

Abstract
Sample preparation for protein quantification by mass spectrometry requires multiple processing steps including denaturation, reduction, alkylation, protease digestion, and peptide cleanup. Scaling these procedures for the analysis of numerous complex biological samples can be tedious and time-consuming, as there are many liquid transfer steps and timed reactions where technical variations can be introduced and propagated. We established an automated sample preparation workflow with a total processing time for 96 samples of 5 hours, including a 2-hour incubation with trypsin. Peptide cleanup is accomplished by online diversion during the LC/MS/MS analysis. In a selected reaction monitoring (SRM) assay targeting 6 plasma biomarkers and spiked β-galactosidase, mean intra-day and inter-day CVs for 5 serum and 5 plasma samples over 5 days were <20%. In a highly multiplexed SRM assay targeting more than 70 proteins, 90% of the transitions from 6 plasma samples repeated on 3 separate days had total CVs below 20%. Similar results were obtained when the workflow was transferred to a second site: 93% of peptides had CVs below 20%. An automated trypsin digestion workflow yields uniformly-processed samples in less than 5 hours. Reproducible quantification of peptides from more than 70 plasma proteins was observed across replicates, days, instruments, and laboratory sites, demonstrating the broad applicability of this approach.
Keywords: liquid chromatography-tandem mass spectrometry (LC/MS/MS), liquid chromatography-selected reaction monitoring (LC-SRM), Automation, protein sample preparation, reproducibility, high throughput
Graphical Abstract
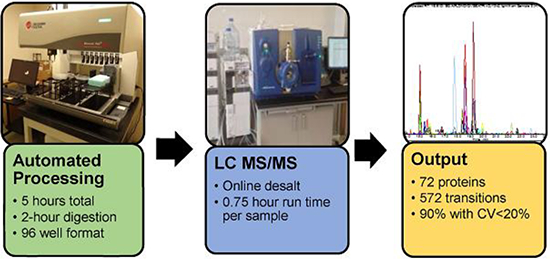
Automated MS sample preparation workflow increases throughput and precision
INTRODUCTION
Biomarker research and development consists of two phases: 1) global protein discovery to identify biomarker candidates and 2) verification and validation to confirm biomarker performance in a larger number of samples1. For proteomic analysis, proteins from complex samples derived from bodily fluids, biopsies, or cultured cells are typically digested into smaller peptides by protease digestion. These proteotypic peptides, as surrogates for the corresponding protein, are more easily measured using a range of liquid chromatography (LC) and mass spectrometry (MS) strategies. Quantitative LC/MS/MS experiments allow researchers to identify differences between the relative amounts of proteins in different sample populations, revealing biomarkers and providing insight into pathophysiology. Global protein discovery requires an in-depth quantitative analysis of the proteome using data-dependent (shotgun)2 or data-independent acquisition (DIA) methods on 10s to 100s of samples. Once putative markers are identfied, targeted proteomic strategies such as selected reaction monitoring mass spectrometry (SRM) are typically used for verification and validation. SRM is the method of choice for targeted biomarker validation because it enables specific, precise quantitation at higher throughput3–5. Regardless of the MS approach, accuracy and precision of the quantitative measurements is critical. Therefore, reproducible sample preparation is fundamental to success. However, throughput, reproducibility, time, and cost remain longstanding barriers to large-scale MS sample processing6–7. The development of a fast, highly reproducible, and completely hands-free MS protein sample preparation workflow would make the entire pathway from biomarker discovery to biomarker validation more robust.
Sample preparation for LC-MS/MS analysis is a multi-step process involving i) protein solubilization and denaturation, ii) disulfide bond reduction and cysteine-blocking to ensure consistent cysteine masses, iii) digestion of proteins into peptides with a site-specific protease (most often trypsin), and iv) clean-up to remove salts, denaturing agents, and other interfering molecules (typically by solid-phase extraction)8–9. Previous studies have shown that sample preparation, particularly the trypsin digestion step, is a major source for variability in LC-MS/MS analysis10–12. Optimal digestion conditions depend on both general and protein-specific factors including the trypsin-to-substrate ratio, buffer composition, protein structure, and the particular amino acid sequence and post-translational modifications adjacent to cleavage sites. It is, therefore, essential to have a highly controlled and standardized digestion method to meet the precision and reproducibility standards required for reliable biomarker verification. To enhance precision and accuracy, each sample preparation step must have accurate liquid transfers, be initiated and stopped at a consistent time, be performed at a controlled temperature and have good mixing for uniform reactions. Automation, and in this case using a 96 well format, can address many of these requirements13. The second aspect for adoption of MS sample preparation automation system are SRM assays of clinical value or the ability to monitor a broader number in a pseudo-discovery mode for research. In this communication, we describe an automated proteomics sample preparation workflow that yields reproducible quantification data on complex proteomic samples and has been implemented in two different laboratories.
EXPERIMENTAL SELECTION
Automated Protein Digestion.
Samples were loaded into a deep 96-well titer plate (Beckman Coulter) with single- or multi-channel pipettes (Eppendorf). The plate was then sealed with X-Pierce™ sealing film (Sigma Aldrich). All other liquid transfers were performed on a Biomek NXP Span-8 Laboratory Automation Workstation operated with Biomek software version 4.1 (Beckman Coulter). The instrument included a Shaking Peltier ALP (Inheco) with a deep well adapter for heating and mixing of samples. The plate was shaken at 1000 RPM for 15 seconds after each reagent addition.
For each reaction, the following were added sequentially: 5 μL plasma, 27.5 μL digestion buffer, 5 μL denaturant, 5 μL internal standards, and 5 μL reducing reagent. The plate was then shaken at 1000 RPM for 60 minutes at 60 °C. Next, 2.5 μL MMTS (200 mM) was added and the plate was shaken for 10 minutes at 1000 RPM. Finally, 10 μL trypsin in 0.1% FA was added and the plate was incubated/shaken at 1000 RPM for 2 hours at 43°C. After this incubation, 10 μl of 10% FA was added to quench the reaction. The plate was centrifuged at 3400 RPM for 5 minutes at 4°C, 10 μL of the supernatant was transferred to 90 μL 2.2 % acetolnitrile (ACN) in 0.1 % FA prior to SRM analysis.
SRM.
Tryptic plasma peptides and internal standards were analyzed on a Prominence UFLCXR HPLC system (Shimadzu, Japan) with a Waters Xbridge Peptide column at 36°C coupled to a QTRAP® 6500 or QTRAP® 5500 MS (SCIEX) with a Turbo V source. Analyst® software (version 1.6.2 for the QTRAP 6500 or 1.5.2 for the QTRAP 5500) was used to control the LC–MS/MS system and for data acquisition. Mobile phase A consisted of 2% ACN, 98% water, and 0.1% FA; and mobile phase B of 95% ACN, 5% water, and 0.1% FA. The flow rate was 200 μL/min for the QTRAP 5500 and 250 μL/min for the QTRAP 6500. After loading the diluted digest (equivalent to 0.05 μL plasma, or 3 μg protein), the column was equilibrated with 5% B for 5 minutes. Peptides were then eluted over 30 minutes with a linear 5% to 35% gradient of buffer B. The column was washed with 98% B for 10 minutes and then returned to 5% B for 5 minutes before loading the next sample. For on-line diversion, a two phase switching valve was used to divert the post-column eluent to waste before it entered the ion source. For trap diversion, a C18 trap column (Phenomenex) was inserted upstream of the Xbridge analytical column and the salt fraction was diverted to waste before entering the analytical column.. Each sample was injected in triplicate into the LC–MS/MS system. All SRM data were processed using MultiQuant™ 2.1 Software (SCIEX). The proteins, peptides, transitions, and MS parameters in the highly-multiplexed SRM assay are listed in supplemental table 1.
Multisite testing.
Laboratory site 1 (Cedars Sinai Medical Center) used a high flow LC-MS system as described above. Laboratory site 2 (SCIEX, Redwood City, CA) used a nanoLCTM 425 system (SCIEX) coupled to a QTRAP® 6500 (SCIEX) with a micro flow mode (5 μL/min) Halopeptide (0.3 ×15 cm) LC column (Eksigent Technology).
Supplemental Materials and Method.
The online supplement identifies the source of reagents, supplies and plasma samples, and provides procedures for manual protein digestion, solid phase extraction, transition validation, and quantification of peptide degradation.
RESULTS AND DISCUSSION
Reduction of technical variation with an automated workflow.
The automated proteomics sample preparation workflow was initially developed and tested with a robust LC-SRM-MS acquisition method targeting serum albumin and β-gal, an exogenous quality control standard. In this assay, stable-isotope labeled (SIL) albumin and β-gal peptides and intact β-gal protein added before the reduction and alkylation reactions are used to detect and normalize for any variability arising from the SRM analysis. For compatibility with an automated workflow, we selected reagents that have negligible non-specific side reactions, are stable in ambient light, are LC-MS/MS friendly, and can be stored as frozen aliquots.
In a preliminary experiment, automated reagent addition and mixing were combined with a 16-hour off-deck trypsin incubation and manual solid phase extraction (SPE). The CVs of the peak area ratios (native/SIL(Stable Isotope-Labeled) peptide signal) for albumin and β-gal were unacceptably high, at 27.1% and 25.6%, respectively (Table 1, set B).
Table 1.
automated digestion workflow evaluation and optimization
| Serum | Control % CV | Control % CV | Control % CV | Clinical % CV | ||||||
| Sample number | 18 (set A) | 14 (set B) | 10 (set C) | 20 (set D) | 12 (set E) | 14 (set F) | 6 (set G) | 177 (set H) | ||
| Processing | manual | Workstation | Workstation | Workstation | Workstation | Workstation | Workstation | Manual | ||
| Digest | Incubator (O/N) | Incubator (O/N) | Incubator (O/N) | Incubator (O/N) | Incubator (O/N) | Incubator (O/N) | Workstation (2hr) | Incubator (O/N) | ||
| Desalting | SPE (HLB) | SPE(HLB) | SPE (HLB) | SPE(HLB) | Online divert | Online Trap/Elute | Online diversion | SPE (HLB) | ||
| Liquid Chromatography | Standard | Standard | Standard | Standard | Standard | Standard | Standard | Standard | ||
| Pool and re-aliquot | none 6500 | None 6500 | Before SPE 6500 | Before LC/MS 6500 | None 6500 | None5500 | None 6500 | None 5500 | ||
| β-galactosidase | WVGYGQDSR | b2 | 12.9 | 23.7 | 14.6 | 4.6 | 9.6 | 10.8 | 2.9 | 21.2 |
| y2 | 12.8 | 5.6 | 9.2 | 10.4 | 4.3 | |||||
| y5 | 15.1 | 5.8 | 7.0 | 16.2 | 2.7 | |||||
| y7 | 15 | 31.6 | 19.0 | 3.9 | 11.6 | 14.3 | 4.5 | 22.2 | ||
| IDPNAWVER | y6 | 27.9 | 24.0 | 10.6 | 5.2 | 11.8 | 9.5 | 7.3 | 18.6 | |
| y7 | 34.3 | 2.8 | 10.0 | 11.4 | 7.0 | |||||
| y7+2 | 12.7 | 23.2 | 15.2 | 3.4 | 10.9 | 5.3 | 8.6 | 20.9 | ||
| GDFQFNISR | y2 | 12.4 | 2.8 | 10.1 | 6.8 | 5.4 | ||||
| y5 | 13 | 3.8 | 9.9 | 7.1 | 5.6 | |||||
| y6 | 12.6 | 3.1 | 10.0 | 8.0 | 6.3 | |||||
| Average | 16.9 | 25.6 | 14.8 | 4.1 | 10.0 | 10.0 | 5.5 | 20.7 | ||
| Standard deviation | 7.7 | 4.0 | 3.4 | 1.1 | 1.4 | 3.4 | 1.9 | 1.5 | ||
| Albumin | LVNEVTEFAK | y6 | 16 | 24.3 | 15.5 | 2.6 | 6.8 | 5.0 | 2.5 | |
| y8 | 17.6 | 24.9 | 10.0 | 2.4 | 6.0 | 5.2 | 3.2 | |||
| DDNPNLPR | y2 | 18.2 | 26.0 | 23.6 | 4.8 | 9.7 | 10.8 | 2.5 | ||
| y5 | 15.9 | 33.2 | 23.9 | 3.5 | 9.5 | 7.1 | 2.3 | |||
| Average | 16.9 | 27.1 | 18.2 | 3.3 | 8.0 | 7.0 | 2.6 | |||
| Standard deviation | 1.2 | 4.1 | 6.7 | 1.1 | 1.9 | 2.7 | 0.4 | |||
CV%s (standard deviation/mean × 100%) were calculated from area under the curve data normalized to data for the corresponding SIL peptide. O/N = overnight trypsin digestion To determine which steps in this procedure contributed substantially to technical variation, digested plasma samples were pooled and then re-aliquoted at different stages in the process. When samples were pooled and re-aliquoted immediately before LC-MS/MS, the average % CV was 3.3% for albumin and 4.1% for β-gal, indicating that the final LC-MS/MS step is highly reproducible (Table 1, set D). In contrast, pooling and re-aliquoting before the SPE desalting step resulted in average % CVs of 18.2% for albumin and 14.8 % for β-gal. Thus, SPE desalting accounted for nearly half of the total variability (Table 1, set C).
Online desalting was investigated as an alternative to SPE, as it provides the potential to reduce technical variation by eliminating the need to manually transfer samples and buffers to the SPE plate, control the vacuum pressure to adjust the flow rate, and dry and re-dissolve the eluted peptides. Two online desalting methods were tested. In the online diversion method, crude peptides were injected onto the C18 analytical column and the 2% organic flow-through was diverted post-column to waste prior to gradient elution of peptides into the MS. The second method used a trap column that was inserted upstream of the analytical column. The flow-through from the trap column was diverted to waste prior to switching the trap column online with the analytical column and performing gradient elution of the peptides. Online diversion is a relatively uncomplicated solution, whereas a trap column can protect the more expensive analytical column from exposure to impurities. The % CVs for albumin and β-gal dropped to 8.0% and 10.0%, respectively, for simple online diversion (Table 1, set E) and to 10.0% and 7.0%, respectively, with a trap column (Table 1, set F). Thus, the online diversion and trap column methods yielded similar results, and both provided a substantial improvement in precision over offline SPE desalting.
Optimizing trypsin digestion conditions.
For the initial optimization tests, the digestion plate was removed from the automated workstation, sealed, and shaken at 1000 RPM for two bench-top incubation steps: 1) denaturation and reduction for 1 hour at 60°C, and 2) trypsin digestion at 37°C. After these incubations, the plate was centrifuged briefly and then returned to the automated workstation for additional processing. The trypsin:substrate ratio was varied by digesting 2 μL, 4 μL or 5 μL plasma with a constant amount of trypsin (25 μg) for 2 hours. Assuming a plasma protein concentration of 70 mg/ml, this corresponds to trypsin:substrate (w/w) ratios of 1:5.6, 1:11.2, and 1:14. All three ratios yielded similar results for albumin and β-gal (Supplemental Figure 1A), so we choose to use 5 μL of plasma as it allowed us to have more material for additional downstream experiments. Next, the digestion time was varied by quenching reactions with formic acid (FA) and then storing them at −80°C until all time points were collected. The signal intensity of all 5 albumin and β-gal peptides was highest after a 2 hour digestion and declined at later timepoints (Supplemental Figure 1B). With a 2-hour digestion at a 1:14 (w/w) trypsin:substrate ratio on the automated workstation, the average % CV for the complete process was further reduced to 2.6% for albumin and 5.5% for β-gal (Table 1, set G). Figure 1 illustrates the schema and protocol for the final automated sample preparation workflow (Figure 1A and 1B).
Figure 1. Automated proteomic sample preparation schema.

A) Workflow and layout of the automated laboratory workstation. B) Outline of the automated digestion protocol. See Materials and Methods for details.
Reproducibility of the automated digestion workflow.
Reproducibility was measured for multiple proteins across multiple days to further validate the automated proteomics sample preparation workflow. Five additional proteins were included in this study: complement C3, alpha-1 antitrypsin, alpha-1-acid glycoprotein 1, hemopexin, and apolipoprotein C-III. These new proteins are well-studied plasma biomarkers (Supplemental Table 2).
To measure reproducibility, five replicate samples of pooled normal human serum and pooled normal human plasma were processed on five different days. Mean intra-day CVs, calculated by comparing the peak areas of five replicate samples run on the same day; ranged from 5.5–8.9% in serum and from 3.9–7.2% for plasma (Table 2, top). Mean inter-day CVs for the seven proteins, calculated by comparing each replicate run on separate days, ranged from 5.8–10.6% in serum and from 3.9–6.0% in plasma (Table 2 and Supplemental Table 3), suggesting that use of automation for sample processing provides good day to day consistency. Interestingly, some intra-day CVs were higher than the corresponding inter-day CVs, suggesting that there is no additional variance in the workflow from multi-day processing. Total CVs, calculated from the square root of the sum of squares of the mean intra- and inter-day CVs, ranged from 9.2–12.3% for serum and 6.4–9.4% for pooled plasma (Table 2, bottom). All of the total CVs were less than 20%, satisfying the best practice acceptance criterion for assay validation of LC-MS/MS protein quantification14.
Table 2.
Automated Proteomics Sample Preparation Workflow Reproducibility
| Intra-day CVa(%) | Day | β-gal WVGYG 2y7 | CO3 ISLPE 2y5 | A1AT SVLGQ 2y7 | A1AG1 TEDTI 2y6 | ALBU LVNEV 2y8 | HEMO NFPSP 2y7 | APOC3 GWVTD +2y7 |
|---|---|---|---|---|---|---|---|---|
| pooled serum | 1 | 1.5 | 4.3 | 6.6 | 2.0 | 5.6 | 5.5 | 6.5 |
| 2 | 4.5 | 7.6 | 6.7 | 4.9 | 6.7 | 5.8 | 6.6 | |
| 3 | 2.4 | 12.0 | 8.4 | 5.5 | 5.7 | 6.8 | 3.8 | |
| 4 | 9.5 | 9.5 | 11.0 | 10.6 | 10.2 | 13.3 | 8.4 | |
| 5 | 9.7 | 7.9 | 11.6 | 7.8 | 7.7 | 9.4 | 10.3 | |
| Mean | 5.5 | 8.3 | 8.9 | 6.1 | 7.2 | 8.1 | 7.1 | |
| pooled plasma K2-EDTA | 1 | 7.6 | 7.3 | 4.7 | 4.9 | 3.8 | 2.3 | 3.6 |
| 2 | 3.0 | 4.5 | 6.9 | 2.4 | 4.7 | 5.8 | 3.8 | |
| 3 | 3.8 | 6.8 | 4.4 | 7.5 | 5.3 | 5.4 | 6.3 | |
| 4 | 2.1 | 8.5 | 8.8 | 5.0 | 7.5 | 6.1 | 7.7 | |
| 5 | 3.0 | 9.1 | 6.3 | 6.6 | 4.9 | 6.8 | 4.7 | |
| Mean | 3.9 | 7.2 | 6.2 | 5.3 | 5.3 | 5.3 | 5.2 | |
| Inter-day CVb(%) | Aliquot | β-gal | CO3 | A1AT | A1AG1 | ALBU | HEMO | APOC3 |
| pooled serum | 1 | 8.4 | 8.6 | 12.7 | 14.0 | 11.5 | 11.0 | 8.7 |
| 2 | 11.9 | 1.2 | 7.5 | 8.4 | 2.6 | 3.9 | 4.9 | |
| 3 | 7.1 | 4.8 | 6.2 | 10.0 | 4.0 | 4.8 | 4.3 | |
| 4 | 6.9 | 9.8 | 7.4 | 11.8 | 6.3 | 6.0 | 7.1 | |
| 5 | 5.9 | 5.2 | 6.1 | 8.9 | 4.7 | 6.5 | 7.1 | |
| Mean | 8.1 | 5.9 | 8.0 | 10.6 | 5.8 | 6.4 | 6.4 | |
| pooled plasma K2-EDTA | 1 | 5.1 | 6.4 | 5.2 | 9.3 | 3.5 | 4.4 | 4.8 |
| 2 | 3.7 | 7.4 | 4.2 | 7.0 | 4.6 | 4.4 | 4.8 | |
| 3 | 3.7 | 4.7 | 5.3 | 4.3 | 3.5 | 4.9 | 1.9 | |
| 4 | 4.8 | 7.5 | 9.6 | 4.6 | 6.8 | 4.3 | 5.9 | |
| 5 | 8.3 | 3.8 | 3.1 | 4.9 | 1.7 | 3.0 | 2.3 | |
| Mean | 5.1 | 6.0 | 5.5 | 6.0 | 4.0 | 4.2 | 3.9 | |
| Total CVc (%) | β-gal | CO3 | A1AT | A1AG1 | ALBU | HEMO | APOC3 | |
| pooled serum | 9.8 | 10.2 | 11.9 | 12.3 | 9.2 | 10.4 | 9.6 | |
| pooled plasma K2-EDTA | 6.4 | 9.4 | 8.3 | 8.0 | 6.6 | 6.7 | 6.5 | |
Results are presented for a representative transition from a proteotypic peptide. Proteins are identified by abbreviated names; peptides are identified by their first five amino acid residues. See supplement table 2 for full protein names, functions, and disease associations; and for complete peptide sequences. Results for 2–4 additional transitions are presented in supplemental table 3.
Intra-day CVs were calculated from the results for five replicate samples prepared on the same day.
Inter-day CVs were calculated from the results over five days of each replicate sample.
Total CV = ((Mean Inter-Day CV)2 + (Mean Intra-Day CV)2)1/2
Reproducibility in a highly-multiplexed SRM assay.
The automated workflow was further evaluated by linking it to a highly-multiplexed assay that targets hundreds of peptides and transitions from 70 or more plasma proteins in a single scheduled 50-minute LC run. The SRM signal intensities ranged from 1×103 to >1×107 cps (Figure 2A, left panel). On average, transitions with greater intensity had higher precision (Figure 2A, right panel). In the highly multiplexed SRM assay, 90% of the transitions had total CVs below 20%, as measured from six replicate automated sample preparations carried out on three separate days (Figure 2B).
Figure 2. Reproducibility of the automated proteomic sample preparation workflow with a highly multiplexed SRM analysis.

Six plasma aliquots were processed three times on separate days. The resulting peptides were analyzed by LC MS/MS with a 30-minute LC gradient and a scheduled SRM method targeting 72 proteins. A) Average intensities (left) and inter-day % CVs (right) of the three independent digests (see supplemental table 5). B) Results stratified by average signal intensity.
Effect of trypsin digestion time on SRM signals.
Having previously observed that the recovery of albumin and β-gal declined after two hours of trypsin digestion, the time-course analysis was extended to more proteins and peptides using the highly-multiplexed assay. The SRM signals of nearly all targeted peptides declined after 2 hours of digestion (Figure 3A). The explanation for this phenomenon was not investigated, but could be due to protease activity in the plasma, aggregation, or adherence to container surfaces. Significantly, peptides derived from the same protein often had different loss rates (Figure 3B). In the intact protein, these peptides are present in identical amounts. Thus, the difference in peptide loss rates must depend upon unique features such as the peptide sequence. This observation highlights the importance of the consistent trypsin digestion times that can be achieved in an automated workflow, and suggests that excessive trypsin digestion times may have led to excessive peptide losses in many previous studies.
Figure 3. Trypsin digestion time course.

Pooled human plasma was digested with trypsin for various times and then analyzed with the highly multiplexed SRM assay. Recoveries for each peptide were normalized to the 2-hour time-point. A) Results for 162 peptides from 70 proteins are plotted individually (left) and collectively (right). B) Peak intensity variations over time for peptides from albumin, alpha-1 antitrypsin, and alpha-1-antichymotrypsin.
Reproducibility between laboratories.
Reproducibility between laboratories has emerged as a major concern impeding wider acceptance of quantitative MS assays15. Eliminating the variability associated with manual sample processing in different environments could improve the transferability of proteomic LC-MS/MS assays. To investigate portability, an identical automated sample preparation workflow was implemented in two different laboratories. Samples were then analyzed by SRM at the same site where they were processed (online supplemental table 4). Overall, more than 93% of the 93 peptides from 44 proteins analyzed at both sites had CVs below 20% (Figure 4A)16–18. Stratification of the CV and SRM intensity data revealed that Site 2 had somewhat lower CVs (Figure 4B).
Figure 4. Inter-lab reproducibility.

The final automated processing method was implemented at Cedars Sinai Medical Center (site 1, n=24) and at SCIEX (site 2, n=48). SRM assay results are presented for 360 transitions from 93 peptides representing 44 proteins that were measured at both sites. A) % CVs for individual peptides. B) Results stratified by % CV ranges.
Reproducibility across plasma samples.
Even with an automated workflow, plasma samples from different individuals might vary in ways that effect accurate MS quantification. To evaluate matrix effects on the accuracy of MS quantification, the automated workflow was tested on 48 human plasma samples. All sample were processed on a single 96-well plate, and the resulting peptides were analyzed with the highly multiplexed SRM assay. In this analysis, data for peptides derived from β-gal that had been spiked into each sample before processing shows the technical variation, which includes effects of the sample matrix on sample processing and analysis (Supplemental Figure 2A). Five β-gal peptides had CVs ranging from 6.7% to 10.5% (Supplemental Figure 2C). Variations in the amounts of peptides derived from endogenous plasma proteins include a combination of technical variation and biological variation in protein levels (Supplemental Figure 2B). Exemplary proteotypic peptides derived from endogenous plasma proteins had CVs ranging from 12.0% to 54.1%, consistent with the expected biological variation between individuals (Supplemental Figure 2C). These results demonstrate that the analytical variance is low enough to enable measurements of the biological variation, at least for the proteins analyzed, across 48 diverse human plasma samples (24 individuals with known coronary artery disease (CAD)19 and 24 age and gender matched controls.
Peptide stability.
Peptides are susceptible to degradation, aggregation, and surface adhesion. Peptide losses during automated sample processing were estimated in six individual plasma samples by quantifying SIL peptides added at the beginning or end of the procedure. Losses for six exemplary SIL peptides ranged from 1.2% to 33.8% (Supplemental Figure 3A). For 97 SIL pepites representing 52 proteins, the mean±s.d. loss was 25.0%±14.7 (Supplemental Figure 3B and Supplemental Table 6). These measurements overestimate the amount of natural peptide loss because the SIL peptides are present throught the entire sample processing workflow whereas natural peptides are not generated until after trypsin addition. Overall, these results confirm previous observations that individual peptides are lost at different rates and should be evaluated accordingly20–21.
Analytical selectivity.
Despite the highly-selective nature of SRM, interference is possible due to the complex and variable composition of plasma. To test for interference, three to five transitions were monitored for each peptide. The best transition, generally a y ion with a high signal and a symmetrical peak, was designated as the quantifier. Other transitions were used for qualification. The relative signal intensities of the qualifier and quantifier transitions should be constant unless there are interferences22–23. Indeed, for six exemplary peptides measured in 48 plasma samples, the qualifier:quantifier ratio for at least one qualifier trasition had a CV of less than 4%. Furthermore, all of the qualifier:quantifier ratios had CVs lower than the 20% standard set by the Clinical and Laboratory Standards Institute (CLSI)24. Thus, results for the processed plasma samples were not compromised by interferences.
CONCLUSION
The automated sample preparation workflow presented in this paper increases throughput and vastly reduces the potential for errors resulting from inconsistent pipetting. When combined with online diversion for the final cleanup, the CV for the entire procedure was reduced to less than 20% for the vast majority (90%) of 572 transitions representing 182 peptides from 72 proteins. The traditional manual method requires at least 20 pipetting steps per sample, which corresponds to 1920 pipetting steps for 96 samples. Analytical errors can be introduced at every step, and are most prominent during the final cleanup (Table 1 set B). In addition, the precise timing of each liquid transfer can also be critical for consistent results, as illustrated by peptide-specific signal losses observed in a highly-multiplexed SRM assay when the trypsin digestion time was extended beyond two hours (Figure 3). Automated sample processing is essential for large scale research studies to determine clinical utility of SRM assays and ultimately in the development of routine clinical MS assays. As well, automated processing can reduce the systematic biases associated with transferring a multistep assay between laboratories, and even between technicians in the same lab, and will therefore facilitate wider future adoption of MS protein assays. This work focuses on a robust sample processing workflow, that will facilitate the development of high confidence clinical SRM assays25.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank David Harrington for providing the information on 24 post-menopausal women donors with known coronary artery disease (CAD) of Wake Forest Baptist Medical Center. Funding from the American Heart Association Challenage grant (JVE), 1R01HL111362–01A1 (JVE), and Erika Glazer endowed chair (JVE).
Abbreviations
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- SRM
selected reaction monitoring
- MMTS
Methyl methanethiosulfonate
- FA
Formic acid
- ACN
acetolnitrile
- β-gal
β-galactosidase
- CV
Coefficient of variation
- SIL peptide
Stable Isotope-Labeled Peptide
- SPE
solid phase extraction
- LC-SRM
liquid chromatograph-selected reaction monitoring
- CAD
coronary artery disease
Footnotes
SUPPORTING INFORMATION
The following files are available free of charge at ACS website http://pubs.acs.org:
REFERENCES
- 1.Anderson L; Hunter CL, Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Molecular & Cellular Proteomics 2006, 5 (4), 573–88. [DOI] [PubMed] [Google Scholar]
- 2.McDonald WH; Yates JR 3rd, Shotgun proteomics and biomarker discovery. Dis Markers 2002, 18 (2), 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toga AW; Foster I; Kesselman C; Madduri R; Chard K; Deutsch EW; Price ND; Glusman G; Heavner BD; Dinov ID; Ames J; Van Horn J; Kramer R; Hood L, Big biomedical data as the key resource for discovery science. J Am Med Inform Assoc 2015, 22 (6), 1126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domon B; Aebersold R, Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol 2010, 28 (7), 710–21. [DOI] [PubMed] [Google Scholar]
- 5.Smith RD, Mass spectrometry in biomarker applications: from untargeted discovery to targeted verification, and implications for platform convergence and clinical application. Clin Chem 2012, 58 (3), 528–30. [DOI] [PubMed] [Google Scholar]
- 6.McIntosh M; Fitzgibbon M, Biomarker validation by targeted mass spectrometry. Nat Biotechnol 2009, 27 (7), 622–3. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy JJ; Abbatiello SE; Kim K; Yan P; Whiteaker JR; Lin C; Kim JS; Zhang Y; Wang X; Ivey RG; Zhao L; Min H; Lee Y; Yu MH; Yang EG; Lee C; Wang P; Rodriguez H; Kim Y; Carr SA; Paulovich AG, Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods 2014, 11 (2), 149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruger T; Lehmann T; Rhode H, Effect of quality characteristics of single sample preparation steps in the precision and coverage of proteomic studies--a review. Anal Chim Acta 2013, 776, 1–10. [DOI] [PubMed] [Google Scholar]
- 9.Dittrich J; Becker S; Hecht M; Ceglarek U, Sample preparation strategies for targeted proteomics via proteotypic peptides in human blood using liquid chromatography tandem mass spectrometry. Proteomics Clin Appl 2015, 9 (1–2), 5–16. [DOI] [PubMed] [Google Scholar]
- 10.Addona TA; Abbatiello SE; Schilling B; Skates SJ; Mani DR; Bunk DM; Spiegelman CH; Zimmerman LJ; Ham AJ; Keshishian H; Hall SC; Allen S; Blackman RK; Borchers CH; Buck C; Cardasis HL; Cusack MP; Dodder NG; Gibson BW; Held JM; Hiltke T; Jackson A; Johansen EB; Kinsinger CR; Li J; Mesri M; Neubert TA; Niles RK; Pulsipher TC; Ransohoff D; Rodriguez H; Rudnick PA; Smith D; Tabb DL; Tegeler TJ; Variyath AM; Vega-Montoto LJ; Wahlander A; Waldemarson S; Wang M; Whiteaker JR; Zhao L; Anderson NL; Fisher SJ; Liebler DC; Paulovich AG; Regnier FE; Tempst P; Carr SA, Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol 2009, 27 (7), 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowenthal MS; Liang Y; Phinney KW; Stein SE, Quantitative bottom-up proteomics depends on digestion conditions. Anal Chem 2014, 86 (1), 551–8. [DOI] [PubMed] [Google Scholar]
- 12.van den Broek I; Romijn FP; Nouta J; van der Laarse A; Drijfhout JW; Smit NP; van der Burgt YE; Cobbaert CM, Automated Multiplex LC-MS/MS Assay for Quantifying Serum Apolipoproteins A-I, B, C-I, C-II, C-III, and E with Qualitative Apolipoprotein E Phenotyping. Clin Chem 2016, 62 (1), 188–97. [DOI] [PubMed] [Google Scholar]
- 13.Zhang YV; Rockwood A, Impact of automation on mass spectrometry. Clin Chim Acta 2015, 450, 298–303. [DOI] [PubMed] [Google Scholar]
- 14.Grant RP; Hoofnagle AN, From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin Chem 2014, 60 (7), 941–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoofnagle AN, Quantitative clinical proteomics by liquid chromatography-tandem mass spectrometry: assessing the platform. Clin Chem 2010, 56 (2), 161–4. [DOI] [PubMed] [Google Scholar]
- 16.Hunter Christie, F. Q, Kowalski Mike, Van Eyk Jennifer E. Automating Protein Digestion for Reproducible Proteomics. SCIEX Protein Digestion Automated Solution using Biomek NXP Laboratory Automated Workstation. Sciex and Beckman Coulter Life Science tech note 2016. (protein_digestion_automation_RUO-MKT-02–2364-A.pdf) [Google Scholar]
- 17.Sajic T; Liu Y; Aebersold R, Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin Appl 2015, 9 (3–4), 307–21. [DOI] [PubMed] [Google Scholar]
- 18.Croft NP; de Verteuil DA; Smith SA; Wong YC; Schittenhelm RB; Tscharke DC; Purcell AW, Simultaneous Quantification of Viral Antigen Expression Kinetics Using Data-Independent (DIA) Mass Spectrometry. Molecular & cellular proteomics : MCP 2015, 14 (5), 1361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrington DM; Reboussin DM; Brosnihan KB; Sharp PC; Shumaker SA; Snyder TE; Furberg CD; Kowalchuk GJ; Stuckey TD; Rogers WJ; Givens DH; Waters D, Effects of estrogen replacement on the progression of coronary-artery atherosclerosis. N Engl J Med 2000, 343 (8), 522–9. [DOI] [PubMed] [Google Scholar]
- 20.Shuford CM; Sederoff RR; Chiang VL; Muddiman DC, Peptide production and decay rates affect the quantitative accuracy of protein cleavage isotope dilution mass spectrometry (PC-IDMS). Molecular & cellular proteomics : MCP 2012, 11 (9), 814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant RP; Hoofnagle AN, From Lost in Translation to Paradise Found: Enabling Protein Biomarker Method Transfer Using Mass Spectrometry. Clin Chem 2014, 60 (7), 941–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kushnir MM; Rockwood AL; Roberts WL; Abraham D; Hoofnagle AN; Meikle AW, Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin Chem 2013, 59 (6), 982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kushnir MM; Rockwood AL; Nelson GJ; Yue B; Urry FM, Assessing analytical specificity in quantitative analysis using tandem mass spectrometry. Clin Biochem 2005, 38 (4), 319–27. [DOI] [PubMed] [Google Scholar]
- 24.Lynch KL, CLSI C62-A: A New Standard for Clinical Mass Spectrometry. Clin Chem 2016, 62 (1), 24–9. [DOI] [PubMed] [Google Scholar]
- 25.Fu Q; Grote E; Zhu J; Jelinek C; Kottgen A; Coresh J; Van Eyk JE, An Empirical Approach to Signature Peptide Choice for Selected Reaction Monitoring: Quantification of Uromodulin in Urine. Clin Chem 2016, 62 (1), 198–207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.