

Supplemental Digital Content is available in the text
Keywords: Osteoarthritis, Autophagy, Apoptosis, Pathogenesis
Abstract
Background:
Plant homeodomain finger protein 23 (PHF23) is a novel autophagy inhibitor gene that has been few studied with respect to orthopedics. This study was to investigate the expression of PHF23 in articular cartilage and synovial tissue, and analyze the relationship between PHF23 and chondrocyte autophagy in osteoarthritis (OA).
Methods:
Immunohistochemical staining and western blot were applied to show the expression of PHF23 in cartilage of different outbridge grades and synovial tissue of patient with OA and healthy control. The normal human chondrocyte pre-treated with rapamycin or 3-methyladenine, treated with interleukin-1β (IL-1β). IL-1β induced expression level of PHF23 and autophagy-related proteins light chain 3B-I (LC3B-I), LC3B-II, and P62, were examined by Western blot. A PHF23 gene knock-down model was constructed with small interfering RNA. Western blot was performed to detect the efficiency of PHF23 and the impact of PHF23 knockout on IL-1β-induced expression of autophagy-related and apoptotic-related proteins in chondrocyte.
Results:
The expression of PHF23 was significantly increased in the high-grade cartilage and synovial tissue of patients with OA. The IL-1β-induced expression of PHF23 was gradually enhanced with time. The level of LC3B-II, P62 changed with time. After knockdown of PHF23, the level of autophagy-related proteins increased and apoptotic-related proteins decreased in IL-1β-induced OA-like chondrocytes.
Conclusions:
The expression of PHF23 increased in human OA cartilage and synovium, and was induced by IL-1β through inflammatory stress. PHF23 can suppress autophagy of chondrocytes, and accelerate apoptosis.
Introduction
Osteoarthritis (OA) is one of the most common degenerative diseases, and is characterized by pain, stiffness, reduced mobility, and joint deformities, and usually leads to disability. OA affects 240 million people globally, and has become a major global public health concern and economic burden.[1] Multiple factors, including mechanics, gene, obesity, and age have been implicated in the pathogenesis of OA, with age being the major risk factor for OA.[2]
Autophagy is an important way of lysosome-dependent degradation of proteins and damaged organelles.[3] It is vital for maintaining cell homeostasis, and is related to the pathogenesis of many aging-related diseases.[4] Many factors, such as abnormal nutritional metabolism, oxidative stress, and aging, can cause autophagy. Studies have shown that a decrease in autophagy is an important cause of many diseases, such as cardiomyopathy, neuro-degeneration, and skeletal system dysplasia.[5] Chondrocyte is the only cell content in cartilage, and when an external stimulus exceeds a certain threshold there is an imbalance of chondrocyte homeostasis, which eventually causes chondrocyte apoptosis and cartilage matrix degradation.[6] Therefore, maintaining the survival and function of chondrocyte plays an important role in preventing and delaying of the occurrence and development of OA.[6] There are many studies to modulate the apoptosis and autophagy of chondrocyte to maintain the balance. Inhibition of mammalian target of rapamycin C1[7] and necrosis factor-κB,[8] microRNA-107 by targeting tumor necrosis factor (TNF) receptor associated factor 3,[9] miRNA-335-5p,[10] miR-140-5p/miR-149 by targeting fucosyltransferase 1[11] were all coordinate the autophagy and apoptosis in articular chondrocytes to prevent OA.
Plant homeodomain finger protein 23 (PHF23) is a novel autophagy inhibitor gene that has never been studied with respect to orthopedics. Thus, we propose the novel hypothesis that the PHF23 gene may provide the link between OA, canonical autophagy, and mitophagy. The purpose of this study was to investigate the expression of PHF23 in articular cartilage and synovial tissue, and analyze the relationship between PHF23 and chondrocyte autophagy in OA.
Methods
Ethical approval
This study was approved by the Ethics Committee of Peking University First Hospital. All the patients included in the study signed the informed consent.
Sample collection
The patients who had chronic mild knee pain and met the American College of Rheumatology guideline of knee OA, were included. The inclusion criteria were as follows: (1) patients had mild knee pain with the visual analog scale scores between 6 and 10; (2) at least 3 months of pain duration, and (3) the severity of radiographic OA was between grade III to IV with the Kellgren-Lawrence grading scale. The exclusion criteria were as follows: (1) known diagnosis of inflammatory or metabolic diseases, such as rheumatoid arthritis, gout, tuberculosis, or septic arthritis; (2) neoplastic diseases of the examined knee; (3) intra-articular bone fracture and ligament rupture of knee joint. There were ten OA patients included. The OA cartilages and synovial tissues were collected during unilateral total knee arthroplasty at the Department of Orthopedic, Peking University First Hospital. The normal synovial tissues were collected from ten patients with meniscus tears during arthroscopic surgery as normal control. The cartilages and synovial tissues were cut for immunohistochemical test and Western blot. The samples for immunohistochemical test were cut mid-sagittal for 4-μm thick section. The sections were preserved at 4°C before use. The samples were preserved at −20°C for Western blot.
Primary normal human chondrocytes clone
Primary normal human chondrocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). The product number is No. 4650. The chondrocytes clone was transported in liquid nitrogen after cryopreservation and stored in liquid nitrogen before use. The chondrocytes were resuscitated and passaged. The third generation of cells was prepared for experiment.
Immunohistochemistry
The cartilage and synovial tissue sections were de-waxed and blocked by 3% H2O2 for 20 min. The rabbit polyclonal antibody against PHF23 (1:100, Abcom plc, China) was applied to the sections and incubated overnight at 4°C. After phosphate buffer saline (PBS) rinsed, the goat anti-rabbit IgG antibody with Horseradish Peroxidase (IgG-HRP) (KeyGEN BioTECH, China) was added and the sections were incubated with the liquid mixture diaminobezidin. After rinsing in water, the sections were dehydrated, cleared, and covered with cover-slips.
Rapamycin or 3-methyladenine (3-MA) treated and interleukin-1β (IL-1β) induced cell culture
The third generation of normal human chondrocytes was cultured in serum-free-Dulbecco's modified Eagle media (DMEM) for 24 h. The rapamycin (CellSignaling Technology, Danvers, MA, USA) pre-treated cells were cultured with 10 nmol/L rapamycin for 2 h, and so did with 3-MA (Sigma, MO, USA). And then the cells were treated with IL-1β (Sigma) at a concentration of 5 ng/mL for 0, 12, 24, and 48 h. The control cells were only treated with IL-1β. Changes of the chondrocytes were observed by optical microscopy.
Small interfering RNA (siRNA) and transfection
A PHF23 gene knock-down model was constructed with siRNA. The sequence of Si-RNA was 5′-GAGGACAUCAUGGUAGAAUTT-3′. Before the transfection, the normal human chondrocytes were cultured in DMEM seeded in 24-well plates for 1 day. Lipofectamine 2000 was used to si-RNA transfection for 6 h at 37°C. After rinsing, the cells were cultured for 48 h, and treated with TNF-α for 30 min. The knock-down efficiency was confirmed by evaluating the expression of PHF23 with Western blot.
Western blot analysis
Twelve percent sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel was prepared. The samples were subjected to SDS-PAGE and then transferred to nitrocellulose membranes after electrophoresis. The membranes were first blocked with 5% non-fat dry milk for 2 h. The primary antibody against PHF23 (1:1000, Abcom plc) was added and incubated at 4°C overnight. After three times of washing, the membrane was incubated with the goat anti-rabbit IgG antibody labeled with HRP at room temperature for 2 h, and was visualized using an enhanced chemiluminescence system. The experiments were carried out on three separate occasions.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA samples were extracted from human chondrocytes with the TRIzol reagent (TIANGEN Biotech, China). RT-PCR was performed using the ABI 7500 RT-PCR System (Applied Biosystems, US). The primers for amplifying PHF23 were 5′-TGGACGAGGACATCATGGTA-3′ and 5′-ACAGAGTGGGGAGGAAGGAT-3′.
Flow cytometry-based annexin V/propidium iodide (PI) staining
The chondrocytes were digested and re-suspended in cold binding buffer at concentrations of 106 cells/mL. One hundred microliters of cell suspension were incubated with 10 μL labeled annexin V on ice for 15 min. The binding buffer and PI solution were added to the cell suspension as the ApoScreen annexin V kit (Beyotime Biotech, Shanghai, China) instruction. Subsequently, the flow cytometer (BD Influx, NJ, USA) counted the number of stained cells.
MitoSOX red dyeing
MitoSOX was added at the final concentration of 5 μmol/L and incubated at 37°C for 30 min. After washing, the fluorescence intensity of MitoSOX was measured by fluorescence spectrophotometer (NanoDrop 2000, Thermo Scientific, MA, USA). The excitation wavelength is 488 nm and the emission wavelength is 580 nm. The shooting conditions of all samples are fixed. MitoSOX fluorescence intensity reflects the amount of superoxide produced in mitochondria.
JC-1 dyeing
The JC-1 dyeing solution was dispensed as the manufacture's protocol. The suspended chondrocytes were washed once with PBS, and added into one hole of the six-well plate, and 1 mL cell culture medium was added. One milliliter JC-1 dyeing solution was added and mixed well. The cells were incubated in incubator at 37°C for 20 min. After centrifugation, the supernatant was removed and washed twice with JC-1 staining buffer (1×). The chondrocytes were suspended by JC-1 staining buffer, and the number of stained cells was assessed via flow cytometer.
Grouping and outcome measurement
For the expression of PHF23 in cartilage and synovial tissue, the cartilage in different Outbridge grades and synovial tissue from patients with OA and healthy controls were used. The number of cells dyed in immunohistochemical staining and the content of protein by Western blot was applied to show the result. For the effect of IL-1β on autophagy and expression of PHF23 in chondrocytes, the human chondrocyte treated with IL-1β was compared with the normal control. The content of PHF23 and autophagy-related proteins light chain 3B-I (LC3B-I), LC3B-II, and P62, were examined by Western blot. A PHF23 gene knock-down model was constructed with siRNA. The content of PHF23, autophagy-related and apoptotic-related proteins in chondrocyte were examined by Western blot in PHF23 gene knock-down chondrocyte and the control group.
Statistical analysis
Data were expressed as mean ± standard deviation. SPSS 19.0 statistical software (SPSS Inc., Chicago, IL, USA) was used for data analysis. One-way analysis of variance was used for intergroup comparisons, while the independent-samples t-test was used for two-sample comparison. A value of P < 0.05 was considered to indicate statistical significance.
Results
Expression of PHF23 in the synovium and cartilage of OA
The immunohistochemistry results revealed that the expression of PHF23 was increased in the synovial tissue of patients with OA compared to normal synovial tissue of patients with acute meniscus tears [Figure 1A]. The expression of PHF23 in Outbridge grade III cartilage was greater than that in Outbridge grade I cartilage [Figure 1B]. The expression of PHF23 detected by Western blot was significantly increased in Outbridge grade III cartilage and OA synovial tissue compared to Outbridge grade I cartilage and normal synovial tissue from patients with acute meniscus tears [Figure 1C–1E].
Figure 1.
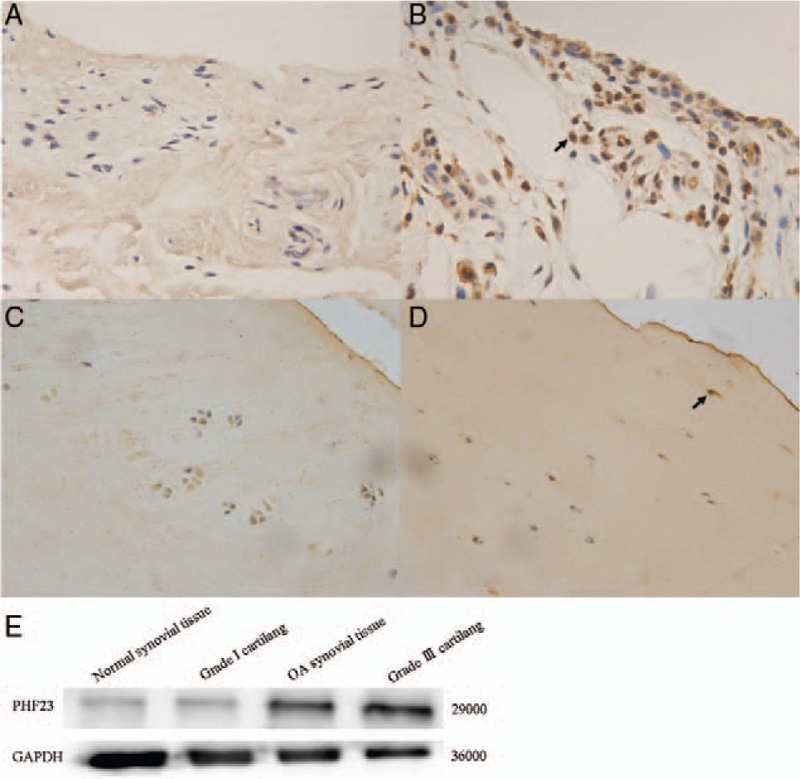
The expression of PHF23 in the synovial tissue and cartilage showed by immunohistochemical staining (original magnification ×200). (A) Normal synovial tissue; (B) OA synovial tissue. (C) Outbridge grade I cartilage; (D) Outbridge grade III cartilage; (E) Western blot for the expression of PHF23 in the synovial tissue and cartilage. The arrow pointed the positive cell. PHF23: Plant homeodomain finger protein 23.
Effect of IL-1β on autophagy and expression of PHF23 in chondrocytes
After stimulation with IL-1β, there was a decrease in the density of chondrocytes, and changes in cell morphology [Figure 2]. The expression levels of autophagy-related proteins LC3B-I, LC3B-II, and P62 were detected by Western blot [Figure 3A]. The ratios of LC3B-II/LC3B-I were 2.13 ± 0.04, 1.83 ± 0.03, and 1.76 ± 0.04 for IL-1β stimulation for 12, 24, and 48 h, respectively [Figure 3B]. The ratios of P62/GAPDH were 0.07 ± 0.01, 0.14 ± 0.02, and 0.20 ± 0.03 for IL-1β stimulation for 12, 24, and 48 h, respectively [Figure 3C]. The expression level of LC3B-II increased and P62 decreased significantly in IL-1β-induced OA-like chondrocytes compared to the control group at 12 h (P < 0.05). And then the level of LC3B-II decreased with P62 increased for 24 and 48 h. This indicated that the autophagy level of chondrocytes was increased significantly in the early stage of IL-1β stimulation (12 h) (P < 0.05). However, the level of autophagy decreased gradually with time (24 and 48 h), while the expression of PHF23 increased significantly over time compared to the control (P < 0.05) [Figure 4A and 4B]. The ratio of PHF23/glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 0.31 ± 0.05, 0.56 ± 0.02, and 0.79 ± 0.05 for IL-1β stimulation for 12, 24, and 48 h, respectively.
Figure 2.

Morphology of chondrocytes with or without IL-1β stimulation observed by optical microscopy (original magnification ×200). IL-1β: Interleukin-1β.
Figure 3.

The expression of autophagy-related proteins in chondrocytes with or without IL-1β stimulation. (A) The expression of LC3B-I, LC3B-II, and p62 were detected by Western blot. (B) Grayscale value of LC3B-II/LC3B-I. (C) Grayscale value of p62/GAPDH. (∗P < 0.05). IL-1β: Interleukin-1β; LC3B: Light chain 3B-I.
Figure 4.
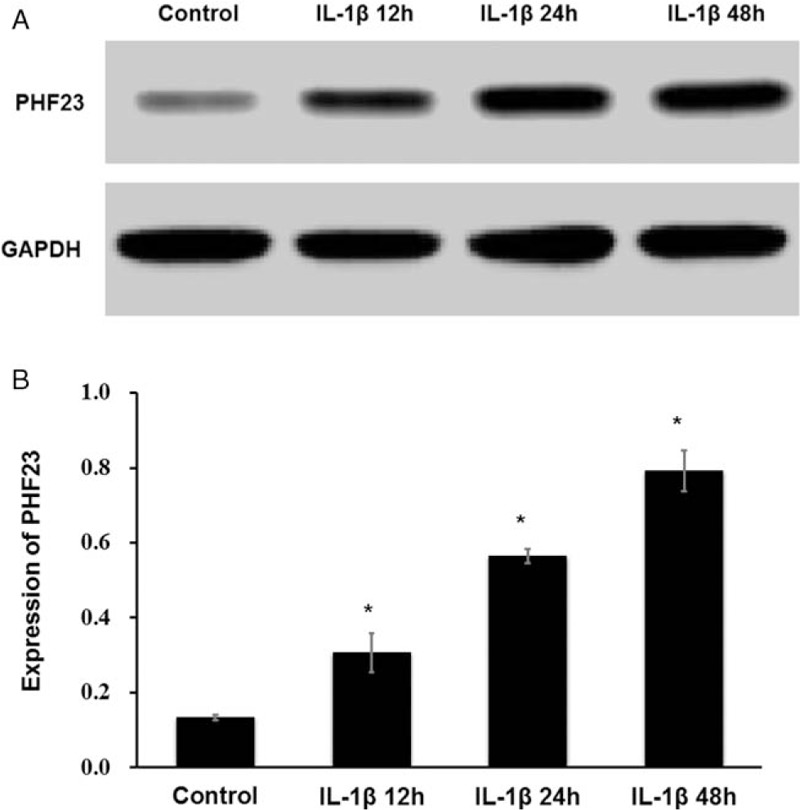
Changes of PHF23 expression in chondrocytes with IL-1β stimulation. The expression of PHF23 increased significantly over time. (A) The expression level of PHF23 was detected by Western blot. (B) Grayscale value of PHF23 (∗P < 0.05). PHF23: Plant homeodomain finger protein 23; IL-1β: Interleukin-1β.
Effect of pre-treatment with rapamycin and 3-MA on IL-1β-induced OA-like chondrocytes
Chondrocyte apoptosis was detected after stimulation with IL-1β by the Annexin V-fluorescein isothiocyante method (flow cytometry). Chondrocyte apoptosis increased with time (12, 24, and 48 h). However, 2 h of pre-treatment with the autophagy enhancer rapamycin or the autophagy inhibitor 3-MA altered the level of apoptosis by exerting positive and negative effects respectively [Supplementary Figure 1A and 1B]. Mitochondrial function was measured by determining the intra-cellular level of reactive oxygen species (ROS) using MitoSOX Red dye, and mitochondrial membrane potential was measured using JC-1 dye, after stimulation with IL-1β. The mitochondrial membrane potential decreased, and the intra-cellular level of ROS increased gradually over time (12, 24, and 48 h), suggesting the impairment of mitochondrial function. However, 2 h of pre-treatment with rapamycin or 3-MA also could reduce and aggravate the IL-1β induced mitochondrial damage, respectively [Supplementary Figure 1A–1D]. The RT-PCR result showed the expression of PHF23 mRNA increased with the time of IL-1β treatment, the relative expression of PHF23 mRNA for the IL-1β induced chondrocyte were 4.73 ± 1.15, 8.67 ± 1.25, and 9.37 ± 1.16 for 12, 24, and 48 h. Two hours of pre-treatment with rapamycin (4.43 ± 1.26, 8.83 ± 0.57, 10.1 ± 0.72) or 3-MA (4.43 ± 0.42, 8.33 ± 0.21, 11.59 ± 1.46) did not affect it at 12 h (P > 0.05) [Supplementary Figure 1E]. However, the expression of PHF23 at 24 and 48 h were different significantly compared to the control (P < 0.05).
Effect of PHF23 knock-down on autophagy and apoptosis in IL-1β-induced OA-like chondrocytes
The third group of chondrocytes was used in the following experiments because it had the lowest PHF23 expression, which confirmed the knock-down efficiency of siRNA [Figure 5].
Figure 5.
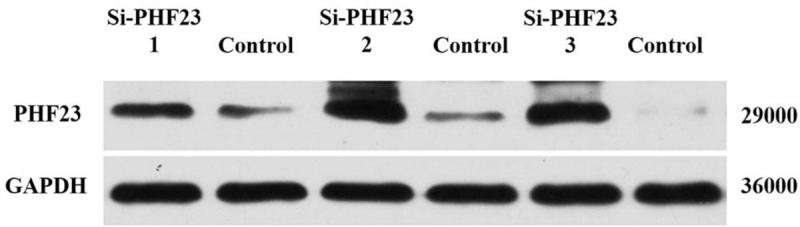
The knock-down efficiency of PHF23 siRNA was detected by Western blot. PHF23: Plant homeodomain finger protein 23; siRNA: Small interfering RNA; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
As shown in Figure 6, after knock-down of PHF23 the expression of LC3B-II and Beclin-1 (BECN1) increased after IL-1β stimulation as compared to the control group, which indicated that PHF23 can specifically inhibit autophagy in chondrocytes. However, after IL-1β stimulation the expression of cleaved-caspased-3 was decreased in the PHF23 knock-down chondrocytes compared to the control group, which indicated that PHF23 can alleviate IL-1β-induced apoptosis in chondrocytes.
Figure 6.

After silencing the expression of PHF23 in chondrocytes, the LC3B-II/LC3B-I ratio increased. With IL-1β stimulation, the expression of caspase-3 and cleaved-caspased-3 increased. IL-1β: Interleukin-1β; LC3B: Light chain 3B-I; PHF23: Plant homeodomain finger protein 23.
Relationship of autophagy and the expression of PHF23 in normal chondrocytes
After construction of high-autophagy level chondrocytes by treating with rapamycin, the expressions of PHF23 protein were 0.89 ± 0.02, 0.84 ± 0.06, 0.80 ± 0.02 for 12, 24, and 48 h, respectively, there was no significant difference compared to the control (P > 0.05) [Figure 7A and 7B], indicating that the level of chondrocyte autophagy did not affect the expression of PHF23.
Figure 7.

The effect of rapamycin on chondrocyte PHF23 expression. (A) The expression level of PHF23 was detected by Western blot. (B) Grayscale value of PHF23. PHF23: Plant homeodomain finger protein 23; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
Discussion
Autophagy is a key mechanism for maintaining cellular homeostasis by recycling dysfunctional cellular organelles and macromolecules, and is associated with many aging-related diseases.[3] Autophagy influences the expression of OA-related genes by regulating apoptosis and the level of ROS.[12]
The PHF23 gene is located on chromosome 17, p13.1, which contains five exons and four introns. PHF23 is a newly discovered autophagy suppressor gene, and is a negative regulator of autophagy.[13] Wang et al[14] reported there was overexpression of PHF23 and low expression of LC3B-II in U2OS and HeLa cells when cultured with low fetal bovine serum DMEM, and that there was a decrease in LC3B-II expression in PHF23 knockout cells. Further experiment demonstrated that PHF23 promoted autophagy inhibition by ubiquitination, and degradation of the E3 ligase, Leucine-rich repeat and sterile alpha motif-containing 1 (LRSAM1).[14] LRSAM1 was reported to have various cellular functions as it modulates the protein aggregation, endosomal sorting machinery and virus egress from the cells.[15] It interacts with both cellular protein degradation machineries and hence it can participate in maintenance of overall cellular proteostasis.[16] A recent study showed that mutations of LRSAM1, which was the downstream gene of PHF23, and the canonical mitophagy gene parkin, both caused similar symptoms of Parkinson disease.[17] So PHF23 may regulate the cell autophagy through LRSAM1. PHF23 has been studied in many diseases, but there are few studies of PHF23 in chondrocytes.
Autophagy and apoptosis of chondrocytes were demonstrated to be important role in the pathogenesis of OA.[18,19] A study showed autophagy of chondrocytes declines in OA, and was accompanied by impairment of mitochondrial function.[2] Almonte-Becerril et al[20] reported that the superficial chondrocytes of normal cartilage tissue expressed high levels of the autophagy proteins BECN1, autophagy related 5, and LC3. In human OA cartilage, the expression of the autophagy-related proteins unc-51 like autophagy activating kinase 1 (ULK1), BECN1, and LC3 were decreased, while the expression of the apoptosis-related protein Poly-ADP(ribose) polymerase (Parp p85) was elevated, as compared with normal cartilage.[21] Zhang et al[22] reported that a decrease of autophagy-related protein expression in OA cartilage was related to the overexpression of mechanistic target of rapamycin kinase (mTOR), which was a key upstream regulator of autophagy. As a regulator of autophagy, we found the overexpression of PHF23 in osteoarthritic chondrocytes and synovium compared to normal chondrocytes and synovium. We used IL-1β to induce chondrocytes to establish an OA model in vitro, and found that the level of autophagy in chondrocyte increased at 12 h, and then decreased gradually with time. At the same time, the expression of PHF23 in chondrocyte increased significantly over time after IL-1β stimulation. Combined with effect of IL-1β on cell,[23,24] our result suggested that IL-1β could induce the overexpression of PHF23 through inflammatory response, and indicated that PHF23 played an important role in the development of OA.
Rapamycin is a macrolide immunosuppressant that inhibits mTOR protein kinase extends lifespan in model organisms.[25] Many novel candidate rapamycin mimetics, including epigallocatechin gallate, isoliquiritigenin (rapamycin), were explored for clinical applications.[26] Carames et al[27] used rapamycin to treat cartilage degeneration in a rat model, and found that rapamycin inhibited the mTOR signaling pathway and as a result the autophagy-related gene LC3 was activated and cartilage degeneration significantly alleviated. He also treated mechanically damaged chondrocytes cultured in vitro with rapamycin, and found the expression of LC3, BECN1, and ULK1 in chondrocytes significantly increased.[28] In our study, we used IL-1β induced apoptosis and mitochondrial damage in chondrocytes, which were pre-treatment with rapamycin or 3-MA. The flow cytometry result suggested that rapamycin reduced the inflammatory-stress damage, while 3-MA aggravated it. The result of PHF23 expression was consistent with the result of apoptosis and mitochondrial damage. But the PHF23 expression was not affected by the pre-treatment with rapamycin or 3-MA. So it suggested that PHF23 might affect the OA chondrocyte apoptosis and mitochondrial damage through autophagy, but be not affected by apoptosis and mitochondrial damage.
In another study using mTOR knockout mice, cartilage autophagy was significantly enhanced, and the degeneration of cartilage, apoptosis, and synovitis were significantly improved.[22] We used siRNA to knockdown PHF23 in chondrocytes, and then treated them with IL-1β. The level of autophagy in the chondrocytes increased significantly, while the level of apoptosis decreased, which confirmed that PHF23 plays a specific negative regulatory role in chondrocyte autophagy. There may be a relationship of PHF23 and mTOR, we would go further on this.
Rapamycin-induced high autophagy level did not alter the expression of PHF23 in normal chondrocytes. Combining with the previous results, we can infer that PHF23 could affect autophagy level adversely without affecting by autophagy levels.
At present, there are still few studies about PHF23 and the mechanism of its effect on autophagy is not very clear, so further studies should be designed to explore the autophagy-related pathogenesis and molecular mechanisms of PHF23 with respect to OA, which may provide a theoretical basis for prevention and new therapeutic targets.
In summary, the expression of PHF23 increased in human OA cartilage and synovium, and was induced by IL-1β through inflammatory stress. It can suppress autophagy of chondrocytes, and accelerate apoptosis.
Conflicts of interest
None.
Supplementary Material
{kind=link}
Footnotes
How to cite this article: Li X, Yang X, Maimaitijuma T, Cao XY, Jiao Y, Wu H, Meng ZC, Liu H, Guan ZP, Cao YP. Plant homeodomain finger protein 23 inhibits autophagy and promotes apoptosis of chondrocytes in osteoarthritis. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000402
References
- 1.Nelson AE. Osteoarthritis year in review 2017: clinical. Osteoarthritis Cartilage 2018; 26:319–325.. doi: 10.1016/j.joca.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 2016; 12:412–420.. doi: 10.1038/nrrheum.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rockel JS, Kapoor M. Autophagy: controlling cell fate in rheumatic diseases. Nat Rev Rheumatol 2016; 12:517–531.. doi: 10.1038/nrrheum.2016.92. [DOI] [PubMed] [Google Scholar]
- 4.Todkar K, Ilamathi HS, Germain M. Mitochondria and lysosome: discovering bonds. Front Cell Dev Biol 2017; 5:106.doi: 10.3389/fcell.2017.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasheghani F, Zhang Y, Li YH, Blati M, Fahmi H, Lussier B, et al. PPAR deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann Rheum Dis 2015; 74:569–578.. doi: 10.1136/annrheumdis-2014-205743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage 2009; 17:971–979.. doi: 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Wen Y, Zhang M, Liu Q, Zhang H, Zhang J, et al. MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint. Autophagy 2019; 21:1–18.. doi: 10.1080/15548627.2019.1606647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Li X, Jin A. Rapamycin inhibits Nf-ΚB activation by autophagy to reduce catabolism in human chondrocytes. J Invest Surg 2019; 4:1–13.. doi: 10.1080/08941939.2019.1574321. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Li H, Wang L. MicroRNA-107 regulates autophagy and apoptosis of osteoarthritis chondrocytes by targeting TRAF3. Int Immunopharmacol 2019; 22:181–187.. doi: 10.1016/j.intimp.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Zhong G, Long H, Ma S, Shunhan Y, Li J, Yao J. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci 2019; 226:164–172.. doi: 10.1016/j.lfs.2019.03.071. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Hu J, Pan Y, Shan Y, Jiang L, Qi X, et al. Correction to: miR-140-5p/miR-149 affects chondrocyte proliferation, apoptosis, and autophagy by targeting FUT1 in osteoarthritis. Inflammation 2019; 42:1515–1516.. doi: 10.1007/s10753-019-01001-5. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum 2012; 64:1920–1928.. doi: 10.1002/art.34323. [DOI] [PubMed] [Google Scholar]
- 13.Reader JC, Leng Q, Rassool FV, Ning Y. Regulation of differentiation by a PHD domain in the NUP98-PHF23 fusion protein. Leuk Res 2010; 34:1094–1097.. doi: 10.1016/j.leukres.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Hu J, Li G, Qu L, He Q, Lou Y, et al. PHF23 (plant homeodomain finger protein 23) negatively regulates cell autophagy by promoting ubiquitination and degradation of E3 ligase LRSAM1. Autophagy 2014; 10:2158–2170.. doi: 10.4161/auto.36439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shy M. LRSAM1 lessons. Ann Neurol 2016; 80:821–822.. doi: 10.1002/ana.24817. [DOI] [PubMed] [Google Scholar]
- 16.Mishra R, Upadhyay A, Prajapati VK, Dhiman R, Poluri KM, Jana NR, et al. LRSAM1 E3 ubiquitin ligase: molecular neurobiological perspectives linked with brain diseases. Cell Mol Life Sci 2019; 76:2093–2110.. doi: 10.1007/s00018-019-03055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aerts MB, Weterman MA, Quadri M, Schelhaas HJ, Bloem BR, Esselink RA, et al. A LRSAM1 mutation links charcot-marie-tooth type 2 to Parkinson's disease. Ann Clin Transl Neurol 2016; 3:146–149.. doi: 10.1002/acn3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musumeci G, Castrogiovanni P, Trovato FM, Weinberg AM, Al-Wasiyah MK, Alqahtani MH, et al. Biomarkers of chondrocyte apoptosis and autophagy in osteoarthritis. Int J Mol Sci 2015; 16:20560–20575.. doi: 10.3390/ijms160920560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YS, Zhang FJ, Zeng C, Luo W, Xiao WF, Gao SG, et al. Autophagy in osteoarthritis. Joint Bone Spine 2016; 83:143–148.. doi: 10.1016/j.jbspin.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, Vega-Lopez MA, Lavalle C, Kouri JB. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of osteoarthritis within an experimental model. Apoptosis 2010; 15:631–638.. doi: 10.1007/s10495-010-0458-z. [DOI] [PubMed] [Google Scholar]
- 21.Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010; 62:791–801.. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis 2015; 74:1432–1440.. doi: 10.1136/annrheumdis-2013-204599. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, Cui W, Song F, Zhai C, Hu H, Zuo Q, et al. Effects of mesenchymal stem cells on interleukin-1β-treated chondrocytes and cartilage in a rat osteoarthritic model. Mol Med Rep 2015; 12:1753–1760.. doi: 10.3892/mmr.2015.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou B, Chen D, Xu H, Zhang X. Proliferation of rabbit chondrocyte and inhibition of IL-1β-induced apoptosis through MEK/ERK signaling by statins. In Vitro Cell Dev Biol Anim 2017; 53:124–131.. doi: 10.1007/s11626-016-0086-1. [DOI] [PubMed] [Google Scholar]
- 25.Arriola Apelo SI, Lamming DW. Rapamycin: an inhibitor of aging emerges from the soil of Easter island. J Gerontol A Biol Sci Med Sci 2016; 71:841–849.. doi: 10.1093/gerona/glw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aliper A, Jellen L, Cortese F, Artemov A, Karpinsky-Semper D, Moskalev A, et al. Towards natural mimetics of metformin and rapamycin. Aging (Albany NY) 2017; 9:2245–2268.. doi: 10.18632/aging.101319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2012; 71:575–581.. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caramés B, Taniguchi N, Seino D, Blanco FJ, D’Lima D, Lotz M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum 2012; 64:1182–1192.. doi: 10.1002/art.33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.