


Abstract
The DEAD-box family of proteins are ATP-dependent, RNA-binding proteins implicated in many aspects of RNA metabolism. Pre-mRNA splicing in eukaryotes requires three DEAD-box ATPases (Prp5, Prp28 and Sub2), the molecular mechanisms of which are poorly understood. Here, we use single molecule FRET (smFRET) to study the conformational dynamics of yeast Prp5. Prp5 is essential for stable association of the U2 snRNP with the intron branch site (BS) sequence during spliceosome assembly. Our data show that the Prp5 RecA-like domains undergo a large conformational rearrangement only in response to binding of both ATP and RNA. Mutations in Prp5 impact the fidelity of BS recognition and change the conformational dynamics of the RecA-like domains. We propose that BS recognition during spliceosome assembly involves a set of coordinated conformational switches among U2 snRNP components. Spontaneous toggling of Prp5 into a stable, open conformation may be important for its release from U2 and to prevent competition between Prp5 re-binding and subsequent steps in spliceosome assembly.
INTRODUCTION
DEAD-box ATPases, named after the conserved tetrapeptide ‘DEAD’ motif in their active sites, are ubiquitous in both prokaryotic and eukaryotic RNA metabolism including RNA processing. They generally possess weak helicase activity and are incapable of translocation along RNA (1,2). Nonetheless, they are essential for a wide-range of biological functions such as the melting of short RNA duplexes, remodeling of RNPs, strand annealing, or RNA binding (1). While only a few DEAD-box enzymes have been studied in biochemical depth, experiments from many laboratories have revealed that their RecA-like ATP-binding domains can exhibit a range of conformations upon crystallization (3). Many of these conformations represent ‘open’ structures in which the ATPase active site formed at the interface of the two RecA-like domains is disrupted. Formation of an ATPase-competent closed complex can be coupled with binding of RNA (1,4). Several DEAD-box proteins are also known to interact with protein cofactors that stimulate or inhibit ATP hydrolysis and RNA unwinding (5–7). In the case of the prototypical Saccharomyces cerevisiae (yeast) DEAD-box ATPase eIF4A, stimulation can occur by perturbation of open/closed conformational transitions upon interaction with the eIF4B and eIF4G cofactors (8,9).
The yeast spliceosomal DEAD-box protein Prp5 (DDX46 in humans) is required for stable association of the U2 snRNP with the intron BS (10). How Prp5 participates in this process is not clear. The ATPase activity of Prp5 is essential in the presence of the U2 snRNP protein Cus2 but not in cus2Δ yeast (11,12). This suggests that Prp5 plays a role in U2 snRNP remodeling. Biochemical and cross-linking experiments have identified a short duplex structure within the U2 snRNA, the BS stem loop (BSL), as a potential RNA binding site for Prp5 (13,14). In addition, Prp5 likely engages in protein-protein interactions with the U2 snRNP protein Hsh155 (SF3B1 in humans) (15,16).
Mutations in the Prp5 SAT sequence (conserved DEAD-box motif III) can decrease ATP hydrolysis by Prp5 in vitro and increase usage of introns containing weak BS (those with low complementarity to the U2 snRNA) in vivo (17). This finding has been interpreted as Prp5 proofreading duplex formation between U2 and the intron by coordinating the rate of pairing with an ATPase-dependent step (17). Alternatively, it has been proposed that proofreading originates from release or retention of Prp5 from the pre-spliceosome, independent of ATPase rate (13). The crystal structure of the helicase core of Prp5 in the absence of RNA revealed an unusual conformation of the RecA-like domains in which they were twisted 183° relative to one another compared to the ATP-bound state of other DEAD-box proteins (Figure 1A) (18). Toggling of Prp5 into and out of this ‘twisted open’ structure may also play a role in BS recognition by U2 (18). However, it is not known if the ‘twisted open’ structure represents the predominant conformation of Prp5 in solution or if the protein undergoes a conformational change upon RNA and ATP binding. We have used smFRET to directly observe structural dynamics of the Prp5 helicase core and their perturbation by ligands and mutations. Our results support formation of a stable ‘twisted open’ conformation in the absence of ligands. Transient closure of the RecA-like domains requires ATP and RNA. Mutations that alter BS usage in yeast change Prp5 dynamics and equilibria between the open and closed states. Our data suggest a mechanism in which a large conformational change of the Prp5 RecA-like domains is associated with U2 snRNP binding during spliceosome assembly. Spontaneous toggling of DEAD-box proteins between open and closed conformations in response to RNA and ATP binding may be used to promote progression along other multi-step reaction pathways in addition to pre-mRNA splicing.
Figure 1.
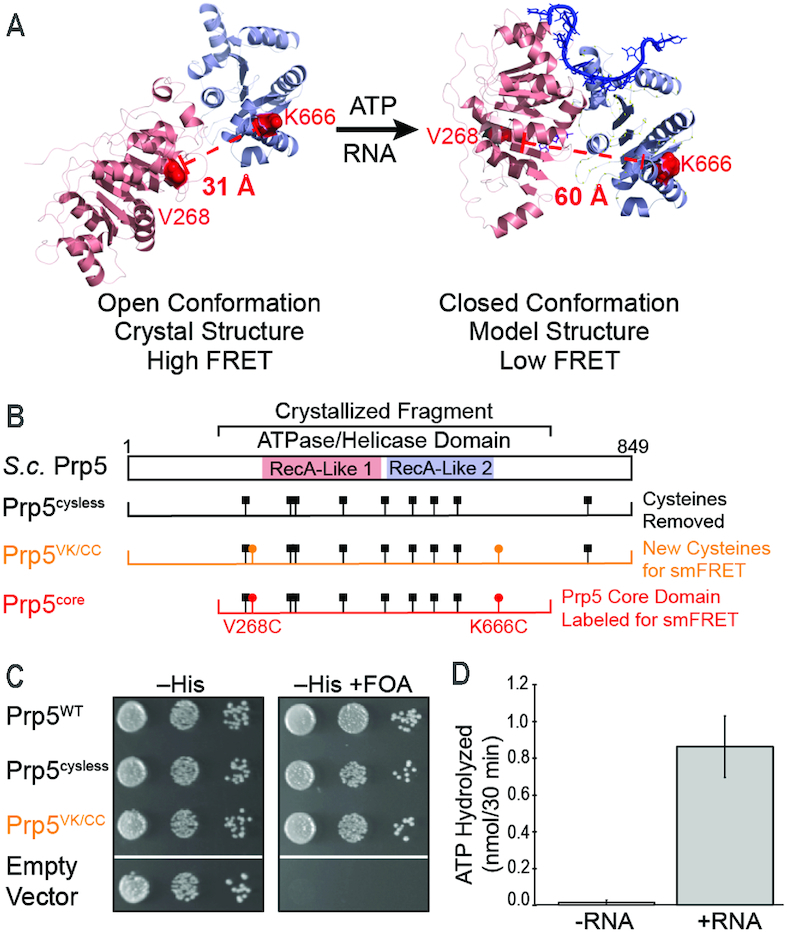
A Variant for Exploring the Conformational Dynamics of the Prp5 RecA-like Domains. (A) Structural model for conformational changes of Prp5 upon binding of ATP and RNA with predicted distances between sites of fluorophore incorporation in Prp5core noted (open structure from 4LK2.pdb; closed model courtesy of Dr Yong-Zhen Xu, Wuhan U.). (B) Mutations incorporated into Prp5 proteins and domain architecture relative to the structures shown in panel (A). (C) Prp5 mutants support yeast viability in the absence of plasmid-borne wildtype PRP5 (+FOA) at 30°C. (D) Addition of poly(A) RNA stimulates the ATPase activity of fluorophore-labeled Prp5core. ATPase assays were carried out in triplicate and the shown value represents the average ±SD.
MATERIALS AND METHODS
Yeast strains and growth
Saccharomyces cerevisiae (yeast) strains used in these studies were derived from yYZX02 [MATa ade2 cup1Δ::ura3 his3 leu2 lys2prp5Δ::loxP trp1, pR316-PRP5(PRP5 URA3 CEN ARS)], a gift from Drs. Charles Query and Yong-zhen Xu (17). Supplementary Tables S1 and S2 contain detailed lists of strains and plasmids. Yeast transformation and growth were carried out using standard techniques and media (19).
Expression, purification, and fluorophore labeling of Prp5core proteins
A fragment of Prp5 corresponding to amino acids 206–699 was cloned from yeast genomic DNA by polymerase chain reaction (PCR) and ligated into the pET28b expression plasmid using NdeI and BamHI restriction sites by standard procedures. The resulting plasmid was sequenced and ultimately coded for Prp5core variants containing a N-terminal histag for initial affinity purification, a TEV protease site for removal of the histag, and a N-terminal AP sequence for biotinylation by the Escherichia coli biotin ligase (9). A list of plasmids used in these studies can be found in Supplementary Table S1.
Recombinant proteins were produced in BL21(DE3) E. coli by co-transforming the bacteria with the pET28b plasmid expressing a Prp5core variant and a plasmid expressing E. coli biotin ligase (pAAH0947). Plasmid-containing cells were selected by growth in liquid or on solid lysogeny broth (LB) media containing 70 μg/ml kanamycin and 100 μg/ml ampicillin. Protein expression was carried out in LB (70 μg/ml kanamycin, 100 μg/ml ampicillin) by first incubating the cells at 37°C with shaking at 225 rpm until an OD600 of ∼0.6 was reached. At that time, protein production was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Cells were then incubated with shaking for an additional 16 h at 16°C. After this time, cells were collected by centrifugation and pellets stored at -80°C.
Prp5core variants were purified by first resuspending the cells in lysis buffer (50 mM BisTris pH 6.5, 500 mM KCl, 5 mM imidazole; 1 ml/g cell pellet) along with 1 U/ml DNase I (RNase Free, Thermo Scientific). Lysozyme (100 mg/ml, Sigma) was then added to a final concentration of 0.1 mg/ml. The cells were then incubated for 1 h while rotating at 23°C before sonication on ice using a microtip (40% power level; 3 s on, 2 s off; 3 cycles of 1 min on-time). The lysate was centrifuged at 18 000 × g for 15 min at 4°C in a Beckman JA-20 rotor. The supernatant was next applied to a Ni-NTA agarose (Qiagen) column (1 ml resin/5 g cell pellet) by gravity flow. The column was washed with high salt wash buffer (50 mM BisTris at pH 6.5, 500 mM KCl, 5 mM imidazole), followed by elution of the protein with imidazole (50 mM BisTris at pH 6.5, 500 mM KCl, 500 mM imidazole).
TEV protease was added to the eluate (0.21 mg TEV/1 ml protein solution, purified in lab), and the solution (3–5 ml) was placed into dialysis using a 20K MWCO Slide-A-Lyzer cassette (Thermo Fisher) for 16 h while stirring at 4°C in dialysis buffer (20 mM BisTris pH 6.5, 100 mM KCl, 10% v/v glycerol, 1 mM TCEP). Following dialysis, the protein was loaded onto a HiTrap™ SP FF cation exchange column (1 ml, GE Healthcare Life Sciences) equilibrated in low-salt buffer (20 mM BisTris pH 6.5, 100 mM KCl, 10% v/v glycerol). Pure protein was eluted using a concentration gradient of 0.1 → 1.0 M KCl over 30 min at a flow rate of 1 ml/min using a NGC™ medium-pressure chromatography system (Bio-Rad). Prp5core-containing fractions were combined, and the protein concentration was determined by measuring the absorbance at 280 nm and the calculated extinction coefficient (ProtParam, https://web.expasy.org/protparam/).
The purified protein was labeled with fluorophores by addition of Cy3 and Cy5 maleimide-derivatized dyes (dissolved in dimethylformamide, GE Healthcare Life Sciences). Typically, protein was present at a concentration of 10 μM and fluorophores were added to a final concentration of 100 μM Cy3 and 120 μM Cy5 in a final volume of 1 ml in labeling buffer (20 mM BisTris pH 6.5, 250 mM KCl, 10% v/v glycerol). This solution was allowed to incubate for 16 h at 4°C while rotating in the dark. After incubation, excess dye was removed and the protein re-purified by cation exchange chromatography as above. Labeled protein was aliquoted (5 μl), frozen in liquid N2, and stored at –80°C.
RNA fragments
An RNA oligonucleotide corresponding to nucleotides 1–90 of the yeast U2 snRNA (lacking post-transcriptional modifications) was purchased from IDT, resuspended in buffer, aliquoted, and stored at –80°C. Poly(A) RNA was purchased from Sigma. RNA concentration was determined by UV-Vis spectroscopy.
Site-directed mutagenesis
Mutations in Prp5 were generated using inverse PCR with Phusion DNA polymerase (New England Biolabs) (15), and all plasmids were confirmed by sequencing.
Preparation of samples for smFRET
Biotinylated, fluorescent Prp5 proteins were immobilized onto PEG/PEG-biotin derivatized quartz slides using streptavidin (20,21). smFRET experiments were carried out in imaging buffer, which replicates conditions that support yeast splicing in vitro and contains an oxygen scavenging system as well as triplet-state quenchers (10 mM HEPES–KOH pH 7.9, 100 mM potassium phosphate pH 7.3, 3% w/v PEG 8000, 2.5 mM MgCl2, 20 mM KCl, 1 mM cyclooctatetraene, 1 mM 4-nitrobenzyl alcohol, 1 mM propyl gallate, 1 mM trolox, 10 mM protocatechuic acid and 1 U/ml protocatechuate dioxygenase) (22–25). When present ATP, ADP or ADPNP were added to the imaging buffer at a final concentration of 1 mM, and U2 RNA was added to a final concentration of 1 μM. Poly(A) RNA was added to a concentration equivalent to 310 μM AMP.
smFRET data acquisition and analysis
smFRET data were collected on a home-built, prism-based total internal reflection fluorescence (TIRF) microscope with laser excitation at 532 and 640 nm (20,26). Excitation by the 640 nm laser (22.5 mW, measured prior to entry into the prism) was used to localize the labeled Prp5 at the beginning and the end of each experiment. The 532 nm laser (10 mW, measured prior to entry into the prism) was used to detect FRET. Data were recorded continuously with a frame rate of 100 ms/frame for a time span of 100 s. Donor and acceptor emission fluorescence were collected simultaneously using a DualView apparatus (DV2, Photometrics) and an EM-CCD camera (Andor). Images were recorded using Metamorph software (Molecular Devices).
Raw images were analyzed using custom MATLAB (Mathworks) software as described (26–28). Briefly, integrated fluorescence intensities for donor (ICy3) and acceptor (ICy5) spots of fluorescence were used to calculate apparent FRET efficiencies using the formula, EFRET = ICy5/(ICy3 + ICy5) along with vbFRET software (29). Transition events and the EFRET values during which they occurred were identified and HaMMy software (30). Lifetimes of the observed EFRET states were determined using maximum likelihood methods and fit to equations containing a single exponential term as described (31) except that fitting was facilitated using AGATHA software developed by our laboratory (https://github.com/hoskinslab/AGATHA). Errors in the fits were determined by bootstrapping (22,27), also implemented in AGATHA (32). Histograms of EFRET values were fit to Gaussian functions using MATLAB.
Temperature sensitivity and FOA-Selection assays
Yeast were grown to mid-log phase, adjusted to OD600 = 0.5 and plated on YPD media in ten-fold serial dilutions. Plates were incubated for 2 days at 30°C or 10 days at 16°C prior to imaging. FOA selection assays were performed in a similar manner, except yeast were grown in liquid media (-His) and plated on solid media (-His) to maintain plasmid selection. When present, FOA (1 mg/ml) was used to select for URA3 loss. Images were taken after 2 days at 30°C.
ACT1-CUP1 copper assays
ACT1-CUP1 reporter 2μ plasmids all contained the LEU2 selection marker and growth assays have been described previously (14,41). Briefly, yeast strains expressing WT or mutant Prp5 proteins and ACT1-CUP1 reporters were grown to mid-log phase in -Leu -His dropout media to maintain selection for the plasmids, adjusted to OD600 = 0.5 using fresh media, and equal volumes were spotted using a 48-pin inoculator onto -Leu -His dropout plates containing 0, 0.025, 0.05, 0.075, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.25 or 2.5 mM CuSO4. Plates were scored for the presence or absence of growth after 3 days at 30°C.
ATPase assay
ATP hydrolysis by Prp5 proteins was determined as described using a charcoal-binding assay (33) except that buffers used matched those in smFRET assays (10 mM HEPES–KOH pH 7.9, 100 mM potassium phosphate pH 7.3, 20 mM KCl, 3% w/v PEG 8000, 2.5 mM MgCl2). Each reaction contained 645 nM labeled Prp5core (WT or variants), 1 mM ATP, and 50 nCi/μl γ-[32P] ATP (Perkin Elmer). 10 μM U2 snRNA (1–90) or 5 μg polyA (equivalent to 310 μM AMP, Sigma) were included in each reaction as noted. Activated charcoal was prepared and resuspended in 20 mM phosphoric acid (34). Reactions (20 μl) were incubated at room temperature for 90 min followed by the addition of 250 μl charcoal suspension. The reactions were chilled on ice for 10 min and then centrifuged to pellet the charcoal. Free [32P]-phosphate was quantified using a scintillation counter.
RESULTS
Fluorophore labeling of the Prp5 RecA-like domains
To create single-molecule probes of Prp5 interdomain distances, we incorporated fluorophores into the helicase core domain of Prp5 at positions in which they are predicted to have high FRET when the protein is in the ‘twisted open’ conformation and lower FRET based on a model of the ATPase-active, closed structure (Figure 1A) (18). First, we constructed a prp5 gene encoding a functional protein lacking cysteines capable of complementing a prp5Δ yeast strain at 30°C (Prp5cysless, Figure 1B, C; Supplementary Tables S1–S3). We then replaced V268 and K666 with cysteines for fluorophore attachment (Figure 1A). This derivative also complemented the prp5Δ strain (Prp5VK/CC, Figure 1B and C).
We expressed and purified from E. coli a fragment of Prp5VK/CC containing the central RecA-like domains and flanking sequences corresponding to the fragment of Prp5 that was crystallized (Prp5core, Figure 1B; Supplementary Figure S1A–C and Table S3) (18). Prp5core also included a genetically encoded biotin acceptor peptide (AP) at the N-terminus of the protein. Co-expression with E. coli biotin ligase resulted in significant biotinylation of the protein (Supplementary Figure S1C), which was used to anchor the protein to microscope slides. Purified Prp5core was labeled with a mixture of Cy3- and Cy5-maleimide, and excess dyes were removed by chromatography (Supplementary Figure S1D). Consistent with previous studies of Prp5 (35), labeled Prp5core possessed very little ATPase activity in the absence of RNA (Figure 1D). When poly(A) RNA was added (310 μM AMP equivalents, ∼30-fold higher than reported Km values for Prp5 for this substrate (18)), the ATP hydrolysis activity was activated (Figure 1D). Thus, labeled Prp5core is an RNA-stimulated ATPase.
smFRET supports formation of an open and stable conformation of Prp5core
We attached biotinylated and Cy3/Cy5-labeled Prp5core to biotin PEG-passivated quartz microscope slides with streptavidin and visualized single Prp5core molecules using a prism-type total internal reflection fluorescence (TIRF) microscope (Figure 2A) (20). We analyzed molecules likely containing both Cy3 and Cy5 fluorophores by illuminating the sample with a 640 nm laser to excite Cy5 and identify molecules containing at least one Cy5 fluorophore. We then monitored smFRET by illumination with a 532nm laser to excite Cy3. Post-acquisition inspection of Cy3- and Cy5-emission and photobleaching from each Cy5-containing molecule identified those containing both fluorophores. For example, molecules containing both Cy3 and Cy5 show an increase in Cy3 fluorescence upon Cy5 photobleaching when illuminated at 532 nm.
Figure 2.
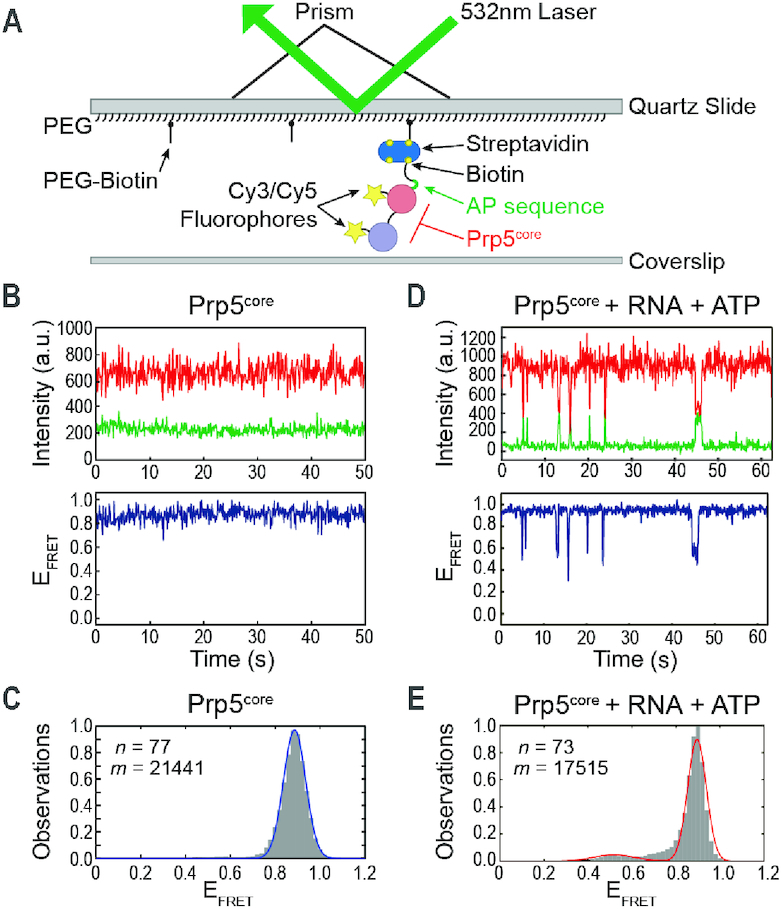
Conformational Switching of Prp5core Requires ATP and RNA. (A) Schematic of the smFRET assay. (B) Top: Cy3 (green) and Cy5 (red) fluorescence trajectories from a single molecule of Prp5core. Bottom: Calculated EFRET from the shown trajectories. (C) Histogram of EFRET values obtained for Prp5core (grey boxes) and a fit of the data (blue line). (D) Data from a single molecule of Prp5core as in panel (B) except in the presence of poly(A) RNA and ATP. (E) Histogram of EFRET values obtained for Prp5core in the presence of poly(A) RNA and ATP as in panel (C) and a fit of the data (red line). n and m represent the number of individual molecules and EFRET data points, respectively, used to construct the histogram. Data fit results are listed in Supplementary Table S4.
Prp5core molecules were uniformly static, exhibiting a stable FRET efficiency (EFRET) of 0.8–0.9 (Figure 2B). Since we failed to observe transitions, this suggests that conformational changes are either very infrequent or that the lifetime of any alternate conformation is much less than our time resolution of 100 ms. Histogram analysis of Prp5core revealed a single peak, which could be fit to a Gaussian distribution centered at EFRET = 0.89 (Figure 2C, Supplementary Table S4). Assuming R0 = 0.54 Å (36), this value yields an approximate distance of 38 Å compared to a 31 Å Cα–Cα distance between V268 and K666 observed in the ‘twisted open’ structure. These results are also in agreement with previous studies of the bacterial YxiN and yeast eIF4A DEAD-box proteins, which both showed unimodal distributions of smFRET in the absence of ligands consistent with the RecA-like domains being open (8,37). Our data indicate that Prp5core exhibits a homogenous, stable conformation in solution that likely corresponds to an open form similar to that observed by crystallography.
RNA and ATP induce transient closure of Prp5core
We next tested the impact of ligands on Prp5core conformation. In the presence of either ATP or poly(A) RNA individually, Prp5core molecules remained static and EFRET distributions continued to show only a single peak near EFRET = 0.88 (Supplementary Figure S2A–D, Table S4). When poly(A) RNA and ATP were simultaneously added, molecules transitioned from high EFRET to a shorter-lived conformation with lower EFRET (Figure 2D). Histogram analysis revealed the appearance of a second, less-populated peak of EFRET at 0.52. The requirement for ATP and RNA to induce Prp5core conformational change is again very similar to observations made with YxiN and eIF4A, both of which require cooperative binding of ATP and RNA to initiate closure of the RecA-like domains (4,8,37,38).
We were able to fit distributions of dwell times for Prp5core in both EFRET states to maximum likelihood equations containing a single exponential term. This yielded characteristic lifetimes for the open (τopen) and closed (τclosed) states of 3.97 ± 0.31 and 0.46 ± 0.04 s, respectively (Supplementary Table S5). These lifetimes are similar to those reported for eIF4A, albeit only in the presence of protein cofactors that induce formation of an alternate half-open conformation of the ATPase (8). Our data are consistent with Prp5core reversibly switching directly between a closed and open state. These transitions require RNA and ATP but, unlike eIF4A, are not dependent on the presence of other protein cofactors or formation of any detectable intermediate structure.
We tested whether or not a fragment of the U2 snRNA (nt 1–90) containing the proposed Prp5 binding site (13) could also trigger Prp5core ATPase and conformational switching. This RNA was able to stimulate both activities, and Prp5core exhibits similar changes in smFRET when exposed to either the poly(A) RNA or U2 fragment (Supplementary Figure S3). For both RNAs under these conditions, the lifetime of the closed conformation is much shorter than that of the open conformation (Supplementary Table S4). Previous work suggested that Prp5 exhibits a substrate preference for the U2 snRNA (35). While we did not test the substrate preference of Prp5core, conformational switching appears to be a general feature of Prp5core in the presence of RNA and ATP.
To further test whether changes in smFRET observed with Prp5core correspond to the proposed open/closed structural transitions (Figure 1A), we constructed an orthogonal reporter in which the protein would exhibit low FRET when open and high FRET when closed (Supplementary Figure S4A). A gene encoding Prp5 with cysteines only at T430C and K543C was also able to complement prp5Δ yeast (Prp5TK/CC, Supplementary Figure S4B and C). The corresponding Prp5 fragment (Prp5core2) exhibited a static distribution of low EFRET in the absence of ligands (Supplementary Figure S4D and E). The fitted EFRET value of 0.37 (Supplementary Table S4) corresponds to an approximate distance of 59 Å, close to the predicted value of 66 Å obtained from the ‘twisted open’ crystal structure. In the presence of U2 RNA and ATP, Prp5core2 also spontaneously switched between low and high EFRET likely corresponding open and closed states, respectively (Supplementary Figure S4F). As with Prp5core, the closed conformation of Prp5core2 was much less abundant than open state (Supplementary Figure S4G). Together data obtained with Prp5core and Prp5core2 support a stable conformation of Prp5 in solution consistent with the ‘twisted open’ crystal structure. Formation of a closed structure is transient under these conditions and requires the presence of both RNA and ATP.
Closure of Prp5core is reversible in the presence of ADPNP
To test the dependence of the closed conformation on ATP, we incubated Prp5core with poly(A) RNA and ADP. Prp5core failed to exhibit any signs of conformational switching in the presence of ADP and only a single, static smFRET state was observed (Supplementary Figure S5A and B). This indicates that ADP cannot substitute for ATP for triggering closed state formation and is consistent with the open structure of Prp5 bound to ADP (18). Non- or slowly hydrolyzable ATP analogs such as ADPNP have been used to trap several DEAD-box proteins in closed conformations for biophysical or structural studies (39–43). We also tested whether ADPNP could similarly trap Prp5core in a closed state by incubating the protein with the analog and U2 RNA and observing changes in smFRET. As expected, the lifetime of the low EFRET (closed) conformation of Prp5core was extended by ADPNP (Supplementary Figure S5C and Table S5). However, individual molecules of the protein still reversibly switched between higher and lower EFRET values. The open conformation was still the predominantly observed species, and it exhibited an ∼3-fold longer lifetime than the closed complex (Supplementary Figure S5D and Table S5). Reversible conformational changes in the presence of ADPNP have previously been reported with yeast eIF4A and are distinct from observations with YxiN, which could be trapped in the closed state (9,43). In the case of eIF4A, this has been interpreted as a less stable closed conformation relative to other DEAD-box family members and failure of ADPNP to provide enough binding energy to fully couple with protein conformational change (9). Prp5, eIF4A, and other DEAD-box proteins may share these properties.
A mutation that decreases weak BS usage also destabilizes the open conformation
The Prp5 E235A mutation has been proposed to disrupt the protein interface that stabilizes the ‘twisted open’ structure (18). This mutation also results in increased ATPase catalytic efficiency (kcat/Km) in vitro and decreased in vivo usage of BS containing mismatches to U2 snRNA (18). To directly test if E235A alters Prp5 conformation, we first incorporated E235A into Prp5VK/CC in yeast to confirm that the mutation changes usage of weak BS in this background. We tested BS usage by monitoring Cu2+-resistance in yeast containing the mutation along with the well-characterized ACT1-CUP1 splicing reporter (Figure 3A) (44). In this assay, increased Cu2+-tolerance correlates with increased splicing of the reporter gene in vivo. As expected, Prp5, Prp5-E235A, Prp5VK/CC and Prp5VK/CC-E235A exhibited similar Cu2+-tolerances when using an ACT1-CUP1 reporter containing a consensus BS (Figure 3B). The E235A mutation resulted in decreased Cu2+ tolerance compared to Prp5WT or Prp5VK/CC when reporters were used containing the U257C weak BS consistent with decreased BS usage and spliced product formation. However, this effect was mitigated when the A258U reporter was used (Figure 3D) suggesting that phenotypes of at least some Prp5 mutants are dependent on the reporter RNA being used as well as the parental Prp5 protein sequence (Prp5WT or Prp5VK/CC).
Figure 3.
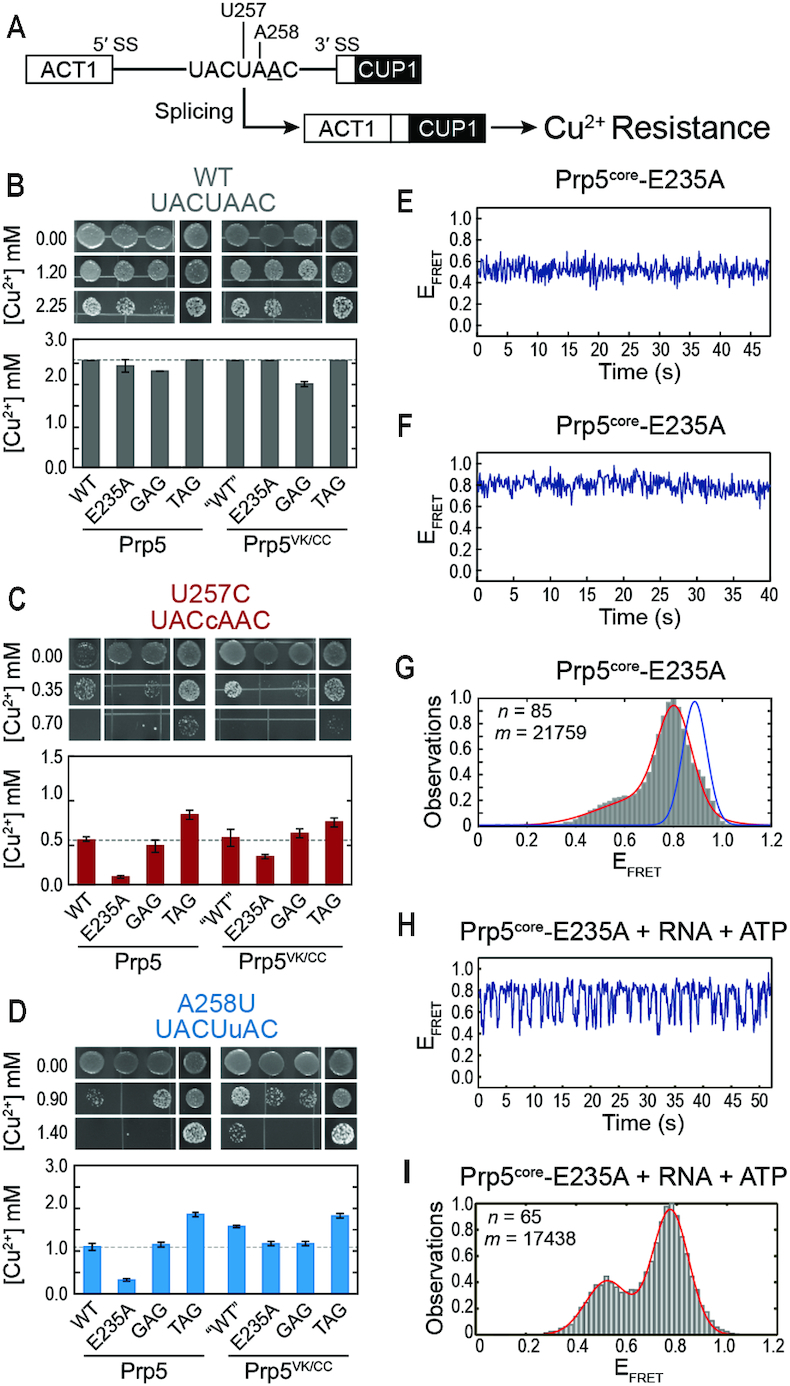
The Prp5-E235A substitution increases splicing fidelity in yeast and conformational dynamics of Prp5core. (A) Schematic of the ACT1-CUP1 reporter pre-mRNA and assay. The BP-A is underlined, and positions of BS mutations are noted. (B–D) Results from Cu2+ growth assays of strains containing the indicated Prp5 mutations and either the consensus, U257C, or A258U ACT1-CUP1 reporter plasmids. Results are the average of quadruplicate experiments ± the standard deviation. (E, F) Representative EFRET trajectories for two molecules of Prp5core-E235A (G) Histogram of EFRET values obtained for Prp5core-E235A (grey boxes) and a fit of the data (red line). Blue line is from Figure 2C. (H) Representative calculated EFRET trajectory for a single molecule of Prp5core-E235A in the presence of poly(A) RNA and ATP. (I) Histogram of EFRET values obtained in the presence of poly(A) RNA and ATP as in panel (G). n and m represent the number of individual molecules and EFRET data points, respectively, used to construct the histogram. Data fit results are listed in Supplementary Table S4.
We incorporated the E235A mutation into Prp5core and then purified and labeled the protein. The E235A mutation significantly increased the ATPase rate of Prp5core (Figure 4D). We carried out smFRET experiments in the absence and presence of RNA and ATP. Molecules of Prp5core-E235A were static in the absence of ligands (Figure 3E and F). However, the distribution of smFRET signals was bimodal with peaks near EFRET = 0.80 and 0.53 (Figure 3G). Inspection of individual smFRET trajectories revealed that while the molecules of Prp5core-E235A were not transitioning, they were apparently locked into one of two conformations corresponding to the peaks in EFRET. Most molecules (89%) occupied the higher EFRET conformation while the remainder (11%) were found to be in the lower EFRET state. Molecules of Prp5core-E235A reversibly switched between these two states once poly(A) RNA and ATP were added (Figure 3H and I). These dynamic molecules were kinetically homogenous, meaning that we were unable to detect sub-populations that could have arisen from the two static populations observed in the absence of ligands. The E235A mutation still permits formation of open and closed structures of Prp5core; however, it also introduces conformational heterogeneity in the absence of added ligands.
Figure 4.
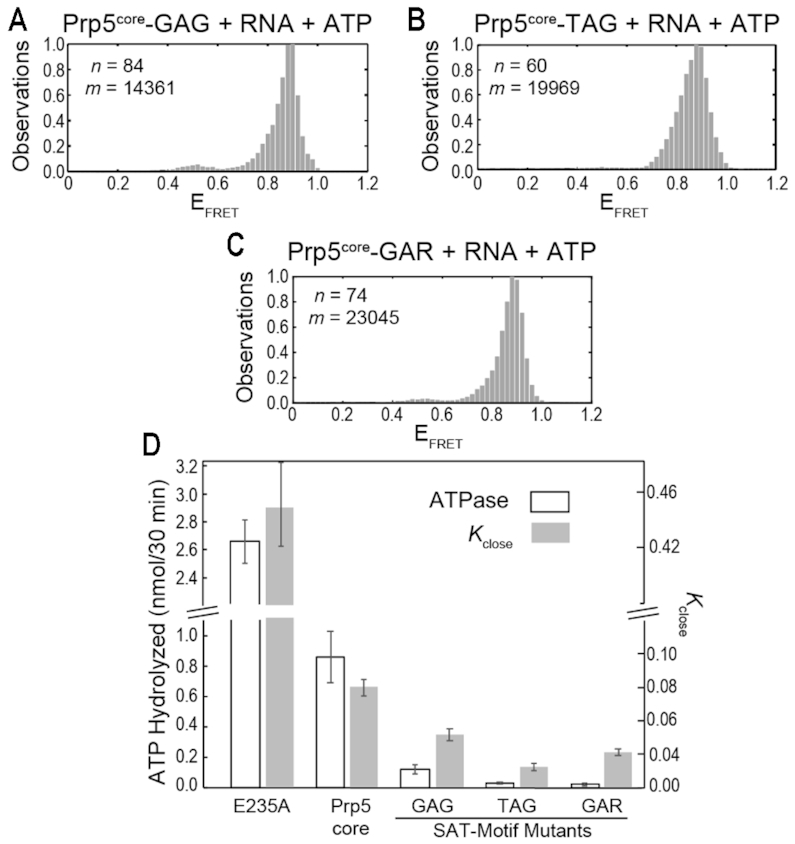
Impact of E235A and SAT-motif Mutations on Prp5core Dynamics. (A–C) Histograms of EFRET values obtained for Prp5core-GAG (A), -TAG (B) and -GAR (C) in the presence of poly(A) RNA and ATP (gray boxes). n and m represent the number of individual molecules and EFRET data points, respectively, used to construct the histogram. (D) Calculated Kclose values and ATPase rates in the presence of poly(A) RNA obtained for Prp5core and the indicated mutants. Histogram fit results, calculated lifetimes, and Kclose values are listed in Supplementary Table S5. ATPase assays were carried out in triplicate and the shown value represents the average ±SD.
Kinetic analysis of ATP- and RNA-stimulated conformational switching of Prp5core-E235A revealed that the mutation destabilized τopen ∼4-fold, while τclose remained unchanged. This agrees with the structure of Prp5 in which E235A alters the domain interface involved in stabilization of the ‘twisted open’ conformation but is not part of the proposed interface formed upon domain closure (18). The increased ATPase catalytic efficiency of Prp5-E235A is largely due to a decrease in the Km for RNA (18). Destabilization of the open conformation could allow for more efficient coupling between RNA binding and closing of the RecA-like domains.
SAT-motif mutants increase weak BS usage but decrease Prp5 dynamics
While the E235A mutation decreases use of weak BS in yeast, several mutations of the DEAD-box SAT motif are known to do the opposite. Mutation of the SAT sequence (Prp5 amino acids 446–448) to TAG or GAG results in increased use of weak BS in vivo as well as decreased rates of ATP hydrolysis in vitro (17). Mutation of this motif to GAR has a more dramatic effect and results in cold sensitivity (cs) and a >95% decrease in ATPase activity (17). These results are consistent with the amino acids of motif III helping to structure the ATP binding cleft formed by the two RecA-like domains in a catalytically competent conformation (1,17,18).
In ACT1-CUP1 assays, yeast expressing the GAG or TAG mutants of Prp5WT or Prp5VK/CC grew similarly to the corresponding parental strain in the presence of a consensus BS reporter (Figure 3B). The TAG mutation increased Cu2+-tolerance of yeast containing either the U257C or A258U reporters in both Prp5WT and Prp5VK/CC backgrounds (Figure 3C and D). Previous studies reported that the GAG mutant causes a slight growth defect in the presence of the WT ACT1-CUP1 reporter and improves growth of yeast relative to WT in the presence of the U257C ACT1-CUP1 reporter, albeit to a lesser extent than the TAG mutant. Using Prp5VK/CC, we were also able to observe the growth defect of the GAG mutant with the WT reporter (Figure 3B); however, we could not observe a significant change in U257C splicing (Figure 3C). Yeast containing the GAR mutation were cs in both Prp5WT and Prp5VK/CC (Supplementary Figure S6). The growth defects of strains with GAR mutations prevent direct comparisons with the GAG and TAG reporters in ACT1-CUP1 assays.
We purified and labeled Prp5core GAG, TAG and GAR mutants. ATPase assays confirmed that the mutants decreased the RNA-stimulated ATPase activity. The GAG mutation had the least impact on activity while the GAR mutation had the greatest. Our ATPase assay results with Prp5core differ from those reported for the full-length protein. For full-length Prp5, the TAG mutant reduced Prp5 ATPase activity by ∼2-fold and was more active than the corresponding GAG mutant (17). For labeled Prp5core, the TAG mutant reduces ATPase activity ∼20-fold and is less active than the GAG mutant. However, our in vivo ACT1-CUP1 assays (Figure 3) agree with previous reports that the TAG mutant increases splicing of RNAs with nonconsensus BS more so than the GAG mutation (17). Thus, in our experiments the TAG mutants have both a more reduced ATPase activity and greater stimulation of splicing using nonconsensus BS when compared to WT or GAG mutant proteins.
We next used smFRET to test whether or not changes in ATPase activity due to SAT-motif mutations also reflect changes in protein conformational dynamics. In the absence of ligands, the GAG, TAG, and GAR mutants showed similar distributions of EFRET compared to Prp5core containing the native SAT-motif (Supplementary Figures S7 and S8). Like Prp5core, molecules containing these mutations were static with EFRET values close to 0.89. The GAR and GAG mutations resulted in slight broadening of histogram distributions towards lower EFRET values (Supplementary Figure S8A and C); however, we were unable to fit these data to more than one Gaussian function. The SAT-motif mutations appeared to have little impact on the open structure of Prp5 consistent with observations that the SAT-motif is involved in binding of ATP at the domain interface after conformational rearrangement.
Each of the Prp5core SAT-motif mutants transitioned between two EFRET states in the presence of ATP and poly(A) RNA (Supplementary Figure S7). However, histogram analysis showed much less accumulation of the lower, closed EFRET state for the mutants relative to Prp5core (Figure 4A–C). Prp5core-TAG and GAR, in particular, showed very little occupancy of this state, and transitions were both infrequent and very short-lived. Differences in proteins due to SAT-motif mutation were much more apparent in the presence of ligands than in their absence, also in agreement with their location near the RecA-like domain interface.
By analyzing τopen and τclose for each Prp5core mutant (Supplementary Table S5), we were able to calculate the equilibrium constants of closing (Kclose) (Figure 4D). At identical concentrations of RNA and ATP, it is apparent that the E235A mutant favors the closed conformation much more than Prp5core or the SAT-motif mutants. The increase in Kclose for the E235A mutation largely stems from a shorter τopen relative to WT (1.09 versus 3.97 s, respectively; Supplementary Table S5). All of the SAT-motif mutations, on the other hand, decreased Kclose as well as the lifetime of the closed state relative to WT. The same trend was observed with both the poly(A) and U2 RNA substrates (Supplementary Figure S9, Table S5). For the TAG and GAR mutants, the decrease in the closed state lifetime was accompanied by an increase in the lifetime of the open state (Supplementary Table S5). Together the data indicate that mutation of the SAT motif can destabilize the closed conformation of Prp5 as well as making it less likely for that closed state to form.
Finally, we compared Kclose values for Prp5core proteins with ATPase measurements acquired at the same concentrations of ATP and poly(A) RNA (Figure 4D). For the E235A mutation, both the ATPase rate and Kclose increased ∼3-fold. However, all of the SAT-motif mutants showed larger-fold changes in ATPase activity than in Kclose. We cannot determine whether or not any one particular FRET transition is associated with hydrolysis of one ATP molecule. However, it is possible that the differences in the magnitude of effects on ATPase and Kclose arise from decoupling between conformational change and ATP hydrolysis. In other words, SAT mutants may toggle conformation without successfully hydrolyzing ATP. This would result in the smaller changes in Kclose relative to ATPase that we observe. In support of this, we note that the GAR mutant has a larger Kclose than the TAG mutant (0.042 vs. 0.024, respectively) despite having a 30% lower ATPase activity.
DISCUSSION
By engineering a version of the yeast DEAD-box Prp5 containing only two cysteines within the ATPase core, we have been able to use smFRET to directly visualize and quantify the structural dynamics of the RecA-like domains. The yeast splicing machinery requires eight well-conserved ATPases for function, three of which are DEAD-box proteins (45). A number of mutations in these ATPases impact splicing fidelity in cells, presumably by altering spliceosome structure or regulating entry into discard pathways leading to spliceosome disassembly (46,47). To our knowledge, this study represents the first real-time analysis of conformational dynamics for any spliceosomal ATPase.
Prp5 switching is ligand dependent and reversible
Our results provide direct evidence for the Prp5 RecA domains undergoing a large-scale, two-state conformational change upon ligand binding. This switch is triggered only by binding of RNA and ATP (or an ATP analog) (Figure 2). Once Prp5 closes, it can spontaneously re-form the open conformation with FRET values similar to those observed in the absence of ligands. This suggests that if Prp5 acts as a conformational switch turned ‘on’ by ligand binding then it can also reversibly transition back to the same ‘off’ conformation observed in the apo enzyme.
We also obtained information about the relative lifetimes of the closed and open states and how these are impacted by mutations. The E235A mutation was previously proposed to destabilize a protein-protein interface in the open state of Prp5, which in turn stimulates ATP hydrolysis. Our smFRET work supports these conclusions and shows that the primary effect of this mutation is on decreasing the open state lifetime without impacting that of the closed state. The impacts of the SAT-motif mutants are more variable. Each of them is able to decrease the lifetime of the closed conformation while TAG and GAR also increase the lifetime of the open conformation. Together our data indicate that Prp5 mutations can potentially have different effects during splicing and at different stages. For example, SAT-motif mutants could inhibit steps during which Prp5 must transition from the open to closed state and/or promote steps in which Prp5 must transition from closed to open. We have not yet deconvoluted how one or all of these activities contribute to increased splicing of pre-mRNAs containing weak BS.
A series of molecular switches for U2 activation and association
Association of the U2 snRNP with the BS is one of the least well-understood steps in spliceosome assembly in part because it involves multiple, coordinated changes in U2 conformation and composition. In addition to the toggling of Prp5 studied here, structural and biochemical data indicate that the large scaffold protein Hsh155 (SF3B1 in humans) must close to dock the U2/BS duplex into its RNA binding cleft (15,48–51). The stem II region of the U2 snRNA must also be readied for spliceosome assembly by adopting the IIa conformation, a process which is stimulated by Cus2 (11,26,52–54). The ATPase activity of Prp5 is dispensable in the absence of Cus2 (12), implying that Prp5 might also promote Cus2 release during this stage (11). Intriguingly, all of these factors are concentrated in one location on the snRNP: Hsh155 contains binding sites for Cus2 (11), Prp5 (15,16), and the U2/BS duplex (55,56). Correct association of U2 with a BS may require multiple conformational switches to be correctly activated on or near Hsh155.
A role for Prp5 conformational switching during splicing
It is likely that protein-protein interactions between Prp5 and Hsh155 (15,16) help localize Prp5 near the U2 snRNA and its site of switching. This may provide a high local concentration of RNA and/or favorable interactions to help Prp5 efficiently toggle to the closed conformation during assembly. During these stages, Prp5 is also known to play a role in BS fidelity and two competing models have been proposed based on ATP hydrolysis activity (17) or affinity of Prp5 for the pre-spliceosome (13). The reversible toggling that we observe between open and closed states could be accommodated by both of these models. It is possible that the open and closed forms of Prp5 have differing affinities for the pre-spliceosome and that spontaneous transitioning from the closed to the open conformation is needed to drive Prp5 release (Figure 5). While ATP is needed to trigger conformational change, our model would suggest that Prp5 conformation rather than ATPase activity per se is the critical determinant of release or retention from the spliceosome. The Prp5 SAT-motif mutants may promote release and splicing by destabilization of the closed complex, while the E235A mutation could inhibit release and splicing by destabilization of the open complex (Figure 4D).
Figure 5.
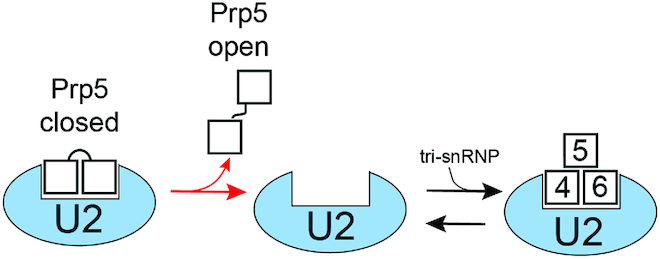
Potential Role for Prp5 Conformational Switching during Spliceosome Assembly. Release of Prp5 from U2 could be accompanied by spontaneous transition from the closed to the open conformation of the Prp5 RecA domains. Since the open conformation is stable in the absence of both RNA and ATP, this could render Prp5 release effectively irreversible (red forward arrow) and facilitate subsequent tri-snRNP binding. For simplicity, we have shown Prp5 and the tri-snRNP with overlapping binding sites on U2. While it is known that Prp5 blocks tri-snRNP addition (13), a structure of Prp5 bound to U2 has not yet been determined, nor is it known if the U2 snRNP undergoes conformational change after Prp5 release.
We also note that spontaneous opening of Prp5 (Figure 2D) during or after its release from U2 can provide an irreversible step during spliceosome assembly (Figure 5). Retention of Prp5 by U2 blocks tri-snRNP addition (13). Spontaneous formation of the conformationally stable (Figure 2B), open Prp5 structure could prevent Prp5 re-association with U2 and interference with tri-snRNP binding. The kinetic pathway for Prp5 closing/opening, ATP hydrolysis, Cus2 release, and U2/BS duplex formation has not yet been determined. Ultimately, the structural and kinetic illumination of this pathway will be important for distinguishing between these and alternate models.
Dynamic properties of DEAD-box proteins
While significant insights have been made into how DEAD-box ATPases bind RNA and promote duplex unwinding (1,2), much less is known about how these activities can be regulated either through the presence or absence of cofactors or by conformational changes. X-ray crystallographic structures of several DEAD-box proteins have revealed a wide range of conformations in the absence of ligands. It has been speculated that this might be an artifact of crystal-packing resulting in isolation of one particular conformation of an otherwise flexible protein in solution (3). Our single-molecule results suggest that this is not the case for Prp5core, which exhibits a single, static EFRET state in solution (Figure 2) consistent with the ‘twisted open’ conformation observed by crystallography (18).
Prp5, YxiN and eIF4A all possess stable open conformations that fail to undergo domain closure in the absence of ligands (8,38,43). We were not able to find evidence for a half-open conformation of Prp5core similar to that previously seen in eIF4A (9). This indicates that formation of an intermediate state with a detectable EFRET signature is not obligatory for domain closure. Both Prp5core and eIF4A are unable to form stable ternary complexes with RNA and ADPNP—consistent with instability of the closed conformations and unlike the stable complexes reported for YxiN (9,43). We speculate that different DEAD-box proteins may fall into distinct functional classes dependent on the stabilities of their open and closed states (4). Some, like eIF4A and Prp5, could depend on relatively unstable closed conformations to limit unproductive RNA binding or ATP hydrolysis. Such DEAD-box proteins could also couple large-scale conformational changes to other events in order to drive multi-step reactions like pre-mRNA splicing forward.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charles Query, Yong-Zhen Xu, and Margaret Rodgers for insightful discussions, manuscript comments, and providing the yeast Prp5 plasmids and coordinates for the modeled Prp5 closed state structure. We thank David Brow, Samuel Butcher, James Keck and Yong-Zhen Xu for their comments on the manuscript.
Author contributions: D.H.B., T.J.C., C.vdF., U.S.T. and A.A.H. designed experiments. D.H.B., T.J.C., C.vdF., J.C.P., N.G. and A.A.H. prepared reagents. D.H.B., T.J.C., C.vdF. and A.A.H. collected and analyzed data. A.A.H. supervised the work and wrote the manuscript with D.H.B. along with input from all authors.
Notes
Present address: Tucker J. Carrocci, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06510, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01 GM112735 to A.A.H., T32-GM08349 to T.J.C.]; a Shaw Scientist Award from the Greater Milwaukee Foundation; a Beckman Young Investigator Award from the Arnold and Mabel Beckman Foundation; startup funding from the University of Wisconsin-Madison, Wisconsin Alumni Research Foundation (WARF); Department of Biochemistry; a Summer Undergraduate Research Fellowship in Biochemistry (DHB); a William H. Peterson Fellowship (to T.J.C.); University of Wisconsin-Madison Biophysics Instrumentation Facility (BIF) for instrument access. Funding for open access charge: NIH R01 GM112735.
Conflict of interest statement. None declared.
REFERENCES
- 1. Putnam A.A., Jankowsky E.. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta. 2013; 1829:884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rudolph M.G., Klostermeier D.. When core competence is not enough: functional interplay of the DEAD-box helicase core with ancillary domains and auxiliary factors in RNA binding and unwinding. Biol. Chem. 2015; 396:849–865. [DOI] [PubMed] [Google Scholar]
- 3. Möhlmann S., Mathew R., Neumann P., Schmitt A., Lührmann R., Ficner R.. Structural and functional analysis of the human spliceosomal DEAD-box helicase Prp28. Acta Crystallogr. D Biol. Crystallogr. 2014; 70:1622–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Samatanga B., Klostermeier D.. DEAD-box RNA helicase domains exhibit a continuum between complete functional independence and high thermodynamic coupling in nucleotide and RNA duplex recognition. Nucleic Acids Res. 2014; 42:10644–10654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rozovsky N., Butterworth A.C., Moore M.J.. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA. 2008; 14:2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noble C.G., Song H.. MLN51 stimulates the RNA-helicase activity of eIF4AIII. PLoS ONE. 2007; 2:e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ballut L., Marchadier B., Baguet A., Tomasetto C., Séraphin B., Le Hir H.. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat. Struct. Mol. Biol. 2005; 12:861–869. [DOI] [PubMed] [Google Scholar]
- 8. Andreou A.Z., Klostermeier D.. eIF4B and eIF4G jointly stimulate eIF4A ATPase and unwinding activities by modulation of the eIF4A conformational cycle. J. Mol. Biol. 2014; 426:51–61. [DOI] [PubMed] [Google Scholar]
- 9. Harms U., Andreou A.Z., Gubaev A., Klostermeier D.. eIF4B, eIF4G and RNA regulate eIF4A activity in translation initiation by modulating the eIF4A conformational cycle. Nucleic Acids Res. 2014; 42:7911–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dalbadie-McFarland G., Abelson J.. PRP5: a helicase-like protein required for mRNA splicing in yeast. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:4236–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Talkish J., Igel H., Hunter O., Horner S.W., Jeffery N.N., Leach J.R., Jenkins J.L., Kielkopf C.L., Ares M.. Cus2 enforces the first ATP-dependent step of splicing by binding to yeast SF3b1 through a UHM-ULM interaction. RNA. 2019; 25:1020–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perriman R., Barta I., Voeltz G.K., Abelson J., Ares M.. ATP requirement for Prp5p function is determined by Cus2p and the structure of U2 small nuclear RNA. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13857–13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang W.-W., Cheng S.-C.. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev. 2015; 29:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perriman R., Ares M.. Invariant U2 snRNA nucleotides form a stem loop to recognize the intron early in splicing. Mol. Cell. 2010; 38:416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carrocci T.J., Zoerner D.M., Paulson J.C., Hoskins A.A.. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017; 45:4837–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang Q., Rodriguez-Santiago S., Wang J., Pu J., Yuste A., Gupta V., Moldón A., Xu Y.-Z., Query C.C.. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 2016; 30:2710–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Y.-Z., Query C.C.. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol. Cell. 2007; 28:838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z.-M., Yang F., Zhang J., Tang Q., Li J., Gu J., Zhou J., Xu Y.-Z.. Crystal structure of Prp5p reveals interdomain interactions that impact spliceosome assembly. Cell Rep. 2013; 5:1269–1278. [DOI] [PubMed] [Google Scholar]
- 19. Amberg D.C., Burke D.J., Strathern J.N.. Methods in Yeast Genetics, 2005 edn. 2005; Cold Spring Harbor: Cold Spring Habor Laboratory Press. [Google Scholar]
- 20. Selvin P.R., Ha T.. 2008; Cold Spring Harbor: Single Molecule Techniques. CSHL Press. [Google Scholar]
- 21. Crawford D.J., Hoskins A.A., Friedman L.J., Gelles J., Moore M.J.. Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:6783–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoskins A.A., Friedman L.J., Gallagher S.S., Crawford D.J., Anderson E.G., Wombacher R., Ramirez N., Cornish V.W., Gelles J., Moore M.J.. Ordered and dynamic assembly of single spliceosomes. Science. 2011; 331:1289–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crawford D.J., Hoskins A.A., Friedman L.J., Gelles J., Moore M.J.. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA. 2008; 14:170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rasnik I., Mckinney S.A., Ha T.. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods. 2006; 3:891–893. [DOI] [PubMed] [Google Scholar]
- 25. Dave R., Terry D.S., Munro J.B., Blanchard S.C.. Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophys. J. 2009; 96:2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodgers M.L., Tretbar U.S., Dehaven A., Alwan A.A., Luo G., Mast H.M., Hoskins A.A.. Conformational dynamics of stem II of the U2 snRNA. RNA. 2015; 22:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friedman L.J., Gelles J.. Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods. 2015; 86:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Feltz C., Hoskins A.A.. Methodologies for studying the spliceosome's RNA dynamics with single-molecule FRET. Methods. 2017; 125:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bronson J.E., Fei J., Hofman J.M., Gonzalez R.L., Wiggins C.H.. Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 2009; 97:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mckinney S.A., Joo C., Ha T.. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006; 91:1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodgers M.L., Didychuk A.L., Butcher S.E., Brow D.A., Hoskins A.A.. A multi-step model for facilitated unwinding of the yeast U4/U6 RNA duplex. Nucleic Acids Res. 2016; 44:10912–10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaur H., Jamalidinan F., Condon S.G.F., Senes A., Hoskins A.A.. Analysis of spliceosome dynamics by maximum likelihood fitting of dwell time distributions. Methods. 2019; 153:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu Y.-Z., Newnham C.M., Kameoka S., Huang T., Konarska M.M., Query C.C.. Prp5 bridges U1 and U2 snRNPs and enables stable U2 snRNP association with intron RNA. EMBO J. 2004; 23:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mo J., Duncan J.A.. Assessing ATP binding and hydrolysis by NLR proteins. Methods Mol. Biol. 2013; 1040:153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Day C.L., Dalbadie-McFarland G., Abelson J.. The Saccharomyces cerevisiae Prp5 protein has RNA-dependent ATPase activity with specificity for U2 small nuclear RNA. J. Biol. Chem. 1996; 271:33261–33267. [DOI] [PubMed] [Google Scholar]
- 36. Lee S., Lee J., Hohng S.. Single-molecule three-color FRET with both negligible spectral overlap and long observation time. PLoS ONE. 2010; 5:e12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karow A.R., Klostermeier D.. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res. 2009; 37:4464–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Theissen B., Karow A.R., Köhler J., Gubaev A., Klostermeier D.. Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andersen C.B.F. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006; 313:1968–1972. [DOI] [PubMed] [Google Scholar]
- 40. Sengoku T., Nureki O., Nakamura A., Kobayashi S., Yokoyama S.. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006; 125:287–300. [DOI] [PubMed] [Google Scholar]
- 41. Bono F., Ebert J., Lorentzen E., Conti E.. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006; 126:713–725. [DOI] [PubMed] [Google Scholar]
- 42. Del Campo M., Lambowitz A.M.. Structure of the Yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol. Cell. 2009; 35:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aregger R., Klostermeier D.. The DEAD box helicase YxiN maintains a closed conformation during ATP hydrolysis. Biochemistry. 2009; 48:10679–10681. [DOI] [PubMed] [Google Scholar]
- 44. Lesser C.F., Guthrie C.. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics. 1993; 133:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Staley J.P., Guthrie C.. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998; 92:315–326. [DOI] [PubMed] [Google Scholar]
- 46. Semlow D.R., Blanco M.R., Walter N.G., Staley J.P.. Spliceosomal DEAH-Box ATPases Remodel Pre-mRNA to Activate Alternative Splice Sites. Cell. 2016; 164:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Semlow D.R., Staley J.P.. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem. Sci. 2012; 37:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Finci L.I., Zhang X., Huang X., Zhou Q., Tsai J., Teng T., Agrawal A., Chan B., Irwin S., Karr C. et al.. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018; 32:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cretu C., Schmitzová J., Ponce-Salvatierra A., Dybkov O., De Laurentiis E.I., Sharma K., Will C.L., Urlaub H., Lührmann R., Pena V.. Molecular architecture of SF3b and structural consequences of Its cancer-related mutations. Mol. Cell. 2016; 64:307–319. [DOI] [PubMed] [Google Scholar]
- 50. Cretu C., Agrawal A.A., Cook A., Will C.L., Fekkes P., Smith P.G., Lührmann R., Larsen N., Buonamici S., Pena V.. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell. 2018; 70:265–273. [DOI] [PubMed] [Google Scholar]
- 51. Carrocci T.J., Paulson J.C., Hoskins A.A.. Functional analysis of Hsh155/SF3b1 interactions with the U2 snRNA/branch site duplex. RNA. 2018; 24:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yan D., Perriman R., Igel H., Howe K.J., Neville M., Ares M.. CUS2, a yeast homolog of human Tat-SF1, rescues function of misfolded U2 through an unusual RNA recognition motif. Mol. Cell Biol. 1998; 18:5000–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perriman R.J., Ares M.. Rearrangement of competing U2 RNA helices within the spliceosome promotes multiple steps in splicing. Genes Dev. 2007; 21:811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hilliker A.K., Mefford M.A., Staley J.P.. U2 toggles iteratively between the stem IIa and stem IIc conformations to promote pre-mRNA splicing. Genes Dev. 2007; 21:821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rauhut R., Fabrizio P., Dybkov O., Hartmuth K., Pena V., Chari A., Kumar V., Lee C.-T., Urlaub H., Kastner B. et al.. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science. 2016; 353:1399–1405. [DOI] [PubMed] [Google Scholar]
- 56. Yan C., Wan R., Bai R., Huang G., Shi Y.. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science. 2016; 353:904–911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.