


Abstract
A large variety of short biologically active peptides possesses antioxidant, antibacterial, antitumour, anti-ageing and anti-inflammatory activity, involved in the regulation of neuro-immuno-endocrine system functions, cell apoptosis, proliferation and differentiation. Therefore, the mechanisms of their biological activity are attracting increasing attention not only in modern molecular biology, biochemistry and biophysics, but also in pharmacology and medicine. In this work, we systematically analysed the ability of dipeptides (all possible combinations of the 20 standard amino acids) to bind all possible combinations of tetra-nucleotides in the central part of dsDNA in the classic B-form using molecular docking and molecular dynamics. The vast majority of the dipeptides were found to be unable to bind dsDNA. However, we were able to identify 57 low-energy dipeptide complexes with peptide-dsDNA possessing high selectivity for DNA binding. The analysis of the dsDNA complexes with dipeptides with free and blocked N- and C-terminus showed that selective peptide binding to dsDNA can increase dramatically with the peptide length.
INTRODUCTION
Binding of proteins and transcription factors (TFs) to DNA is a fundamental and crucial step for gene regulation (1). These proteins specifically bind DNA to control the complex system of genome expression. However, many mechanisms of DNA recognition and specific binding by TFs are still unknown or not completely understood (2,3). It is known that double-stranded DNA (dsDNA) in classic B-form interacts with different ligands, including low molecular weight organic compounds, peptides, and globular proteins (4). Interactions may occur both in the major and minor grooves of dsDNA. Binding mechanisms can be divided into two categories: where the ligand recognizes a unique pattern of the DNA bases (base readout) and those where the ligand recognizes a sequence-dependent DNA shape (shape readout) (5). Base readout interactions occur due to the formation of hydrogen bonds and hydrophobic contacts between the side chains of amino acids and functional groups of the bases (2). The B-form is the most biologically common form of DNA that has deep major and minor grooves capable of binding different ligands including proteins. dsDNA grooves have significant differences in their hydrogen bond acceptor/donor patterns. (6). DNA interactions are sequence-specific only in the major groove, where ligands can recognize all base pairs. In the minor A/T and T/A grooves, or G/C and C/G, base pairs have the same ligands recognition patterns preventing sequence-specific binding. Shape refers to the recognition of the structural features of the DNA binding site. The main mechanism for reading the DNA shape is a match between the width of the groove and the electrostatic potential of DNA receptor and corresponding characteristics of a ligand, while shape reading in the DNA minor groove is normally carried out by recognizing its geometry with positively charged amino acids (Arg+, Lys+ and His+) (2).
DNA is the target for a wide range of anti-cancer, antimicrobial and antiviral small organic compounds. Covalent binding to DNA is irreversible and causes permanent arrest of transcription leading to cell death. Non-covalent interaction is usually reversible and could be separated in such types as minor groove binders, intercalators, backbone binders, and major groove binders. Many the DNA-targeting ligands (polypyrroles, polyamides, bisbenzimidazoles and bisamidines) bind dsDNA in the minor groove with the preference for A/T-rich regions (7). However, the antineoplastic and antibiotic natural products such as mithramycin and chromomycin bind G/C-rich DNA sequences in the minor groove. Biomacromolecules such as proteins preferably interact with the major groove of dsDNA via hydrogen bond interactions (7). There are also some natural products of DNA major groove binders such as pluramycins, aflatoxins, azinomycins, leinamycins, aminosugars, neocarzinostatins that can specifically bind dsDNA. However, these ligands interact primarily with DNA via intercalation between base pairs (6,7).
Since specific ligand–DNA interactions in the major groove are necessary for inhibition of DNA–protein interactions it is important to understand how small molecules such as short peptides could specifically interact with dsDNA. Certainly, natural products have always been attractive for biomedical applications as leading compounds (6).
Interactions of short peptides with dsDNA are not well studied. PDB currently contains many structures of dsDNA complexes with low molecular weight organic compounds and globular proteins. However, there are only a few complexes of dsDNA with short peptides in the PDB, namely PDB: 2EZF, 2EZD, 2EZE and 2EZG (8). Given high chemical diversity of short peptides and many of their known interactions with globular proteins, it is difficult to imagine that evolution found no effective way to utilize the interactions of short peptides with dsDNA in the living cell.
The reason for such a situation is not completely clear. There is a large variety of short biologically active peptides possessing antibacterial, antitumor, anti-inflammatory and antioxidant activities, involved in the regulation of neuro-immuno-endocrine system, digestive processes, appetite and blood pressure and having analgesic and anti-ageing, pancreo- and nephro-bronchoprotective effects (9–14). Stable complexes of Arg+- and Lys+-rich di- and oligopeptides with dsDNA have been observed since the beginning of 1980s by X-ray analysis (15,16). It was observed that the side chains of Arg+ and Lys+ can bind via electrostatic interactions with the phosphate groups of the DNA main chains stabilizing the B-form conformation. Recently such complexes of dsDNA with a number of dipeptides were also observed (17,18). Therefore, studies of the mechanisms of their biological activity are attracting increasing attention in molecular biology, biochemistry, biophysics and medicine (19).
The overwhelming majority of short peptides existing in cell are products of protein hydrolysis by endo- and exopeptidases. In addition, there are hundreds of short biologically active peptides that are encoded in small open reading frames (smORFs), which biological function remain unknown (20). It was shown in recent studies that the transcriptomes of flies, mice and humans contain thousands of smORFs of different classes that are actively transcribed and encode peptides with unknown functions. It is assumed the existence of several functional classes of smORFs, ranging from inert DNA sequences to transcribed and translated cis-regulators of translation and peptides with a propensity to function as regulators of membrane-associated proteins, or as components of ancient protein complexes in the cytoplasm (21).
Many short peptides easily penetrate through the cell membranes in both directions (22,23). In addition, peptides have large chemical diversity, high specificity in protein binding and low toxicity due to their safe metabolites. Despite peptides are attractive for drug discovery, there are restrictions of their usage due to their high propensity to be rapidly metabolized and low oral bioavailability. There are many works aimed for search of stable peptides that exist in nature and to increase stability of synthetic peptides (24).
Computational methods to construct peptides with the maximum possible stability of the target biologically active conformation and biological activity have been developed and are successfully applied in blocking RecA and SOS-response in bacteria (25).
Synthesis of peptides is relatively easy and inexpensive. These circumstances cause an ever-growing interest to peptides not only on the part of the scientific community and fundamental research, but also the pharmaceutical and food industries, actively developing new drugs and food additives based on short peptides (26). Therefore, in this work, we attempted to systematically analyze the ability of short peptides to bind dsDNA in classic B-form using computational methods of molecular docking and molecular dynamics.
MATERIALS AND METHODS
Preparation for docking
Spatial structures of all possible dipeptides, both with free N- and C-termini and with protective acetyl and amide groups at the N- and C-terminus respectively, consisting of 20 standard amino acid residues, were generated with ICM-Pro software package (Molsoft LLC, USA).
Structures of dsDNA consisting of all possible combinations of 4 bp having unique spatial structures (totally 136 sequences) were generated in the central part of dodecamers flanked by four AT nucleotide pairs at both 3′ and 5′ termini. The resulting dsDNA structures were energy minimized in the ICMFF force field of the ICM-Pro software package using default set of energy parameters for van der Waals, electrostatic, hydrogen bonding, torsion energy interactions and solvation free energy (27).
Virtual ligand screening
The virtual screening of the ligands in target dsDNA binding pocket was performed using the ICM-Dock method, the ICMFF force field and ICM standard protocols for docking of flexible ligands as implemented in the DockScan utility of ICM-Pro software package (Molsoft LLC) (27,28). The calculations were done using SPbPU supercomputing cluster (totally 560 computational cores of ‘Tornado’ supercomputer). The search for peptide conformations in DNA–ligand complexes was carried out with the highest thoroughness corresponding to the number of free torsion angles of ligand (thorough = 30), which was selected on preliminary tests of the reproducibility of the docking results. The most energetically favorable poses of all ligands in every receptor under consideration were selected for further analysis. The complete set of docking results are described in Supplementary Data.
Molecular dynamics
All-atom model protein/DNA complexes were solvated in TIP3P water molecules within a cube box, ensuring a solvent shell of at least 12 Å around the solute. The solute was neutralized with K+ ions and then sufficient K+Cl− ion pairs were added to reach a salt concentration of 150 mM. The ions were initially placed at random, but at least 5 Å away from DNA and 3.5 Å away from one another.
Molecular dynamics (MD) simulations were performed with the AMBER 16 suite (University of California, San-Francisco) of programs (29) using ff14SB force field (30) parameters and the bsc1 modifications for the solute and Joung/Cheatham ion parameters for the surrounding ions running on the multiprocessor cluster of SPbPU (‘Tornado’). MD simulations involve several standard steps including: (a) creation of topology file for all peptide–DNA complexes and preparation of input data for AMBER 16 using the TLEAP tool; (b) construction of hydration models for the protein complex under investigation in a periodic water box with a minimal distance to the water box border of 12 Å; (c) energy minimization and thermodynamic equilibration of the hydrated peptide–DNA complexes and surrounding solvent; (d) MD simulations at constant temperature.
A thermodynamically equilibrated system was used to perform MD simulations at 300 K using the Langevin thermostat with constant pressure (1 atm) and the MD duration of 100 ns with time steps of 2 fs. The model system states were recorded after every 10 ps of MD time for analysis. Neighbor searching was performed at every 10 steps. The PME algorithm was used for electrostatic interactions with the cutoff of 1.0 nm as implemented in AMBER. The cut-off of 1.0 nm was used for van der Waals interactions. SHAKE algorithm was used to constrain the bonds involving hydrogen.
For analyzing of peptide/dsDNA complexes stability first DNA conformations were fitted to their initial МD conformation and then time dependence of the no fit Root Mean Square Deviation (RMSD-NOFIT) of the peptide backbone (heavy atoms only) was used.
Electrophoretic mobility shift assays
A double-stranded 34 bp fluorescein-labelled duplex DNA oligonucleotides were used in electrophoretic experiments. The oligonucleotide sequences were as follows FAM-TCACCAATGAAACCATCGATAGCAGCACCGTAAT and AGTGGTTACTTTGGTAGCTATCGTCGTGGCATTA. The labeled DNA was used at 21 μM (in total nucleotides) in DNA binding reactions containing 10 mM MES (pH 6.4), 10 mM MgCl, 5% (w/v) glycerol. The aforementioned components were incubated at 25°C with 4 mg/ml concentrations of the peptide. After 10 min, 18 μl of each reaction were loaded onto a native 14% polyacrylamide gel and subjected to electrophoresis in TBE buffer (50 mM Tris–borate and 10 mM EDTA). Images were captured using a Bio-Rad ChemiDoc MP system with UV transillumination mode and processed with ImageLab 5.2 software.
RESULTS AND DISCUSSION
DNA receptor characterization
Since dsDNA in its classic B-form is a symmetric double helix molecule, it is obvious that for any particular DNA sequence there is another ‘mirror’ sequence with exactly the same spatial structure. Therefore, we can reduce the computational efforts by eliminating ‘mirror’ sequences from the DNA set under consideration.
In this work docking of dipeptides was done in the central part of dsDNA segment consisting of all possible combinations of four pairs of nucleotides flanked by four pairs of nucleotides with ATAT sequence on both DNA termini (see Figure 1). This is due to the size of the central part of 4 nucleotide pairs in B-form, which is found to have approximately the same size as dipeptide backbone.
Figure 1.
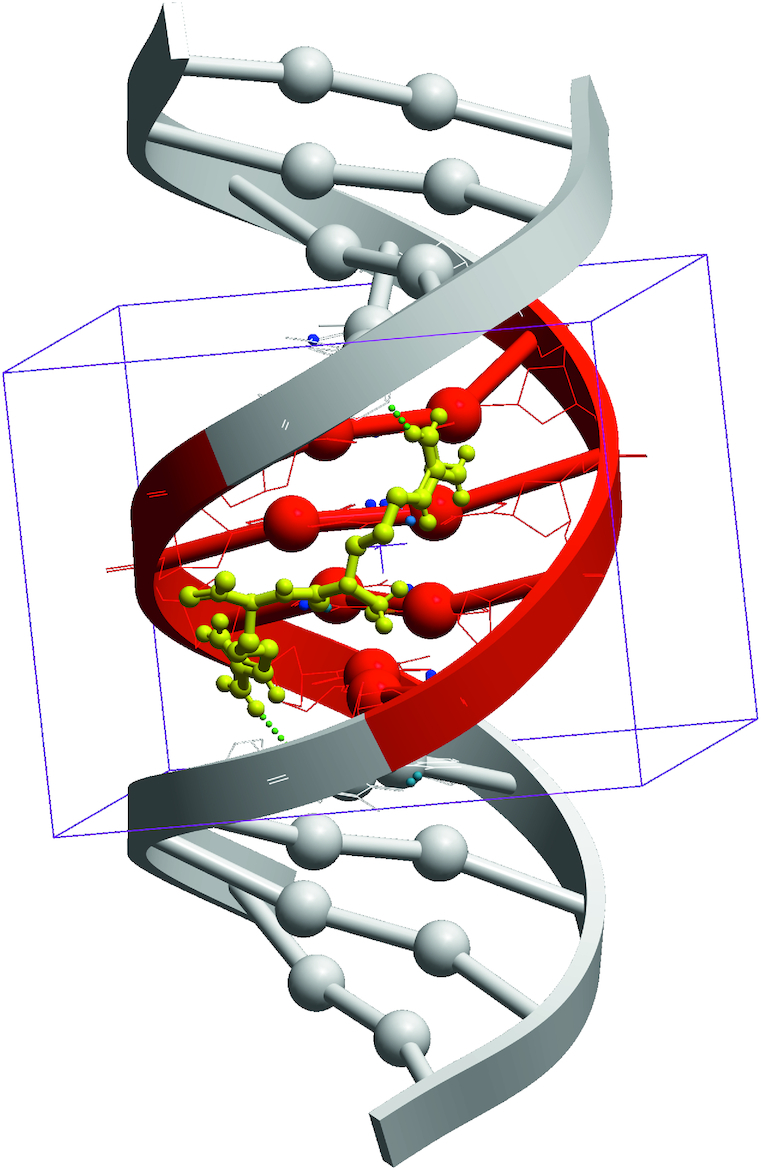
Spatial model of di-Arg+ peptide docked in four pairs of nucleotides (ACGA) of the central part of dsDNA (shown in red).
Based on the symmetry and complementarity of DNA strands in B-form we developed a computer program for the analysis of possible repetitions of the DNA spatial structure in dsDNA segments of four nucleotides length. This analysis showed that there are 136 DNA segments consisting of four pairs of nucleotides generating unique spatial dsDNA structures. The complete list of the sequences is shown in Table 1.
Table 1.
List of all possible dsDNA sequences of four nucleotides length used in this studya
| 1 | AAAA | 18 | ACAC | 35 | AGCG | 52 | ATGA | 69 | GAAC | 86 | GGGG | 103 | TAGG | 120 | TGCG |
| 2 | AAAC | 19 | ACAG | 36 | AGCT | 53 | ATGC | 70 | GAAG | 87 | GGTG | 104 | TATA | 121 | TGGA |
| 3 | AAAG | 20 | ACCA | 37 | AGGA | 54 | ATGG | 71 | GACC | 88 | GTAC | 105 | TATC | 122 | TGGC |
| 4 | AAAT | 21 | ACCC | 38 | AGGC | 55 | ATGT | 72 | GACG | 89 | GTAG | 106 | TATG | 123 | TGGG |
| 5 | AACA | 22 | ACCG | 39 | AGGG | 56 | ATTA | 73 | GAGC | 90 | GTCC | 107 | TCAC | 124 | TGTC |
| 6 | AACC | 23 | ACGA | 40 | AGGT | 57 | ATTC | 74 | GAGG | 91 | GTCG | 108 | TCAG | 125 | TGTG |
| 7 | AACG | 24 | ACGC | 41 | AGTA | 58 | ATTG | 75 | GATC | 92 | GTGC | 109 | TCCC | 126 | TTAA |
| 8 | AACT | 25 | ACGG | 42 | AGTC | 59 | CAAG | 76 | GATG | 93 | GTGG | 110 | TCCG | 127 | TTAC |
| 9 | AAGA | 26 | ACGT | 43 | AGTG | 60 | CACG | 77 | GCAG | 94 | GTTG | 111 | TCGA | 128 | TTAG |
| 10 | AAGC | 27 | ACTA | 44 | ATAA | 61 | CAGG | 78 | GCCG | 95 | TAAA | 112 | TCGC | 129 | TTCA |
| 11 | AAGG | 28 | ACTC | 45 | ATAC | 62 | CATG | 79 | GCGC | 96 | TAAC | 113 | TCGG | 130 | TTCC |
| 12 | AAGT | 29 | ACTG | 46 | ATAG | 63 | CCGG | 80 | GCGG | 97 | TAAG | 114 | TCTC | 131 | TTCG |
| 13 | AATA | 30 | AGAA | 47 | ATAT | 64 | CGCG | 81 | GCTG | 98 | TACA | 115 | TCTG | 132 | TTGA |
| 14 | AATC | 31 | AGAC | 48 | ATCA | 65 | CGGG | 82 | GGAG | 99 | TACC | 116 | TGAC | 133 | TTGC |
| 15 | AATG | 32 | AGAG | 49 | ATCC | 66 | CTAG | 83 | GGCC | 100 | TACG | 117 | TGAG | 134 | TTGG |
| 16 | AATT | 33 | AGCA | 50 | ATCG | 67 | CTCG | 84 | GGCG | 101 | TAGA | 118 | TGCA | 135 | TTTC |
| 17 | ACAA | 34 | AGCC | 51 | ATCT | 68 | CTGG | 85 | GGGC | 102 | TAGC | 119 | TGCC | 136 | TTTG |
aA - adenine, T - thymine, G - guanine, C - cytosine.
After the refinement of the unique segments of dsDNA, the spatial structures of the 136 dsDNA receptors were generated in the form of dodecamers consisting of two repetitive nucleotide pairs (first and last four pairs, AT-rich sites) and alternating sequences of the binding sites listed in Table 1 using specially developed software scripts in the ICM-Pro program language. The 136 receptor spatial structures were minimized in the force field of the ICM-Pro software package (ICMFF) using standard protocols. The binding site for DockScan docking computer software was defined as four central pairs of nucleotides in the dsDNA molecules.
Reproducibility of the docking results
Since all the available docking methods of small flexible ligands in the active sites of receptors of different nature are based on stochastic approaches such as global energy minimization, molecular dynamics (MD) and Monte-Carlo, one of their main problems is the docking results reproducibility. In this work we used the ICM-Dock, one of the most effective and widely used docking methods for virtual screening of flexible ligands, the force field ICMFF and standard parameter set as implemented in ICM-Pro software package. One of the main parameters affecting reproducibility of the docking results by the DockScan algorithm is the thoroughness factor that controls the number of efforts in docking algorithm (recommended range from 1 to 20). At the first step of the work, we investigated the effect of the thoroughness parameter on docking reproducibility using the docking of a test set of 36 dipeptides differing in the number of rotating bonds in 30 different dsDNA structures in classic B-form in order to reproduce the results of docking at least in 90% of the cases. The probability of the docking non-reproducibility with 5 repeated starts of the virtual screening is shown in Figure 2.
Figure 2.
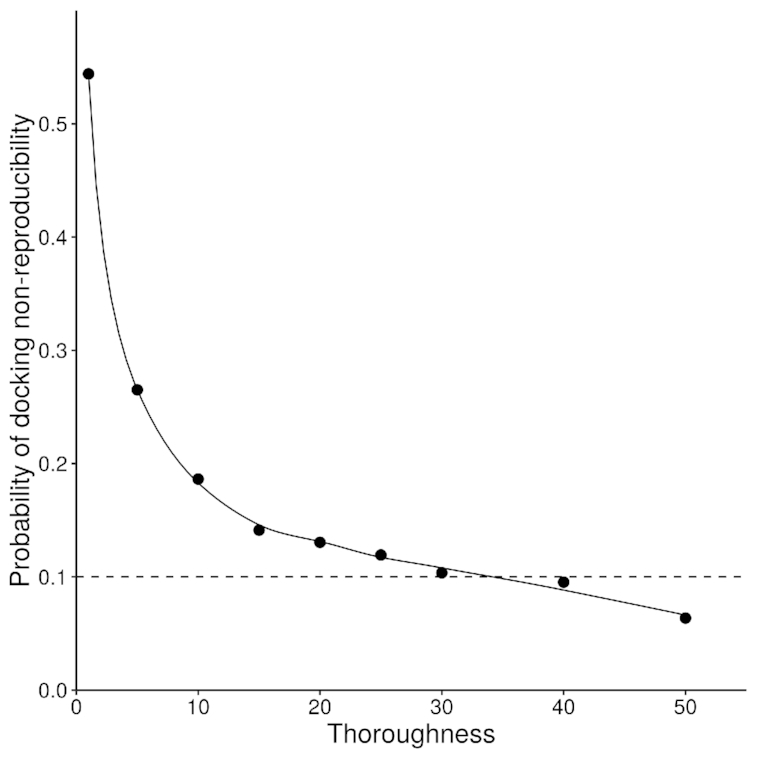
Dependence of the error probability of the docking of a representative test set of dipeptides in 30 different dsDNA structures on the thoroughness parameter of the DockScan algorithm. The dashed line shows the minimum level of the docking results reproducibility used in this work.
Figure 2 shows that the increase of Thoroughness indeed gradually decreases the level of non-reproducibility of the docking results. We set thoroughness parameter to 30 based on the analysis of the plot above, since reproducibility error of 10% is acceptable for our study. In this way, only 10% of all ligand conformations had a standard deviation of the coordinates of heavy atoms of more than 1Å from the conformation with the lowest ICM-Score.
Four hundred dipeptides both with free and blocked termini were docked in the 136 variants of the central parts of dsDNA. In total, 108 800 complexes of dipeptides in dsDNA were analysed. The results of the calculations for all complexes are listed in the Appendix. Figure 3 shows the probability distribution of ICM-Score for the complexes of dsDNA with dipeptides.
Figure 3.

Probability distribution of ICM-Score for the obtained dsDNA–dipeptide complexes. The mean and standard deviations are shown in the figure legend.
The probability distribution functions shown in Figure 3 have a non-symmetric unimodal distribution. The medians of the ICM-Score distributions are −18.5 and −19.2 for dipeptides with free and blocked termini respectively. The non-parametric Mann–Whitney U test show that two scores distributions are not equal (P = ∼0.000002) indicating that the observed differences of the parameters are statistically significant.
Generally, the lower ICM-score, the better the binding energy for small drug-like molecules and proteins under consideration, while the minimum requirement for a binder was found to be −32 (31). Therefore, we used −32 as a threshold score to filter peptides and its conformations in DNA-peptide complexes. As one can see from the ICM-Score distributions, the vast majority of the tested dipeptides and their complexes with dsDNA are higher than −32. However, ∼6.8% of dsDNA-peptides complexes with free peptide termini and ∼4.7% of complexes with blocked peptide termini in this study showed ICM-score ≤ −32, while for some complexes it was even better than −60. Meanwhile, out of 400 peptides 30.8% and 32.5% are capable of binding different dsDNA sequences in its classic B-form. As expected, the lowest ICM-Score values (∼–62) were found for the complexes of different dsDNA with positively charged peptides containing Arg+ and Lys+, such as RR/ACGA shown in Figure 1. Interestingly, that all dipeptide conformations selected by the score are bound in the DNA minor groove.
The complete map of the dsDNA-dipeptide interactions for both dipeptides with free and protected termini is shown in Figure 4. The dipeptides were divided into 25 groups, according to physico-chemical similarity of their amino acid residues in corresponding positions.
Figure 4.
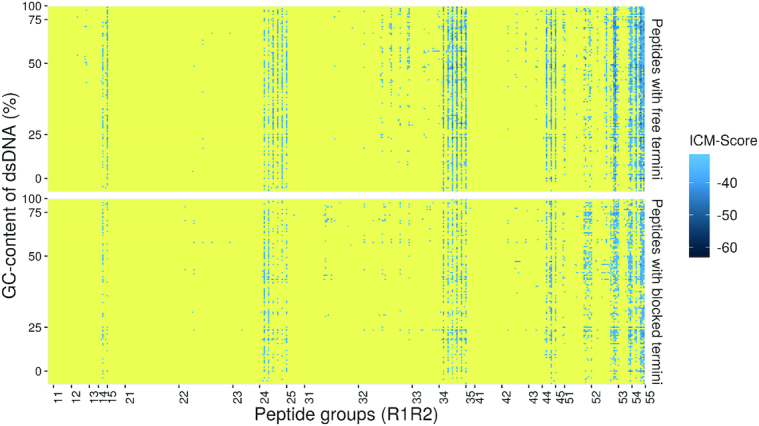
Complete map of the dsDNA-dipeptide interactions for dipeptides with free and protected termini. The complexes with ICM-scores >–32 are shown in yellow, while those with ICM-Scores ≤ −32 are in gradation of blue. DNA sequences are sorted according to their GC-content. All peptides are sorted into 25 groups according to their amino acid composition: 1 – negatively charged (Asp−, Glu−), 2 – uncharged aliphatic (Gly, Ala, Val, Ile, Leu, Pro), 3 – uncharged polar (Ser, Thr, Cys, Met, Asn, Gln), 4 – uncharged aromatic (Phe, Trp, Tyr), 5 – positively charged (Lys+, His+, Arg). The complete set of the data (Supplementary Tables S1 and S2) is available in Supplementary Data.
The legend of the abscissa axis of Figure 4 shows the codes of all the dipeptide groups used in these calculations, the first number is an N-terminal amino acid group number, the last number is a C-terminal amino acid group number. The indexes of the peptide groups denote the amino acid group numbers of the N- and C-terminal amino acid residues. The ordinate axis shows all the 136 dsDNA sequences considered in this work in the order of their GC-content increase. The ICM-Score is indicated in blue: the darker the colour, the lower the ICM-Score value of the dsDNA-dipeptide complex and the higher its stability. Yellow color indicates complexes with ICM-Scores ≥−32 which are considered to be nonbinders.
Based on the obtained distributions of the stable complexes of different peptide groups, it can be concluded that the dipeptides may have different affinity for dsDNA and ability for selective dsDNA binding. As expected, binding to dsDNA is the most common for dipeptides containing positively charged amino acids from the group # 5 due to dsDNA being negatively charged. Non-selective DNA binding of poly-Arg+ and poly-Lys+ has been observed experimentally (18). In our calculations binding of positively charged dipeptides is observed to almost every dsDNA sequence as indicated in practically uninterrupted blue strips of the 25, 35, 45, 52, 53, 54 and 55 groups seen in Figure 4. Sparse blue strips indicate peptides with composition without positively charged amino acids but with polar uncharged (group # 3) and aromatic amino acids (group # 4) and higher selectivity. There are also dipeptides with high selectivity containing amino acids from other groups.
In total, these calculations identified 28 and 29 cases of selective dipeptides having only one specific dsDNA sequence of four nucleotide pairs with docking score lower than −32 for peptides with free and blocked termini, respectively. The selective pairs of peptides and corresponding DNA sequences are listed in Table 2.
Table 2.
Selective complexes of dipeptide with dsDNA
| Peptides with free termini | Peptides with blocked termini | |||||
|---|---|---|---|---|---|---|
| # | Peptide | dsDNA | ICM-Score | Peptide | dsDNA | ICM-Score |
| 1 | KE | TCGA | −35.8 | WT | TGTG | −37.2 |
| 2 | MQ | GTCG | −35.1 | ND | TGCG | −37.2 |
| 3 | TT | ACCA | −33.6 | YY | TCCG | −35.5 |
| 4 | KA | GGGG | −33.4 | WH | CATG | −35.0 |
| 5 | GN | AAGA | −33.4 | KV | TCGA | −34.9 |
| 6 | QP | GCGG | −33.4 | TY | TTGA | −34.6 |
| 7 | SY | CACG | −33.4 | TF | TTGA | −34.0 |
| 8 | HS | TCGA | −33.4 | TA | AGGG | −33.9 |
| 9 | YW | GCGG | −33.3 | HF | TTGA | −33.6 |
| 10 | NF | TTCC | −33.2 | CH | ACTG | −33.6 |
| 11 | YY | GCGG | −33.2 | KA | TGGA | −33.5 |
| 12 | YS | GCGG | −33.1 | QH | GGCC | −33.4 |
| 13 | QM | TTGA | −33.1 | NY | GGGG | −33.3 |
| 14 | WN | TGAG | −33.1 | DK | TACA | −33.1 |
| 15 | KP | GCGG | −32.9 | KD | TCGA | −33.0 |
| 16 | TF | CACG | −32.8 | LQ | CAGG | −32.9 |
| 17 | FK | CACG | −32.7 | VQ | CAGG | −32.8 |
| 18 | VQ | GTCG | −32.7 | QG | TCCG | −32.8 |
| 19 | TG | TCCG | −32.7 | IQ | CAGG | −32.7 |
| 20 | HF | TTCC | −32.5 | GH | ACTG | −32.4 |
| 21 | QS | TCAG | −32.5 | HH | GCTG | −32.3 |
| 22 | GQ | TTCG | −32.3 | SP | GTCG | −32.2 |
| 23 | QT | TCCG | −32.3 | WD | CGGG | −32.2 |
| 24 | HC | TCCG | −32.2 | AH | ACTG | −32.2 |
| 25 | DK | TCAG | −32.2 | GY | TTGA | −32.2 |
| 26 | LQ | GTCG | −32.2 | GN | ACTG | −32.1 |
| 27 | HW | TCGA | −32.2 | YS | CCGG | −32.1 |
| 28 | PK | TGCC | −32.1 | MY | TTGA | −32.1 |
| 29 | QV | GGCC | −32.0 | |||
One can see from the data that most of the selective peptides are uncharged and, in most cases, contain polar and aromatic amino acid residues. However, there are a few cases of positively as well as negatively charged peptides. Interestingly, although some peptides listed in the table contain positively charged Lys+, none of them include Arg+ in their sequence. Most probably this effect is due to the too high affinity of Arg+ to the negatively charged DNA backbone. DNA sequences of the selective complexes have significantly elevated GC contents, 67.8% and 63.4% on average in the case of free and blocked dipeptide termini, respectively. Generally, ICM-Scores of the complexes are in the range from −32 to −37 indicating a moderate level of peptide affinity in the complexes.
Many of found selective peptides listed in Table 2 have not yet been identified in human tissues and have no physiological or cell-signaling effects according to the literature data. However, some of them have biological activity. It is of particular interest to experimentally verify their ability to specifically bind dsDNA.
For example, the KE peptide has the highest affinity to dsDNA among the selective charged dipeptides and different biological activities. In particular, the administration of the KE peptide causes a 2-fold suppression of HER-2/neu (human breast cancer) gene expression in transgenic mice. This suppression is accompanied by a reliable reduction of the tumour diameter (32). It also contributes to a reduced incidence of tumours and a general increase of mean lifespan in animals (33). The effect of the KE peptide on the expression of 15 247 murine heart and brain genes before and after peptide administration was studied with DNA-microarray technology. It was shown that the KE peptide upregulates the expression of 157 genes and downregulates the expression of 23 genes (34). Also, it has been observed that the KE peptide is capable of regulating gene expression of Interleukin-2 in blood lymphocytes, indicating its immunomodulating activity (35). The KE peptide also regulates expression of СD4, CD5 and CD8 glycoproteins in thymic cell culture and induces immune cell differentiation (36). In addition, the KE peptide was found to be capable to activate heterochromatin in the cell nuclei in senile patients and facilitate the “release" of genes suppressed as a result of heterochromatinization of chromosome euchromatin areas (14). Thus, the KE peptide has geroprotective effect in immune cells. Therefore, the KE peptide is a promising immunoprotector, geroprotector and antitumour substance and the molecular mechanism of its biological activity is very important.
The results of the calculations show that the general pattern of dsDNA–dipeptide interactions is similar in both dipeptides with free and blocked termini, but the number of predicted stable complexes of dipeptides with blocked termini is much smaller than that in the case of peptides with peptide free termini. It is noteworthy that standard capping groups at N- (CH3-CO-NH-) and C- (-CO-NH2) peptide termini used in the study reproduce the protein backbone imposing additional conformational constraints when binding to dsDNA. This is the most probable explanation of the remarkable increase of the binding selectivity of the dipeptides with blocked termini. Another possibility is that blocking groups eliminate a positively charged amine at the peptide N-terminus that may be involved in non-specific interactions with DNA phosphates.
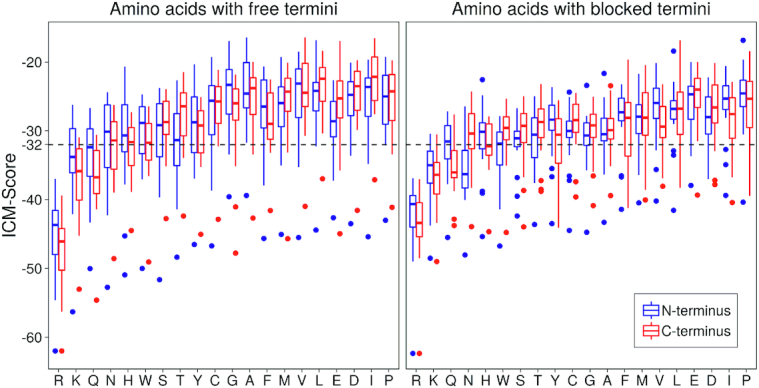
In this study we also investigated the possible position effect of 20 standard amino acids on the ICM-Score of their complexes with dsDNA. Figure 5 shows the best values of the ICM-Score for each dipeptide with charged and blocked termini coded in the colour scheme as in Figure 4. N- and C-terminal amino acid residues of the 400 dipeptides under consideration are represented in the legend of the abscissa and ordinate axis respectively.
Figure 5.
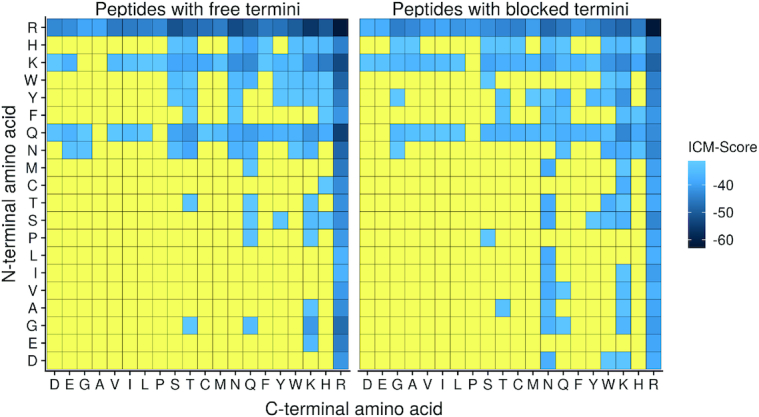
Maps of the best ICM-Score values for each dipeptide. The data are listed in the Supplementary Tables S3 and S4 which are available in Supplementary Data.
As expected from the asymmetry of the maps, it can conclude the inequivalent role of the N- and C-terminal amino acid positions in the dipeptide binding to dsDNA. Only Arg+ is equally favorable in both N- and C-terminal positions. Meanwhile, Lys, Asn, Gln are a bit less favorable than Arg+. Gly, Ser, Thr and aromatic residues also show some binding potential to dsDNA in dipeptides.
According to our calculations, the majority of the dipeptides (∼70%) are not capable of dsDNA binding at all. Amino acid sequences of the dipeptides for which DNA binding seems to be possible are dominated by the positively charged amino acids. However, these peptides generally are not selective. Nevertheless, there are relatively few selective dipeptides containing polar and aromatic residues but their binding constants to dsDNA are not expected to be very high. Our results also show that both the binding selectivity and the constants may quickly improve with the increase in peptide length. Further calculations using longer peptides will help to verify this hypothesis. In addition, many of the dipeptides which showed selective binding to dsDNA may be potentially interesting leads for the development of biologically active ligands.
The charge and amino acid position in dipeptides are important factors affecting their binding abilities dsDNA. Figure 6 shows the ICM-Score distributions for dipeptides. One can see that peptides with positively charged Arg+ and Lys+ have much lower ICM-Scores and therefore are better binders than other amino acids. Also, the positively charged Arg+ and Lys+ residues have better Scores in C-terminal positions, while negatively charged Asp and Glu are more preferable in N-terminal positions. There are some binding possibilities for dipeptides with polar and aromatic residues in N-terminal positions.
Figure 6.
The ICM-Score distributions for dipeptides having 20 standard amino acids at N- and C-terminal positions.
In general, statistical analysis of the data showed that Arg, Lys, Gln are more preferable at C-termini while Asn, Trp, Ser, Thr, Cys, Gly, Ala, Phe, Met, Val, Leu, Glu, Asp, Ile and Pro at N-termini of the dipeptides without blocking groups. The presence of blocking and peptide's termini may totally change this pattern, indicating an important role of charged peptide termini in binding dsDNA.
MD test of stability of peptides/DNA complexes
Based on the analysis of docking scores of dipeptide/DNA complexes one can come to the following general conclusions. Generally, positively charged amino acids of dipeptides increase the conformational stability of dipeptide/DNA complexes and Arg+ is the best DNA binder. N- or C-terminal positions in dipeptides are not equally preferable for different amino acids. Although generally positively charged dipeptides have high affinity to dsDNA they also have low selectivity. While uncharged dipeptides have relatively low affinity and high selectivity to dsDNA. A combination of two amino acid types may lead to a balance between affinity and selectivity to dsDNA in short peptides.
Analysis of the docking results indicated peptides with positively charged residues bind DNA in such a way that positively charged groups remain fixed in the minor groove of dsDNA, while the negatively charged groups tend to be turned out from dsDNA. However, despite this similarity different complexes possess very different ICM-scores and hence different level of conformational stability.
In order to independently verify these observations, series of equilibrium MD simulations in periodic water box were carried out on a representative set of di- and tripeptides complexes with dsDNA. Those include the peptides with positively charged (Arg+, Lys+), negatively charged (Asp–) amino acid residues as well as its combinations. DNA sequences obtained in this work, namely: RR/ACGA (ICM-Score = −62.0), DR/GTCG (-43.5), RD/TTCG (–41.6), DD/ACCC (–21.1), KK/ACGG (-42.1), DK/TCAG (-32.2) and KD/TGAG (–28.7).
Figure 7 shows time dependence of RMSD-NOFIT of the peptide backbone as a measure of the deviation of the ligands from their initial position obtained by docking as well as impact of amino acid position on the conformational stability of the complexes.
Figure 7.
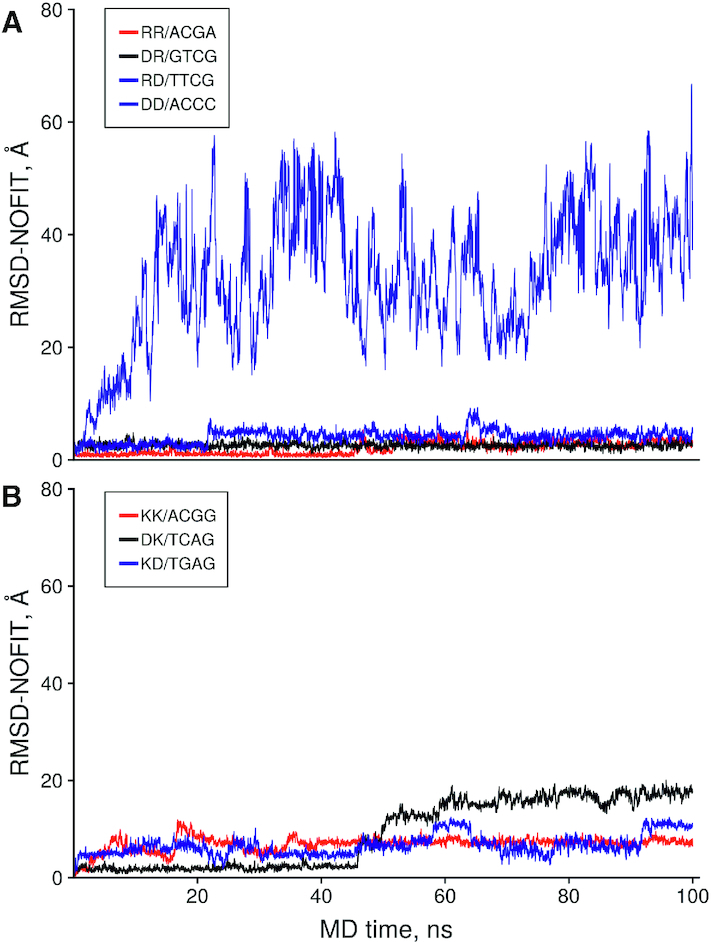
The time dependence of the no fit Root Mean Square Deviation (RMSD) of the Cα, C, N, O atoms of the dipeptide in the representative set of the peptides with negatively and positively charged amino acids with dsDNA. Peptide and DNA sequences of the complexes are shown in the legend.
The results of MD simulations show that, with the possible exception of DD/ACCC having the worst ICM-score, all other complexes are well equilibrated within the simulation time scale. All DNA complexes with peptides containing Arg+ are conformationally stable. As expected, RR/ACGA with the lowest ICM-Score of −62 exhibits only minimal conformational transitions from its initial position. On the other hand, DNA complexes with peptides containing Lys+ show significantly higher RMSD values indicating much less efficiency in stabilizing peptide-DNA complexes as compared to Arg+.
Position of amino acid residues within a peptide also affects conformational stability of its complex with DNA. DR/GTCG and RD/TTCG complexes showed similar to RR/ACGA level of conformational stability. However, minor change of the dipeptide position within DNA binding site is observed after 20 ns of the MD simulations of RD/TTCG complex as opposed to DR/GTCG that showed stability over 100 ns. Similarly, DK/TCAG complex showed higher conformational stability and better ICM docking scores than KD/TGAG complex. This leads to a conclusion that positively charged Arg+ at C-terminal position may be a good way to stabilize highly selective peptides with low conformational stability.
To verify this idea dipeptide/dsDNA complexes were selected from uncharged peptides obtained in our analysis of docking results, namely: QQ/GTCG (–40.4) and MQ/GTCG (–35.1) and results of their MD simulation were compared with that of peptides with additional C-terminal Arg+ on the same dsDNA. Figure 8 shows time dependence of RMSD-NOFIT of peptide backbone as a measure of the deviation of the ligands from their initial position obtained by docking and modification.
Figure 8.
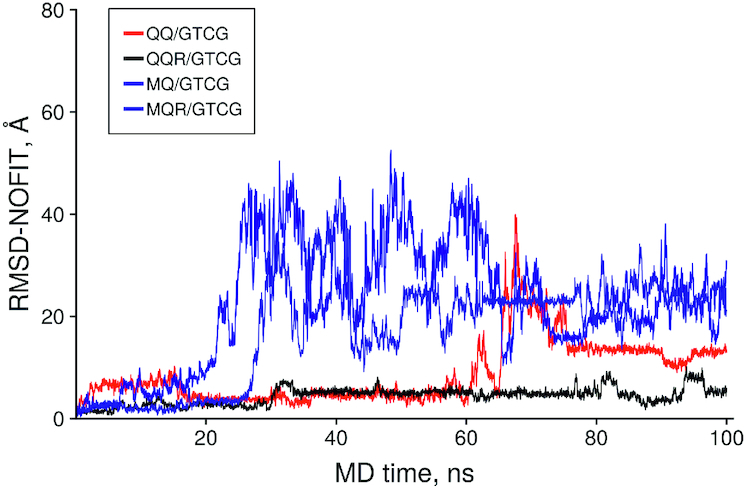
The time dependence of the no fit Root Mean Square Deviation (RMSD) of the peptide heavy atoms in uncharged and modified peptides.
It is of interest that complex QQ/GTCG showed remarkable stability of its initial conformation during the first 60 ns of its MD trajectory, however, dipeptide slightly changed initial conformation at around 18 ns of MD and then returned to the starting conformation. Adding of Arg+ to C-terminal lead to the conformation stabilization of the QQ-part in the initial position. MQ/GTCG complex was less stable than QQ but addition of Arg+ lead to stabilization of this complex as well.
The results indicate that conformational stability of peptide/DNA complexes in MD simulation depends on the presence of positively charged amino acids and position of amino acids in peptides, although it seems that in some cases uncharged peptides could bind dsDNA to form the stable complexes, however, adding a positively charged amino acid leads to increased stabilization of the complexes. This is not surprising since, unlike globular proteins, DNA is a highly negatively charged receptor.
However, its ICM-scores and expected conformational stability generally are rather low. Since the charge is important for stability of dipeptide/DNA complexes, MD simulations in octahedral water box were carried out on a series of selective complexes from Table 2 containing positively charged amino acids, namely: KE/TCGA (–35.8), KA/GGGG (–33.4), HS/TCGA (–33.4), FK/CACG (–32.7), HW/TCGA (–32.2). Figure 9 shows results of the MD simulations.
Figure 9.
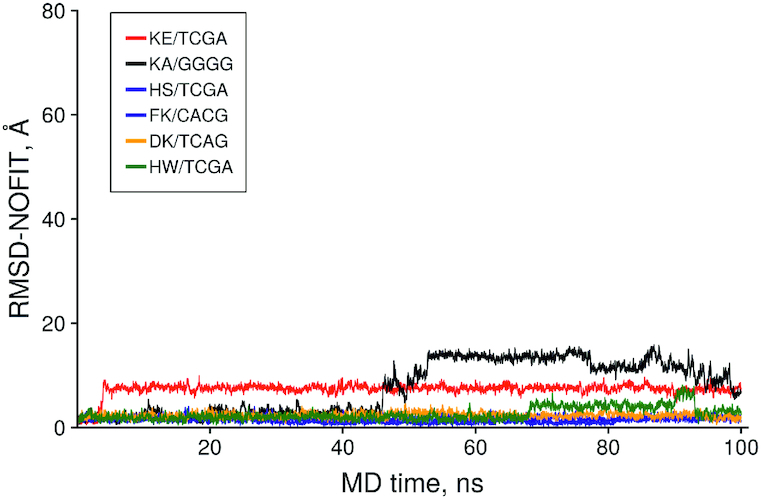
The time dependence of the no fit Root Mean Square Deviation (RMSD) of the peptide heavy atoms in selective dipeptide/DNA complexes.
The DNA complexes with dipeptides HS, FK and HW showed remarkable stability of their initial conformation during 100 ns of their MD trajectories indicating of high potential for selective DNA binding. It is of interest that the KE/TCGA complex changed its initial conformation and found a more stable position at 5 ns MD also indicating a potentially high binding constant. The KA/GGGG complex was stable for 45 ns which could be potentially interesting for binding DNA of high G/C content.
Thus, our results show that generally only a small fraction of all dipeptides is capable for effecient DNA binding. The most preferable binders are those containing positively charged groups of amino acids. Only 57 dipeptides out of 800 could selectively bind dsDNA. The high stability of some complexes has been confirmed with MD simulations in periodical water box. For others stability of their complexes with DNA can be improved by adding C-terminal Arg+. It is of interest that at least one dipeptide with a series of known biological activities was found in our calculations among selective dsDNA binders. This peptide is known to have various immunoprotector, geroprotector, anti-inflammatory and antitumour activities in vivo. However, the precise molecular mechanisms of these biological activities are still unknown. Many of other selective peptides may also possess biological activity due to their ability to specifically bind dsDNA. However, this interesting hypothesis requires experimental verification.
Although ICM-scores of the DNA-peptide complexes and recommended threshold (-32) to discriminate nonbinders are generally confirmed by the MD simulation of conformational stability of the complexes there were a few cases when complexes with good binding score did not show high stability in MD simulations, for example: KK/ACGG (–42.1), QQ/GTCG (–40.4), KE/TCGA (–35.8), MQ/GTCG (–35.1), KA/GGGG (–33.4). It could be attributed to a drawback of the ICM-score parametrization for DNA–proteins complexes.
Binding test using electrophoretic mobility shift assays
In order to verify our in silico observations, we selected several peptides for the experimental validation of their binding the dsDNA using the electrophoretic mobility shift assay. The peptides include KE and DR having at least one dsDNA sequence with better than −32 ICM docking score and AE and ED with no binding sites in the dsDNA were used in this experiment. Figure 10 shows DNA sequences of the binding sites for DR and KE peptides in red and blue, respectively. Totally there are 8 sites (4 at each DNA strand) with better than −32 ICM-docking score for the DR peptides, namely: TTCA/–39.2, GGTG/–36.2, GATG/–34, 2 ACCA sites/–33.7, TCAC/–32.8, AATG/–32.2 and TTGG/–32.0 and only one site for the KE peptide (TCGA/–35.8). Binding sites of the DR peptide overlap in some cases, so the peptide cannot bind all calculated sites simultaneously.
Figure 10.
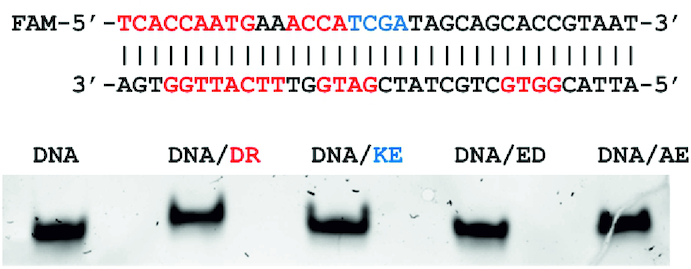
Electrophoretic mobility shift assay of dsDNA/dipeptides complexes. The binding sites of dipeptides DR and KE are shown in the dsDNA sequences in red and blue, respectively.
The experiment was repeated three times showing similar results. As expected the DR peptide having the best ICM-docking scores and highest number of binding sites in the dsDNA showed the most significant mobility shift as compared to other peptides. The KE peptide is the second in this ranking. The AE and ED peptides are at the third and fourth places, respectively, in general agreement with the results of our calculations.
CONCLUSIONS
It is known that protein binding to dsDNA is a fundamental factor of regulatory gene processes. The mechanisms of DNA sequence recognition by proteins are based on specific hydrogen bonds and hydrophobic contacts between proteins and dsDNA in their major groove. The mechanisms of such recognition have been widely investigated since the early 1980s. There have been many motifs found in dsDNA–protein complexes. However, all of them require globular proteins capable of maintaining a particular conformation of the protein DNA-binding elements and therefore generally are not applicable to short flexible peptides. Therefore, at the moment it is not clear what the minimal peptide size is required for specific recognition of the DNA sequences.
Short peptides play a very important role in various functions of the living cell. In this work, we for the first time systematically analysed the ability of all possible dipeptides to bind all possible combinations of tetra-nucleotides in the central part of dsDNA in the classic B-form using molecular docking and molecular dynamics. Our results demonstrate that ∼7% of all dipeptides are capable of selective binding to dsDNA, although with moderate affinity only. As expected, there are many others, mostly positively charged dipeptides capable of high affinity binding to dsDNA but with low selectivity. This is the most probable explanation for almost total absence of crystal structures of short peptide complexes with dsDNA in the Protein Data Bank. It is of interest that the affinity and selectivity of dsDNA binding quickly increases with additional blocking groups. This suggests that tri- or tetra-peptides may be enough for that purpose. However, in order to verify this hypothesis further calculations and experiments are required.
Supplementary Material
ACKNOWLEDGEMENTS
The results were obtained using the computing resources of Supercomputer Center, the Peter the Great's St. Petersburg Polytechnic University (www.scc.spbstu.ru). The authors would like to thank Dr Olga N. Mikhailova and Dr Georgii Pobegalov for proofreading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Foundation for Basic Research grant [# 19-015-00142 to N.K. and M.P.] and grant support from “Kurchatov genome center" to D.B. Funding for open access charge: Russian Scientific Center of Radiology and Surgical Technologies.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chiu T.-P., Rao S., Mann R.S., Honig B., Rohs R.. Genome-wide prediction of minor-groove electrostatic potential enables biophysical modeling of protein–DNA binding. Nucleic Acids Res. 2017; 45:12565–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li J., Sagendorf J.M., Chiu T.P., Pasi M., Perez A., Rohs R.. Expanding the repertoire of DNA shape features for genome-scale studies of transcription factor binding. Nucleic Acids Res. 2017; 45:12877–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Branden C.-I., Tooze J.. Introduction to Protein Structure. 1999; 2nd ednNY: Garland Pub. [Google Scholar]
- 5. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamilton P.L., Arya D.P.. Natural product DNA major groove binders. Nat. Prod. Rep. 2012; 29:134–143. [DOI] [PubMed] [Google Scholar]
- 7. Bhaduri S., Ranjan N., Arya D.P.. An overview of recent advances in duplex DNA recognition by small molecules. Beilstein J. Org. Chem. 2018; 14:1051–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huth J.R., Bewley C.A., Nissen M.S., Evans J.N., Reeves R., Gronenborn A.M., Clore G.M.. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat. Struct. Biol. 1997; 4:657–665. [DOI] [PubMed] [Google Scholar]
- 9. Sánchez A., Vázquez A.. Bioactive peptides: a review. Food Qual. Saf. 2017; 1:29–46. [Google Scholar]
- 10. Kraskovskaya N.A., Kukanova E.O., Lin’kova N.S., Popugaeva E.A., Khavinson V.K.. Tripeptides restore the number of neuronal spines under conditions of in vitro modeled Alzheimer's disease. Bull. Exp. Biol. Med. 2017; 163:550–553. [DOI] [PubMed] [Google Scholar]
- 11. Zamorskii I.I., Shchudrova T.S., Lin’kova N.S., Nichik T.E., Khavinson V.K.. Nephroprotective effect of EDL peptide at acute injury of kidneys of different genesis. Bull. Exp. Biol. Med. 2017; 163:389–393. [DOI] [PubMed] [Google Scholar]
- 12. Khavinson V., Linkova N.S., Kukanova E.O., Bolshakova A.V., Gainullina A.N., Tendler S.M., Morozova E., Tarnovskaia S.I., Vinki D.S.P., Bakulev V. et al.. Neuroprotective effect of EDR peptide in mouse model of huntington's disease. J. Neurol. Neurosci. 2017; 8:166. [Google Scholar]
- 13. Khavinson V.K., Tendler S.M., Kasyanenko N.A., Tarnovskaya S.I., Linkova N.S., Ashapkin V.V., Yakutseni P.P., Vanyushin B.F.. Tetrapeptide KEDW interacts with DNA and regulates gene expression. Am. J. Biomed. Sci. 2015; 7:156–169. [Google Scholar]
- 14. Anisimov V.N., Khavinson V.. Peptide bioregulation of aging: results and prospects. Biogerontology. 2010; 11:139–149. [DOI] [PubMed] [Google Scholar]
- 15. Fita I., Campos J.L., Puigjaner L.C., Subirana J.A.. X-ray diffraction study of DNA complexes with arginine peptides and their relation to nucleoprotamine structure. J. Mol. Biol. 1983; 167:157–177. [DOI] [PubMed] [Google Scholar]
- 16. Azorin F., Vives J., Campos J.L., Jordan A., Lloveras J., Puigjaner L., Subirana J.A., Mayer R., Brack A.. Interaction of DNA with lysine-rich polypeptides and proteins. The influence of polypeptide composition and secondary structure. J. Mol. Biol. 1985; 185:371–387. [DOI] [PubMed] [Google Scholar]
- 17. Parveen S., Arjmand F., Mohapatra D.K.. Zinc(II) complexes of Pro-Gly and Pro-Leu dipeptides: synthesis, characterization, in vitro DNA binding and cleavage studies. J. Photochem. Photobiol. B. 2013; 126:78–86. [DOI] [PubMed] [Google Scholar]
- 18. Solovyev A.Y., Tarnovskaya S.I., Chernova I.A., Shataeva L.K., Skorik Y.A.. The interaction of amino acids, peptides, and proteins with DNA. Int. J. Biol. Macromol. 2015; 78:39–45. [DOI] [PubMed] [Google Scholar]
- 19. Khavinson V., Tendler S.M., Vanyushin B.F., Kasyanenko N.A., Kvetnoy I.M., Linkova N.S., Ashapkin V.V., Polyakova V.O., Basharina V.S., Bernadotte A.. Peptide regulation of gene expression and protein synthesis in bronchial epithelium. Lung. 2014; 192:781–791. [DOI] [PubMed] [Google Scholar]
- 20. Andrews S.J., Rothnagel J.A.. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014; 15:193–204. [DOI] [PubMed] [Google Scholar]
- 21. Couso J.P., Patraquim P.. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017; 18:575–589. [DOI] [PubMed] [Google Scholar]
- 22. Guidotti G., Brambilla L., Rossi D.. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci. 2017; 38:406–424. [DOI] [PubMed] [Google Scholar]
- 23. Fedoreyeva L.I., Kireev I.I., Khavinson V., Vanyushin B.F.. Penetration of short fluorescence-labeled peptides into the nucleus in HeLa cells and in vitro specific interaction of the peptides with deoxyribooligonucleotides and DNA. Biochemistry (Mosc.). 2011; 76:1210–1219. [DOI] [PubMed] [Google Scholar]
- 24. Foltz M., van Buren L., Klaffke W., Duchateau G.S.. Modeling of the relationship between dipeptide structure and dipeptide stability, permeability, and ACE inhibitory activity. J. Food Sci. 2009; 74:H243–H251. [DOI] [PubMed] [Google Scholar]
- 25. Yakimov A., Pobegalov G., Bakhlanova I., Khodorkovskii M., Petukhov M., Baitin D.. Blocking the RecA activity and SOS-response in bacteria with a short α-helical peptide. Nucleic Acids Res. 2017; 45:9788–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feyzizarnagh H., Yoon D.Y., Goltz M., Kim D.S.. Peptide nanostructures in biomedical technology. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016; 8:730–743. [DOI] [PubMed] [Google Scholar]
- 27. Arnautova Y.A., Abagyan R.A., Totrov M.. Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling. Proteins. 2011; 79:477–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abagyan R., Totrov M.. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994; 235:983–1002. [DOI] [PubMed] [Google Scholar]
- 29. Case D.A., Cheatham T.E. 3rd, Darden T., Gohlke H., Luo R., Merz K.M. Jr, Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015; 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abagyan R., Orry A., Raush E., Totrov M.. ICM-Pro User Guide v.3.8. 2018; USA: MolSoft LLC. [Google Scholar]
- 32. Khavinson V.K., Malinin V.V.. Gerontological Aspects of Genome Peptide Regulation. 2005; Basel; NY: Karger. [Google Scholar]
- 33. Khavinson V.K., Anisimov V.N., Zavarzina N.Y., Zabezhinskii M.A., Zimina O.A., Popovich I.G., Shtylik A.V., Malinin V.V., Morozov V.G.. Effect of vilon on biological age and lifespan in mice. Bull. Exp. Biol. Med. 2000; 130:687–690. [DOI] [PubMed] [Google Scholar]
- 34. Anisimov S.V., Khavinson V., Anisimov V.N.. Elucidation of the effect of brain cortex tetrapeptide Cortagen on gene expression in mouse heart by microarray. Neuro Endocrinol. Lett. 2004; 25:87–93. [PubMed] [Google Scholar]
- 35. Khavinson V.K., Morozov V.G., Malinin V.V., Kazakova T.B., Korneva E.A.. Effect of peptide Lys-Glu on interleukin-2 gene expression in lymphocytes. Bull. Exp. Biol. Med. 2000; 130:898–899. [PubMed] [Google Scholar]
- 36. Sevostianova N.N., Linkova N.S., Polyakova V.O., Chervyakova N.A., Kostylev A.V., Durnova A.O., Kvetnoy I.M., Abdulragimov R.I., Khavinson V.H.. Immunomodulating effects of vilon and its analogue in the culture of human and animal thymus cells. Bull. Exp. Biol. Med. 2013; 154:562–565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.