


Abstract
Chemical similarity searching is a basic research tool that can be used to find small molecules which are similar in shape to known active molecules. Despite its popularity, the retrieval of local molecular features that are critical to functional activity related to target binding often fails. To overcome this limitation, we developed a novel machine learning-based chemical binding similarity score by using various evolutionary relationships of binding targets. The chemical similarity was defined by the probability of chemical compounds binding to identical targets. Comprehensive and heterogeneous multiple target-binding chemical data were integrated into a paired data format and processed using multiple classification similarity-learning models with various levels of target evolutionary information. Encoding evolutionary information to chemical compounds through their binding targets substantially expanded available chemical-target interaction data and significantly improved model performance. The output probability of our integrated model, referred to as ensemble evolutionary chemical binding similarity (ensECBS), was effective for finding hidden chemical relationships. The developed method can serve as a novel chemical similarity tool that uses evolutionarily conserved target binding information.
INTRODUCTION
Most chemical similarity scores consider the overall structural similarity based on predefined and equally weighted structural features (i.e., molecular fingerprints) (1–3). Although these methods have been widely used to search for similar compounds owing to their fast processing speed and ease of use (4–7), the simplicity often hinders the detection of meaningful hits where only a few chemical features are important for target-binding (6,8,9).
Machine-learning methods have been extensively studied in the field of cheminformatics. Many sophisticated models have been proposed using quantitative structure–activity relationship (QSAR) data for consideration of complex target-binding molecular features and have been successfully applied to many biological areas (4,9–12). QSAR models are usually built on active and non-active compound sets defined for a target protein, and the models have been effective for extracting important spatial features, despite the two-dimensional nature of some molecular descriptors (13). However, even with the advantages of QSAR methods, the pairwise molecular relationships cannot be defined because they are mostly limited to a predefined target and it is difficult to consider multiple chemical-target relationships.
Similarity-learning techniques provide context-dependent pairwise similarity measures using machine-learning methods (14–16). Similar to other classification problems, paired data sets are labeled as similar or dissimilar and used to build a model or function which can decide if new paired data is similar. Metric learning and classification similarity-learning are two common approaches that are used to address the similarity-learning problem. Metric-learning techniques focus their attention on learning a similarity measure that satisfies the mathematical properties of a metric distance (17), whereas classification similarity-learning techniques produce a score, rather than a metric, that effectively classifies similar/dissimilar objects (16,18). Classification similarity-learning is particularly useful when the overall objective is to rank data according to similarity relationship and the metric properties are not strictly required to output similarity scores (14).
In our previous study, classification similarity-learning was applied to small molecule drugs and showed promising results with regards to finding unknown drug-target interactions and novel pharmacological effects (11). However, despite the initial success, many key issues such as insufficient chemical-target binding data, unlabeled chemical-target interactions, ambiguous target relationships, and lack of general-purpose chemical similarity remain unsolved.
Herein, we present a novel evolutionary chemical binding similarity (ECBS) method using a classification similarity-learning framework defined with paired chemical data and target's evolutionary relationship (Figure 1). The ECBS method is designed to encode molecular features enriched in evolutionarily conserved chemical-target binding relationships, and formulated by the likelihood of chemical compounds binding to identical targets. The inclusion of evolutionary information linked to chemical compounds through their binding targets (Figure 1A and B) is a unique property of the ECBS method that enables substantial expansion of available chemical-target interaction data, contributing to significant improvement of model performance. Details regarding the model construction and performance evaluation are described in the following sections.
Figure 1.

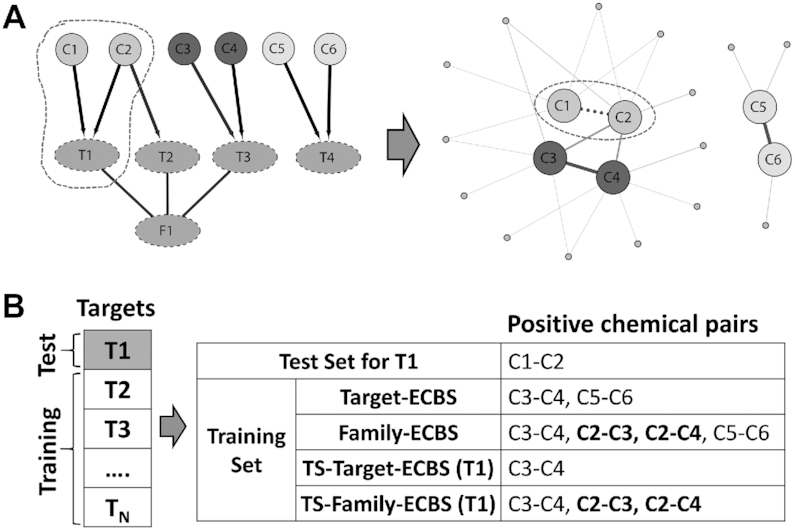
Overview of evolutionary chemical binding similarity (ECBS) method. (A) The simplified chemical-target-evolutionary relationship is used as an example for a schematic description of different ECBS models. The ‘C’ in the first layer means a chemical compound, the ‘T’ means a target, and the ‘F’ represents arbitrary evolutionary information (e.g. family). The C-T connection is defined by the direct binding between C and T, and T-F is defined by the evolutionary class information F of the target T. The chemical pairs are classified based on the evolutionary relationship of binding targets. For example, C1–C2, C3–C4 and C5–C6 are evolutionarily related chemical pairs (ERCPs) by the common binding targets, and C1–C3, C1–C4, C2–C3 and C2–C4 are also ERCPs defined by F1. The other unrelated chemical pairs are considered as negative data. (B) The conceptual classification scheme for different ECBS models is depicted using the example in (A). In the QSAR-like model, active molecules are defined for the target T1 and the classification model (dotted line) clusters them together in chemical feature space. In the Target-ECBS model, the ERCPs defined by the multiple targets (T1, T2 and T3) will be clustered by the model, but the ERCPs by T1 and T2 are likely to have closer distances in chemical feature space because of the similar molecular features for binding to the evolutionarily conserved binding pocket defined by F1. On the other hand, the Family-ECBS model starts to consider the ERCPs defined by evolutionary information of the targets (chemical pairs in the green background). These additional ERCPs will make an enhancement effect to locate the F1-related chemical compounds in close proximity by presenting evolutionarily conserved features more evidently. The TS-Family-ECBS model is a target-specific Family-ECBS model that only considers the ERCPs defined from the targets evolutionarily related to the predefined target T1 (square box). Therefore, the model construction procedure is identical to the Family-ECBS model except that C5–C6 is excluded because the target T3 has no evolutionary relationship to T1 or T2. Compared to the target-specific model, the Target-ECBS and Family-ECBS are categorized as a unified model, because it considers multiple and heterogeneous chemical-target binding information altogether.
MATERIALS AND METHODS
Collection of chemical-target binding data
Chemical structures and target-binding information were collected from the DrugBank (19) and BindingDB (20) databases. In the DrugBank database, drug-target interaction data (28 July 2017) were retrieved only for ‘polypeptide’ targets and used to obtain Structure Data Format (SDF) files for the drugs. In the BindingDB database, the 2D SDF file was downloaded (BindingDB_All_terse_2D_2018m3.sdf updated on 1 April 2018) and parsed to obtain binding affinity data represented by Ki, IC50, Kd and EC50 values. To exclude low-affinity promiscuous binding, interactions were considered only when the affinity determined by any of the measurements was below 100 nM. As a result, the total number of small molecules, targets, and interactions were 6671, 4283 and 16 587 in DrugBank, and 587 693, 5425 and 1 018 895 in BindingDB, respectively. The two databases were integrated after removing redundant small molecules by comparing InChIKey (21) for the construction of target-specific ECBS models.
Definition of evolutionarily related chemical pairs
Homologous target proteins usually have evolutionarily conserved ligand binding sites and perform similar biological functions (22). In the present study, it is assumed that the structural and functional similarity between homologous proteins can be transferred to their binding chemical compounds because they are likely to share common three-dimensional (3D) pharmacophore features to bind similar structural environments in the conserved pocket (23). Accordingly, the chemical compounds that bind identical or homologous targets are considered as ‘evolutionarily related’, and the chemical pairs which have a common evolutionary target, domain, family, or superfamily annotation in their binding targets are defined as ‘evolutionarily related chemical pairs’ (ERCPs) (Figure 1A). The evolutionary information of targets can vary according to the definition of homology between targets.
Multiple annotations of evolutionary information to target genes
To avoid mislabeling in proteomic-scale evolutionary annotations for target genes, we incorporated evolutionary motif, domain, family and superfamily information defined in highly qualified and curated protein databases. The databases used in this study were UniProtKB (24), PFAM (25), SMART (26), PRINT (27), Gene3D (28), TIGRFAM (29), FAMILY (30) and SUPERFAMILY (30). Identifiers for target genes were unified by UniProtKB entry name. The InterPro (31) database (protein2ipr.dat) was used to map UniProtKB entry names to the protein databases.
The Superfamily (1.75) server provided hidden Markov models (HMMs) pre-built for 2478 sequenced genomes, which enabled flexible structural protein domain annotation for the target genes using the SCOP family and superfamily ID. The HMM library (http://supfam.org/SUPERFAMILY/downloads/license/supfam-local-1.75/) in the Superfamily database was applied to all target sequences using the script ‘superfamily.pl’ (downloaded from the Superfamily server) with default options to annotate family and superfamily-level description to targets. In summary, target genes were annotated by diverse evolutionary information such as a sequence-based motif, domain, family, and structure-based family and superfamily information.
Feature vector generation for representing a chemical pair
Structural information (formatted by the SDF file) for each chemical compound was converted to chemical binary fingerprints using ChemmineR and ChemmineOB cheminformatics packages in R (32). A fingerprint is a collection of predefined features regarding a local fragment found within a structure and is typically represented by a bit-string where 1 and 0 indicate ‘existence’ and ‘absence’ of each feature. MACCS (256 bits) and FP4 (512 bits) fingerprints available in the ChemmineOB package were concatenated to represent each chemical compound using a 768-bit numeric vector. The fingerprints with empty values for all drugs in DrugBank were discarded to reduce the dimension of feature space, which eventually generated a 386-bit feature vector representing an individual chemical compound. The feature vector for a chemical pair was subsequently generated by element-wise summation of the chemical fingerprints as follows.
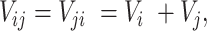 |
where Vi is a fingerprint for chemical i and Vj for chemical j.
The element-wise summation of Vi and Vj generated Vij, a feature vector for a chemical pair, where the elements 0, 1 and 2 indicate ‘none’, ‘different’, and ‘common’ features, respectively.
Classification similarity-learning for modeling chemical binding similarity
The collected chemical pair, target, and evolutionary data (Figure 1A) were used to build ECBS models by classification similarity-learning. Specifically, an ECBS model was designed to classify ERCPs from ‘unrelated chemical pairs’ and so the output value by the ECBS model represented a chemical similarity score prioritizing the selection of ERCPs. The model was trained as follows:
Training data: 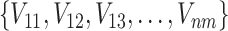 , where Vnm is a feature vector for a chemical pair consisted of Vn and Vm.
, where Vnm is a feature vector for a chemical pair consisted of Vn and Vm.
The data label 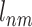 for Vnm is defined as follows:
for Vnm is defined as follows:
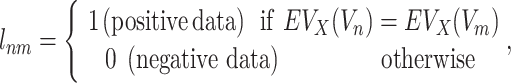 |
where 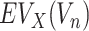 represents an evolutionary annotation X for the targets of a chemical compound Vn. Accordingly, the data label can be defined in many different ways according to the evolutionary information used to train ECBS models. For example, in the Target-ECBS model (Figure 1B), ERCPs (positive data) are defined by the identity of binding targets, whereas in Family-ECBS model, ERCPs are defined by the chemicals that have common ‘Family’ annotation in the binding targets.
represents an evolutionary annotation X for the targets of a chemical compound Vn. Accordingly, the data label can be defined in many different ways according to the evolutionary information used to train ECBS models. For example, in the Target-ECBS model (Figure 1B), ERCPs (positive data) are defined by the identity of binding targets, whereas in Family-ECBS model, ERCPs are defined by the chemicals that have common ‘Family’ annotation in the binding targets.
Target-specific ECBS model is designed to overcome the data size limitation of the unified ECBS model. The model requires a predefined target (e.g. T1 for TS-Family-ECBS in Figure 1B) and the positive data (ERCPs) are only defined for the targets evolutionarily related to the predefined target. Thus, in the target-specific model, for a given target T, the label 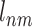 for Vnm is defined as follows:
for Vnm is defined as follows:
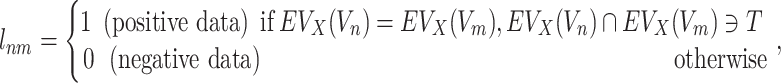 |
where 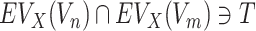 means that the predefined target T should have the common evolutionary annotation X for the targets of a chemical compound Vn and Vm. Schematic model description for each ECBS variant is shown in Figure 1. Details are in the Results section.
means that the predefined target T should have the common evolutionary annotation X for the targets of a chemical compound Vn and Vm. Schematic model description for each ECBS variant is shown in Figure 1. Details are in the Results section.
Sampling unrelated chemical pairs
Sampling negative data can be important to determine the model quality (33,34), because the current chemical-target binding data is highly imbalanced, with much larger amounts of unrelated chemical pairs (negative data). Thus, a procedure for sampling unrelated chemical pairs was designed to balance with the positive data (ERCPs) and to avoid possible overfitting towards the abundant negative data.
Specifically, six negative chemical pairs for each positive pair were generated by sampling chemical pairs which are structurally similar but evolutionarily unrelated to the positive pair. The chemical database was first scanned to find chemical compounds structurally similar but evolutionarily unrelated to a positive chemical pair Pa–Pb. Next, three molecules (Na1, Na2 and Na3) most similar to Pa were paired with Pb, resulting in three negative chemical pairs Pb–Na1, Pb–Na2 and Pb–Na2. An identical procedure for Pb generated another three negative chemical pairs Pa–Nb1, Pa–Nb2 and Pa–Nb2. The generated negative data were excluded if 2D structure similarity was too high (Tanimoto coefficient > 0.85) or evolutionarily related by at least one of the evolutionary databases.
Random forest classifier for ECBS model generation
Various machine learning techniques have been successfully applied in bio- and cheminformatics. However, in this study, available methods were limited because a large amount of paired chemical data (positive pairs: NC2 + negative pairs: 6 x NC2) hindered efficient parameter tuning and model training. Therefore, we chose to use ‘ranger’, a fast implementation of random forest classifier, because it features less-adjustable parameters, fast runtime, and efficient memory usage particularly suited for high-dimensional data (35). For training the ECBS models, ranger parameters were set with the following options: num.trees = 200 or 500, save.memory = TRUE, and down-weighting negative samples by 0.35 with the case.weights option. Feature vectors for ERCPs (positive data) and unrelated pairs (negative data) were generated and trained to predict the data labels according to different evolutionary information of target genes, each of which resulted in X-ECBS model where X represented arbitrary evolutionary information used to train the model.
Generation of an ensemble ECBS model
An ensemble ECBS (ensECBS) classifier integrating all X-ECBS models was built by ranger package based on the output scores from the individual X-ECBS models (Figure 2). The ensemble model was trained as follows:
Figure 2.
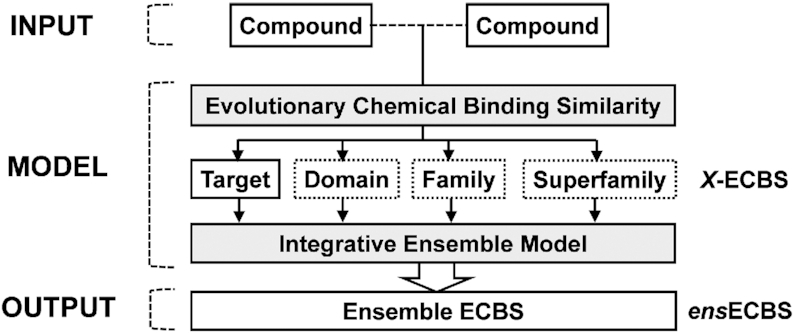
The model structure of the ensemble ECBS model. An input chemical pair is scored by the individual X-ECBS models defined by different evolutionary relationship (‘X’ represents target, domain, family, and superfamily in this case). The scores from all X-ECBS models are integrated by the ensemble model to generate a final chemical binding similarity score.
Training data: {X1-ECBSnm, X2-ECBSnm,..., Xj-ECBSnm}, where Xj-ECBSnm represents a similarity score for a chemical pair Vn and Vm calculated by the Xj-ECBS model trained by evolutionary information Xj. The label 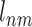 for a chemical pair Vn and Vm is defined as follows:
for a chemical pair Vn and Vm is defined as follows:
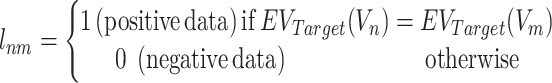 |
where 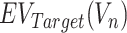 represents a target identity for a chemical compound Vn.
represents a target identity for a chemical compound Vn.
The predicted scores for the cross-validation test set by each X-ECBS model were used to train the ensECBS model. Specifically, all the test scores from the cross-validation procedures were combined and split into five sets. One set (20%) was only used to train the model to avoid possible overfitting problem, and the rest (80%) was tested for model validation. The procedure was repeated five times with the identical ranger parameters used to train X-ECBS models. Furthermore, the trained model was further validated with another independently generated dataset (test set generation is described in the following section). The test results for the cross-validation set and the independent set were used to estimate the prediction performance of the ensemble ECBS models (Table 1). To check the robustness of the ensemble models, the whole test procedure was repeated 100 times and the stability of the model performance was confirmed (Supplementary Figure S1).
Table 1.
Summary of ECBS model performance according to different model type and test set
| Test set | Cross-validation set | Independent set | |||
|---|---|---|---|---|---|
| Performance (AUC) | PR | ROC | PR | ROC | |
| Ensemble models | TS-ensECBS | 0.8555 | 0.9672 | 0.8181 | 0.9316 |
| (avg. TS-X-ECBS*) | (0.7013) | (0.9337) | (0.5962) | (0.8169) | |
| ensECBS | 0.6964 | 0.8872 | 0.7704 | 0.8895 | |
| (avg. X-ECBS*) | (0.6545) | (0.8778) | (0.7350) | (0.9064) | |
| Single evolutionary models | Target-ECBS+ | 0.5609 | 0.8086 | 0.7307 | 0.8839 |
| Pfam-ECBS | 0.5891 | 0.8331 | 0.6909 | 0.888 | |
| Family-ECBS | 0.6487 | 0.8762 | 0.7026 | 0.8979 | |
| Superfamily-ECBS | 0.6438 | 0.8745 | 0.6937 | 0.8915 | |
| TS-Target-ECBS | 0.6663 | 0.9047 | 0.5332 | 0.7536 | |
| TS-Pfam-ECBS | 0.6904 | 0.9281 | 0.4396 | 0.6930 | |
| TS-Family-ECBS | 0.6933 | 0.9305 | 0.4088 | 0.6319 | |
| TS-Superfamily-ECBS | 0.5689 | 0.8604 | 0.4011 | 0.6787 | |
| TS-SMART-ECBS | 0.6086 | 0.8557 | 0.4600 | 0.7355 | |
| TS-PRINT-ECBS | 0.6083 | 0.8558 | 0.4423 | 0.7002 | |
| TS-TIGR-ECBS | 0.3465 | 0.6875 | 0.3095 | 0.6329 | |
| TS-Gene3D-ECBS | 0.4950 | 0.8069 | 0.4049 | 0.6740 | |
| Ligand-based structure similarity | LIGSIFT (ShapeSim) | 0.2187 | 0.5604 | 0.3733 | 0.6135 |
| LIGSIFT (ChemSim) | 0.2204 | 0.5571 | 0.3794 | 0.6178 | |
| Lisica (2D) | 0.3314 | 0.6500 | 0.5395 | 0.7437 | |
| Lisica (3D) | 0.3244 | 0.6490 | 0.5534 | 0.7531 | |
| 2D structure similarity | 0.2537 | 0.5716 | 0.4865 | 0.7225 | |
*All single ECBS scores are averaged to compare with the ensemble models.
+Reference original method, Park et al. (2011).
The target-specific ensemble model (TS-ensECBS) integrating all target-specific ECBS (TS-X-ECBS) and unified ECBS (X-ECBS) models was built (Supplementary Figure S2) and tested in the same manner. One difference was that the TS-ensECBS model included additional data size information into the training data to reduce target-specific bias. The training data for a given target T were defined as follows.
Training data: {TS-X1-ECBSnm,...,TS-Xi-ECBSnm, X1size,...,Xisize, X1-ECBSnm,...,Xj-ECBSnm}, where TS-Xi-ECBSnm represents a similarity score for a chemical pair Vn and Vm calculated by the TS-Xi-ECBS model built for a target T, Xisize represents a trained data size of the TS-Xi-ECBS model, and Xj-ECBSnm represents a similarity score for a chemical pair Vn and Vm calculated by the unified Xj-ECBS model.
The inclusion of the amount of evolutionary information (Xisize) in the feature vector was to down weight the scores calculated by the TS-X-ECBS models which were built on a very small amount of evolutionary data. Each type of evolutionary information contributed very differently to the model's accuracy, which correlated to the amount of available information in the training dataset (Supplementary Figure S3). The output scores (Xj-ECBSnm) from the unified X-ECBS models were included to compensate for possible defects of the TS-X-ECBS models caused by the limited usage of chemical-target binding information (See the Results section for a comparison between the target-specific and unified model).
Cross-validation test set for evaluating ECBS models
A test chemical pair dataset was generated by splitting all binding targets into 12 sets. Among them, 11 sets were used to train the model, and the remaining one was used to validate model performance (12-fold cross-validation). For example, in Figure 3, a target T1 is selected as a test set, so the chemical pair C1–C2 (dotted line in Figure 3A) which commonly binds to T1 is assumed to be unknown (test chemical pair). On the other hand, the other chemicals such as C3, C4, C5 and C6 are used to train ECBS models by defining ERCPs (Figure 3B) based on the chemical-target-evolutionary relationships in Figure 3A.
Figure 3.
Cross-validation test set for evaluating the ECBS models (A) The simplified chemical-target-evolutionary relationships similar to Figure 1A are defined to schematically show how the ECBS models are differently trained and how the test chemical pairs are generated by splitting the target set. For clarity, the ERCPs (positive data in training) and their relationships are also represented by a network where the blue edge represents the ECRPs defined by target identity, the green edge represents the ERCPs defined by evolutionary information F1, and the dotted edge is the test chemical pair. Arbitrary unrelated chemical pairs (negative data) are shown by grey nodes and edges. (B) For the cross-validation, binding targets are split into test and training set (1:11). In the example where T1 is selected as the test target, all ERCPs defined by T1 (i.e. C1–C2) are considered as a blind test set common for all ECBS models. To predict the test data, each ECBS model differently organizes the training set by the different definition of ECRPs (Figure 1B). For example, Target-ECBS only considers C3–C4 and C5–C6 as ECRPs, whereas Family-ECBS additionally considers C2–C3 and C2–C4 through the F1 information. On the other hand, the target-specific models exclude C5–C6 in the training set because T4 is not annotated by F1.
According to the model type and definition of evolutionary information (Figure 1B), each ECBS model differently uses the chemical-target-evolutionary relationships in the training procedure. For instance, Target-ECBS only considers C3–C4 (by T3) and C5–C6 (by T4) as positive data (ERCP) for training, whereas Family-ECBS additionally considers C2–C3 and C2–C4 as positive data by F1 family annotation (Figure 3B). On the other hand, target-specific (TS-) ECBS models only consider the targets evolutionarily related to the predefined target T1 to define ERCPs, which is why TS-Target-ECBS for T1 only uses C3–C4 as positive data without C5–C6. The negative data are generated by sampling the chemical pairs which are structurally similar but evolutionarily unrelated to the corresponding ERCPs.
The cross-validation by splitting targets instead of chemical pairs was to test the effectiveness of evolutionary information by assuming that all direct chemical binding information for a test target is unknown. Without direct target binding information, chemical similarity scores will be estimated only through the indirect evolutionary information, which is very hard to achieve by traditional QSAR methods.
Independent test set for evaluating ECBS models
Another test set was independently generated by using drug-target binding data recently updated in DrugBank. The newly updated data in the file downloaded in December 2018 (the previous version was retrieved in July 2017) included 360 drugs, 202 targets, and 915 drug-target interactions. Among them, only the targets whose TS-X-ECBS model was built with more than two chemical compounds were considered for comparison. As a result, the independent test set contained 1538 ERCPs defined by target identity (positive data). The fourfold unrelated chemical pairs (negative data) were generated in the same way used to build the cross-validation set. Any redundant chemical pairs to the cross-validation or training set were deleted from the test set.
Performance evaluation by precision-recall curve
Area under the curve (AUC) values in precision-recall (PR) and receiver operating characteristic (ROC) curves were calculated to estimate the prediction performance. It is well-known that the ROC (true positive rate vs. false positive rate) is inappropriate to test highly imbalanced data with a much higher amount of negative samples, because the false positive rate is significantly affected by high true negative (TN) values. However, the PR curve is negligibly affected by a large number of negative samples, because precision depends on the true positive (TP) and false positive (FP) values (36).
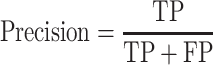 |
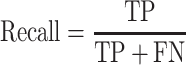 |
The higher sensitivity of the PR curve towards positive samples makes it more suitable for the evaluation of model performance by focusing on positive samples. The ‘PRROC’ R package was used to calculate AUC values in the PR and ROC curves (37).
Chemical structure similarity
The 386-bit fingerprint representing a chemical pair was identically used to calculate 2D chemical structure similarity using the Tanimoto coefficient (i.e. ratio of intersection-bits over union-bits). LiSiCA (Ligand Similarity using Clique Algorithm) (38) and LIGSIFT (39) were used with default options to calculate conformational ligand shape similarity. Because both methods are sensitive to 3D conformations of chemical structures, conformers for each chemical compound were generated using BEST method in Discovery Studio software (Dassault Systemes BIOVIA, Discovery Studio Modeling Environment, Release 4.5, 2015) with default options (RMSD cut-off: 0.2 Å). At the same time, the energy-minimized structure by CHARMm force field was also obtained for each compound. For a chemical pair Vn and Vm, the low energy 50 conformers of Vn were compared to the energy-minimized structure of Vm by LIGSIFT and LiSiCA, and the maximum value was considered as a representative similarity score for the chemical pair. For LIGSIFT, ShapeSim and ChemSim scores were used to calculate similarity scores, and for LiSiCA, 2D and 3D options were separately applied with -d option.
Virtual screening by ECBS and in vitro kinase binding assay
To screen new inhibitory chemical compounds for serine/arginine protein kinase 1 (SRPK1) and SRPK2, we used the TS-ensECBS models built for the targets to screen MarinLit (40) and Maybridge chemical database (screening collection). The ECBS scores between all chemicals in the database and NCC007 (41) were calculated using the respective TS-ensECBS model. The NCC007 is a recently found chemical compound initially discovered to have dual inhibition activities for casein kinase Iα (CKIα) and Iδ (CKIδ) but also show unexpected inhibition for SRPK1 and SRPK2 (41). The candidate molecules most similar to NCC007 by the ECBS scores were retrieved and tested by in vitro kinase binding assay.
The KINOMEscan kinase assay provided by DiscoverX was used to measure competitive binding strength of the candidate molecules for SRPK1 and SRPK2. In the assay, high-affinity chemical compounds that prevent SRPK binding to the immobilized ligand reduce the amount of SRPK on the solid support, whereas low-affinity compounds have no effect. Thus, a higher percentage of SRPK dissociated from the immobilized ligand implies higher binding affinity. Details for the assay is described on the company website.
RESULTS
The underlying principle of evolutionary chemical binding similarity
In general, machine learning-based classification is designed to transform labeled samples originally located in a hardly-distinguishable feature space into a more clearly-separable space by adjusting the sample distances according to classification boundaries (14,42). Similarly, well-trained classification similarity-learning models should maintain close distances between identically labeled (similar) samples compared to those of differently labeled (dissimilar) samples by extracting effective distinguishing features (14,43). Moreover, formulation of similarity in a classification similarity-learning framework should allow a group of samples sharing common critical features to be clustered together at close distances in a transformed feature space even for mislabeled or weakly related samples (16,44). In the present study, the intrinsic property of classification similarity-learning was used to define target-centric chemical binding similarity.
We implemented the classification similarity-learning models in various formats according to the input data type, model type, and evolutionary information (Figure 1B). The details of different ECBS models will be discussed with the simplified chemical-target-evolutionary relationships shown in Figure 1A. In addition, the schematic figure is shown together for each ECBS model (Figure 1B) to represent how the classification similarity-learning conceptually works for the chemical pairs to calculate ECBS scores. In Figure 1A, the chemical-target relationship is defined by direct binding and the target-evolutionary relationship by annotated groups of homologous targets that have a common ancestry. Because of the diverse definition of evolutionary information, multiple ECBS models were built and labeled as X-ECBS model, where X represented arbitrary evolutionary information used to train the model.
QSAR-like
A QSAR model is typically built by collecting active and inactive chemical compounds for a predefined target. The activity can be determined by either phenotypic profile or direct target-binding, and the classification model is constructed to prioritize active compounds sharing pharmacophore features for the target. Thus, the resulting QSAR model is expected to group the active compounds together in chemical feature space (Figure 1B). The input data type of QASR model is a set of chemical compounds, and the model is built for a predefined single target (i.e. target-specific) without consideration of evolutionary information.
Target-ECBS
In contrast to QSAR models, the input data type of all ECBS models are chemical pairs required to apply classification similarity-learning. In the ECBS models, chemical pairs are categorized into positive and negative pairs, where the former represents ERCPs that bind either identical or evolutionarily related target, and the latter represents evolutionarily unrelated chemical pairs. The Target-ECBS model defines the positive pairs only by target identity. For example, in Figure 1A, the chemical pairs, C1–C2 (by T1), C3–C4 (by T2) and C5–C6 (by T3) are considered as positive pairs and are likely to be grouped together in arbitrary feature space by the classification similarity-learning model. Because target T1 and T2 are evolutionarily related by F1 (family-level annotation), C1–C2 and C3–C4 will likely share evolutionarily conserved molecular features necessary for target binding, and thus, it should be straightforward for a similarity-learning classifier to cluster C1, C2, C3 and C4 in close proximity (Figure 1B). The C5–C6 defined by T3 will be clustered by themselves in isolated feature space with coupled negative data. As a result, chemical compounds from T1 and T2, and those from T3, respectively, will have high chemical binding similarity.
Accordingly, the Target-ECBS model can consider multiple targets at the same time through the unified classification similarity-learning framework, which ‘intrinsically’ reveals hidden ERCPs such as C1–C3, C1–C4, C2–C3 and C2–C4 without specifying F1 information (target identity is the only information used to train the model). The Target-ECBS model is used as a reference method to test the effectiveness of evolutionary information.
Family-ECBS
The Family-ECBS model starts to explicitly consider evolutionary information to estimate chemical binding similarity. In Figure 1A, the chemical pairs defined by evolutionarily related targets are C1–C3, C1–C4, C2–C3 and C2–C4 (by F1). Because the Family-ECBS model uses Family information to define ERCPs, the four chemical pairs are additionally considered as positives (shown in the green background in Figure 1B), which results in bigger cluster consisted of all F1-related chemical compounds by the classification similarity-learning model. Thus, the additional ERCPs defined by Family information are expected to reinforce the clustering effect in chemical feature space by presenting conserved molecular features for target-binding more evidently and by enriching the chemical-target binding information at multiple levels of evolutionary information. The ERCPs in Figure 1B (C1–C3, C1–C4, C2–C3 and C2–C4) are likely to share an abstract form of evolutionarily conserved molecular features, and this additional information can be useful to improve ECBS model performance when combined with the target-based molecular features.
TS-Family-ECBS
The TS-Family-ECBS model is a target-specific Family-ECBS model where positive chemical pairs are defined only from the targets evolutionarily related to a predefined target. For example, in Figure 1A where T1 is specified as the predefined target, the TS-Family-ECBS model only defines the chemical pairs evolutionarily related to T1 as ERCPs. Thus, all the chemical pairs defined by C1, C2, C3 and C4 are considered as ERCPs by target (T1, T2) or family (F1) but C5–C6 is excluded.
Although the TS-Family-ECBS model is target-specific, it is still applicable to the case that chemical binding information for a predefined target is completely unknown because ERCPs can be also defined from evolutionarily related targets (e.g. C3–C4 by T2). The strengths and weakness of the unified (X-ECBS) and target-specific (TS-X-ECBS) models will be discussed in the following section.
The unified and target-specific ECBS models
All X-ECBS models were based on drug-target binding information in DrugBank because incorporating BindingDB data caused a practical memory problem during the model training procedure due to the quadratic growth of the paired chemical data. The number of available chemical-target interactions in DrugBank was 16 587 but increased to 1 018 895 by combining with BindingDB (ca. 60-fold increase). The big chemical dataset was not well tolerated by the unified X-ECBS models, and so the target-specific X-ECBS (TS-X-ECBS) model was proposed to overcome the data size limitation.
Because the TS-X-ECBS models were built for a subset of targets, the ERCP data size could be significantly reduced without losing a lot of information regarding potentially related chemical pairs, and so the incorporation of BindingDB data became feasible. Plenty of chemical-target binding data for natural products and synthetic compounds in BindingDB other than drugs (45) was expected to improve the performance of ECBS models. Moreover, because of the reduced training data size, supplementary evolutionary information defined by SMART (26), PRINT (27), Gene3D (28), and TIGRFAM (29), could be adopted to maximize the use of evolutionary relationship data and diversify target-specific evolutionary information in addition to the PFAM, Family and Superfamily information. However, the prior target-specification can be a weakness of TS-X-ECBS models. If a predefined target lacks evolutionary information and has no homologous targets, the TS-X-ECBS model will be almost identical to the typical QSAR model.
On the other hand, in the construction of ensemble model, all X-ECBS models are considered to have equivalent weight, so the output scores from each X-ECBS model are directly used as training data for constructing the secondary ensemble model (ensECBS) (Figure 2). However, the reliability of the output scores from each TS-X-ECBS model can vary according to a predefined target. For instance, if the predefined target is not annotated by evolutionary information Xj, the prediction accuracy of the TS-Xj-ECBS model will be very low for the target, because of the small amount of training data. To overcome the problem, information on the size of training data and output scores from the unified X-ECBS models for each target was additionally included in the training data to construct the ensemble model of target-specific ECBS models (TS-ensECBS) (Supplementary Figure S2).
Hidden evolutionary chemical relationship can be revealed by ECBS models
The principal assumption of the ECBS models is that classification similarity-learning for the unified paired chemical data derived from heterogeneous target-binding information will compulsively cluster similar target-binding chemical compounds, maintain close distances, and eventually provide an effective chemical binding similarity score. The clustering effect is also expected to enable detection of novel ERCPs.
To examine the assumption, we tested whether the basal Target-ECBS model could detect unknown ERCPs. Specifically, all-versus-all ECBS calculations for 1778 approved drugs (1 579 753 pairs) were performed using Target-ECBS and 2D structure similarity, respectively. Then, we focused on the drug pairs that had no common binding targets but showed high ECBS scores (i.e. false positives by the definition of Target-ECBS model) and checked how many of them bind to evolutionarily related targets. The drug pairs whose targets are not identical but evolutionarily related in at least one of the evolutionary databases were assumed to be a potential binding molecule for the respective target, because of the evolutionarily conserved structure-function relationship in homologous proteins. All drug pairs used to train the Target-ECBS model were excluded from the analysis.
The results showed that the drugs pairs binding to evolutionarily related targets had higher similarity scores than unrelated pairs with a significant population difference (P-value < 2.2e–16 by Student's t-test) by both Target-ECBS and 2D structure similarity (Supplementary Figure S4). When the detection ratio for ERCP was compared for the high-rank drug pairs, the Target-ECBS model detected ERCPs with a significantly higher ratio (e.g. 88% for top 100 pairs and 74% for top 1000), whereas 2D structure similarity showed low detection ratio (42% for the top 100 and 50% for the top 1000) (Figure 4). Overall results suggested that the assumption for the clustering effects between evolutionarily related chemical compounds is valid and effective by the classification similarity-learning models.
Figure 4.
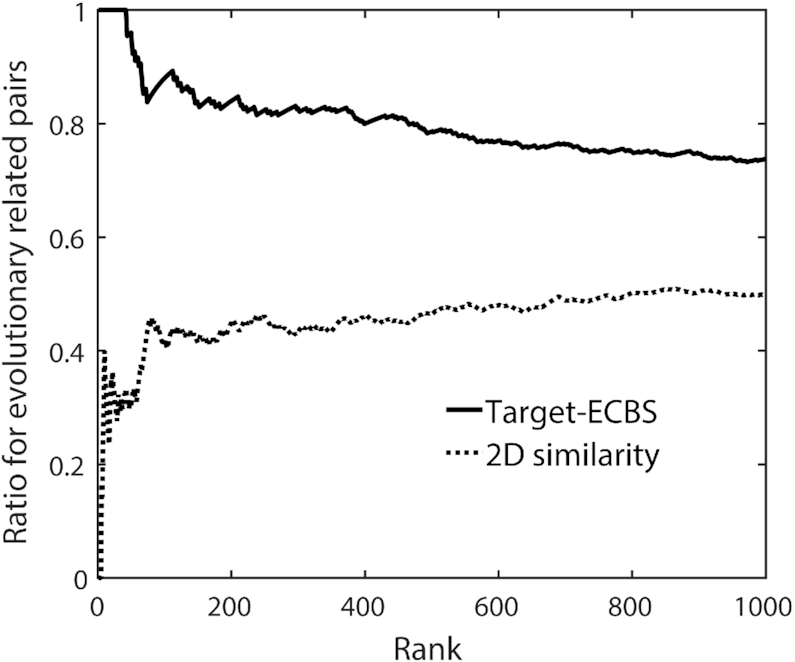
Hidden evolutionary chemical relationship revealed by Target-ECBS model. The ratios detecting ERCPs (whose targets are not identical but evolutionarily related) are compared between Target-ECBS and 2D chemical structure similarity. It is assumed that the ERCPs are potential target-binding molecules even though it is not experimentally validated. The chemical pairs are sorted by each similarity score and the ratio of ERCPs within ranks is plotted. The Target-ECBS model better describes the potential target-binding chemicals with high ERCP ratio.
ECBS model performance for cross-validation and independent test set
Two test sets were separately generated to estimate the prediction performance of ECBS models. One is a cross-validation set (Figure 3) and another is an independent test set created by extracting the recently updated drug-target information in DrugBank. The former is a comprehensive test set covering most chemical pairs in the database, and the latter is a much smaller test set that consists of completely unseen data. Overall model performance was estimated and compared by AUC values in the PR curve.
The test results for the cross-validation set (Figure 5A) showed that ECBS models clearly outperformed the methods based on global structure similarity, such as LIGSIFT, LiSiCA and 2D structure similarity. Among the ECBS models, both ensECBS and TS-ensECBS showed better performance than Target-ECBS and TS-Target-ECBS, respectively, suggesting that evolutionary information was effective to improve the model performance. The TS-ensECBS model showed the best performance and clear separation of ERCPs (Supplementary Figure S5), likely thanks to the richest training information including X-ECBS models.
Figure 5.

Test performance of ECBS models for (A) the cross-validation set and (B) the independent set are shown by the PR curve. The methods based on chemical structure similarity (LIGSIFT, LiSiCA and 2D structure similarity) are shown for comparison.
The single evolutionary ECBS models other than Target-ECBS or TS-Target-ECBS model were designed to be ensembled with the target-based models, not to be used solely. As shown in Figure 3, the evolutionary models were trained to predict ERCPs as well as the common target-binding chemical pairs. However, the identical test set with the Target-ECBS model was used to evaluate the model for comparison (Table 1). This extended definition of ERCPs in the evolutionary ECBS models might hinder the accurate prediction of the chemical pairs binding to common targets but the ensemble procedure was expected to resolve these issues by integrating all the information.
Another test using the independent set showed comparable results despite the smaller data size (Figure 5B). One difference was the low performance of the TS-Target-ECBS model, suggesting that the performance of target-specific models can vary according to the selection of targets. However, the unified X-ECBS and the ensemble ECBS models consistently showed reasonable performance. The AUC values in PR and ROC curves for all individual and ensemble ECBS models are summarized in Table 1.
Virtual screening using ECBS
Recently, we identified a novel dual inhibitor, NCC007, for CKIα and CKIδ, and demonstrated its functional role to control circadian rhythms through the CKI inhibition (41). Interestingly, comprehensive kinase profiling of NCC007 unexpectedly suggested that NCC007 also binds to SRPK1 and SRPK2, a potential therapeutic target for neovascular eye disease (46). Inspired by the finding, a virtual screening procedure was devised using ECBS and applied for SRPK1 and SRPK2 as another blind test. The TS-ensECBS models were built for SRPK1 and SRPK2, respectively, each of which was used to scan a virtual chemical library to find new chemical compounds evolutionarily related to NCC007. A total of five chemical compounds selected by TS-ensECBS were ordered and tested by in vitro kinase binding assay.
The assay results revealed that the TS-ensECBS model successfully discovered new SRPK inhibitors with high accuracy (Figure 6). Out of five tested compounds, three for SRPK1 and one for SRPK2 showed promising binding affinity. Because none of the chemicals are known for SRPK inhibitor so far, they might serve as a novel lead compound for neovascular eye disease in the future.
Figure 6.
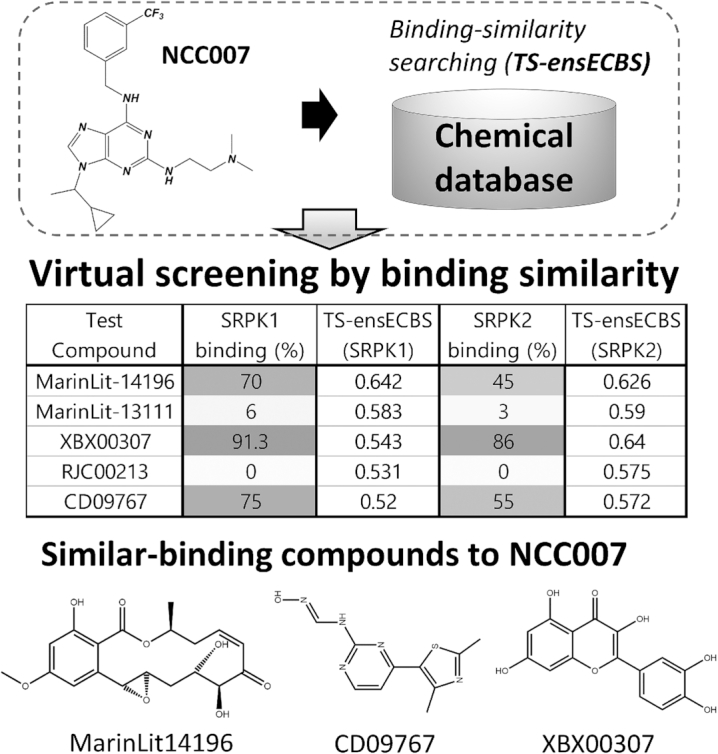
Blind test by virtual screening for SRPK1 and SRPK2 using TS-ensECBS. NCC007 is used to screen a virtual chemical library to identify novel compounds similar-binding to NCC007. The TS-ensECBS models built for SRPK1 and SRPK2, respectively, are used to calculate chemical similarity scores. The chemical compounds with high ECBS score are further validated by in vitro kinase binding assay. Three (MarinLit-14196, CD09767, XBX00307) for SRPK1 and one (XBX00307) for SRPK2 are confirmed to have considerable binding affinity.
DISCUSSION
Unique characteristics of different evolutionary information
We find that evolutionary information has the following distinctive features: (i) different hierarchical levels defining an evolutionary class (e.g. motif, domain, family, and superfamily), (ii) source information used to extract evolutionary relationship (e.g. sequence or structure-based) and (iii) unique target annotation coverage.
Specifically, evolutionary information of target genes has been defined at multiple levels such as a motif, domain, family, and superfamily. A motif is the smallest unit to define evolutionary relationships between targets, because it is mostly represented by a short nucleotide or amino acid pattern mediating a common function. A family represents a group of proteins that descend from a common ancestor and therefore are likely to share evolutionarily conserved structure, function, and sequence that possibly consist of multiple domains. A superfamily is a high level classification that the common ancestry is inferred by 3D structure similarity, even if no sequence similarity is evident. One superfamily often contains several protein families. Therefore, motif, domain, family, and superfamily information can provide a variety of chemical-target binding features which are evolutionarily conserved at different hierarchical levels.
In the present study, seven protein databases, PFAM, SMART, TIGRFAM, PRINT, Gene3D, Family, and Superfamily, were used to represent diverse evolutionary features for targets. Because there is the possibility of generating unclear evolutionary relationships with single motif information, the PRINT database, which characterizes a family based on protein fingerprints (a group of conserved motifs), was only used to represent motif information. PFAM, TIGRFAM and SMART were used to provide a sequence-based annotation of domain or family, whereas Gene3D and Superfamily 2.0 databases were to provide structural information at the SCOP family/superfamily and CATH domain level.
In addition, the target annotation coverage varied according to the annotation coverage of each database. For example, the PFAM, Family, and Superfamily information annotated more than 74% of targets, whereas SMART, TIGRFAM, PRINT and Gene3D showed <50% target annotation coverages (Supplementary Figure S6). It seemed that the target annotation coverage of each of evolutionary information was related to the performance of each X-ECBS model. For instance, the TS-TIGR-ECBS model based on TIGRFAM information showed the lowest performance for both test sets (Table 1), which is likely due to the least evolutionary information annotated for targets.
Contribution of evolutionary information
The contribution of each evolutionary information was quantitatively estimated by analysing variable importance (Gini index) during the cross-validation procedure which was performed to test the TS-ensECBS model. The variable importance (a total decrease in node impurity) measures how effectively the output scores from each ECBS model contribute to classifying the training data in the construction of the ensemble model. The node impurity values were retrieved from the iterative cross-validation procedures and summarized in Figure 7A. In the result, Family, Target, and PFAM information consistently represented high importance regardless of the model type. In contrast, TS-SMART, TS-TIGR, TS-PRINT and TS-Gene3D (G3DSA) showed low importance probably due to the low model performance (Table 1) and target coverage (Supplementary Figure S6). Interestingly, the variable importance of superfamily information varied according to the model type. The Superfamily-ECBS model represented higher importance than the TS-Superfamily-ECBS model, which might be related to the higher model performance of the Superfamily-ECBS model for both test sets (AUC 0.64 versus 0.57 for the cross-validation set and 0.69 versus 0.40 for the independent set). The reason for the performance difference is not clear but we think that superfamily-based evolutionary annotation generates ambiguous target relationships irrelevant to chemical binding property (one superfamily is often linked to many motifs, domains, and families). This noisy data might decrease the performance of the TS-Superfamily-ECBS model. The unified models such as Superfamily-ECBS could be more robust to the noisy information because they take full advantage of the clustering effect shown in Figure 1B by considering all chemical-target binding information together.
Figure 7.
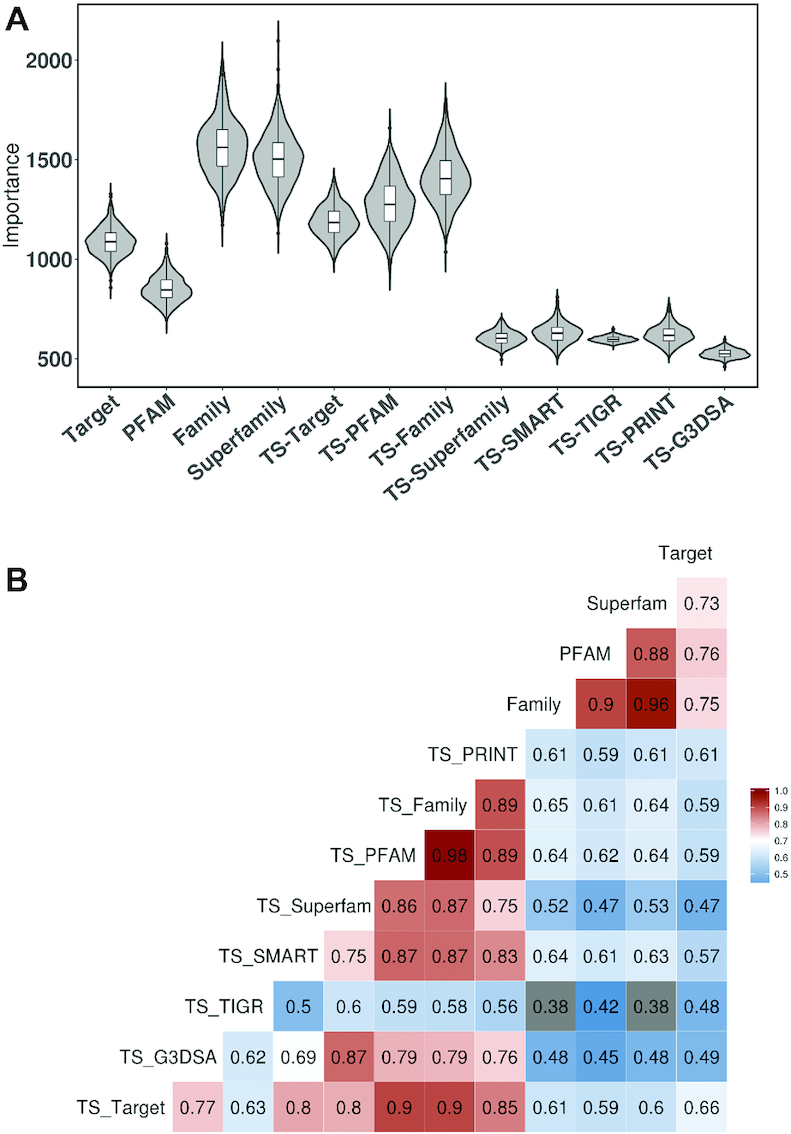
Contribution of evolutionary information in the construction of the ensemble ECBS models. (A) The contribution of each evolutionary information is estimated by calculating variable importance (Gini index) in the construction of the TS-ensECBS model. (B) The correlations between different ECBS models are represented by calculating Pearson's r for the predicted scores for the cross-validation test set.
Complementarity of the unified and target-specific ECBS model
The unified (X-ECBS) and target-specific (TS-X-ECBS) model differently encode evolutionary information to the output similarity scores by the distinctive model design (Figure 1B). To check the complementarity of the ECBS models, pairwise Pearson correlation coefficients between all ECBS scores were calculated using the predicted scores for the cross-validation test set (Figure 7B).
Mostly, the correlations within the same model type (i.e. either unified or target-specific) are much higher than those between different types. Among the unified ECBS models, PFAM-ECBS, Family-ECBS and Superfamily-ECBS have a high correlation (>0.88), whereas Target-ECBS has a relatively different score distribution compared to others. It suggested that domain, family, and superfamily information should be distinct from the target information, which might help to improve model performance as shown in the variable importance analysis (Figure 7A). In contrast to the unified models, TS-Target-ECBS is highly correlated to the other evolution-based target-specific models, probably because of the restriction of evolutionary information to a predefined target.
In summary, various ECBS models encoded different evolutionary information, which showed distinct score distributions, except for a few cases mentioned above. Especially, all unified ECBS models showed high variable importance in the construction of the TS-ensECBS model, suggesting their complementarity to the target-specific ECBS models.
Evolutionarily related chemical pairs highlight the conserved molecular binding features
A target ‘Cholera enterotoxin subunit B’ (ctxB, Uniprot ID P01556) was used to show how ERCPs help to improve model quality. The ctxB was selected because both TS-Target-ECBS and TS-ensECBS showed high accuracy in the cross-validation test set (AUC 0.72 and 0.96 for TS-Target-ECBS and TS-ensECBS, respectively) with a moderate number of test and training data (14 drugs in the blind test set and 7 drugs in the training set). The test and trained chemical pairs for ctxB were categorized into ‘test’ (dotted in red), ‘trained by target’ (solid blue line), and ‘trained by evolution’ (solid green line) in Figure 8.
Figure 8.

Enriched chemical features by evolutionarily related chemical pairs (ERCPs). All the chemical compounds used to predict the chemical pairs commonly binding to ‘cholera enterotoxin subunit B’ (ctxB) are represented by the network as in Figure 3A. In the network, the blue edges represent the ERCPs defined by target identity, the green edges represent the ERCPs defined by evolutionary information (motif, domain, family, and superfamily are mixed for clarity), and the red dotted edges are the blind test set which commonly binds to ctxB. The 2D chemical structures for the nodes are shown together, and the arbitrary unrelated chemical pairs are represented by grey nodes and edges.
Because all drugs binding to ctxB were included as a blind test set (see Figure 3 for the test set generation), similarity scores for the ‘test’ pairs would be inferred only by the indirect information related to ctxB. For example, two known targets of DB02213 are ctxB and heat-labile enterotoxin B chain (eltB, UniProt ID: P32890), both of which are evolutionarily related by the common PFAM (PF01376, Enterotoxin_b), PRINT (PR00772, enterotoxin B), Family (50204, Bacterial AB5 toxins B-subunits) and Superfamily (50203, Bacterial enterotoxins) although the source organisms are different (ctxB is from Vibro cholerae serotype O1 and eltB from Escherichia coli). In this case, the TS-Target-ECBS model would consider the ‘trained by target’ chemical pairs defined by eltB as ERCPs for model training (e.g., DB03421, DB04040, DB03446, DB03242 and DB04396).
However, the TS-ensECBS model would consider additional ERCPs such as DB02213-DB08501, DB02213-DB04465 and many others for DB03077, DB03721 and DB02379, depending on their evolutionary relatedness. The targets of DB03077, DB03721 and DB02379 are cholera enterotoxin B-subunit (Q57193), enterotoxin type B (P01552), and Shiga-like toxin 1 subunit B (P69178), respectively, all of which belong to the same bacterial enterotoxin superfamily (50203) without a common target. The evolutionary information contributed to generate more ERCPs (‘trained by evolution’ pairs), which resulted in the densely connected network that consisted of similar-binding chemical compounds (Figure 8). The number of ERCPs significantly increased by consideration of superfamily information, and so the important molecular features should be enriched and emphasized in the model training procedure. As expected, the ERCPs shared a few functional moieties such as the benzamide or galactopyranosyl groups representing the conserved molecular features for target-binding.
In summary, the target-binding information of ctxB can be highly limited if the target identity is only considered to build an ECBS model. The inclusion of many ERCPs will make the relationships (or distances) between similar-binding chemical compounds much closer by providing densely connected ERCPs and therefore highlighting the conserved molecular binding features.
CONCLUDING REMARKS
Categorization of paired chemical data based on evolutionary target-binding information enabled the use of classification similarity-learning method to develop novel chemical binding similarities. Two major factors are closely related to the performance improvement of the ensemble ECBS models compared with the original Target-ECBS that only used target-binding information of drugs. Incorporating more chemical-target binding data from BindingDB was very effective to improve the model performance, suggesting the possibility of further improvement with future data generation. More importantly, collecting targets’ evolutionary information from a variety of curated databases was critical, as it reflected the diverse evolutionary relationship of targets at multiple levels such as a motif, domain, family, and superfamily.
The target-binding information of chemical compounds is valuable to infer functional activity, because chemical-target interactions are directly linked to biological functions but are difficult to predict from a chemical structure alone. Evolutionarily related targets contain broad functional information thanks to the well-maintained structure-function relationship, particularly in the evolutionarily conserved binding pockets in homologous proteins (47). It has been consistently discussed that the evolution rate in the functionally important regions, such as enzyme active site, is much slower than the surface region (23). The ECBS models encode this evolutionary target-binding information into pairwise chemical relationships, where the similarity scores can compare more complicated binding properties between chemical compounds. Accordingly, we expect that ECBS can be widely used with applications such as large-scale ligand-based screening, target-specific ligand identification, drug-repositioning, and general chemical binding similarity calculations by modeling functional similarity.
DATA AVAILABILITY
The script for ensECBS is available in the GitHub repository (https://github.com/keunwan-kist/ECBS) and the pre-built models are deposited in the Zenodo (https://sandbox.zenodo.org/record/297093).
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Lee for helpful discussion on virtual screening. The model building was facilitated through the use of KIST Server Farm infrastructure.
Author contributions: K.P. conceived the study and developed the method. K.P., D.P. and Y.J.K. evaluated the results. D.P. and C.H.P. assisted in algorithm design. K.P. wrote the manuscript with input from all authors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Oceans and Fisheries, Korea [20170488]; KIST institutional grant [2E29562]. Funding for open access charge: Ministry of Oceans and Fisheries, Korea [20170488]; KIST institutional grant [2E29562].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bajusz D., Racz A., Heberger K.. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations. J. Cheminformatics. 2015; 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muegge I., Mukherjee P.. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Dis. 2016; 11:137–148. [DOI] [PubMed] [Google Scholar]
- 3. Cereto-Massague A., Ojeda M.J., Valls C., Mulero M., Garcia-Vallve S., Pujadas G.. Molecular fingerprint similarity search in virtual screening. Methods. 2015; 71:58–63. [DOI] [PubMed] [Google Scholar]
- 4. Geppert H., Vogt M., Bajorath J.. Current trends in ligand-based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010; 50:205–216. [DOI] [PubMed] [Google Scholar]
- 5. Ripphausen P., Nisius B., Bajorath J.. State-of-the-art in ligand-based virtual screening. Drug Discov. Today. 2011; 16:372–376. [DOI] [PubMed] [Google Scholar]
- 6. Sheridan R.P., Kearsley S.K.. Why do we need so many chemical similarity search methods. Drug Discov. Today. 2002; 7:903–911. [DOI] [PubMed] [Google Scholar]
- 7. Willett P., Barnard J.M., Downs G.M.. Chemical similarity searching. J. Chem. Inf. Comp. Sci. 1998; 38:983–996. [Google Scholar]
- 8. Yu X., Geer L.Y., Han L.Y., Bryant S.H.. Target enhanced 2D similarity search by using explicit biological activity annotations and profiles. J Cheminformatics. 2015; 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo M., Wang X.S., Tropsha A.. Comparative analysis of QSAR-based vs. chemical similarity based predictors of GPCRs binding affinity. Mol. Inform. 2016; 35:36–41. [DOI] [PubMed] [Google Scholar]
- 10. Ma J.S., Sheridan R.P., Liaw A., Dahl G.E., Svetnik V.. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015; 55:263–274. [DOI] [PubMed] [Google Scholar]
- 11. Park K., Kim D.. Drug-drug relationship based on target information: application to drug target identification. BMC Syst. Biol. 2011; 5:S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang L., Tan J.J., Han D., Zhu H.. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov. Today. 2017; 22:1680–1685. [DOI] [PubMed] [Google Scholar]
- 13. Bender A., Mussa H.Y., Glen R.C., Reiling S.. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J. Chem. Inf. Comput. Sci. 2004; 44:1708–1718. [DOI] [PubMed] [Google Scholar]
- 14. Lopez-Inesta E., Grimaldo F., Arevalillo-Herraez M.. Classification similarity learning using feature-based and distance-based representations: a comparative study. Appl Artif Intell. 2015; 29:445–458. [Google Scholar]
- 15. Chen Y.H., Garcia E.K., Gupta M.R., Rahimi A., Cazzanti L.. Similarity-based classification: concepts and algorithms. J. Mach. Learn. Res. 2009; 10:747–776. [Google Scholar]
- 16. Lopez-Inesta E., Grimaldo F., Arevalillo-Herraez M.. Learning similarity scores by using a family of distance functions in multiple feature spaces. Int. J. Pattern Recogn. 2017; 31:1750027. [Google Scholar]
- 17. Hua K., Yu Q., Zhang R.. A guaranteed similarity metric learning framework for biological sequence comparison. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016; 13:868–877. [DOI] [PubMed] [Google Scholar]
- 18. Lopez-Inesta E., Grimaldo F., Arevalillo-Herraez M.. Combining feature extraction and expansion to improve classification based similarity learning. Pattern Recogn. Lett. 2017; 93:95–103. [Google Scholar]
- 19. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. et al.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gilson M.K., Liu T.Q., Baitaluk M., Nicola G., Hwang L., Chong J.. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016; 44:D1045–D1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heller S., McNaught A., Stein S., Tchekhovskoi D., Pletnev I.. InChI - the worldwide chemical structure identifier standard. J. Cheminform. 2013; 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lichtarge O., Bourne H.R., Cohen F.E.. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996; 257:342–358. [DOI] [PubMed] [Google Scholar]
- 23. Echave J., Spielman S.J., Wilke C.O.. Causes of evolutionary rate variation among protein sites. Nat. Rev. Genet. 2016; 17:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. The UniProt, C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Letunic I., Bork P.. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018; 46:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Attwood T.K., Coletta A., Muirhead G., Pavlopoulou A., Philippou P.B., Popov I., Roma-Mateo C., Theodosiou A., Mitchell A.L.. The PRINTS database: a fine-grained protein sequence annotation and analysis resource–its status in 2012. Database. 2012; 2012:bas019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis T.E., Sillitoe I., Dawson N., Lam S.D., Clarke T., Lee D., Orengo C., Lees J.. Gene3D: extensive prediction of globular domains in proteins. Nucleic Acids Res. 2018; 46:D1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haft D.H., Selengut J.D., White O.. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003; 31:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson D., Madera M., Vogel C., Chothia C., Gough J.. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res. 2007; 35:D308–D313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finn R.D., Attwood T.K., Babbitt P.C., Bateman A., Bork P., Bridge A.J., Chang H.Y., Dosztanyi Z., El-Gebali S., Fraser M. et al.. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017; 45:D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cao Y., Charisi A., Cheng L.C., Jiang T., Girke T.. ChemmineR: a compound mining framework for R. Bioinformatics. 2008; 24:1733–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park Y., Marcotte E.M.. Revisiting the negative example sampling problem for predicting protein-protein interactions. Bioinformatics. 2011; 27:3024–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng Z.Z., Huang K., Wang Y., Liu H., Guan J.H., Zhou S.G.. Selecting high-quality negative samples for effectively predicting protein-RNA interactions. BMC Syst. Biol. 2017; 11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wright M.N., Ziegler A.. ranger: a fast implementation of random forests for high dimensional Data in C plus plus and R. J. Stat. Softw. 2017; 77:1–17. [Google Scholar]
- 36. Gu Q., Zhu L., Cai Z.H.. Evaluation measures of the classification performance of imbalanced data sets. Comm. Com. Inf. Sci. 2009; 51:461–471. [Google Scholar]
- 37. Grau J., Grosse I., Keilwagen J.. PRROC: computing and visualizing precision-recall and receiver operating characteristic curves in R. Bioinformatics. 2015; 31:2595–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lesnik S., Stular T., Brus B., Knez D., Gobec S., Janezic D., Konc J.. LiSiCA: a software for ligand-based virtual screening and its application for the discovery of butyrylcholinesterase inhibitors. J. Chem. Inf. Model. 2015; 55:1521–1528. [DOI] [PubMed] [Google Scholar]
- 39. Roy A., Skolnick J.. LIGSIFT: an open-source tool for ligand structural alignment and virtual screening. Bioinformatics. 2015; 31:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dabb S., Blunt J., Munro M.. MarinLit: database and essential tools for the marine natural products community. Abstr. Pap. Am. Chem. S. 2014; 248:1–34. [Google Scholar]
- 41. Lee J.W., Hirota T., Ono D., Honma S., Honma K., Park K., Kay S.A.. Chemical control of mammalian circadian behavior through dual inhibition of casein kinase 1 alpha and delta. J. Med. Chem. 2019; 62:1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ponzoni I., Sebastian-Perez V., Requena-Triguero C., Roca C., Martinez M.J., Cravero F., Diaz M.F., Paez J.A., Arrayas R.G., Adrio J. et al.. Hybridizing feature selection and feature learning approaches in QSAR modeling for drug discovery. Sci. Rep. 2017; 7:2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scholkopf B. The kernel trick for distances. Adv Neur In. 2001; 13:301–307. [Google Scholar]
- 44. Eick C.F., Rouhana A., Bagherjeiran A., Vilalta R.. Using clustering to learn distance functions for supervised similarity assessment. Lect. Notes Artif. Int. 2005; 3587:120–131. [Google Scholar]
- 45. Chen X., Yan C.C., Zhang X., Zhang X., Dai F., Yin J., Zhang Y.. Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinform. 2016; 17:696–712. [DOI] [PubMed] [Google Scholar]
- 46. Batson J., Toop H.D., Redondo C., Babaei-Jadidi R., Chaikuad A., Wearmouth S.F., Gibbons B., Allen C., Tallant C., Zhang J.X. et al.. Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem. Biol. 2017; 12:825–832. [DOI] [PubMed] [Google Scholar]
- 47. Park K., Kim D.. A method to detect important residues using protein binding site comparison. Genome Inform. 2006; 17:216–225. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The script for ensECBS is available in the GitHub repository (https://github.com/keunwan-kist/ECBS) and the pre-built models are deposited in the Zenodo (https://sandbox.zenodo.org/record/297093).