


Abstract
Fine-tuning of gene expression is desirable for a wide range of applications in synthetic biology. In this context, RNA regulatory devices provide a powerful and highly functional tool. We developed a versatile, robust and reversible device to control gene expression by splicing regulation in human cells using an aptamer that is recognized by the Tet repressor TetR. Upon insertion in proximity to the 5′ splice site, intron retention can be controlled via the binding of TetR to the aptamer. Although we were able to demonstrate regulation for different introns, the genomic context had a major impact on regulation. In consequence, we advanced the aptamer to develop a splice device. Our novel device contains the aptamer integrated into a context of exonic and intronic sequences that create and maintain an environment allowing a reliable and robust splicing event. The exon-born, additional amino acids will then be cleaved off by a self-cleaving peptide. This design allows portability of the splicing device, which we confirmed by demonstrating its functionality in different gene contexts. Intriguingly, our splicing device shows a high dynamic range and low basal activity, i.e. desirable features that often prove a major challenge when implementing synthetic biology in mammalian cell lines.
INTRODUCTION
A main objective of synthetic biology is the specific control of cellular behaviour to understand fundamental genetics. Precise, reversible and temporary control of gene expression is necessary and can be achieved at the transcriptional, translational and post-translational level (1,2). RNA is not only an intermediate for the conversion of gene expression to protein synthesis but also provides an attractive molecular scaffold for the design of genetic control elements. By interacting with ligands, RNA can alter its shape to act as a switch inside the cell (3,4). Moreover, RNA-based systems allow fast regulatory responses, genetic modularity and portability, i.e. features that make them suitable for complex platforms to achieve a broad spectrum of regulatory outputs (5,6).
Several successful attempts have demonstrated conditional gene expression with synthetic RNA devices (7). Among others, RNA-binding proteins (RBPs) were used to predictably influence gene expression that both increased the number of cellular functions available for regulation while also enhancing the precision of the regulation (3). One example of a protein-responsive RNA switch that controls translation in mammalian cells is based on the interaction between the ribosomal protein L7Ae and the box C/D kink-turn (8). In other studies, the L7Ae and the bacteriophage coat protein MS2 were used to design microRNA high- and low-sensors to engineer complex circuits in mammalian cells (9). In another case, an RBP device was developed to control alternative splicing. There, RNA structures recognized by proteins involved in NFkB and Wnt signalling were used to control cell fate through exon skipping. Thus, the expression of herpes simplex virus-thymidine kinase that confers sensitivity to a pro-apoptosis drug was controlled (10). Another interesting example of an RBP-based switch makes use of Pumilio and its derivatives. Here, the PUF domain has been fused with a splicing regulatory domain to regulate gene expression, primarily by controlling exon skipping (11,12).
Alternative splicing of pre-mRNA in mammals is one of the most important cellular processes and primarily responsible for the diversity of the human proteome. The accuracy of the splicing process involves the recognition of short sequences at the 5′ splice site (5′SS) and the 3′ splice site (3′SS) within the pre-mRNA that delimit the exon–intron boundaries. Nearly 90% of human genes are subjected to alternative splicing and disruption of the splicing machinery may lead to genetic diseases and cancer (13). Reprogramming of aberrant splicing could provide a novel approach for the development of gene therapies to tackle disease phenotypes. For this purpose, it is necessary to engineer tools that allow precise and timely control of the splicing process. Despite the extraordinary importance of alternative splicing for gene regulation, the number of synthetic splicing devices is rather limited (10,14,15) and suggests that the full potential for their development has been far from realized to date.
In this study, we designed a versatile and highly efficient TetR Splicing Device (TSD) for the conditional control of gene expression in human cells that makes use of an RNA aptamer recognized by the bacterial Tet Repressor TetR (16). The TetR aptamer folds into a stem-loop structure with an internal loop that displays the protein-binding site. The affinity of the TetR aptamer complex is extremely high and corresponds to that of the TetR bound to the tetO operator DNA (17). Binding of tetracycline to TetR leads to conformational changes within the protein resulting in DNA and RNA release, respectively, and consequently allows ligand-dependent reversible binding (17–19). The applicability of the TetR aptamer system for the control of gene expression has been already confirmed by various approaches in different organisms. The aptamer was first used to activate TetR-controlled transcription in Escherichia coli by displacing TetR from its DNA-binding site (16). Next, portability and broader applicability of the system was documented with its successful use in the protozoon Plasmodium falciparum and in yeast (20,21). Further, an additional layer of regulation was added to the TetR aptamer system with the design of a theophylline-responsive TetR aptamer that proved functional (22). We recently used the TetR aptamer in human cells to control miRNA biogenesis (23). These approaches highlight the universal nature and potential of the TetR aptamer regulatory system.
Here, we describe the development of a universally applicable splicing device for the control of gene regulation via intron retention. For this, the TetR aptamer was placed near the 5′SS in such a way that it interferes with initial steps of spliceosomal assembly when it is bound by TetR, thus resulting in intron retention. Repression is fully relieved by the addition of doxycycline (dox) that leads to the release of TetR from the RNA. We demonstrated regulation for different introns and target genes. However, success of regulation was context-dependent, i.e. intron control with the TetR aptamer did vary depending on the specific intron. Therefore, we created a splicing device consisting of an aptamer-controlled intron along with defined flanking exonic sequences that allow reliable regulation. The engineered device is able to predictably control the genetic output of any gene of interest by placing the 292 nt long module upstream of the coding sequence. Amino acids that result from the additional exonic sequences will be removed by a self-cleaving P2A peptide. With this device, we were able to demonstrate applicability for several reporter and human genes. With a high regulatory dynamic range of up to ∼10 fold, the novel regulatory device allows reversible and efficient control of gene expression depending on the presence of a small-molecule ligand. In comparison with existing RNA splicing devices in mammals, our device is thus a considerable improvement.
MATERIALS AND METHODS
Cell culture
HEK293 and HeLa cells (DSMZ, No. ACC-305 and ACC-57, respectively) and HF1-3 cells ‘Flp-In Host Cell Line’ (24) were maintained at 37°C in a 5% CO2 humidified incubator and cultured in Dulbecco's Modified Eagle Medium (DMEM, Sigma Aldrich) supplemented with 10% fetal bovine serum (FBS Superior, Biochrom), 100 U/ml penicillin (PAA, the Cell Culture Company), 100 μg/ml streptomycin (PAA, the Cell Culture Company) and 1 mM sodium pyruvate (PAA The Cell Culture Company). For HF1-3 cells, 150 μg/mL zeocin (Invitrogen) was additionally supplemented to the medium, whereas the medium of the HF1-3 cells harbouring the integrated constructs P2A_GFP control and TSD_GFP contained 200 μg/ml hygromycin (Invitrogen).
Plasmid construction
Plasmids were constructed by standard cloning techniques using overlap extension PCR with Q5 Polymerase (NEB) and restriction and ligation reactions with HF restriction enzymes (NEB) and T4-DNA ligase (NEB), respectively. All vector maps and PCR primers are available upon request. Custom oligonucleotides were synthesized by Sigma Aldrich. FLuc was driven by a CMV promoter and terminated by a bgl polyA site. For stable integration, the gfp gene was cloned into pcDNA5/FRT vector using the unique restriction sites for BamHI and NotI, resulting in pcDNA5/FRT_GFP. The exon from CI4T5, the complete intron with TetR-aptamer and six nucleotides from the second exon were cloned into pcDNA5/FRT_GFP by unique restriction sites for SpeI and MluI, resulting in the construct SP. All modifications of SP were done by overlap extension PCR and amplicon insertion by unique restriction sites for SpeI and MluI. Further, the mCherry or MAX genes were cloned into SP_cr by unique restriction sites for BamHI and NotI, resulting in TSD_mCherry and TSD_MAX-GFP. TetR protein was expressed under the CMV promoter. TetR was modified at the N-terminus with a nuclear localization signal from c-MYC (NH2-CCGGCCGCGAAACGCGTGAAACTGGAT-COOH). The construct expressing TetR-mCherry was cloned by insertion of the mCherry gene by unique restriction sites for KpnI and AgeI.
Genomic integration
HF1-3 cells were transfected with the plasmids pcDNA5/FRT_GFP and pOG44 (recombinase expression plasmid, Invitrogen) at a molar ratio of 1:9 using Lipofectamine® 2000 (Life Technologies) according to manufacturer instructions. The medium was changed 24 h after transfection to DMEM. Cells were selected for stable integration by adding 200 μg/ml hygromycin. After two weeks of selection with hygromycin, cells were sorted by the S3e™ Cell Sorter (Bio-Rad) for GFP positive cells and analysed for stable integration by genomic PCR and sequencing.
Luciferase reporter assay
A total of 60 000 HeLa and 120 000 HEK293 cells per well were seeded into a 24-well plate. These cells were transfected with 50 ng reporter DNA and 100 ng TetR plasmid using 1 μl Lipofectamine® 2000 (Life Technologies) per well according to manufacturer instructions. The medium was changed 2 h after transfection to DMEM (without phenol red) with or without 50 μM doxycycline (Sigma-Aldrich). Luciferase activity was measured after 24 h using the Dual-Glo® (Promega) system. Luminescence values were recorded using the microplate reader Infinite® M200 (Tecan). For each well, the ratio between the FLuc and Renilla luminescence values was calculated. Mean values and standard deviations were calculated from triplicates and normalized to the values of the corresponding vector without aptamer. Each experiment was repeated three times.
Western blotting
For western blot analyses, cells were transfected with the respective plasmids and grown overnight with or without dox. From these cells, either a whole cell extract was prepared or fractions were split into cytosolic and nuclear extract as reported earlier (25). Protein concentration was measured with the Bradford method (Bio-Rad) according to the instructions provided by the manufacturer. Ten microgram protein was loaded onto precast gels (Any kD™ Mini-PROTEAN® TGX™, Bio-Rad) and blotted onto PVDF membranes (Bio-Rad). Primary antibodies targeting TetR (C. Berens, FAU Erlangen) were used in a 1:5000 dilution and anti-actin (Sigma-Aldrich) in a 1:7000 dilution. Horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Jackson Immunoresearch) were used as secondary antibodies. Blots were developed with the ECL system (Bio-Rad). Images were detected using the ChemiDoc Imaging System (Bio-Rad) and quantified using Image Lab Software (Bio-Rad). GFP blots were developed using a LI-COR Odyssey system (Biosciences). Anti-GFP (Sigma-Aldrich) was used as primary antibody in a 1:1000 and anti-actin (I-19:sc-1616, Santa Cruz Biotechnology) in 1:7000 dilution. IRDye® 800CW Donkey anti-Mouse IgG (Santa Cruz Biotechnology) and IRDye® 800CW Donkey anti-Goat IgG (Santa Cruz Biotechnology) for GFP and actin, respectively, were used as a secondary antibodies. Images were detected using the Odyssey Imaging System (Image Studio Lite). Three independent experiments were performed.
RT-PCR analysis
A total of 240,000 HEK293 cells per well were seeded into a 12-well plate. These cells were transfected with 100 ng reporter DNA and 300 ng TetR plasmid using 2 μl of Lipofectamine® 2000 (Life Technologies) per well according to manufacturer instructions. Then, 2 h after transfection, the medium was changed to DMEM with or without 50 μM dox, and the cells were incubated at 37°C and 5% CO2 for 24 h. RNA was isolated using TRIzol® (Life Technologies). Contaminating DNA was removed with the TURBO DNA-free kit (Life Technologies), and the RNA was quality-checked on a 1% (w/v) agarose gel. Next, 1 μg RNA was reverse-transcribed by MuLV (Applied Biosystems) using random hexamers (Fermentas) with the supplied buffers (10 min at 20°C, 15 min at 42°C, 5 min at 99°C). Then, 50 ng cDNA was PCR amplified using Taq polymerase (New England Biolabs, initial denaturation 2 min at 96°C, 30 s at 96°C, 30 s at 54°C, 30 s at 72°C, 35 cycles) and analysed on a 3% (w/v) agarose gel. The amplified products were cloned (CloneJET PCR Cloning Kit, Thermo Scientific) and sequenced for verification. Each RT-PCR was repeated in three independent experiments. Primers are listed in Supplementary Table S1.
qPCR analysis
For qPCR analysis, the Fast SYBR Green Master Mix (Applied Biosystems) was used and the samples were analysed on a StepOnePlus Real-Time PCR machine (Applied Biosystems). The total volume of 20 μL of a standard mix for one reaction was 10 μl SYBR Green Master Mix (2×), 5 μl cDNA and 1 μl of a primer mix of the respective two gene specific primers (10 μM). PCR program for SYBR Green-based qPCR was 20 s at 95°C, 3 s at 95°C, 30 s at 60°C, 40 cycles and for the melting curve: 15 s at 95°C, 60 s at 60°C, 15 s at 95°C. Primers are listed in Supplementary Table S2. Analysis was performed with samples from three independent experiments in technical replicates. Results were calculated according to the ΔΔCt method (26).
Flow cytometry
A total of 120 000 HEK293 or 60 000 HeLa cells per well were seeded into a 24-well plate. The cells were transfected with 50 ng reporter DNA and 150 ng TetR plasmid using 1 μl Lipofectamine® 2000 (Life Technologies) per well according to manufacturer instructions. The medium was changed 2 h after transfection to DMEM (without phenol red) with or without 50 μM doxycycline (Sigma-Aldrich). The GFP and mCherry expression was measured after 24 h using flow cytometry (CytoFlex S, Beckman Coulter). Mean values and standard deviations were calculated from triplicates and normalized to the TetR-mCherry. Each experiment was repeated three times. GFP and mCherry were excited using a 488 nm laser and a 550/30 filter and 561 nm laser and a 610/20 filter, respectively. Cells were analysed using FlowJo (TreeStar Inc., Ashland, OR), and populations were selected by gating out the GFP background signal of untransfected cells. In the case of stable integrated cells, a total of 60 000 HF1-3 cells expressing P2A_GFP control or TSD_GFP construct per well were seeded into a 12-well plate. The cells were transfected with 300 ng TetR plasmid using 2 μl Lipofectamine® 2000 (Life Technologies) per well according to manufacturer instructions. The medium was changed 4 h after transfection to DMEM (without phenol red) with or without 50 μM doxycycline. The GFP and mCherry expressions were measured after 72 h using flow cytometry. Populations were selected by gating out the GFP background signal of untransfected cells with TetR-mCherry.
RESULTS
The TetR aptamer controls intron retention
The 53 nt long TetR aptamer was used to set up a switching device to control intron retention (Figure 1A). Our intention was to place the aptamer close to the 5′SS in a way that binding of TetR to the aptamer efficiently inhibits initial steps of spliceosomal assembly. Consequently, the intron will be retained. Addition of dox then released TetR from the aptamer freeing the 5′SS, which resulted in correct splicing (Figure 1).
Figure 1.
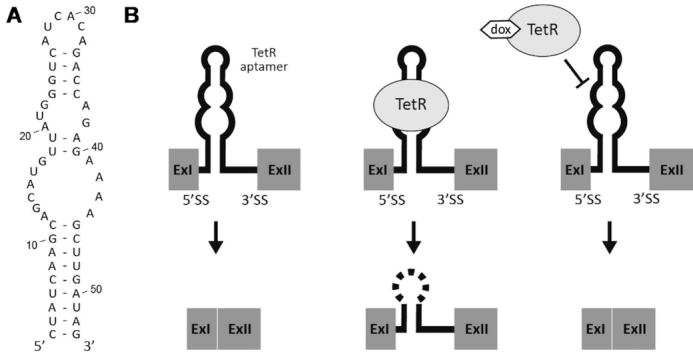
TetR aptamer controls intron retention. (A) Secondary structure of the TetR aptamer. The internal loop (nt 12–22 and 27–44) represents the TetR binding site (16). (B) Sketch of the proposed model. (left) In absence of TetR, the intron is spliced out. (middle) Binding of TetR to the aptamer leads to the intron retention event. (right) Splicing can be restored by addition of dox, which leads to a conformational change of TetR freeing the aptamer. Exons are displayed as grey boxes and the intron with the TetR aptamer as a line.
An intronless firefly luciferase (FLuc) gene was used to establish the system. We introduced a chimeric intron (CI) composed of the 5′SS from the first intron of human β-globin gene and the 3′SS from the intron of an immunoglobulin gene heavy chain variable region (Supplementary Figure S1, (27)). The CI intron was designed in a way that FLuc activity only occurs when the mRNA is correctly spliced. Unspliced mRNA should not be exported. If it does escape from the nucleus, premature stop codons in every reading frame in the intron sequence would either lead to rapid mRNA degradation via the nonsense-mediated decay pathway or to translation of a truncated protein. First, we examined the influence of the position of the intron on FLuc expression. Thirteen different positions mimicking exonic splice features were selected within the FLuc cDNA, which resulted in the constructs CI1-13 (Supplementary Figure S2A). HEK293 cells were transiently transfected with CI1-13. The FLuc activity was monitored 24 h after transfection. We observed only a modest influence of the CI position on FLuc expression. Increased distance of CI from the 5′ end of FLuc resulted in a slight increase of FLuc activity. The addition of 50 μM dox had no influence of FLuc activity (Supplementary Figure S2A).
To analyse if the TetR aptamer can influence 5′SS recognition, the aptamer was inserted into the 13 different constructs CI1-CI13 six nucleotides behind the 5′SS, resulting in the constructs CI1T-CI13T (Figure 2A). HEK293 cells were transiently transfected with these constructs together with a plasmid expressing TetR. TetR itself was expressed from a strong CMV promoter and modified at its N-terminus with a nuclear localization signal (NLS) from c-MYC that significantly increased its localization in the nucleus (Supplementary Figure S3). The insertion of the TetR aptamer into CI influenced FLuc activity to a different degree (Supplementary Figure S2B). The expression of TetR led to significant reduction of FLuc activity in most constructs, yet upon addition of dox FLuc activity was completely restored (exemplified for CI4T5 in Figure 3A). The dox-dependent regulation of luciferase activity (2- to 15-fold) is displayed in Figure 2A.
Figure 2.
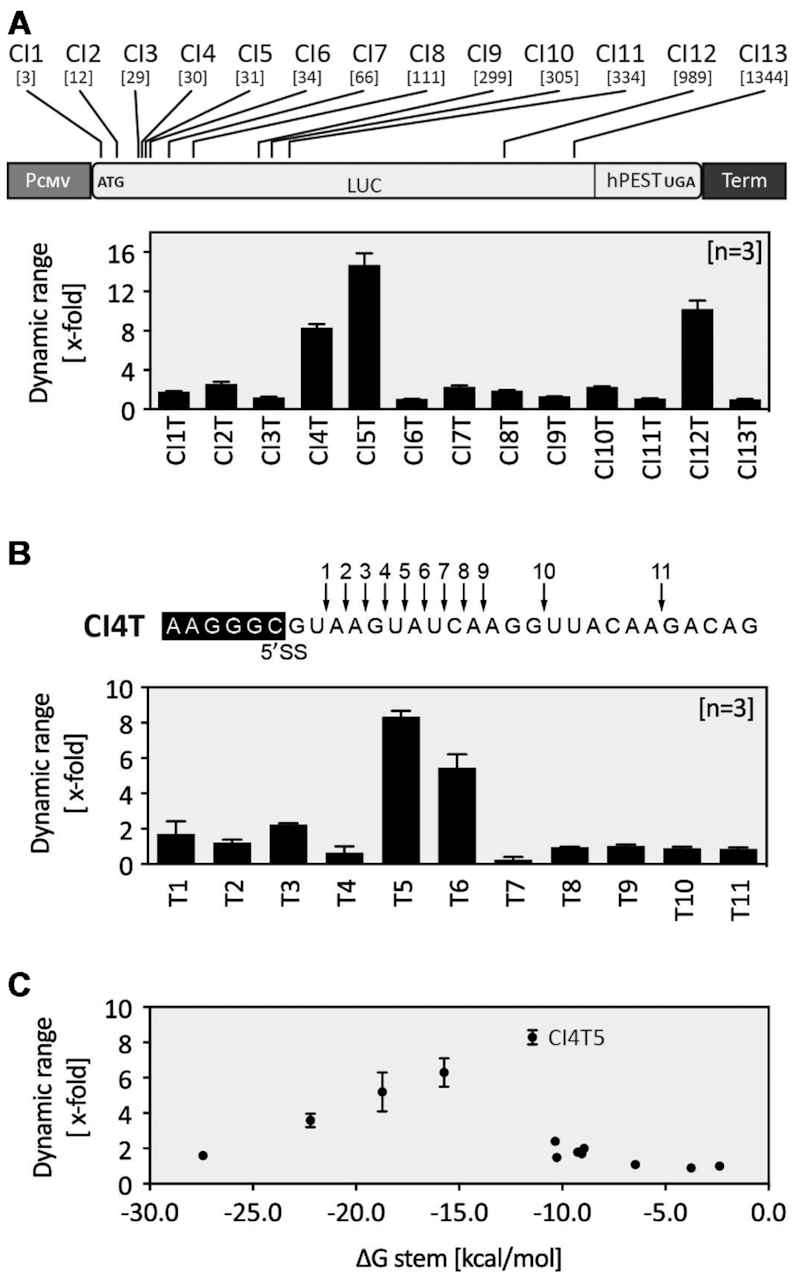
Optimization of the TetR aptamer-controlled intron retention. (A) Displayed are the positions of the intron within FLuc (positions shown in nucleotides relative to the start codon). The effect of the aptamer insertion on the dynamic range is displayed. (B) Based on CI4T, the relative positioning of the aptamer within the intron relative to the 5′SS was analysed. (A and B) Constructs CI1T-CI13T were co-transfected with a plasmid expressing TetR. FLuc activity was measured 24 h after transfection in the absence or presence of 50 μM dox. (C) Dynamic range of regulation depending on stem stability (different ΔG (kcal/mol)). Based on CI4T5, the length and the stability of the aptamer closing stem was modified (sequences of the different closing stems are given in Suppl. Figure S4). All experiments were performed in triplicates and repeated three times. Error bars represent the standard deviation from the means from three independent experiments.
Figure 3.

Characterization of TetR aptamer-controlled intron retention. (A) FLuc activity of CI4T5 in all three states of the proposed model – see Figure 1B. Cells were co-transfected with the construct CI4T5 together with TetR. FLuc activity was measured 24 h after transfection in the absence or presence of 50 μM dox. (B) Quantification of mRNA level for CI4 and CI4T5. Cells were transiently co-transfected with the constructs CI4 or CI4T5 and TetR and treated with or without dox for 24 h. Total RNA was prepared and used for qPCR for quantification. Results were normalized as 2−ΔΔCt. (C) Dose-response curve for the induction of CI4T5 with dox with EC50 as the half maximal effective concentration. The system is half-induced by 80 nM dox and to 90% induced at 0.7 μM dox (D). Aptamer mutations A15U and A20C destroy TetR binding (16), thus leading to a complete loss of regulation. All experiments were performed in triplicates and repeated three times. Error bars represent the standard deviation from the means from three independent experiments.
Only three out 13 tested intron positions (CI4T, CI5T and CI12T) exhibited a significant dynamic range of regulation (8.3-, 14.7- and 10.2-fold, respectively). These constructs have both a high dynamic range but also a low basal activity in the OFF state. Unfortunately, the constructs CI5T and CI12T also showed a very low overall expression level in the ON state. Therefore, we selected the construct CI4T for further characterization. We analysed whether the distance of the TetR aptamer to the 5′SS site had an influence on the dynamic range of regulation. Starting from the position immediately next to the conserved GU nucleotides of the 5′SS, we shifted the aptamer nucleotide-wise downstream, thus generating the constructs T1-11 (Figure 2B, Supplementary Figure S2C. In both figures, the numbers behind the T correspond to the different aptamer insertion sites within the intron). The best regulation was observed for T5 (CI4T5, i.e. intron insertion site 4, aptamer insertion site 5), which by chance was the original insertion site. TetR aptamers placed 9 nt (or more) downstream of the 5′SS had no influence, probably due to free accessibility of the splice site. The insertion of the aptamer six nucleotides behind the 5′SS (position T5) appears to be the best insertion site for regulation, as other introns could also be controlled when the aptamer was placed precisely there (Supplementary Figure S4). We also modified the length and the stability of the aptamer closing stem (Figure 2C and Supplementary Figure S5). Only stem stabilities between −11 and −16 kcal/mol allow efficient regulation. Again, CI4T5 with a ΔG of −11.45 kcal/mol was revealed as the construct with the best switching properties.
Finally, after completion of all optimization steps, the construct CI4T5 emerged as the switch with the best properties, so that we proceeded with a more detailed characterization (Figure 3). We quantified mRNA levels for the construct CI4T5 and compared it to its corresponding construct without aptamer (CI4). The mRNA levels of CI4T5 showed the same 8-fold increase as documented for reporter gene activity, whereas no increase was detectable for CI4 (Figure 3B). Moreover, the system was activated up to 90% with only 0.7 μM dox (Figure 3C). We confirmed that dox had no influence on cell viability at the concentration used (Supplementary Figure S6). We further demonstrated that TetR can also influence the responsiveness of the system in a dosage-dependent manner (Supplementary Figure S7). Finally, the introduction of the two mutations A15U and A20C that destroy the TetR binding (16) led to a complete loss of regulation, indicating that switching is indeed mediated by TetR–aptamer interaction (Figure 3D).
In sum, we developed a TetR aptamer-based splice switch that is able to control intron retention in a FLuc reporter gene in mammalian cells. We optimized the location of the aptamer in respect to splice site, aptamer stability and the location of the intron within the mRNA. Finally, we arrived at a splice switch with an 8-fold dynamic range.
From switch to device: engineering a universal TetR splicing device (TSD)
Under cellular conditions, synthetic regulatory devices are influenced by the sequence environment and/or additional cellular factors, also described as intrinsic and extrinsic noise. Therefore, it is always challenging to transfer regulatory devices from one context into another (28–30). The integration of the TetR aptamer-controlled intron CI from FLuc into a similar position of a GFP gene resulted in only 2-fold regulation (data not shown). This prompted us to develop the sequences of CI4T5 essential for efficient splicing into a regulatory device that can in principle be applied to any gene of interest (GOI) with constant efficiency irrespective of its (genomic) context.
First, we had to define a minimal sequence of CI4T5 important for efficient and consistent switching. Due to the small size of the first exon (30 nt), we decided to take the complete first exon from CI4T5, the intron with the TetR aptamer and the first six nucleotides of the second exon (marked in Figure 4A) and placed it in front of a GFP gene. Thereby the AUG of the GFP gene was deleted and the AUG from the first exon was used instead. The resulting fusion construct was named SP (Supplementary Figure S8A). HEK293 cells were transiently transfected with the construct SP together with a plasmid expressing a TetR-mCherry fusion protein. GFP and mCherry expressions were monitored 24 h after transfection in the absence or presence of dox using flow cytometry. mCherry expression was used to normalize for variation in transfection efficiency. The construct exhibited similar expression levels as the luciferase in the ON and OFF state (18% and 3%, respectively), as well as a dynamic range of 6.6 (Figure 4B).
Figure 4.
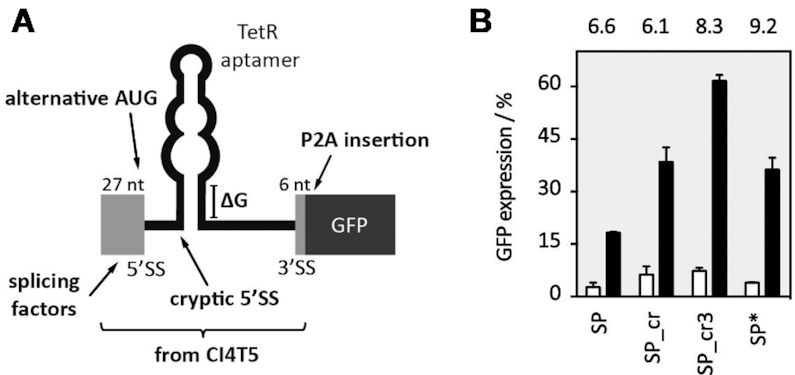
Engineering a universal TSD. (A) Sketch presenting TSD optimization in the context of a GFP reporter gene. The exonic parts taken from the original construct CI4T5 are displayed as light grey, the GFP open reading frame as a dark grey box (B) Optimized constructs were co-transfected with TetR-mCherry. GFP and mCherry expression was measured 24 h after transfection in the absence or presence of 50 μM dox with flow cytometry and mCherry expression was used to normalize for variation in transfection efficiency. A GFP gene with the additional exon-born nucleotides that remain after splicing together with the P2A sequence was set as 100%. GFP fluorescence values are displayed without (white bars) and with dox (black bars). The dynamic range is given above each construct. All experiments were performed in triplicates and repeated three times. Error bars represent the standard deviation from the means from three independent experiments.
We recognized a cryptic 5′SS in the stem of the aptamer that could be responsible for the overall expression level being so low (Supplementary Figure S8A). We mutated this site (G37C and C87G), thus generating the construct SP_cr. GFP expression in the new construct doubled, yet the dynamic range remained nearly unchanged (Figure 4B). We repeated the assessment whether and to what extent the stability of the aptamer influenced regulation and designed two constructs with shorter and one with a longer closing stem (SP_cr1-3, Supplementary Figure S9A). Whereas destabilization of the stem decreased the dynamic range (comparable to the results obtained for FLuc, see Figure 2), stabilization of the stem resulted in improved switching activity (SP_cr3: 8.3-fold) and higher GFP expression of the ON state compared to the SP_cr (Figure 4B). However, the GFP expression of the construct was still only 50% compared to wild type GFP expression.
We speculated that the weak intron removal may be due to exonic splicing silencers (ESS) located in the first exon. RT-PCR data indicated an accumulation of pre-mRNA and low level of mRNA (data not shown). Bioinformatics analyses proposed the existence of putative binding motifs for ESS (31). Mutational analysis exhibited significantly higher GFP expression, both in the OFF and ON state (up to 100% intron removal), supporting indeed the existence of additional splice regulatory sites. However, none of the constructs showed increased regulation (data not shown).
The additional nucleotides from exons 1 and 2 of the construct SP_cr that remain after removal of the intron at the 5′ end of the open reading frame corresponds to additional 13 amino acids attached to the N-terminus of the protein of interest, which may not be tolerated by several proteins. A first attempt to remove these nucleotides was to simply move the start codon towards the 5′SS, but this was not successful (data not shown). Therefore, we inserted a self-cleaving peptide between the device and the start codon to remove the additional exon-born amino acids (construct SP*, Figure 4B). We used the porcine teschovirus-1 2A (P2A) peptide with the sequence GSGATNFSLLKQAGDVEENPGP. The cleavage site of the peptide is located between its last two amino acids (32,33). Consequently, only one proline is added to the N-terminus of the protein of interest. We expect that the majority of proteins will tolerate this minimally invasive change in their protein sequence. To confirm this for GFP, we created a control construct with the P2A sequence attached to the N-terminus of GFP (P2A_GFP). This construct was expected to produce GFP with an additional N-terminal proline after successful cleavage of the peptide (see Supplementary Figure S10). The addition of the P2A domain slightly decreased the GFP expression, yet resulted in an increase of the dynamic range to 9.2-fold (Figure 4B).
Finally, SP* as the best construct was chosen as TSD_GFP (Figure 4B). The complete sequence of the device (292 nt) that can be transferred upstream of the coding sequence of any gene of interest is given in Supplementary Figure S10, which also includes a schematic overview of the different constructs. We have carried out a careful characterization of TSD_GFP. We analysed the switching capability at the mRNA level. Intron retention was visualized by RT PCR using oligonucleotides binding to exons 1 and 2 and quantified it by qPCR (Figure 5A). In addition, the protein level was detected by western blot (Figure 5B). The qPCR data were consistent with GFP expression measured by flow cytometry supported by the western blot experiment. Furthermore, we stable integrated TSD_GFP using the HeLa Flp-In system. Stable integrated TSD_GFP showed the same regulation as in the transient situation (Figure 5C). Finally, we were able to show that TSD_GFP was functional in HEK293, HeLa, A549 and CHO cell lines (transient data, data not shown).
Figure 5.
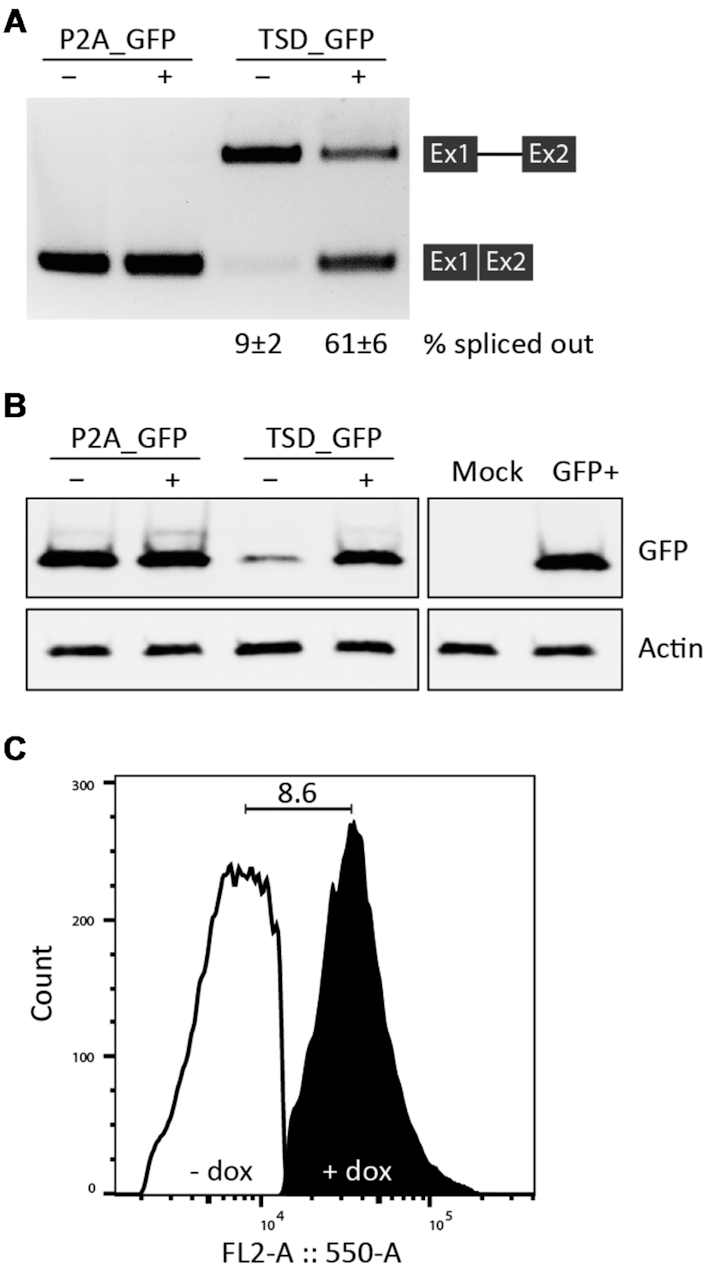
Validation of the TSD. (A) Visualization and quantification of intron retention by RT-PCR and qPCR, respectively. Cells were transiently transfected with the constructs P2A_GFP and TSD_GFP and treated with (+) or without (–) 50 μM dox for 24 h. Total RNA was prepared and used for RT-PCR with primer pairs binding to both exons. qPCR values are given as % spliced out. (B) The protein expression of P2A_GFP and TSD_GFP constructs was analysed by western blot analysis. Anti-GFP was used for GFP expression and anti-Actin was used as a loading control. Mock corresponds to cells transfected with TetR-mCherry, GFP labels the sample of the reporter protein without P2A domain. Experiments were repeated three times. (C) The constructs P2A_GFP and TSD_GFP were stable integrated into HeLa HF1-3 cell line using the Flp-In system. Afterwards, cell lines expressing these constructs were transiently transfected with TetR-mCherry and treated with or without 50 μM dox for 72 h. The histogram shows the results for TSD_GFP. GFP and mCherry were excited using a 488 nm laser and a 550/30 filter and 561 nm laser and a 610/20 filter, respectively. Populations were selected by gating out the GFP background signal of untransfected cells with TetR-mCherry by FlowJo software.
In sum, we have developed a TSD that efficiently controls gene expression in a dox-dependent manner. The insertion of a self-cleaving peptide sequence allows the removal of the additional regulatory sequences.
Dual control of translation and splicing
A prerequisite for the design of synthetic RNA-based regulators is that the combined building elements are sufficiently modular and functional in a different genetic context. We tested if our TSD can be combined with other RNA-based regulation, such as the control at translation level. Translational regulation constitutes an important point of post-transcriptional control of gene expression, allowing the cell to rapidly change the level of a gene product. Previous studies have shown that the introduction of the TetR aptamer into the 5′ untranslated region (UTR) of an mRNA in yeast can enable regulation (20). We first assessed the capacity of TetR aptamer to control translation also in human cells. The aptamer was placed within the 5′ UTR of the GFP reporter gene in a way that TetR binding to the aptamer efficiently inhibits ribosomal scanning (T1 and T2, Supplementary Figure S11A and B). Consequently, the TetR aptamer in the 5′ UTR induces gene expression with a dynamic range of 6.7-fold (construct T2) in the presence of dox.
The construct T2 and the SP* construct were cloned together (Figure 6). The resulting construct T2-SP* showed a significant increase in the dynamic range from 8.4- to 14.7-fold. Therefore, the combination of the regulatory devices resulted in an additive effect on the dynamic range of regulation.
Figure 6.
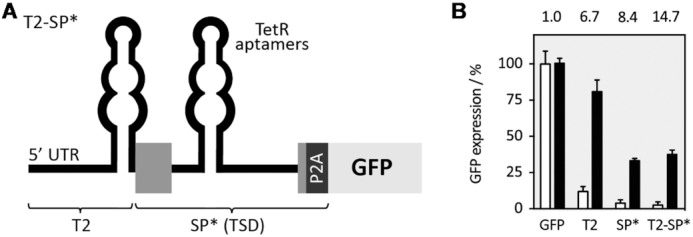
Dual control of splicing and translation. (A) A second TetR aptamer was placed upstream of the switching part of the SP* construct within the 5′ UTR to confer additional control at the level of translation. (B) Constructs T2, SP* and T2-SP* were co-transfected with TetR-mCherry. GFP and mCherry expression was measured 24 h after transfection in the absence or presence of 50 μM dox with flow cytometry and mCherry expression was used to normalize for variation in transfection efficiency. GFP fluorescence values are displayed without (white bars) and with dox (black bars). The dynamic range is given above each construct. All experiments were performed in triplicates and repeated three times. Error bars represent the standard deviation from the means from three independent experiments.
Portability of the TetR splicing device
In a final series of experiments, we assessed portability of the TSD in the context of another reporter gene mCherry and the human transcription factor MAX. The MAX gene was fused with GFP as a reporter at its C-terminus to allow detection of MAX protein expression by flow cytometry. In analogy to the GFP context, the TSD replaced the AUG of the coding sequence of both genes, generating TSD_mCherry and TSD_MAX-GFP, respectively. The constructs were co-transfected with the plasmid expressing TetR. mCherry and (MAX)-GFP expression were measured 24 h after transfection in the absence or presence of 50 μM dox using flow cytometry. The TSD_mCherry construct exhibited 10-fold and MAX-GFP a 5.4-fold regulation (Figure 7). The dynamic range of MAX-GFP was slightly lower compared to the TSD_GFP or TSD_mCherry constructs, maybe due to the fusion of MAX to GFP. In sum, these results clearly demonstrate that our TSD was able to control the expression of several gene of interests, with splicing regulation directly translating into protein output. This once again highlights the genetic modularity and robustness of the device.
Figure 7.

TSD portability. (A) General sketch of the TSD design. The TSD element is located in front of a gene of interest (GOI). Exons are displayed as boxes and the intron as a line, the self-cleaving domain P2A is displayed as a dark grey box. (B) TSD controlling mCherry expression. Cells were co-transfected with plasmids expressing (TSD)_mCherry and TetR. mCherry expression was measured 24 h after transfection in the absence or presence of 50 μM dox with flow cytometry. mCherry fluorescence values are displayed without (white bars) and with dox (black bars) and the dynamic range is given above each construct. (C). TSD controlling MAX transcription factor. MAX was additionally fused with GFP as a reporter at its C-terminus to allow detection of MAX expression by flow cytometry. Cells were co-transfected with plasmids expressing (TSD)_MAX-GFP and TetR-mCherry. mCherry and GFP expression were measured 24 h after transfection in the absence or presence of 50 μM dox. GFP fluorescence values are displayed without (white bars) and with dox (black bars) and the dynamic range is given above each construct. All experiments were performed in triplicates and repeated three times. Error bars represent the standard deviation from the means.
DISCUSSION
Synthetic biology has pioneered transformative genetic devices that enable the study of cellular and molecular biology in cells. Mammalian regulatory devices use diverse mechanisms to allow flexible, precise, and comprehensive control over gene expression and cellular development (34). Fine-tuning of gene expression is critical for many synthetic biology applications, and for this particular purpose, the use of conditional promoters is still the most popular way (35–37). Although bacterial promoters are relatively easy to manipulate, their mammalian counterparts require more complex transcriptional machinery that varies among different cell types and states, thus limiting a general applicability and practical utility of synthetic promoters. However, synthetic promotors may also lead to metabolic burdens and pleiotropic effects. More recently, RNA-based regulatory devices have been explored for modulating mammalian gene expression (6,15,38). Synthetic biology is advancing the design of devices to modulate splicing mechanisms in response to diverse classes of molecules to alter protein sequence, diversity and cellular behaviour.
Motivated by the extraordinary importance of splicing in mammalian cells, we attempted to develop a robust and modular synthetic RNA device for conditional control of gene expression via the control of intron retention for the application in mammalian cells. Splicing control with aptamers has already been successfully demonstrated in mammalian cells, although with modest regulatory dynamic ranges (10,14,15). In this study, we have developed a new system for direct and transcript-specific control of protein expression achieved by control of splicing. No knowledge of transcriptional regulation is required, a small number of defined, genetically-encoded components suffices. In other words, we present a versatile and highly efficient device. A reversible and robust control using the TetR aptamer allowed us to establish the first genetic device to control intron retention. Induction of the TSD is achieved using inexpensive, cell-permeable and non-toxic dox.
Modularity is an essential concept in engineering fields that can be applied to synthetic biology. However, engineered devices may not actually exhibit modular behaviour and often a device's features may change under different conditions (30,39,40). Designing the TSD, we observed an influence of the environment surrounding the aptamer cis-elements on its switching behaviour. First, a cryptic 5′SS in the aptamer closing stem was responsible for low expression of both FLuc and GFP genes. RT-PCR analysis confirmed significant accumulation of pre-mRNA and low mRNA levels for these constructs. Furthermore, inhibition of intron removal was also caused by binding of exonic splicing silencers directly upstream of the 5′SS. Mutations in both the cryptic 5′SS and the predicted ESS motifs exhibited considerably higher GFP expression. However, loss of the ESS binding motifs were associated with weakened regulation properties. Thus, our findings provide considerable additional insight to inform and optimize design of future artificial systems for controlling transgene expression in mammalian cells.
To demonstrate the scope of our results, we took another step and confirmed that our switching module can be easily transferred into any gene of interest. The proper performance of the switching module was assessed in the context of different reporter genes like FLuc, GPF and mCherry and the human transcription factor MAX. The engineered switch shows a high dynamic range up to 10-fold and low basal activity in the repressed state, i.e. favourable traits that have rarely been achieved in mammalian synthetic biology. The dynamic range of regulation obtained in our study represents a major improvement compared to existing RNA-based splicing devices in mammals (10,14,15). Splicing regulation by RNA-based systems have been demonstrated in yeast and mammalian cells. Kim and co-workers have shown splicing inhibition through theophylline-sequestering of branch point sequence and the 3′SS (14,41). In an in vitro splicing assay, a 4-fold reduction of gene expression was observed upon addition of theophylline. In HeLa cells, the effect was not strong enough to influence a switch in splicing site choice. Vogel et al. demonstrated the control of exon skipping in HeLa and HEK293T cells by blocking a 3′SS using a tetracycline-binding aptamer (15). Culler and co-workers have used the MS2 system to control cell fate through exon skipping to regulate the expression of herpes simplex virus-thymidine kinase in human cells (10). However, the regulatory effects of these devices were modest (∼2- to 4-fold). A similar approach has been developed for application in plants (42). In addition, Weigand and co-workers showed control of the 5′SS using the tetracycline-binding aptamer in yeast with up to 16-fold regulation, although transfer attempts into human cells lines to date have been unsuccessful. In this system, the regulation was also significantly increased when the control of splicing was combined with the control of translation (43).
In conclusion, we developed a robust, reversible, modular and portable genetic device for the efficient control of gene expression by intron retention in mammalian cells. We optimized this device for application in the chimeric intron although in principle, it is possible to use the device to control any intron of interest. However, adaptation and optimization of context will be required for each individual intron. Such aptamer-controlled regulation may also be applied to gain insight into diverse cellular mechanisms, such as pre-mRNA splicing, to study the impacts on cellular behaviour. Additionally, the TSD can be easily combined with other regulatory systems to control gene expression at different levels, thus achieving tighter control of gene expression compared to either individual module as demonstrated here by dual control at the level of splicing and translation. In sum, the novel splicing device introduced here will in all likelihood be widely employed for control of gene regulation and for the construction of synthetic circuits in mammalian cells.
Supplementary Material
ACKNOWLEDGEMENTS
Thank you to Anne Caterina Emmerich for her help with western blotting using the LI-COR Odyssey system and Dr Gábor Balázsi for the idea to use the P2A domain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [SFB902(A2)]; European Union's Horizon 2020 research and innovation MetaRNA programme under the Marie Skłodowska-Curie [642738]; LOEWE CompuGene. Funding for open access charge: DFG.
Conflict of interest statement. None declared.
REFERENCES
- 1. Purnick P.E.M., Weiss R.. The second wave of synthetic biology: From modules to systems. Nat. Rev. Mol. Cell Biol. 2009; 10:410–422. [DOI] [PubMed] [Google Scholar]
- 2. Cameron D.E., Bashor C.J., Collins J.J.. A brief history of synthetic biology. Nat. Rev. Microbiol. 2014; 12:381–390. [DOI] [PubMed] [Google Scholar]
- 3. Groher F., Suess B.. Synthetic riboswitches - a tool comes of age. Biochim. Biophys. Acta - Gene Regul. Mech. 2014; 1839:964–973. [DOI] [PubMed] [Google Scholar]
- 4. Hallberg Z.F., Su Y., Kitto R.Z., Hammond M.C.. Engineering and In Vivo Applications of Riboswitches. Annu. Rev. Biochem. 2017; 86:515–539. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y.Y., Galloway K.E., Smolke C.D., Endy D., Rebatchouk D., Daraselia N., Narita J., Knight T., Kouprina N., Larionov V. et al.. Synthetic biology: advancing biological frontiers by building synthetic systems. Genome Biol. 2012; 13:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKeague M., Wong R.S., Smolke C.D.. Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 2016; 44:2987–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berens C., Groher F., Suess B.. RNA aptamers as genetic control devices: The potential of riboswitches as synthetic elements for regulating gene expression. Biotechnol. J. 2015; 10:246–257. [DOI] [PubMed] [Google Scholar]
- 8. Saito H., Fujita Y., Kashida S., Hayashi K., Inoue T.. Synthetic human cell fate regulation by protein-driven RNA switches. Nat. Commun. 2011; 2:160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wroblewska L., Kitada T., Endo K., Siciliano V., Stillo B., Saito H., Weiss R.. Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat. Biotechnol. 2015; 33:839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Culler S.J., Hoff K.G., Smolke C.D.. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010; 330:1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y., Cheong C.G., Tanaka Hall T.M., Wang Z.. Engineering splicing factors with designed specificities. Nat. Methods. 2009; 6:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooke A., Prigge A., Opperman L., Wickens M.. Targeted translational regulation using the PUF protein family scaffold. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:15870–15875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scotti M.M., Swanson M.S.. RNA mis-splicing in disease. Nat. Rev. Genet. 2015; 17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim D.-S., Gusti V., Dery K.J., Gaur R.K.. Ligand-induced sequestering of branchpoint sequence allows conditional control of splicing. BMC Mol. Biol. 2008; 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vogel M., Weigand J.E., Kluge B., Grez M., Suess B.. A small, portable RNA device for the control of exon skipping in mammalian cells. Nucleic Acids Res. 2018; 46:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hunsicker A., Steber M., Mayer G., Meitert J., Klotzsche M., Blind M., Hillen W., Berens C., Suess B.. An RNA aptamer that induces transcription. Chem. Biol. 2009; 16:173–180. [DOI] [PubMed] [Google Scholar]
- 17. Steber M., Arora A., Hofmann J., Brutschy B., Suess B.. Mechanistic basis for RNA aptamer-based induction of TetR. ChemBioChem. 2011; 12:2608–2614. [DOI] [PubMed] [Google Scholar]
- 18. Tiebel B., Aung-Hilbrich L.M., Schnappinger D., Hillen W.. Conformational changes necessary for gene regulation by Tet repressor assayed by reversible disulfide bond formation. EMBO J. 1998; 17:5112–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tiebel B., Radzwill N., Aung-Hilbrich L.M., Helbl V., Steinhoff H.J., Hillen W.. Domain motions accompanying Tet repressor induction defined by changes of interspin distances at selectively labeled sites. J. Mol. Biol. 1999; 290:229–240. [DOI] [PubMed] [Google Scholar]
- 20. Goldfless S.J., Belmont B.J., De Paz A.M., Liu J.F., Niles J.C.. Direct and specific chemical control of eukaryotic translation with a synthetic RNA-protein interaction. Nucleic Acids Res. 2012; 40:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldfless S.J., Wagner J.C., Niles J.C.. Versatile control of Plasmodium falciparum gene expression with an inducible protein–RNA interaction. Nat. Commun. 2014; 5:5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ausländer D., Wieland M., Ausländer S., Tigges M., Fussenegger M.. Rational design of a small molecule-responsive intramer controlling transgene expression in mammalian cells. Nucleic Acids Res. 2011; 39:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Atanasov J., Groher F., Weigand J.E., Suess B.. Design and implementation of a synthetic pre-miR switch for controlling miRNA biogenesis in mammals. Nucleic Acids Res. 2017; 45:e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berens C., Lochner S., Löber S., Usai I., Schmidt A., Drueppel L., Hillen W., Gmeiner P.. Subtype selective tetracycline agonists and their application for a two-stage regulatory system. ChemBioChem. 2006; 7:1320–1324. [DOI] [PubMed] [Google Scholar]
- 25. Kemmerer K., Fischer S., Weigand J.E.. Auto- and crossregulation of the hnRNPs D and DL. RNA. 2017; 24:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu G.J., Wang R.Y., Han D.S., Alter H.J., Shih J.W.. Characterization of the humoral and cellular immune responses against hepatitis C virus core induced by DNA-based immunization. Vaccine. 1999; 17:3160–3170. [DOI] [PubMed] [Google Scholar]
- 28. Kærn M., Elston T.C., Blake W.J., Collins J.J.. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005; 6:451–464. [DOI] [PubMed] [Google Scholar]
- 29. Balázsi G., Van Oudenaarden A., Collins J.J.. Cellular decision making and biological noise: From microbes to mammals. Cell. 2011; 144:910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roßmanith J., Narberhaus F.. Modular arrangement of regulatory RNA elements. RNA Biol. 2017; 14:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rimoldi V., Soldà G., Asselta R., Spena S., Stuani C., Buratti E., Duga S.. Dual Role of G-runs and hnRNP F in the Regulation of a Mutation-Activated Pseudoexon in the Fibrinogen Gamma-Chain Transcript. PLoS One. 2013; 8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y., Wang F., Wang R., Zhao P., Xia Q.. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Sci. Rep. 2015; 5:16273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Ruth Vaseghi H., Qian L., Liu J.. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017; 7:2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mathur M., Xiang J.S., Smolke C.D.. Mammalian synthetic biology for studying the cell. J. Cell Biol. 2017; 216:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tornoe J., Kusk P., Johansen T.E., Jensen P.R.. Generation of a synthetic mammalian promoter library by modification of sequences spacing transcription factor binding sites. Gene. 2002; 297:21–32. [DOI] [PubMed] [Google Scholar]
- 36. Lienert F., Lohmueller J.J., Abhishek G., Silver A.P., Lienert F., Lohmueller J.J., Garg A., Silver P.A.. Synthetic biology in mammalian cells: Next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 2014; 15:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Segall-Shapiro T.H., Sontag E.D., Voigt C.A.. Engineered promoters enable constant gene expression at any copy number in bacteria. Nat. Biotechnol. 2018; 36:352–358. [DOI] [PubMed] [Google Scholar]
- 38. Kobori S., Yokobayashi Y.. Analyzing and tuning ribozyme activity by deep sequencing to modulate gene expression level in mammalian cells. ACS Synth. Biol. 2018; 7:371–376. [DOI] [PubMed] [Google Scholar]
- 39. Del Vecchio D., Ninfa A.J., Sontag E.D.. Modular cell biology: Retroactivity and insulation. Mol. Syst. Biol. 2008; 4:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pantoja-Hernández L., Martínez-García J.C.. Retroactivity in the context of modularly structured biomolecular systems. Front. Bioeng. Biotechnol. 2015; 3:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim D.-S., Gusti V., Pillai S.G., Gaur R.K.. An artificial riboswitch for controlling pre-mRNA splicing. RNA. 2005; 11:1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hickey S.F., Sridhar M., Westermann A.J., Qin Q., Vijayendra P., Liou G., Hammond M.C.. Transgene regulation in plants by alternative splicing of a suicide exon. Nucleic Acids Res. 2012; 40:4701–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weigand J.E., Suess B.. Tetracycline aptamer-controlled regulation of pre-mRNA splicing in yeast. Nucleic Acids Res. 2007; 35:4179–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.