same patients, which might not truly represent normal tissue.
In conclusion, these data indicated that reduction or over-expression of RFC3 could attenuate or increase the invasion and migration of lung adenocarcinoma cells, respectively. In addition, this study revealed that RFC3 regulated lung adenocarcinoma biological behavior potentially by inducing EMT via the Wnt/β-catenin pathway, and RFC3 expression was closely associated with the clinical outcome of patients with lung adenocarcinoma. These findings suggested that RFC3 may provide a potential anticancer strategy for the treatment of metastasis of advanced lung adenocarcinoma.
Supplementary Data
Acknowledgments
Not applicable.
Funding
This study was funded by the PhD Research Fund of China Medical University.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
SG and QZ designed the experiments. SG, XQ, SY, SZ and PL performed the experiments, and SG, SY and PL analyzed the data. SG and SZ wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All experimental procedures involving human tissue conformed to the ethical standards of The First Affiliated Hospital of China Medical University. This study was approved by the Institutional Research Ethics Committee of China Medical University and written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, et al. Non-small cell lung cancer, version 6.2015. J Natl Compr Canc Netw. 2015;13:515–524. doi: 10.6004/jnccn.2015.0071. [DOI] [PubMed] [Google Scholar]
- 4.Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637–658. doi: 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 5.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K, Yang J, DeMayo FJ, Whitsett JA, Parmacek MS, Morrisey EE. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet. 2008;40:862–870. doi: 10.1038/ng.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo YZ, Xie XL, Fu J, Xing GL. SOX9 regulated proliferation and apoptosis of human lung carcinoma cells by the Wnt/β-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22:4898–4907. doi: 10.26355/eurrev_201808_15626. [DOI] [PubMed] [Google Scholar]
- 9.Ding L, Yao W, Lu J, Gong J, Zhang X. Upregulation of circ_001569 predicts poor prognosis and promotes cell proliferation in non-small cell lung cancer by regulating the Wnt/β-catenin pathway. Oncol Lett. 2018;16:453–458. doi: 10.3892/ol.2018.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Li N, Zhang H. Knockdown of homeobox B5 (HOXB5) inhibits cell proliferation, migration, and invasion in non-small cell lung cancer cells through inactivation of the Wnt/β-catenin pathway. Oncol Res. 2018;26:37–44. doi: 10.3727/096504017X14900530835262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberg RA. Mechanisms of malignant progression. Carcinogenesis. 2008;29:1092–1095. doi: 10.1093/carcin/bgn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Z, He J, Xing X, Li P, Zhang W, Tong Z, Jing X, Li L, Liu D, Wu Q, Ju H. Mn12Ac inhibits the migration, invasion and epithelial-mesenchymal transition of lung cancer cells by downregulating the Wnt/β-catenin and PI3K/AKT signaling pathways. Oncol Lett. 2018;16:3943–3948. doi: 10.3892/ol.2018.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B, Sun L, Li J, Jiang R. miR-577 suppresses cell proliferation and epithelial-mesenchymal transition by regulating the WNT2B mediated Wnt/β-catenin pathway in non-small cell lung cancer. Mol Med Rep. 2018;18:2753–2761. doi: 10.3892/mmr.2018.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akiri G, Cherian MM, Vijayakumar S, Liu G, Bafico A, Aaronson SA. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 2009;28:2163–2172. doi: 10.1038/onc.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uhlmann F, Cai J, Flores-Rozas H, Dean FB, Finkelstein J, O'Donnell M, Hurwitz J. In vitro reconstitution of human replication factor C from its five subunits. Proc Natl Acad Sci USA. 1996;93:6521–6526. doi: 10.1073/pnas.93.13.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venclovas C, Colvin ME, Thelen MP. Molecular modeling-based analysis of interactions in the RFC-dependent clamp-loading process. Protein Sci. 2002;11:2403–2416. doi: 10.1110/ps.0214302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 18.Shen H, Cai M, Zhao S, Wang H, Li M, Yao S, Jiang N. Overexpression of RFC3 is correlated with ovarian tumor development and poor prognosis. Tumour Biol. 2014;35:10259–10266. doi: 10.1007/s13277-014-2216-2. [DOI] [PubMed] [Google Scholar]
- 19.Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR, III, Menssen A, Hermeking H. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle. 2007;6:205–217. doi: 10.4161/cc.6.2.3742. [DOI] [PubMed] [Google Scholar]
- 20.Xiong S, Wang Q, Zheng L, Gao F, Li J. Identification of candidate molecular markers of nasopharyngeal carcinoma by tissue microarray and in situ hybridization. Med Oncol. 2011;28(Suppl 1):S341–S348. doi: 10.1007/s12032-010-9727-5. [DOI] [PubMed] [Google Scholar]
- 21.Arai M, Kondoh N, Imazeki N, Hada A, Hatsuse K, Matsubara O, Yamamoto M. The knockdown of endogenous replication factor C4 decreases the growth and enhances the chemosensi-tivity of hepatocellular carcinoma cells. Liver Int. 2009;29:55–62. doi: 10.1111/j.1478-3231.2008.01792.x. [DOI] [PubMed] [Google Scholar]
- 22.Niu G, Wang D, Pei Y, Sun L. Systematic identification of key genes and pathways in the development of invasive cervical cancer. Gene. 2017;618:28–41. doi: 10.1016/j.gene.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Srihari S, Kalimutho M, Lal S, Singla J, Patel D, Simpson PT, Khanna KK, Ragan MA. Understanding the functional impact of copy number alterations in breast cancer using a network modeling approach. Mol Biosyst. 2016;12:963–972. doi: 10.1039/C5MB00655D. [DOI] [PubMed] [Google Scholar]
- 24.Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer. 2007;43:415–432. doi: 10.1016/j.ejca.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He ZY, Wu SG, Peng F, Zhang Q, Luo Y, Chen M, Bao Y. Up-regulation of RFC3 promotes triple negative breast cancer metastasis and is associated with poor prognosis Via EMT. Transl Oncol. 2017;10:1–9. doi: 10.1016/j.tranon.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao Z, Hu K, Huang H, Xu S, Wang Q, Zhang P, Yang P, Liu B. shRNA-mediated silencing of the RFC3 gene suppresses hepatocellular carcinoma cell proliferation. Int J Mol Med. 2015;36:1393–1399. doi: 10.3892/ijmm.2015.2350. [DOI] [PubMed] [Google Scholar]
- 27.Shen H, Xu J, Zhao S, Shi H, Yao S, Jiang N. ShRNA-mediated silencing of the RFC3 gene suppress ovarian tumor cells proliferation. Int J Clin Exp Pathol. 2015;8:8968–8975. [PMC free article] [PubMed] [Google Scholar]
- 28.Lockwood WW, Thu KL, Lin L, Pikor LA, Chari R, Lam WL, Beer DG. Integrative genomics identified RFC3 as an amplified candidate oncogene in esophageal adenocarcinoma. Clin Cancer Res. 2012;18:1936–1946. doi: 10.1158/1078-0432.CCR-11-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeng S, Kim GJ, Choi EJ, Yang HO, Lee DS, Sohn YC. 9-Cis-retinoic acid induces growth inhibition in retinoid-sensitive breast cancer and sea urchin embryonic cells via retinoid X receptor α and replication factor C3. Mol Endocrinol. 2012;26:1821–1835. doi: 10.1210/me.2012-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobin LH, Gospodarowicz MK, Wittekind C, editors. International Union Against Cancer (UICC): TNM Classification of Malignant Tumours. 8th edition. Wiley-Blackwell; Oxford: 2017. [Google Scholar]
- 31.Kim YR, Song SY, Kim SS, An CH, Lee SH, Yoo NJ. Mutational and expressional analysis of RFC3, a clamp loader in DNA replication, in gastric and colorectal cancers. Hum Pathol. 2010;41:1431–1437. doi: 10.1016/j.humpath.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Pelengaris S, Khan M. The many faces of c-MYC. Arch Biochem Biophys. 2003;416:129–136. doi: 10.1016/S0003-9861(03)00294-7. [DOI] [PubMed] [Google Scholar]
- 33.Steiner P, Philipp A, Lukas J, Godden-Kent D, Pagano M, Mittnacht S, Bartek J, Eilers M. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 1995;14:4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berns K, Hijmans EM, Bernards R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene. 1997;15:1347–1356. doi: 10.1038/sj.onc.1201280. [DOI] [PubMed] [Google Scholar]
- 35.Nusse R. Wnt signaling. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011163. pii: a011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghahhari NM, Babashah S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. Eur J Cancer. 2015;51:1638–1649. doi: 10.1016/j.ejca.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 37.He W, He S, Wang Z, Shen H, Fang W, Zhang Y, Qian W, Lin M, Yuan J, Wang J, et al. Astrocyte elevated gene-1(AEG-1) induces epithelial-mesenchymal transition in lung cancer through activating Wnt/β-catenin signaling. BMC Cancer. 2015;15:107. doi: 10.1186/s12885-015-1124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database Issue):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013;41(Web Server Issue):W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.