


Abstract
The rapid RNase H1-dependent mislocalization of heterodimer proteins P54nrb and PSF to nucleoli is an early event in the pathway that explains the effects of most toxic phosphorothioate ASOs (PS-ASOs). Using a recently developed NanoLuciferace (NLuc)-based structural complementation reporter system which allows us to observe ASO/protein interactions in real time in live cells, we have determined that safe and toxic PS-ASOs associate with these proteins with kinetics and impact on subcellular localization that differ. Toxic PS-ASOs interact in a complex that includes RNase H1, P54nrb and PSF; but RNase H1/P54nrb complexes were observed in only the cells treated with toxic, but not safe PS-ASOs. In addition, experiments performed in vitro suggest that RNA is also a required component of the complex. The protein–protein interaction between P54nrb and RNase H1 requires the spacer region of RNAse H1, while the P54nrb core domains are required for association with RNase H1. In addition, we have determined that PS-ASOs bind P54nrb via RRM1 and RRM2, while they bind RNase H1 primarily via the hybrid binding domain, however catalytic domain interactions also contribute to overall affinity. These ASO–protein interactions are highly influenced by the chemistry of the PS-ASO binding environment, however little correlation between affinity for specific proteins and PS-ASO toxicity was observed.
INTRODUCTION
The broad and increasing use of oligonucleotides of various types as research tools and platforms for drug discovery and development demand a more thorough understanding of how these pharmacological agents interact with various proteins and how chemical modifications, sequence and structure influence interactions with proteins. We recently reported that protein-binding contributes profoundly to the toxic potentials of PS-ASOs, with toxic PS-ASOs binding more cellular proteins with higher affinity than non-toxic PS-ASOs (1). These sequence and chemistry dependent ASO–protein interactions were shown to correlate with toxic potentials of cEt-modified PS-ASOs in vitro and in vivo altering the stability, function, or distribution of many cellular proteins and resulting in significant toxicity. Toxic, but not safe cEt PS-ASOs, caused rapid mislocalization of the paraspeckle proteins P54nrb and PSF to nucleoli, resulting in nucleolar stress and fragmentation, upregulation of P21 mRNA and activation of caspase activity, and ultimately, apoptotic cell death. In animals, because PS-ASOs accumulate in high concentrations in the liver, the most common manifestation of the toxicity is the death of hepatocytes which results in liver failure most easily demonstrated by increases in liver enzymes, aminoalanine transferase (ALT) and aspartate aminotransferase (AST) and histological changes. Furthermore, in the same manuscript, the substitution of a 2′-methoxy nucleotide at position 2 of the DNA ‘gap’ portion of the PS-ASOs was demonstrated to ablate or greatly ameliorate the toxicity without meaningful loss of potency. Given the breadth of use of antisense technology and drugs and the potential impact of these insights on the therapeutic index of PS-ASO drugs, it is important to better understand how these proteins interact with PS-ASOs and identifying the nucleic acid sequences (and possibly structures) required to assemble these complexes is vital to understanding the role these complexes play in inducing cellular toxicity.
Proteins interact with DNA and RNA through electrostatic interactions, hydrogen bonding, hydrophobic interactions and base stacking (2–5). These forces contribute in varying degrees to proteins binding in a structure and sequence specific or non-sequence specific manner (6). We recently reported the development of a highly sensitive and high throughput BRET affinity assay which relies on the transfer of light energy from a Nanoluc luciferase (Nluc) tagged binding protein acting as the BRET donor, to a fluorescently tagged ASO acting as the BRET acceptor (7). In the current work we use this assay to define to domains of P54nrb with which PS-ASOs interact. We further characterize the contribution of PS-ASO chemistry to these interactions as well as that of the binding environment.
We have also developed a NLuc-based structural complementation reporter system which enables observation of ASO–protein interactions, in real time in live cells (1). Using this assay, we elucidate the kinetics and subcellular localization of ASO–protein interactions for toxic and safe cEt PS-ASOs. In addition, using a variation of the same assay, we follow the association of various proteins with the P54nrb–PS-ASO complex. We show that this complex is made up of not only P54nrb and PSF, but significantly includes RNase H1 in the presence of toxic, but not safe PS-ASO. We finally characterize the interaction of PS-ASOs with RNase H1as well as the protein–protein interaction of P54nrb and RNase H1.
MATERIALS AND METHODS
Preparation of antisense oligonucleotides
Synthesis and purification of phosphorothioate oligonucleotides was performed as described previously (7). All ASOs were ‘gapmers’ 16–20 nucleotides in length with 2′-Fluoro (2′-F), 2′-O-methoxyethyl (MOE) or constrained ethyl (cEt) (8) substitutions at the positions indicated in in Table 1. Standard phosphodiester and phosphorothioate deoxy and ribo oligonucleotides were obtained from Integrated DNA technologies (Coralville, IA, USA).
Table 1.
Sequence of ASOs used in study
| ASO | Length | Sequence | Chemistry | Conjugate |
|---|---|---|---|---|
| N/A | 20 | CTGCTAGCCTCTGGATTTGA | PO DNA/PS DNA | none |
| N/A | 20 | CUGCUAGCCUCUGGAUUUGA | PO RNA/PS RNA | none |
| 766638 | 20 | CTGCTAGCCTCTGGATTTGA | PS 2′-F gap-mer | 3′ Alexa Fluor 594 |
| 766634 | 20 | CTGCTAGCCTCTGGATTTGA | PS MOE gap-mer | 3′ Alexa Fluor 594 |
| 766636 | 20 | CTGCTAGCCTCTGGATTTGA | PS cEt gap-mer | 3′ Alexa Fluor 594 |
| 936532 | 16 | GTCTGTGCATCTCTCC | PS cEt gap-mer | 3′ Alexa Fluor 594 |
| 936533 | 16 | GGCTACTACGCCGTCA | PS cEt gap-mer | 3′ Alexa Fluor 594 |
| 978780 | 16 | GTCTGTGCATCTCTCC | PS cEt gap-mer | 3′ SmBiT peptide |
| 978671 | 16 | GGCTACTACGCCGTCA | PS cEt gap-mer | 3′ SmBiT peptide |
| 464917 | 16 | GTCTGTGCATCTCTCC | PS cEt gap-mer | none |
| 549148 | 16 | GGCTACTACGCCGTCA | PS cEt gap-mer | none |
| 982034 | 16 | GTCTgTGCATCTCTCC | PS cEt gap-mer | none |
| 558807 | 16 | GCATGTTCTCACATTA | PS cEt gap-mer | none |
| 936053 | 16 | GCATgTTCTCACATTA | PS cEt gap-mer | none |
| 482050 | 16 | ATCATGGCTGCAGCTT | PS cEt gap-mer | none |
| 982037 | 16 | ATCAtGGCTGCAGCTT | PS cEt gap-mer | none |
| 508031 | 16 | TGAGGTCCTGCACTGG | PS cEt gap-mer | none |
| 895155 | 16 | TGAGgTCCTGCACTGG | PS cEt gap-mer | none |
| N/A | 20 | CTGCTAGCCTCTGGATTTGA | PO DNA | 3′ Alexa Fluor 594 |
| N/A | 20 | UCAAAUCCAGAGGCUAGCAG | PO RNA compliment | none |
Sequence of ASOs used in study. Substitution with chemically modified nucleotides in gap-mer wings is indicated by bold type. 2′-O-Methyl modified bases are indicated by lower case. Insertion of modified nucleotides is indicated by bold type.
The synthesis of 3′-amino PS-ASOs was performed on an ÄKTAoligopilot synthesizer (GE Healthcare Bioscience) in 40-μmol scale. The oligonucleotide was synthesized on 3′-amino-modifier C7 CPG (Glen Research Inc.) solid support. A solution of phosphoramidites in acetonitrile (0.1 M), and standard sulfurization reagent, coupling reagents and capping reagents were used. For each of the modified analogs 4-fold excess of nucleoside 3′-phosphoramidite were delivered with a 12-min coupling time. The 5′-end dimethoxytrityl group was left on to facilitate purification. Post-synthetically, oligonucleotide bearing solid support was treated with 50% piperidine in DMF for 1 h to remove Fmoc from 3′-amino group, and treated with 1:1 triethylamine: acetonitrile to remove cyanoethyl protecting groups from the phosphorothioate linkages. Subsequently, solid support was treated with aqueous NH4OH (28–30 wt%) at room temperature for 4 h. 10% (v/v) of 40% methylamine in water was added then incubated at room temperature for additional 14 h to cleave ASO from support and remove all protecting groups. Oligonucleotide was purified by ion-exchange chromatography using a NaBr gradient across a column packed with Source 30Q resin. Pure fractions were desalted using HPLC on a reverse phase column. Purity and mass of oligonucleotides were determined using ion-pair LCMS analysis.
SmBiT peptides were conjugated to 3′-amino PS-ASOs as described previously (9) (Supplementary Figure S1). 3′ Amino ASO (1 umol) in 0.1 M sodium tetraborate buffer, pH 8.5 (0.15 ml) was combined with a solution of BCN N-hydroxysuccinimide ester 2 (2 umol, (1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-ylmethyl N-succinimidyl carbonate) in DMSO (0.04 ml) and the reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with water (3 ml) and desalted by HPLC on reverse phase column to yield the3′- BCN carbamate ASO conjugate in 87–90% yield. A solution of 1.5 umol SmBiT peptide (Val-Thr-Gly-Tyr-Arg-Leu-Phe-Glu-Glu-Ile-Leu-Gly-Gly-Ser-Gly-Gly-Lys(N3)-NH2) containing an azide group at the C-terminus (CPC Scientific, 1245 Reamwood Ave, Sunnyvale, CA, USA) in DMSO (0.5 ml) was added and the reaction mixture was stirred at room temperature for 5 h. The reaction mixture was diluted with water (13 ml) and purified by HPLC on a strong anion exchange column (GE Healthcare Bioscience, Source 30Q, 30 μm, 2.54 × 8 cm, A = 100 mM ammonium acetate in 30% aqueous CH3CN, B = 1.5 M NaBr in A, 0–60% of B in 60 min, flow 14 ml min–1). The fraction containing full length conjugates (assessed by LC–MS analysis) were pooled together and concentrated. The residue dissolved in water and desalted by HPLC on reverse phase column to yield the peptide conjugated ASOs in an isolated yield of 75–80%. The PS-ASO conjugates were characterized by ion-pair-HPLC–MS analysis with Agilent 1100 MSD system.
Construction, expression and purification of fusion proteins
Amino-terminal NLuc fusions were created using the vector pFN31K Nluc CMV-neo (Promega). Briefly, P54nrb and RNase H1 were amplified from plasmids RC206688 and RC200595 (Origene) respectively using PCR primers complementary to the full length cDNAs. The forward PCR primer was comprised of sequence complimentary to the sequence following the AUG start codon preceded by an XhoI site for cloning in frame with NLuc, whereas the reverse primer was complementary to the sequence preceding the stop codon followed by an EcoRI site. The PCR amplified product was digested with XhoI and EcoRI then ligated into the pFN31K Nluc CMV-neo vector prepared with the same enzymes using standard techniques. A 6X HIS-tag (CAT CAT CAT CAC CAC CAC) was inserted upstream of the NLuc cassette by site directed mutagenesis using a Q5 Site-Directed Mutagenesis Kit (New England BioLabs) according to the manufacturers protocol. Sequences of PCR primers can be found in Supplementary Table S1. The LgBiT fusions were created by replacing the existing NLuc with LgBiT in the fusion plasmid. The NLuc cassette was excised by digestion with NheI and XhoI. LgBiT was amplified from pBiT1.1-C[TK/LgBiT] vector (Promega) with a forward primer consisting of NheI site Kozak sequence and sequence complimentary to the sequence following the AUG start codon (gcat tcga GCTAGCGCT CACC ATG GTC TTC ACA CTC GAA GAT TTC) and a reverse primer complementary to the sequence preceding the stop codon followed by an XhoI site (GTT CCG AGT AAC CAT CAA CAG T ctcgag gcat tcga).
Deletion mutants, inserts and point mutations were also generated by site directed mutagenesis of the parent NLuc fusion plasmids using a Q5 Site-Directed Mutagenesis Kit (New England BioLabs). Primers were designed to delete the indicated amino acids using the NEBaseChanger tool (Supplementary Table S2).
Gene editing for the generation of the LgBiT-P54nrb HeLa cell line has been described previously (1).
NanoBRET/NanoBiT assays for binding affinity
Fusion proteins were expressed by transfecting the plasmids into 6 × 105 HEK 293 cells using Effectene transfection reagent according to the manufacturer's protocol (Qiagen). Following a 24-h incubation, cells were removed from the plate by trypsinization, washed with PBS, then resuspended in 250 μl Pierce IP Lysis Buffer (Thermo Scientific). Lysates were incubated 30 min at 4°C while rotating, then debris pelleted by centrifugation at 15,000 rpm for 5 min. The fusion protein was purified by adding 20 μl HisPur Ni-NTA Magnetic Beads (Thermo Scientific) and 10 mM imidazol then incubating at 4°C for 2 h. Beads were then washed 4 times with PBS + 10 mM imidazol and 0.01% Tween-20. Fusion protein was eluted from the beads in 100 μl PBS + 200 mM imidazol, followed by dilution with of 200 μl IP buffer.
BRET assays were performed in white 96-well plates as previously described (7). Alexa-linked ASOs at the indicated concentrations were incubated at room temperature for 15 min in 1× binding buffer with 106 RLU/well of Ni-NTA purified NLuc fusion protein. For the PO DNA/RNA heteroduplex, 200 uM of each strand was first annealed in 1× PBS by slow cooling from 95°C. Following the incubation, NanoGlo substrate (Promega) was added at 0.1 μl/well. Readings were performed for 0.3 s using a Glomax Discover system using 450 nm/8 nm band pass for the donor filter, and 600 nm long pass for the acceptor filter. BRET was calculated as the ratio of the emission at 600/450 (fluorescent excitation emission/RLU). For competitive binding assays the Alexa-linked PS-ASO was added at approximately the KD and the unconjugated competing ASO added at the indicated concentrations in 50 μl water. 106 RLU/well of purified fusion protein or whole cell lysate well was then added in 50 μl 2× binding buffer for a final volume of 100 μl. After incubation at room temperature for 15 min, substrate addition and BRET readings were carried out as detailed above. P54nrb 1× binding buffer: 100 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM EDTA, 0.1% NP-40. RNase H1 1 X binding buffer: 50 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM McCl2, 1 mM TCEP.
Western blot analysis
Western blots were carried out as detailed previously (7). Cells in 6 cm plates were transfected with NLuc-fusion plasmids (500 ng) using Effectene transfection reagent (Qiagen). Following a 24-h incubation, cells were removed from the plate by trypsinization, pelleted, washed 1× with PBS, then resuspended in IP Lysis buffer (Pierce) supplemented with 1× Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific), quantitated using a BCA protein assay (Pierce), and separated on a 4–12% gradient SDS-PAGE gel. Proteins were transferred to a nitrocellulose membrane using iBlot Gel Transfer Device (Thermo Fisher Scientific). The membranes were blocked at room temperature for 30 min with blocking buffer containing 5% (w/v) nonfat dry milk in 1× PBS and incubated with primary antibodies in blocking buffer at room temperature for 2 h or 4°C overnight. After washing three times with washing buffer (0.1% Tween-20 in 1× PBS) for 5 min each wash, membranes were incubated with secondary antibodies in blocking buffer at room temperature for 1 h. After washing three times with washing buffer for 5 min each wash, proteins were detected based on enhanced chemiluminescence (Abcam).
Immunofluorescence (IF)
Cells seeded on glass-bottom culture dishes (MatTek) were treated as specified. Cells were fixed at room temperature for 30 min with 4% formaldehyde and permeabilized for 5 min with 0.1% Triton X-100 in 1× PBS. Fixed cells were then blocked in blocking buffer (1 mg/ml BSA in 1× PBS) at room temperature for 30 min, and incubated in blocking buffer with primary antibody NanoLuc pAb, Promega; P54nrb pAb, ab70335 ABCAM) at room temperature for 2 h. After washing three times with washing buffer (0.1% Tween-20 in 1× PBS; 5 min per wash), cells were incubated in blocking buffer with anti Rb Alexa-488 secondary antibody (Abcam ab 150073) at room temperature for 1 h. Finally, cells were washed three times in washing buffer for 5 min each. Images were generated using an LV200 Inverted Microscope (Olympus) fitted with ND10, 472/30 and 520/35 filters and analyzed using cellSens Dimension v1.15 (Olympus).
Live cell imaging of ASO/NanoBiT interactions
Cells seeded on glass-bottom culture dishes (MatTek) were transfected with LgBiT fusion plasmids using Effectene transfection reagent according to the manufacturer's protocol. The following day cells were transfected with SmBiT conjugated ASO at 100 nM in 1 ml Leibovitz's L-15 Medium, no phenol red (Thermo Fisher Scientific) with 6 ug/ml Lipofectamine 2000 and 1 μl Nano-Glo Live cell EX-5455 substrate (Promega). Bright-field (ND1 and ND6 filters) and luminescent images (460/60 filter) were collected from 0 to 2 h at 3-min intervals using an LV200 Luminoview bioluminescent imaging system (Olympus) and processed using cellSens Dimension v1.15 software (Olympus).
RIP-Seq
To construct RIP-seq libraries 5 × 106 HeLa cells were transfected with pHIS-SmBiT-H1 or with HIS-SmBiT-RNaseH1 and LgBiT-P54nrb together using Effectene reagent. The following day cells were transfected with 100 nM PS-ASO 464917 in OptiMEM media with 6ug/μl Lipofectamine 2000. Cells were collected by trypsinization and the pellet lysed in 250 μl IP buffer for 30 min at 4°C. RNA–protein complexes were purified by affinity pull-down with Ni-NTA magnetics beads then the RNA extracted using an RNeasy Mini kit (Qiagen). The purified RNA was then used to generate libraries using a NEXTflex Rapid Directional qRNA-Seq kit according to the manufacturer's protocol (BIOO Scientific). Following 14 rounds of PCR amplification, libraries were size selected using 2% Pippin Prep gel collecting amplicons between 250 and 550 bp. The purified and size selected libraries were then sequenced on an Illumina NextSeq 550 System.
RESULTS
Interaction of ASOs with P54nrb is highly dependent on chemistry and binding environment
P54nrb and PSF, are essential for multiple cellular processes, including transcription, splicing, polyadenylation, nuclear retention, translation and DNA repair (10,11). The structure of the human P54nrb/PSF heterodimer encompasses two RNA binding domains: RRM1, RRM2, as well as NOPS, and coiled-coil core domains. P54nrb and PSF form homo- and heterodimers via their core domains (12) and have been shown to interact with nucleic acids through their RRM domains (11). Although the RRM1 domain of P54nrb is canonical, containing four aromatic residues at conserved positions that are typically essential for RNA binding (13), RRM2 is considered noncanonical with three conserved residues substituted to Thr, Lys and Ile, implying that either the RRM2s do not bind RNA, or that they bind in an unexpected manner.
To investigate the binding interaction of PS-ASOs with P54nrb, we utilized a previously described ASO/protein affinity assay (7) which relies on the transfer of light energy from a Nano luciferase (Nluc) tagged binding protein acting as the BRET donor, to a fluorescently conjugated ASO acting as the BRET acceptor. N-terminal NLuc fusions were constructed for full-length P54nrb along with truncated proteins consisting of only the RNA binding domains (RRM1 or RRM2) or the core domains (NOPS and coiled-coil) (Figure 1A, Supplementary Figure S2A). 106 RLU/well of the immunoprecipitated P54nrb fusion protein were incubated in a 96-well plate with PS-modified, 5-10-5 ‘gap-mer’ PS-ASOs containing 10 deoxyribonucleotides in the middle flanked at both ends by five 2′-constrained ethyl (cEt), 2′-fluoro (2′-F), or 2′-O-methoxyethyl (MOE) conjugated at either the 3′ end with Alexa 594 (Table 1) at concentrations ranging from 10 pM to 1 μM. NLuc substrate was then added and BRET ratios determined as detailed in Materials and Methods. PS-ASOs were determined to bind the full-length protein with a KD of ∼3 nM, however binding to the full-length protein was not strongly influenced by the wing chemistry of the PS-ASO gap-mer (Figure 1B/C, red lines). In contrast, binding to the individual RRM domains was highly influenced by the chemistry of the PS-ASO. For the 2′-F PS-ASO (766638) binding affinity to RRM1 (green lines) was approximately half that of the full-length protein, whereas binding of the cEt (766636) or MOE (766634) PS-ASO to RRM1 was 7–8-fold weaker than to the full-length protein. The amplitude of the BRET signal for binding of RRM2 (blue lines) was much lower than to the full-length protein or to the RRM1 domain. However, BRET efficiency is a measure of distance between the donor and the acceptor, which varies according to an inverse sixth power of the distance between the two molecules (14). It is possible that the tertiary structure of RRM2 alone is such that the distance between the ASO binding site and the NLuc donor is greater than to RRM1 alone or the full-length protein. When binding was normalized for BRET amplitude (Supplementary Figure S2B), it was apparent that binding affinity to RRM2 was significantly greater for the MOE and cEt gap-mers than for the 2′-F. Essentially no binding was observed to the core domains (black lines).
Figure 1.

(A) NLuc-P54nrb fusion constructs. NLuc was fused in frame to the amino-terminus of the P54 cDNA. RNA binding motifs RRM1 and RRM2, and core domain were cloned separately beginning and ending at the indicated amino acid positions. Expression of the fusion protein was confirmed by Western blot (Supplementary Figure S2A). (B) PS-ASOs bind P54nrb primarily via the RRM1 and RRM2 domains in a chemistry dependent manner. NLuc-P54nrb fusion proteins were immunopurified as detailed in Online Methods and subsequently incubated with Alexa 594 conjugated 5-10-5 2′-F, cEt, or MOE gap-mer PS-ASO at concentrations ranging from 10 pM to 1 μM. BRET ratios were determined for full-length P54nrb-NLuc (red), RRM1 (green), RRM2 (blue), or NOPS/coiled-coil (black). Concentration response curves and KD’s ± SEM (nM) are representative of two to three independent experiments. Data normalized to maximum BRET signal may be found in Supplementary Figure S2B. (C) KD’s (nM) are shown in tabular form with green = highest affinity; red = lowest affinity. (D) Competitive binding of PS and PO RNA and DNA ASOs to P54nrb. Unconjugated competing ASOs at 3–10 000 nM were combined with 10 nM Alexa-linked cEt gapmer PS-ASO 766636, then binding to p54nrb evaluated by NanoBRET. (E) Salt dependence of PS-ASO binding to P54nrb domains. KD’s (nM) for binding of cEt gap-mer ASO to p54nrb in 25–400 mM NaCl. Concentration response curves can be found in Supplementary Figure S4.
To evaluate the contribution of the PS linkage and sugar to the interaction of ASOs with P54nrb, a competitive binding experiment was performed with full PS or full PO ASOs containing either ribose or deoxyribose sugar moieties. Competitive binding to full length P54nrb was maximal for the PS DNA ASO and ∼2 orders of magnitude less for the PS RNA ASO (Figure 1D, dashed lines). No competitive binding was detected for either the PO DNA or PO RNA ASOs (solid lines). The results were nearly identical for binding to the RRM1 domain alone (Supplementary Figure S3A) and imply that the PS linkage is essential for ASO binding to P54nrb. In fact, binding affinity appears to be directly proportional to the number of PS linkages in the ASO (Supplementary Figure S3B).
We next evaluated the effect of sodium concentration on the interaction of the cEt gap-mer PS-ASO with P54nrb. Binding of the cEt gap-mer PS-ASO 766636 was evaluated to the full-length P54nrb protein as well as to the RRM1 and RRM2 domains alone in binding buffer with 25–400 mM NaCl. For the full-length P54nrb and the RRM2 domain only, PS-ASO binding was strongly affected by salt concentration with the affinity decreasing as the salt concentration was increased (Figure 1E, Supplementary Figure S4), whereas PS-ASO binding to RRM1 was only slightly affected by changes in salt concentration. These data suggest that ionic interactions are a strong contributor to the binding interaction between RRM2 and PS-ASOs, while affinity for RRM1 may involve other forces, such as base stacking or hydrophobic interactions.
Evaluation ASO/protein interactions and kinetics in live cells
Since binding to P54nrb and other proteins has been shown to significantly affect the toxic potentials of PS-ASOs we sought to develop a NLuc-based structural complementation reporter system which would enable elucidation of the kinetics and localization of protein interactions interactions with toxic and safe cEt PS-ASOs, as well as protein–protein interactions in real time in live cells (1). NanoLuc Binary Technology (NanoBiT) is a two-subunit system based on NLuc that was initially developed for intracellular detection of protein–protein interactions (15). In this assay, Large BiT (LgBiT; 17.6 kDa) and Small BiT (SmBiT; 11 amino acids) NLuc subunits are fused to proteins of interest, and when expressed, the protein–protein interaction brings the subunits into close proximity to form a functional enzyme that generates a bright, luminescent signal. CRISPR was used to generate a stable cell line in which the LgBiT polypeptide was fused in-frame at the amino-terminus of the endogenous P54nrb gene. Expression and localization of the fusion protein was confirmed by immunofluorescent staining of the fixed cells with both NLuc and P54nrb antibodies (Supplementary Figure S5A) and was consistent with the previously reported nuclear distribution of this protein (11).
To confirm the utility of the LgBiT-P54nrb fusion, we constructed an amino-terminal SmBiT fusion to PSF which is known to form a heterodimer with P54nrb (16). A control HeLa cell line and the cell line harbouring the P54nrb-LgBiT fusion were transfected with pSmBiT-PSF. The following day Nluc signal was evaluated using a bioluminescent imaging microscope. No Nluc signal was detected in cells expressing SmB-PSF or in the untransfected LgB-P54nrb cell line however, a strong nuclear NLuc signal was detected in the LgBiT-P54nrb cell line in which SmBiT-PSF was co-expressed (Figure 2A). The kinetics and localization of PS-ASO interaction with the P54nrb/PSF complex were next evaluated. Again, the LgBiT-P54nrb cell line was transfected with pSmBiT-PSF. The following day the cells were transfected with Alexa 594-linked safe (936533) or toxic (936532) cEt PS-ASOs, then visualized at 3-min intervals for a total of 2.5 h. Again, formation of the LgBiT-P54nrb/SmBiT-PSF heterodimer resulted in a clear nuclear NLuc BRET donor signal (Figure 2B, green: Supplementary Figure S5B, video). Fluorescent signal (red) from both of the unbound PS-ASOs was observable in the cytoplasm ∼60 min post-transfection. From the cytoplasm the PS-ASOs traveled almost immediately to the nucleus whereupon association with the P54nrb/PSF complex was detected by BRET emission (yellow). Consistent with previous observations (1), the toxic PS-ASO, 936532, was then localized in the nucleolus by 90 min (Figure 2B; Supplementary Figure S5C, video), while the safe PS-ASO, 936533, remained weakly associated with P54nrb in the nucleoplasm.
Figure 2.

P54nrb-PSF NanoBiT assay. (A) Evaluation of LgBiT-P54nrb expression in CRISPR cell line. HeLa CRISPR LgBiT-P54nrb or HeLa control cells seeded on glass-bottom culture dishes then transfected with a plasmid expressing a SmBiT-PSF fusion protein (pSmB-PSF). The following day NanoGlo substrate was added to the cells and luminescence visualized using an Olympus LV200 Inverted microscope. Left panel, HeLa LgBiT–P54 fusion; center, control HeLa cells transfected with pSmB-PSF; right, HeLa LgB-P54 cells transfected with pSmB-PSF. (B) Safe/Toxic PS-ASO association with P54nrb-PSF heterodimer in live cells. The LgBiT-P54nrb HeLa cell line was transfected with pSmBiT-PSF. The following day cells were treated with Alexa595-linked safe ASO 936533 (left panels) or toxic ASO 936532(right panels) at 100 nM. Cells were then visualized using a bioluminescent imaging microscope at 3-min intervals for a total of 2.5 h collecting brightfield, ASO fluorescence (red) and NLuc bioluminescence (green). Lower panels; brightfield removed. This figure is representative of numerous fields and was repeated 3–4 times. For videos see Supplementary Figure S5B/C. (C) Substitution with 2′-methoxy nucleotide at position 2 of the DNA ‘gap’ portion of toxic PS-ASOs prevents nucleolar accumulation of P54nrb/PSF heterodimer. The LgBiT-P54nrb HeLa cell line was transfected with pSmBiT-PSF. The following day cells were treated at 200 nM for 2.5 h with toxic PS-ASOs (upper panels) or ASOs of the same sequence substituted at gap position 2 with 2′-methoxy (lower panels). Cells were then visualized using a bioluminescent imaging microscope collecting brightfield and NLuc bioluminescence (yellow).
Since the substitution of a 2′-methoxy nucleotide at position 2 of the DNA ‘gap’ portion of toxic PS-ASOs has been demonstrated to greatly reduce toxicity without meaningful loss in potency (1), we also tested several pairs of unmodified and 2′-methoxy substituted cEt gap-mers for localization of the PS-ASO interaction with the P54nrb/PSF in live cells. Again, the LgBiT-P54nrb cell line was transfected with pSmBiT-PSF. The following day the cells were transfected with toxic cEt PS-ASOs with and without 2′-methoxy nucleotide at position 2 (Table 1) followed by addition of NLuc substrate and microscopy. Once again treatment with each of the known toxic unmodified PS-ASOs resulted localization of the LgBiT-P54nrb/SmBiT-PSF heterodimer to the nucleoli (Figure 2C, upper panel), whereas in cells treated with 2′-methoxy-substituted ASOs of the same sequence the LgBiT-P54nrb/SmBiT-PSF heterodimer remained evenly distributed throughout the nucleus (lower panel).
To monitor the interaction of ASOs with proteins more directly, PS-ASOs were synthesized with the SmBiT peptide tethered to the 3′ end. HeLa-LgBiT-P54nrb cells were transfected with safe (978671) or toxic (978780) SmBiT-linked cEt gap-mer PS-ASOs and cells visualized for 1.5–2.5 h at 3-min intervals. For the safe PS-ASO, luminescent association with P54nrb was detected in the nucleus as early as 60 min post-transfection (Figure 3A left panel, video Supplementary Figure S6A). Association of the toxic PS-ASO was also not observed until ∼ 60 min post-transfection, however the P54nrb complex with the toxic PS-ASO was concentrated in the nucleoli rather than in the nucleoplasm. (Figure 3A right panel, video Supplementary Figure S6B). To confirm the nucleolar localization of the toxic PS-ASO, cells were transfected with a GSP fusion plasmid overexpressing the nucleolar marker protein nucleophosmin 1 (NPM1), which was determined to be co-localized with the PS-ASO/P54nrb complex (Supplementary Figure S6C). The kinetics and subcellular localization of several other PS-ASOs previously identified as safe or toxic (1) were also evaluated with essentially identical results (data not shown).
Figure 3.

(A) Kinetics of cEt gap-mer ASO association with P54 in live cells. The LgBiT-P54nrb HeLa cell line was treated with 100 nM safe SmBiT-conjugated cEt ASO 978671 or toxic SmBiT-conjugated cEt ASO 978780. Cells were then visualized using a bioluminescent imaging microscope at 3-min intervals for a total of 2.25 h collecting brightfield and NLuc bioluminescence (blue). Arrows identify nucleolar localization of P54nrb–ASO complex. For video see Supplementary Figure S6A/B. (B) Toxic cEt gap-mer ASO association with P54nrb in live cells treated with siRNAs targeting known ASO interacting proteins. The LgBiT-P54nrb HeLa cell line was treated with an siRNAs targeting RNAse H1, P32, FUS or KPNB1 at 10 nM. Forty eight hours later cells were treated with toxic SmBiT-conjugated cEt ASO 978780 at 200 nM for 2H. Following addition of NanoGlo cells were imaged collecting substrate brightfield and NLuc. These figures are representative of numerous fields and were repeated 2–4 times. Kinetic imaging for ASO 978780 in RNAse H1 reduced cells from 0 to 2 H can be found in Supplementary Figure S7B.
RNase H1 is required for the formation of the nucleolar P54nrb/ASO complex
HeLa-LgBiT-P54nrb cells were next treated 48 h with siRNAs targeting RNase H1, P32, FUS and KPNB1 all proteins which have previously been demonstrated to bind and alter activity of PS-ASOs (1,17,18). Reduction of the targeted protein was confirmed by qRT/PCR and western blot (Supplementary Figure S7A). The cells were treated toxic SmBiT-linked PS-ASO 978780 for 2 h then NLuc emission form the ASO-P54nrb complex visualized. As observed previously, the toxic PS-ASO/P54nrb complex was localized in the nucleolus while the safe PS-ASO was associated with P54nrb throughout the nucleus of cells treated with the control siRNA (Figure 3B). Reduction of P32 or FUS had little effect on the localization of the P54nrb–ASO complex for either PS-ASO. In contrast, when KPNB1 was reduced little P54nrb–ASO association was observed. KPNB1 functions in nuclear protein import, either in association with an adapter protein, such as importin-alpha subunit, or by acting as autonomous nuclear transport receptor (19). These data suggest that PS-ASOs may enter the nucleus via a nuclear receptor after which they may then associate with other proteins such as P54nrb. Interestingly, P54nrb–ASO complex formation was observed after RNase H1 knockdown, however, rather than the toxic PS-ASO complex being localized in the nucleolus, it was dispersed throughout the nucleoplasm. The kinetics of toxic PS-ASO P54nrb interactions were also evaluated in RNase H1 reduced cells and found to be similar to those of the safe PS-ASO (compare videos, Supplementary Figures S6A/S7B).
These data suggest that RNase H1 is required for nucleolar accumulation of the P54nrb–ASO complex. To explore this, we constructed a RNase H1-SmBiT fusion plasmid. HeLa-LgBiT-P54nrb cells were transfected with pH1-SmBiT and treated with safe (549148) or toxic (464917) cEt gap-mer PS-ASOs the following day (Table 1). NanoLuc luminescence from the association of P54nrb with RNase H1 was monitored for 90 min at 3-min intervals. Initially, no NLuc signal was detected with either PS-ASO suggesting that there is little association of P54nrb and RNase H1 in the absence of PS-ASO (Figure 4, video Supplementary Figure S8A). At ∼30 min, NLuc was detected in the nucleoli with the toxic PS-ASO (464917) and maximal association of the PS-ASO with the P54nrb/RNase H1 was reached at ∼60 min. However, only a very slight nuclear signal was detectable at the same time with the safe PS-ASO (549148).
Figure 4.
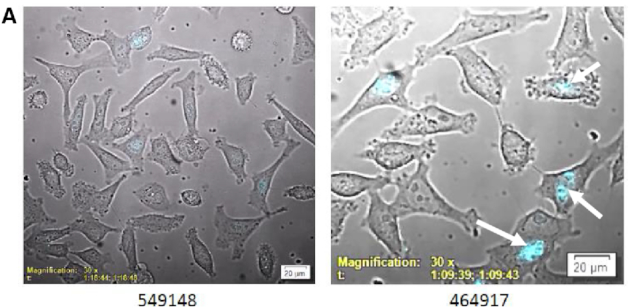
Nucleolar P54nrb-RNAseH1 complex formation occurs in the presence of toxic, but not safe ASO. (A) LgBit-P54nrb HeLa cells were transfected with SmBiT RNAse H1 plasmid. The following day cells were treated with safe (549148) or toxic (464917) ASOs at 200 nM capturing brightfield and bioluminescent NLuc signal (blue) for 1.5 h at 3-min intervals. Arrows identify nucleolar localization of P54nrb/RNAseH1-ASO complex. For video, see Supplementary Figure S8A.
An in vitro NanoBiT assay was used to further explore the interaction between P54nrb and RNase H1. HeLa cells were transfected with HIS-LgBiT-P54nrb or with SmBiT-RNaseH1 and HIS-LgBiT-P54nrb together. The following day cells were transfected with toxic cEt PS-ASO for 2 h then lysates prepared and the P54nrb/RNase H1 complex purified using Ni-NTA beads. In this assay, NLuc luminescence can only be generated when RNase H1 is closely associated with the HIS-tagged P54nrb. As expected, very little luciferase signal was detected with the HIS-LgBiT-P54nrb or SmBiT-RNaseH1 plasmids, however co-expression with of the HIS-LgBiT-P54nrb with SmBiT-RNaseH1 resulted in a significant increase in NLuc signal (Figure 5A, red bars). Furthermore, treatment of the lysates with RNase A reduced the NLuc signal substantially (blue bars), suggesting that RNA may also be a component of the P54nrb/RNaseH1 complex. Experiments in which the HIS-tag was placed on the SmBiT-RNase H1 rather than the LgBiT-P54nrb also produced a significant NLuc signal only when the plasmids were co-expressed (data not shown).
Figure 5.

RNA is associated with the P54nrb-RNaseH1-ASO complex. (A) Treatment with RNase A disrupts P54-RNase H1 association. HeLa cells were transfected with HIS-LgBiT-P54nrb or with SmBiT-RNaseH1 and HIS-LgBiT-P54nrb together. The following day cell lysates were prepared. 1/2 of each lysate was treated with RNase A at 1 μg/ml for 30 min. Following RNase A treatment the P54nrb/RNase H1 complex purified using Ni-NTA beads. Following 3 washes, bound material was eluted with 200 mM imidazole. NanoGlo substrate was then added and NLuc luminescence assayed. Red bars control; blue bars +RNase A. (B) RNAP I/II inhibitors prevent formation of P54-RNase H1 complex. HeLa cells were transfected with LgBiT-P54nrb or with HIS-SmBiT-RNaseH1 and HIS-LgBiT-P54nrb together. The following day cells were treated for 6 h with RNAP inhibitors. Following treatment cell lysates were prepared and the P54nrb/RNase H1 complex purified and quantitated as above red bars, no inhibitor; blue bars, 1 μg/ml Actinomycin D; green bars, 1 μM triptolide; grey bars, 0.5 μM flavopiridol. (C) Treatment of cells with RNAP I inhibitor CX-5461 inhibits formation of P54nrb-RNaseH1-ASO complex. Experiment was carried out as in B except following plasmid transfection cells were treated overnight with 300 nM CX-5461. (D) Identification of P54/RNase H1 associated RNAs by RIP-Seq. HeLa cells were transfected with HIS-SmBiT-RNaseH1 or with HIS-SmBiT-RNaseH1 and LgBiT-P54nrb. The P54nrb/RNase H1 complex was isolated the following day and RNA extracted. Directional, strand-specific libraries were prepared for NEXT-Seq analysis. Genes represented by more than 5 times the number of reads in RNaseH1/P54nrb vs RNaseH1 only are shown.
To identify the type of RNA associated with the P54nrb/RNaseH1 complex, cells were treated with various RNA polymerase (RNAP) inhibitors following co-transfection with HIS-SmBiT-RNaseH1 and LgBiT-P54nrb. Once again, very little NLuc signal was detected in cells transfected with LgBiT-P54nrb only, whereas in the presence of SmBit-H1 a significant increase in signal was observed (Figure 5B, red). Treatment of the cells with 1 ug/ml actinomycin D (blue), 1 uM triptolide (green) or 0.5 uM flavopiridol (gray) each significantly reduced the amount of P54nrb/RNase H1 association as measured by NLuc intensity. Since Act D and triptolide inhibit both RNAP I and II transcription, and flavopiridol inhibits RNAP II elongation and RNAP I processing (20), it is possible that the RNA(s) associated with the complex are either RNAP I or II-dependent. We therefore repeated the experiment, treating the cells with CX5461, a RNAP I-specific inhibitor which effectively reduced formation of the LgBiT-P54nrb/SmBit H1 complex (Figure 5C). We then attempted to identify specifically associated RNAs by RIP-Seq. Again, cells were transfected with HIS-SmBiT-RNaseH1 or with HIS-SmBiT-RNaseH1 and LgBiT-P54nrb together and the P54nrb/RNase H1 complex isolated the following day. RNA was then purified from the complex and directional, strand-specific libraries prepared for NEXT-Seq analysis as detailed in Materials and Methods. Many RNAP II transcripts were identified which generated 5X or more reads in the LgBiT-P54nrb/SmBit H1 library relative to HIS-SmBiT-RNase H1 alone (Figure 5D). However, no individual RNAP II RNA was found to be predominantly associated with the P54nrb/RNase H1 complex. In contrast, 5.8S ribosomal RNA (highlighted) was detected at very high levels and increased ∼2-fold in the presence of both the p54nrb and RNase H1 plasmid. The association of 5.8S RNA with the P54nrb–RNaseH1 complex is consistent with the results of RNAP inhibition and the nucleolar association of toxic PS-ASO/P54nrb/RNase H1 complex.
The association of RNase H1 with the P54nrb–toxic ASO complex was also evaluated by affinity pull-down. HeLa cells were co-transfected with plasmids expressing HIS tagged P54nrb-NLuc or RNaseH1. The following day cells were transfected with safe or toxic PS-ASO for 2 h. Lysates of the cells were then prepared and protein complexes pulled down via the 6X-HIS tagged P54nrb, separated by SDS-PAGE, and transferred to nitrocellulose. Membranes were incubated then probed with antibodies to RNase H1 or P54nrb. A similar amount of P54nrb was pulled down with or without PS-ASO treatment (Supplementary Figure S8B, left panel). In contrast, in the absence of PS-ASO treatment, a weak band corresponding to P54nrb-associated RNase H1 was detected (Supplementary Figure S8B, right panel). In agreement with the live-cell NanoBiT assay, the intensity of the band was significantly increased with administration of the toxic PS-ASO. Interestingly, levels of P54nrb-associated RNase H1 pulled down were slightly reduced in cells treated with the safe PS-ASO as compared to the control. This may be the result of the safe ASO binding RNase H1 without promoting the association with P54nrb, effectively reducing the amount of RNase H1 capable of interacting with P54nrb.
Taken together, these data suggest that the nucleolar accumulation of the toxic PS-ASO may be mediated by increased interactions with RNaseH1 and P54nrb relative to the safe PS-ASO. In fact, the in vitro binding affinity as determined by NanoBRET for the toxic PS-ASO 936532 was 10-fold greater to P54nrb than the affinity for the safe PS-ASO 936533, and ∼2-fold greater to RNase H1 (Supplementary Figure S9A). However, no difference in RNase H1 binding affinity was observed between toxic PS-ASOs and safe 2′-methoxy nucleotide substituted ASOs of the same sequence although one ‘safe’ ASO (982037) did show a 2–3-fold reduction affinity to P54nrb relative to the unmodified parent ASO (482050) (Supplementary Figure S9B).
P54nrb and RNAse H1 interact via specific domains
To determine which domains of RNase H1 are required for the formation of the complex, RNase H1 deletion mutants were generated (Figure 6A). HeLa cells were then co-transfected with pLgBiT-P54nrb and the pSmBiT-H1 deletions. The following day cells were transfected with toxic cEt PS-ASO 936532 and NLuc signal imaged 2H post-transfection. As previously observed, the P54nrb/RNase H1 complex was localized to the nucleolus upon treatment with the toxic PS-ASO (Figure 6B). Deletion of the hybrid binding domain (dHBD) did not affect P54nrb/RNaseH1 complex formation or localization, however no P54nrb/RNase H1 association was detected with deletion of the C-terminal region (dCTD) of RNase H1. A deletion mutant in which only the spacer domain was deleted (dSPCR) also was not able to form a complex with P54nrb suggesting that the spacer region is the point of contact of RNase H1 with P54nrb.
Figure 6.

(A) SmBiT/NLuc-RNAse H1 fusion constructs. SmBiT/NLuc was fused in frame to the amino-terminus of the RNAse H1 cDNA. Deletions were generated by site-directed mutagenesis as indicated in Supplementary Table S2. For the dCTD clone, amino acids 81–286 were deleted, for the spacer- clone amino acids 73–135 were deleted, and for the dHBD clone 1–80 were deleted. Expression of the fusion protein was confirmed by Western blot probed with NLuc pAB. Expected size of NLuc fusions are full length, 52kD; H1-dCTD, 29kD; H1-dHBD, 37.8kD; and dSPC-, 45.7kD. (B) RNAse H1 associates with P54nrb via the spacer domain. The LgBiT-P54nrb HeLa cell line was transfected with pSmBiT-RNAseH1 and with pSmBiT-RNAseH1 domain mutants as depicted in 5A. The following day cells were treated with toxic ASO 936532 at 200 nM for 2 h. Cells were then visualized collecting brightfield and NLuc bioluminescence (blue). (C) P54nrb associates with RNAse H1 via the NOPs/coiled-coil domains. HeLa cells were co-transfected with pSmBiT-RNAseH1 and with pLgBiT-P54nrb or with the indicated pLgBiT-P54nrb domain deletion mutants. The following day cells were treated with toxic ASO 936532 at 200 nM for 2 h. Cells were then visualized as in panel B. Images are representative of numerous fields and were repeated 3–4 times.
A similar set of experiments was performed by generating LgBiT-P54nrb deletion mutants which were co-expressed in HeLa cells with the full length SmBiT RNase H1 then transfected with toxic cEt PS-ASO 936532. Once again, full-length P54nrb was associated with RNase H1 and the PS-ASO in the nucleolus (Figure 6C). Deletion of RRM1, RRM2 or the coiled-coil domains had no effect on the formation or localization of the P54nrb/RNase H1 complex. However, deletion of the NOPS domain reduced the number of P54nrb/RNase H1 complexes significantly and when both the NOPS and coiled-coil domains were deleted, no P54nrb/RNase H1 association was detected. Taken together these data imply that RNase H1 and P54nrb interact with one another via the RNase H1 spacer and the P54nrb core domains, while PS-ASOs appear to interact with this complex via the RRM1 or RRM2 domain of P54nrb.
Evaluation of PS-ASO binding interactions with RNase H1
The interaction of PS-ASOs with RNase H1 was also evaluated. ASO NanoBRET assays were performed with affinity purified NLuc_RNaseH1fusion protein as well as the deletion mutants depicted in Figure 6A. Affinity for the full-length protein was similar for the 2′-F, MOE and cET gap-mers with KD’s ranging between 2 and 4 nM (Figure 7A, red lines). Deletion of the spacer domain had no effect on binding of the 2′-F and increased affinity for cEt and MOE PS-ASOs only slightly (blue lines). Binding to the HBD-only (green lines) was ∼10-fold weaker than to the full-length protein, whereas PS-ASO affinity for the catalytic domain (black lines) was 4–5-fold less than for the HBD. These data suggest that PS-ASOs bind optimally to the full-length protein, but that interactions at the HBD contribute more to overall PS-ASO affinity than do those at the CAT domain. The observations also suggest that the binding isotherm for the full-length protein likely requires interactions with two binding sites, one in the HBD and one in the catalytic site and that affinities of the two sites are within 10-fold of each other as has been reported using other assays (21).
Figure 7.

(A) ASOs bind RNAse H1 at the HBD and catalytic domains, but do not interact with the spacer domain. NLuc-RNAse H1 fusion proteins were immunopurified as detailed in Online Methods and subsequently incubated with Alexa 594 conjugated 5-10-5 cEt gap-mer ASO at concentrations ranging from 10 pM to 1 μM. BRET ratios were determined for full-length NLuc-RNAse H1 (red), Spacer- (blue), HBD-only (green) or CAT-only (black). Concentration response curves and KD’s ± SEM (nM) are representative of 2–3 three independent experiments. B) PS-ASOs interact with the RNAse H1 HBD differently than does the native RNA/DNA heteroduplex. ASO/BRET assays were carried out with purified NLuc-RNAse H1 HBD fusion protein and with the indicated HBD mutant proteins. The purified fusion proteins were incubated with Alexa 594 conjugated 5-10-5 cEt gap-mer ASO or with DNA/RNA heteroduplex of the same sequence with Alexa 594 on the DNA strand at concentrations ranging from 10 pM to 3 μM. Concentration response curves and KD’s ± SEM (nM) are representative of two independent experiments. (C) PS-ASOs and PO heteroduplex interact with the RNase H1 catalytic domain similarly. ASO/BRET assays were carried out with purified NLuc-RNase H1 CAT fusion protein and with the indicated CAT domain mutant proteins. The purified fusion proteins were incubated with Alexa 594 conjugated 5-10-5 cEt gap-mer ASO or with DNA/RNA heteroduplex as above at concentrations ranging from 10 pM to 1 μM. Concentration response curves and KD’s ± SEM (nM) are representative of 2 independent experiments.
We next explored the effect of the binding environment on the interaction of the cEt gap-mer PS-ASO with RNase H1. Binding of 766636 was evaluated to the full-length RNase H1 protein as well as to the HBD and CAT domains alone in binding buffer with 25–400 mM NaCl. For the full-length protein little effect of salt concentration was observed (Supplementary Figure S10). Similarly, binding to the HBD was largely unaffected by salt concentration except at 400 nM NaCl at which a loss of 4-fold in affinity was observed. In contrast, ASO binding to the catalytic domain was found to be highly affected by salt concentration with affinity varying over 50-fold. These similarities in binding between the full-length protein and the HBD further suggest that PS-ASOs interact primarily with the HBD, but it is also likely that CAT domain interactions contribute to PS-ASO affinity as well.
We also explored the binding interaction of PS-ASOs relative to the native PO DNA/RNA heteroduplex. As it has previously been shown that the conserved Trp43, Lys59 and Lys60 residues constitute the binding surface for the HBD of human RNase H1 (22), three mutants were prepared in which alanine was substituted for the conserved lysine residues at positions 59 and 60 (K59A, K60A), the conserved tryptophan at position 43 (W43A), and an additional mutant were generated in which residues Phe58, Lys59 and Lys60 were deleted (dFKK). Binding affinity of the PO heteroduplex as determined by NanoBRET of the affinity purified mutant proteins, was significantly affected the K59A and K60A mutations with a nearly 10-fold reduction in affinity relative to the native RNase H1 HBD, while binding was completely ablated by the W43A substitution and the FKK deletion (Figure 7B, left). In contrast, the FKK deletion had no effect on binding of the PS-ASO and only a 2-fold diminution in affinity was observed with the Trp43, Lys59 and Lys60 to Ala substitutions (right panel).
Similarly, several amino acids have been identified as important contributors to the interaction of the RNase H1 catalytic domain with the PO heteroduplex (23). C-terminal domain only mutants were prepared in which alanine was substituted for Phe at position 151 (N151A), Arg at position 179 (R179A), Asn at position 182 and Gln at position 183 (N182A, Q183A), Phe at position 213 (F213A), Trp at positions 221 and 225 (W221A,W225A), and Thr at position 232 and Ser at position 233 (T232A, S233A). Again, NanoBRET assays were performed to determine the ASO binding affinity of the mutant proteins relative to the parent. Mutations N151A And R179A had the greatest effect on binding of the PO heteroduplex reducing affinity by more than 7-fold and 2-fold respectively (Figure 7C). Other substitutions had little effect on the binding of the heteroduplex to the catalytic domain. A similar effect was observed for binding of the PS gap-mer ASO which was strongly reduced by the N151A and R179A mutations, but not other substituted amino acids. Taken together, these data suggest that the PS-ASO and PO heteroduplex interact similarly, with the catalytic domain, but that the interaction of PS-ASOs in the HBD may involve additional interactions relative to the PO heteroduplex.
DISCUSSION
Recent work from our laboratory (17,24) has shown the PS-ASO’s interact with >50 cellular proteins and that interactions with cellular proteins are critical to cellular uptake and distribution as well as pharmacological activities and toxicities. The interactions of PS-ASOs with many proteins are influenced by sequence, chemistry, length and where in a PS-ASO modifications are placed (7). We recently reported that protein-binding contributes profoundly to the toxic potentials of PS-ASOs, with toxic PS-ASOs binding more cellular proteins with higher affinity than non-toxic PS-ASOs (1). These sequence and chemistry dependent ASO–protein interactions were shown to correlate with toxic potentials of cEt-modified PS-ASOs in vitro and in vivo altering the stability, function, or distribution of many cellular proteins and resulting in significant toxicity. Toxic, but not safe cEt PS-ASOs, caused rapid mislocalization of the paraspeckle proteins P54nrb and PSF to nucleoli, resulting in nucleolar stress and fragmentation, upregulation of P21 mRNA and activation of caspase activity, and ultimately, apoptotic cell death. Given these observations, a more detailed understanding of the sites of PS-ASOs interactions with proteins and the chemical characteristics of those interactions that meaningfully influence the behaviors of PS-ASO’s and the cellular responses to the PS-ASOs is essential. This is the first report of our efforts to answer these important questions.
Interaction of ASOs with P54nrb
Here we present an analysis of PS-ASO interactions with P54nrb and associated proteins in an attempt to better understand the contribution of PS-ASO/protein interactions to differences in cellular toxicity at the molecular level. Our data show that ASOs must contain PS linkages in order to bind P54nrb (Figure 1D, Supplementary Figure S3A) and that the affinity of an ASO for P54nrb is directly proportional to the number of PS linkages in in the ASO (Supplementary Figure S3B). This is the first publication to show that there are two independent binding sites in P54nrb for PS-ASOs. Binding interactions occur primarily via the RRM1 and RRM2 domains, but strikingly, the interaction can be influenced by the chemistry of the PS-ASO (Figure 1C, Supplementary Figure S2B/C). Specifically, PS-ASOs with the 2′-F modification in the wings bind P54nrb primarily via RRM1 interactions, while for PS-ASOs with MOE or cEt modifications, RRM1 and RRM2 domain interactions contribute nearly equally to overall binding. The strength of PS-ASO affinity for RRM2 was also found to be highly influenced by the sodium concentration of the binding buffer, while binding to RRM1 was not strongly affected by changes in sodium concentration (Figure 1E, Supplementary Figure S4). The salt-dependence of a bimolecular association is indicative of the contribution of charge–charge interactions to the free energy of binding. Therefore, the increased effect of added sodium chloride on binding to RRM2 as compared to RRM1 suggests that cEt PS-ASO interactions with RRM2 are primarily electrostatic in nature, involving contacts between basic residues on the RRM2 domain and the phosphorothioate groups in the PS-ASO while interactions with RRM1 may be mediated by hydrophobic or other forces. This is consistent with the observation that the RRM1 interaction appears to dominate the overall binding to the full-length protein for the more hydrophobic 2′-F gap-mer, while RRM1 and RRM2 interactions contribute more equally to overall P54nrb binding of the MOE and cEt gap-mers.
Evaluation ASO/protein interactions and kinetics in live cells
In an attempt to understand differences in the interaction of safe and toxic ASOs with proteins in real time in live cells we used a NanoBiT complementation assay which relies on the association of a SmBiT-linked ASO and LgBiT-linked protein to generate a detectable luminescent signal. A stable cell line was generated in which the LgBiT polypeptide was fused in-frame to P54nrb (Figure 2A). When these cells were treated with a SmBiT peptide conjugated PS-ASO, the binding interaction between the PS-ASO and P54nrb protein could be visualized using a bioluminescent imaging microscope (Figure 3, Supplementary Figure S6). For the safe PS-ASO, association was observed in the nucleus, while for the toxic PS-ASO the interaction was limited to the nucleolus, consistent with previously reported observations (1,25). Following the association in real time, we determined that PS-ASOs can be observed binding P54nrb as early as 45 min after addition to the cells (Figure 3, Supplementary Figure S6). The kinetics observed are consistent with our previously reported data for PS-ASO activity (26).
These observations were further confirmed by monitoring the BRET interaction of fluorescently tagged PS-ASOs with the LgBit-P54nrb/SmBit-PSF complex. Again, the safe ASO was found to interact with the P54nrb/PSF heterodimer throughout the nucleus beginning ∼45–60 min after addition to the cells (Supplementary Figure S5B). Similarly, the toxic ASO was also observed to bind to the P54nrb/PSF heterodimer throughout the nucleus 1 h after addition to the cells, however, within 15 min the complex began localizing to the nucleoli and by 2 h virtually all toxic PS-ASO bound heterodimer was contained within the nucleoli (Supplementary Figure S5C). These results were extended using with a series a series of unmodified and 2′-methoxy substituted cEt gap-mers. Insertion of a single 2′ methoxy at position 2 in the oligodeoxynucleotide gap in PS-ASO’s has previously been demonstrated to ameliorate or ablate PS-ASO toxicity (1,25). Again, all of the toxic ASOs were localized with the P54nrb/PSF heterodimer in the nucleolus, while ASOs of the same sequence made safe as a result of a single 2′-methoxy substitutions remained bound throughout the nucleus (Figure 2C).
RNase H1 is specifically associated with the nucleolar toxic PS-ASOs complex with P54nrb/PSF
In an effort to understand the biochemical nature of the nucleolar complex of toxic PS-ASOs with P54nrb, HeLa-LgBiT-P54nrb cells treated with siRNAs targeting a number of proteins previously demonstrated to bind and alter activity of PS-ASOs (1,17,18). In cells with reduced RNAse H1, the association of the toxic PS-ASO with P54nrb was nuclear rather than concentrated in the nucleoli, resembling that of a safe PS-ASO (Figure 3A, Supplementary Figure S7B). This suggests that toxic PS-ASOs may uniquely interact in a complex that includes RNase H1, P54nrb and PSF. To determine nature of this interaction, RNase H1 was fused with SmBiT peptide, which was co-transfected into HeLa cells with LgBiT P54nrb. Cells were then treated with safe or toxic cEt PS-ASOs. Little association of RNaseH1 and P54nrb was observed in the presence of either the safe or toxic PS-ASO at early timepoints, however after 90 min RNase H1/P54nrb complexes were observed in only the cells treated with the toxic PS-ASO (Figure 4). Deletion of various domains of P54nrb and RNase H1 indicates that the RRM2/NOPS domain of P54nrb and the spacer region RNase H1 are required for the association of these proteins in the presence of toxic PS-ASO (Figure 6). These data imply that RNase H1 plays an important role in ASO induced toxicity, not related to RNA cleavage, but rather spacer domain interactions with the P54nrb complex, thus explaining why knock out of RNase H1 virtually ablates the hepatotoxicity of toxic PS-ASOs (27).
Together these data suggest a model in which P54nrb and RNAseH1 associate via binding interactions with toxic PS-ASOs to form a complex which may be stabilized by interactions with cellular RNA, including 5.8S rRNA (Figure 5), and other proteins such as PSF. On the other hand, while safe PS-ASOs may bind either P54nrb or RNAse H1, they do not appear to support the formation of a complex. Our data also suggest that the phenotype observed with the toxic PS-ASO may be mediated by increased interactions with P54nrb and/or RNaseH1 relative to the safe PS-ASO. Although PS-ASOs can bind both the hybrid binding and catalytic domains of RNase H1, our data suggest that PS-ASOs have unique interactions with the RNase H1 HBD relative to a PO DNA/RNA heteroduplex (Figure 7) which may contribute to the formation of the toxic PS-ASO complex. Indeed, the toxic PS-ASO, 936532, was demonstrated to bind to both RNAse H1 and P54nrb with greater affinity than the safe PS-ASO, 936533 (Supplementary Figure S9A). However, except for 1 pair, there was little difference in binding affinity to P54nrb or RNAse H1 for toxic PS-ASOs and safe PS-ASOs of the same sequence with 2′-methoxy at gap position 2 as measured by NanoBRET affinity assay (Supplementary Figure S9B). While we believe that the difference in affinity for these proteins is a contributing factor to PS-ASO toxicity, other ASO protein interactions may also contribute to the toxic potential of PS-ASOs.
On a global scale we have previously shown that protein binding was significantly less for safe as compared to toxic ASOs (1,25). That there was not a strong correlation between binding to a specific protein and toxicity is consistent with the observation that toxic PS-ASOs interact with multiple proteins and protein complexes (7). In the cell, the NanoBiT assay assesses a very complex binding interaction. Given the fact that the toxic complex includes P54nrb, PFS, RNase H1, 5.8S RNA and perhaps other components, it is unsurprising that there is little direct correlation between toxicity and affinity of PS-ASOs to a single protein component such as P54nrb or RNase H1. The central roles that P54nrb and RNAse H1 play in the much more complex interactions that occur in the cell that are more clearly reflected by the NanoBiT assay than by binding to the purified proteins. Our current focus is to better characterize the participants in this important interaction and to learn more about the transient interactions with other proteins (and possibly RNAs) that may occur enroute to the nucleolus or in the nucleolus itself.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Liang Liang, Wen Shen and Ying Wang for helpful discussion, and Tracy Reigle for help with preparation of the figures.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ionis Pharmaceuticals, Inc. The funder provided support in the form of salaries for authors but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Funding for open access charge: Ionis Pharmaceuticals, Inc.
Conflict of interest statement. None declared.
REFERENCES
- 1. Shen W., Hoyos C.L.D., Migawa M.T., Vickers T.A., Sun H., Low A., Rahdar M., Bell M., Riney S., Murray S.F. et al.. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019; 37:640–650. [DOI] [PubMed] [Google Scholar]
- 2. Luscombe N.M., Laskowski R.A., Thornton J.M.. Amino acid–base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001; 29:2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jayaram B., Jain T.. The role of water in Protein-DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 2004; 33:343–361. [DOI] [PubMed] [Google Scholar]
- 4. Tolstorukov M.Y., Jernigan R.L., Zhurkin V.B.. Protein–DNA hydrophobic recognition in the minor groove is facilitated by sugar switching. J. Mol. Biol. 2004; 337:65–76. [DOI] [PubMed] [Google Scholar]
- 5. Koh Y.Y., Wang Y., Qiu C., Opperman L., Gross L., Tanaka Hall T.M., Wickens M.. Stacking interactions in PUF–RNA complexes. RNA. 2011; 17:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rhodes D., Burley S.K.. Protein–nucleic acid interactions: Editorial overview. Curr. Opin. Struct. Biol. 2000; 10:75–77. [DOI] [PubMed] [Google Scholar]
- 7. Vickers T.A., Crooke S.T.. Development of a quantitative BRET affinity assay for nucleic acid-protein interactions. PLoS One. 2016; 11:e0161930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Kinberger G., Migawa M.T., Gaus H., Bhat B. et al.. Design, synthesis and evaluation of constrained methoxyethyl (cMOE) and Constrained Ethyl (cEt) nucleoside analogs. Nucleic Acids Symp. Ser. 2008; 52:553–554. [DOI] [PubMed] [Google Scholar]
- 9. Østergaard M.E., Yu J., Kinberger G.A., Wan W.B., Migawa M.T., Vasquez G., Schmidt K., Gaus H.J., Murray H.M., Low A. et al.. Efficient synthesis and biological evaluation of 5′-GalNAc conjugated antisense oligonucleotides. Bioconjug. Chem. 2015; 26:1451–1455. [DOI] [PubMed] [Google Scholar]
- 10. Patton J.G., Porro E., Galceran J., Tempst P., Nadal-Ginard B.. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993; 7:393–406. [DOI] [PubMed] [Google Scholar]
- 11. Shav-Tal Y., Zipori D.. PSF and p54nrb/NonO – multi-functional nuclear proteins. FEBS Lett. 2002; 531:109–114. [DOI] [PubMed] [Google Scholar]
- 12. Fox A.H., Bond C.S., Lamond A.I.. P54nrb Forms a Heterodimer with PSP1 That Localizes to Paraspeckles in an RNA-dependent Manner. Mol. Biol. Cell. 2005; 16:5304–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Christophe M., Cyril D., H.‐T. A.F.. The RNA recognition motif, a plastic RNA-binding platform to regulate post‐transcriptional gene expression. FEBS J. 2005; 272:2118–2131. [DOI] [PubMed] [Google Scholar]
- 14. Chen H., Puhl H.L., Koushik S.V., Vogel S.S., Ikeda S.R.. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 2006; 91:L39–L41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dixon A.S., Schwinn M.K., Hall M.P., Zimmerman K., Otto P., Lubben T.H., Butler B.L., Binkowski B.F., Machleidt T., Kirkland T.A. et al.. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016; 11:400–408. [DOI] [PubMed] [Google Scholar]
- 16. Zhang W.W., Zhang L.X., Busch R.K., Farrés J., Busch H.. Purification and characterization of a DNA-binding heterodimer of 52 and 100 kDa from HeLa cells. Biochem. J. 1993; 290:267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang X.-h., Sun H., Shen W., Crooke S.T.. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015; 43:2927–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey J.K., Shen W., Liang X.-h, Crooke S.T.. Nucleic acid binding proteins affect the subcellular distribution of phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 2017; 45:10649–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marfori M., Mynott A., Ellis J.J., Mehdi A.M., Saunders N.F.W., Curmi P.M., Forwood J.K., Bodén M., Kobe B.. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 2011; 1813:1562–1577. [DOI] [PubMed] [Google Scholar]
- 20. Bensaude O. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity. Transcription. 2011; 2:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu H., Lima W.F., Crooke S.T.. Investigating the structure of human RNase H1 by Site-directed mutagenesis. J. Biol. Chem. 2001; 276:23547–23553. [DOI] [PubMed] [Google Scholar]
- 22. Lima W.F., Wu H., Nichols J.G., Prakash T.P., Ravikumar V., Crooke S.T.. Human RNase H1 uses one tryptophan and two lysines to position the enzyme at the 3′-DNA/5′-RNA terminus of the heteroduplex substrate. J. Biol. Chem. 2003; 278:49860–49867. [DOI] [PubMed] [Google Scholar]
- 23. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
- 24. Shen W., Liang X.-h., Sun H., Crooke S.T.. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015; 43:4569–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Migawa M.T., Shen W., Wan W.B., Vasquez G., Oestergaard M.E., Low A., Hoyos C.L.D., Gupta R., Murray S., Tanowitz M. et al.. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019; 47:5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vickers T.A., Crooke S.T.. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015; 43:8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lima W.F., Murray H.M., Damle S.S., Hart C.E., Hung G., De Hoyos C.L., Liang X.-H., Crooke S.T.. Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 2016; 44:5299–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.