


Abstract
Many bacteria acquire dissemination and virulence traits in G1-phase. CtrA, an essential and conserved cell cycle transcriptional regulator identified in the dimorphic alpha-proteobacterium Caulobacter crescentus, first activates promoters in late S-phase and then mysteriously switches to different target promoters in G1-phase. We uncovered a highly conserved determinant in the DNA-binding domain (DBD) of CtrA uncoupling this promoter switch. We also show that it reprograms CtrA occupancy in stationary cells inducing a (p)ppGpp alarmone signal perceived by the RNA polymerase beta subunit. A simple side chain modification in a critical residue within the core DBD imposes opposing developmental phenotypes and transcriptional activities of CtrA and a proximal residue can direct CtrA towards activation of the dispersal (G1-phase) program. Hence, we propose that this conserved determinant in the CtrA primary structure dictates promoter reprogramming during the growth transition in other alpha-proteobacteria that differentiate from replicative cells into dispersal cells.
INTRODUCTION
Tight regulation of gene expression during the cell cycle is paramount to ensure proper timing and coordination of DNA replication, chromosome segregation and cell division, often concurrently with morphological changes. Caulobacter crescentus is a rod-shaped and dimorphic alpha-proteobacterium that undergoes an asymmetric cell division into two unequally sized and polarized daughter cells: a motile and non-replicative dispersal (swarmer, SW) cell residing in G1-phase and a capsulated and replicative (stalked, ST) cell. In C. crescentus, cell cycle progression is intimately tied to polar remodeling via a circuit of transcriptional activators that direct sequential gene expression programs (1,2). The G1-phase program is implemented in the SW daughter cell that inherits the new cell pole where the flagellum and adhesive pili are located. By contrast, the old pole is inherited by the replicative ST cell that is engaged in DNA-replication. As the cell cycle proceeds, the ST cell prepares for division, expresses the late S-phase program and polarizes before dividing asymmetrically into a SW and ST daughter cell (Figure 1A). The transcriptional programs are not only temporally ordered, but also spatially confined during cytokinesis, with the G1-phase program being activated in the nascent SW chamber during cytokinesis, but not in the ST cell chamber (1,2).
Figure 1.

ctrA401 as gain of function mutation. (A) Schematic of Caulobacter capsulation (blue) along cell cycle and regulatory interactions that controls Caulobacter transcriptional switch between late S-phase and G1-phase. CtrA controls activity of late S- and G1-phase promoters. SciP which is expressed in G1-phase under the control of CtrA negatively regulates S-phase genes. During cell cycle, capsulation is regulated through expression of hvyA that prevents capsulation in SW cell and under the control of the transcriptional regulators CtrA. (B) Immunoblots showing steady-state levels of HfsJ and SpmX in WT, ΔspoT, ΔpleC, ctrA401 and derivatives in exponential and stationary phase. CCNA_00163 serves as a loading control. (C) Genome wide occupancies of CtrA on the Caulobacter WT, ctrA401 and ctrA401-SS genome as determined by ChIP-Seq. The x-axis represents the nucleotide position on the genome (bp), whereas the y-axis shows the normalized ChIP profiles in read per million (rpm). (D) ChIP-Seq traces of CtrA, CtrA401 (T170I) and CtrA401-SS (T168I/T170I) on different CtrA target promoters. Genes encoded are represented as boxes on the upper part of the graph, gene names and CCNA numbers gene annotation are indicated in the boxes or above. (E, F) Schemes showing the regulatory interactions happening at the late S- and G-phase promoters based on C, D and Table 1.
Cell cycle analyses are facile with C. crescentus because the non-capsulated G1-phase (SW) cells can be separated from capsulated S-phase (ST) cells by density gradient centrifugation (3). The acquisition of replicative functions marks the obligate G1→S-phase transition that morphologically manifests with the differentiation from SW to ST cells. Pili and the flagellum are lost from the old cell pole, followed by the onset of stalk outgrowth from the vacated site (1). Concurrently, the polysaccharide-based capsule is synthesized which increases the cellular buoyancy (4), and DNA synthesis initiates bidirectionally from a single origin of replication (Cori, Figure 1A) on the circular chromosome. A multicomponent genetic circuit of global transcriptional activators and repressors temporally and spatially coordinates these events at the molecular level (1,2).
The late S-phase and G1-phase programs are both activated by CtrA, a highly conserved member of the OmpR family of DNA-binding response regulators (5). CtrA is essential for viability and proper cell cycle control in C. crescentus (5) and in many other alpha-proteobacteria (1). CtrA switches from activating the late S-phase promoters before cell division to inducing G1-phase promoters in the nascent SW cell chamber at cytokinesis. While CtrA also binds Cori and prevents the initiation of DNA replication in G1-phase (5–7), it is degraded by the ClpXP protease during the G1→S transition (8–10). It is re-synthesized in late S-phase and again degraded in the ST compartment during cytokinesis, while being maintained in the SW compartment (Figure 1A). The conserved target sequence motif (CtrA box: 5′-TTAA-N7-TTAA-3′) is present in both promoter classes and recognized by the C-terminal DNA binding domain (DBD) of CtrA. At the N-terminus, CtrA harbors a receiver domain (RD) with a phosphorylation site at a conserved aspartate (at position 51, D51). Phosphorylation at D51 stimulates DNA binding and is required for viability. The hybrid histidine kinase CckA directs a multi-component phosphoryl-transfer reaction to D51 of CtrA (11–14). Though loss of CckA is lethal, missense mutations in the CtrA RD were isolated in unbiased selection for mutant derivatives that can support viability of C. crescentus cells lacking CckA (15).
Mutations in the DBD domain of CtrA that are critical for viability have also been isolated. In the landmark study by Quon et al, ctrA was uncovered as an essential gene in C. crescentus [as the ctrA401 mutant allele, encoding CtrA (T170I)] in a two-step genetic selection. First, based on earlier evidence that the fliQ (class II) flagellar assembly gene is transcriptionally de-repressed in late S-phase, the authors selected for mutants with elevated fliQ promoter (PfliQ) activity at 28°C. Next, those mutants were retained that also exhibit thermosensitive growth at 37°C compared to wild-type (WT), yielding the ctrA401 mutant (5). Since PfliQ activity is elevated at 28°C, but strongly impaired at 37°C in ctrA401 cells, it was concluded that CtrA acts positively and negatively at PfliQ (and likely other late S-phase promoters).
How CtrA switches its specificity from late S-phase promoters to G1-phase promoters is unclear. Determinants in CtrA that are specific for each promoter class have not been identified. At least two different negative regulators, one targeting the late S-phase promoters and another acting on G1-phase promoters (15–17), reinforce the promoter switch. The conserved helix-turn-helix protein SciP specifically inhibits late S-phase promoters that are activated by CtrA. SciP is restricted to G1-phase due in part to its synthesis from a CtrA-activated promoter (PsciP) that fires in G1-phase and in part due to a short half-life imparted by the Lon protease (18). Thus, SciP couples the activation of G1-phase promoters with the shutdown of late S-phase promoter activity, but it cannot act as the trigger for the promoter switch. The repressor paralogs MucR1 and MucR2 negatively regulate the CtrA-activated promoters that fire in G1-phase. While MucR1/2 abundance and (steady-state) promoter occupancy does not change during the cell cycle, the ability of MucR1/2 to exclude competing proteins from target promoters is reduced in G1-phase (19). Interestingly, MucR orthologs regulate virulence gene expression in other alpha-proteobacteria and bind orthologous promoters (15). These free-living alpha-proteobacteria express virulence traits in G1-phase (20–22), suggesting that the mechanism of restricting promoter firing to G1-phase by MucR is conserved.
In addition to its cell cycle function in C. crescentus, CtrA also plays an important role in regulating the terminal differentiation of the alpha-proteobacterium Sinorhizobium meliloti into bacteroids during their symbiotic interaction with plants (23). Moreover, CtrA has been implicated in regulating development during a growth phase transition in obligate intracellular alpha-proteobacteria from the order Rickettsiales (24). Akin to the developmental cycle in other obligate intracellular bacteria, the dimorphic rickettsial pathogen Ehrlichia chaffeensis differentiates into infectious dispersal (dense-cored) cells following the rapid intracellular expansion of the replicative (reticulate) cells (25,26), suggesting a nutritional trigger may underlie development. Rickettsia do not encode MucR or SciP orthologs in their genomes, but CtrA peaks during the late stages of growth when the transition from reticulate to dense-cored cells occurs. In stationary phase, a starvation stress response is triggered in C. crescentus cells by the alarmone (p)ppGpp (guanosine tetraphosphate and guanosine pentaphosphate) that is produced by a bifunctional RelA-SpoT Homologue (RSH) enzyme SpoT (27). (p)ppGpp suppresses DNA replication and maintains CtrA abundance to arrest cells predominantly in G1 phase and to a lesser extent in the predivisional phase (28–30). Whether CtrA is also involved in transcriptional re-programming during this growth phase transition in C. crescentus cells has not been investigated.
Here, we unearth a molecular determinant within the DBD of CtrA that is required to execute the switch from late S- to G1-phase promoters and to reprogram CtrA in stationary C. crescentus cells accumulating (p)ppGpp. A highly conserved triad centered in the core DBD dictates whether CtrA can switch G1-phase promoters and use it to control the cell cycle during a growth phase transition induced by (p)ppGpp in response to starvation stress. CtrA activity can be maintained in cells expressing a mutant form of RNA polymerase that no longer depends on (p)ppGpp, indicating that transcriptional regulation coordinates growth phase transitions regulated by CtrA.
MATERIALS AND METHODS
Growth conditions
Caulobacter crescentus NA1000 and derivatives were grown at 30°C in PYE (peptone yeast extract). Antibiotic used for C. crescentus include kanamycin (solid: 20 μg/ml; liquid: 5 μg/ml), tetracycline (1 μg/ml), gentamycin (1 μg/ml) and nalidixic acid (20 μg/ml). When needed, d-xylose or sucrose was added at 0.3% final concentration, glucose at 0.2% final concentration and vanillate at 500 μM final concentration. Escherichia coli S17–1 λpir and EC100D (Epicentre Technologies, Madison, WI, USA) were grown at 37°C in LB. Swarmer cell isolation, electroporations, biparental mattings and bacteriophage ϕCr30-mediated generalized transductions were performed as previously described (3). Plasmids for β-galactosidase assays were introduced into C. crescentus by electroporation.
Bacterial strains, plasmids, and oligonucleotides
Bacterial strains, plasmids, and oligonucleotides used in this study are listed and described in supplementary material and methods (Supplementary Tables S8–S10).
Motility suppressors of ctrA401 mutant cells
Spontaneous mutations that suppress the motility defect of the ctrA401 mutant cells appeared as ‘flares’ that emanated from nonmotile colonies after ∼4–5 days of incubation at 30°C. Ten isolates (MS 1–10) were selected and tested for the excision of the MGE (mobile genetic element) by PCR using primers amplifying the CCNA_00477 (presence of the MGE) and attB (MGE excised) fragments. Two ctrA401 MS (MS 2 and 4) were subjected to whole genome sequencing. The Nextera kit from illumina was used for library preparation with 50 ng of DNA as input. The Library molarity and quality was assessed with the Qubit and Tapestation using a DNA High sensitivity chip (Agilent Technologies). Libraries were loaded on a HiSeq 4000 single-read Illumina flow cell. Read length of 50 bases were generated. The nine other suppressors were then PCR sequenced by Sanger sequencing for the CCNA_03998 (amplified using primers 3998_NdeI and 3998_EcoRI). Table 1 summarizes the mutations obtained in the suppressors. To confirm that these mutations were responsible for the suppression of the motility defect of the ctrA401 mutant cells, ΔMGE ctrA401 and Δ03998 ctrA401 double mutants were constructed through ϕCr30 mediated generalized transduction of the ctrA401 allele. The transducing phage stock is a lysate of ctrA401 MS2 cells in which plasmid pNPTS138-ctrA-ds had been integrated (ctrA401::pNPTS138-ctrA-ds). PYE plates containing sucrose (3%) were then used to isolate clones in which the pNPTS138-ctrA-ds plasmid had been eliminated by homologous recombination and were kanamycin susceptible. Sanger sequencing of PCR fragments was used to verify the integrity of the mutation using primers ctrA_NdeI and ctrA_XbaI.
Table 1.
Mutations identified in the ctrA401Motility suppressors
| Strains | CCNA_03998 | Nucleotide or Ins change | MGE |
|---|---|---|---|
| MS 1 | Ins G93Stop | Ins 475797 | + |
| MS 2 | Ins V217Stop | Ins 476105 | + |
| MS 3 | W61Stop | G 475713 A | + |
| MS 4 | / | / | - |
| MS 5 | / | / | - |
| MS 6 | / | / | - |
| MS 7 | / | / | - |
| MS 8 | Ins C311Stop | C Ins 476340 | + |
| MS 9 | Ins C311Stop | C Ins 476340 | + |
| MS 10 | P32H | C 465626 A | + |
List of genetic changes in motility suppressor mutants (MS1–10) derived from the ctrA401 strain. The mutations were determined by Illumina sequencing and verified by PCR-based Sanger sequencing. Nucleotide numbers refer to the genome nucleotide coordinates from Caulobacter crescentus NA1000 (CP001340) https://www.ncbi.nlm.nih.gov/nuccore/CP001340. Ins denotes nucleotide insertion and MGE refers to the presence of the mobile genetic element.
Motility suppressors of NA1000 ΔptsP ΔspoT mutant cells
Spontaneous mutations that suppress the motility defect of the ΔptsP ΔspoT mutant cell appeared as ‘flares’ that emanated from nonmotile colonies after ∼4–5 days of incubation at 30°C. One isolate (UG5591) was subjected to whole genome sequencing, and mutation in the rpoB (rpoBH559Y) gene was found. In this suppressor mutant, the histidine codon at position 559 in rpoB was changed to one encoding tyrosine (change of nt 549505 from C to T relative to the NA1000 genome sequence). To confirm that this mutation was responsible for improving the motility defect of the ΔptsP ΔspoT mutant cells, we backcrossed the mutation rpoBH559Y into WT cells. To this end, a pNPTS138-rpoC-ds, conferring kanamycin resistance, was integrated by homologous recombination downstream of the rpoC gene in the ΔptsP ΔspoT rpoBH559Y mutant. Next, ϕCr30 mediated generalized transduction was used to transfer the rpoBH559Y allele into WT cells. PYE plates containing sucrose (3%) were then used to select for elimination of the pNPTS138-rpoC-ds plasmid by homologous recombination and clones were screened for kanamycin susceptibility. Sanger sequencing of PCR fragments was used to verify the integrity of the mutation using primers rpoB-seq1 and rpoB-seq2.
ϕCr30-mediated transduction of the ctrA401 allele
ctrA401 transducing phage stock is a lysate of the generalized transducing phage ϕCr30 that was grown on strain ctrA401 MS2 ctrA401::pNPTS138-ctrA-ds.
Genome-wide transposon mutagenesis coupled to deep sequencing (Tn-Seq)
NA1000 ctrA401 MS2 pilA::PpilA-nptII Tn-Seq
Transposon mutagenesis of C. crescentus ctrA401 MS2 pilA::PpilA-nptII cells was done by intergeneric conjugation from E. coli S17–1 λpir harbouring the himar1-derivative pMAR2xT7 (31) and a Tn-library of >200,000 gentamicin- and kanamycin-resistant clones was collected, grown overnight in PYE and chromosomal DNA was extracted. Genomic DNA was used to generate barcoded Tn-Seq libraries and submitted to Illumina HiSeq 4000 sequencing (Fasteris SA). Tn insertion-specific reads (150 bp read length) were sequenced using the himar-Tnseq2 primer (5′-AGACCGGGGACTTATCAGCCAACCTGT-3′). Specific reads attesting to an integration of the transposon on a 5′-TA-3′ specific DNA locus were sorted (Rstudio_V1.1.442) from the tens of million reads generated by sequencing, and then mapped (Map_with_Bowtie_for_Illumina_V1.1.2) to the C. crescentus NA1000 genome (NC_011916.1) using the web-based analysis platform Galaxy (https://usegalaxy.org). Using Samtool_V0.1.18, BED file format encompassing the Tn insertion coordinates were generated and then imported into SeqMonk V1.40.0 (www.bioinformatics.babraham.ac.uk/projects/) to assess the total number of Tn insertion per chromosome position (Tn-insertion per millions of reads count) or per coding sequence (CDS). For the CDS Tn-insertion ratio calculation, SeqMonk datasets were exported into Microsoft Excel files (Dataset Supplementary Table S6) for further analyses and used to generate Supplementary Figure S4B, as described previously (32). Briefly, to circumvent ratio problems for a CDS Tn-insertion value of 0 and CDS with insufficient statistical power, an average value of all CDS-Tn insertions normalized to the gene size was calculated, and 1% of this normalized value was used to correct each CDS-Tn insertion value.
NA1000, NA1000 ctrA401 and ctrA (T170A) Tn-Seq
Overnight WT (NA1000), ctrA401 (T170I) and ctrA(T170A) cell cultures were grown in PYE and mutagenized with a himar1 transposon (Tn). Transposon mutagenesis of C. crescentus WT (NA1000), ctrA401 and ctrA(T170A) was done by mobilizing the himar1 transposon (KanR) encoded on plasmid pHPV414 (33) from Escherichia coli S17–1 λpir into WT (NA1000), ctrA401 and ctrA(T170A) strain by selecting for kanamycin-nalidixic acid-resistant C. crescentus clones. For the Tn-libraries of WT, ctrA401 and ctrA(T170A), >200,000 kanamycin-resistant clones were collected, grown overnight in PYE and chromosomal DNA was extracted. Genomic DNA was used to generate barcoded Tn-Seq libraries and submitted to Illumina HiSeq 2000 sequencing (Fasteris SA). Tn-insertion specific reads (50 bp long) were sequenced using the himar-Tnseq1 primer (5′-AGACCGGGGACTTATCAGCCAACCTGT-3′) and yielded several million reads that were mapped to Caulobacter crescentus NA1000 (NC_011916.1) as previously described (32). CDS Tn-insertions calculation is presented in Supplementary Table S7 and were used to generate Figure 3D and Supplementary Figure S6.
Figure 3.
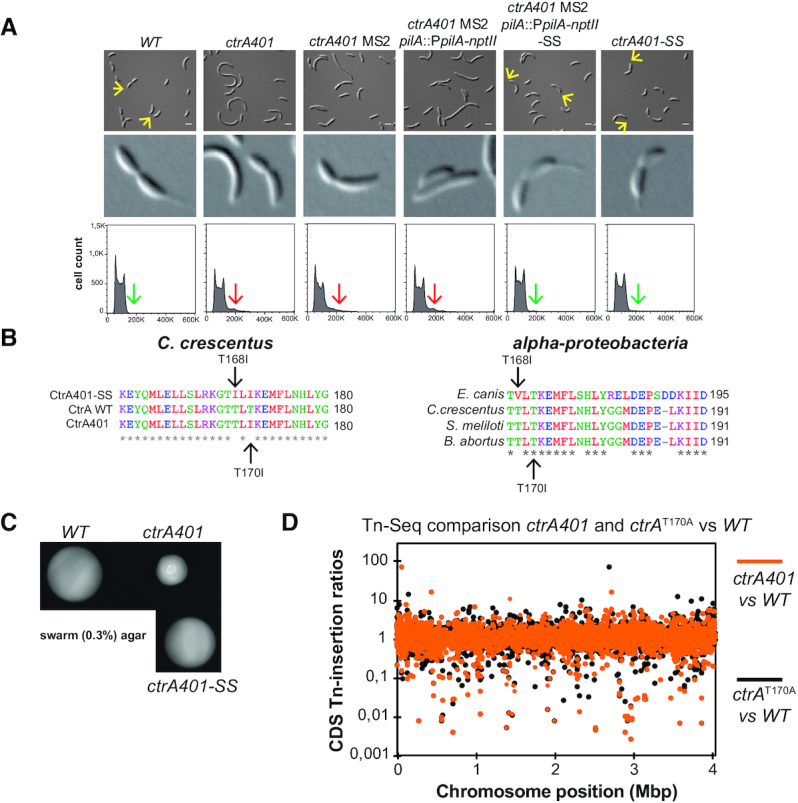
Intragenic suppressor mutations of ctrA401 alter DNA-binding. (A) DIC microscopy, 8X zoom images and FACS analysis of the WT, ctrA401 and derivatives carrying the pilA::PpilA-nptII transcriptional reporter. Microscopy pictures and DNA content (FL1-A channel) quantification were performed during exponential growth in PYE. ctrA401 MS2 pilA::PpilA-nptII cells resistant to 40 μg/ml of kanamycin (kan) were screened by microscopy for the presence of stalk (yellow arrow). DIC images and FACS analysis of the ctrA401 and ctrA401- MS2 show cells that are elongated, accumulating more than 2n chromosomes (red arrow) and stalkless in exponential phase. Selection of ctrA401 MS2 pilA::PpilA-nptII cells on PYE plates supplemented with 40 μg/ml of kanamycin and screening by DIC microscopy for the presence of stalk (yellow arrow, 8X zoom) lead to the identification of the ctrA401-SS (T170I/T168I) mutation. ctrA401-SS cells do not elongate and do not accumulate extra-chromosome (green arrow). (B) On the left, the alignment of the 28 amino acids around the T170 and the T168 shows the amino acid replacements in ctrA401, ctrA401-SS compared to WT. On the right, the alignment of CtrA protein sequences from several members of the alpha-proteobacteria phylum is shown. (C) Motility (0.3% agar) plates inoculated with WT, ctrA401, ctrA401-SS cells. The T168I amino acid change restores motility to ctrA401 cells. (D) Tn insertion bias in coding sequences (CDS) of ctrA401 (T170I) cells relative to WT cells (orange dots) and ctrA (T170A) cells relative to WT (black dots) cells as determined by Tn-Seq. The abscissa shows position as function of genome position, and the ordinate provides Tn-insertion ratios. Peaks show CDSs with the highest number of Tn insertions. Noncoding sequences are not included.
Stalk suppressors of ctrA401 cells
To isolate mutations that restore polarity of ctrA401 MS2 mutant cells, we integrated the transcriptional reporter PpilA-nptII at the chromosomal pilA locus and selected for spontaneous suppressor mutants that grow on PYE agar plates supplemented with 40 μg/ml of kanamycin. Kanamycin resistant clones were then screened by DIC microscopy for the presence of the stalk (SS mutants). Five mutants had stalks and their ctrA gene was PCR sequenced to exclude potential revertants. Sanger sequencing instead revealed a second mutation within ctrA in all the five isolates in which the codon of threonine (ACC) at residue 168 was exchanged for one encoding an isoleucine (ATC) in addition to the T170I mutation. To confirm that this mutation was responsible for the restoration of stalk synthesis, we backcrossed the alleles harboring mutations (T170I and T168I) into WT cells. To this end, we used a ctrA401 mutant that we selected for by growth at 37°C and that had stalks, since the ctrA(T168I/T170I) allele permits growth at 37°C. The pNPTS138-ctrA-ds plasmid, conferring kanamycin resistance, was integrated by homologous recombination downstream of the ctrA gene in this ctrA(T168I/T170I) mutant. Next, ϕCr30-mediated generalized transduction was used to transfer the ctrA(T168I/T170I) allele into WT cells. PYE plates containing sucrose (3%) were then used to select for elimination of the pNPTS138-ctrA-ds plasmid by homologous recombination and the resulting clones were screened for kanamycin susceptibility. Sanger sequencing was used to verify the integrity of the mutation using primers ctrA_NdeI and ctrA_XbaI and the resulting mutants were probed for the presence of the stalk by phase contrast microscopy.
β-Galactosidase assays
β-Galactosidase assays were performed at 30°C. Exponential phase cells (50–200 μl) at OD660 nm = 0.1–0.5 were lysed with chloroform and mixed with Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl and 1 mM MgSO4 heptahydrate) to a final volume 800 μl. Two hundred μl of ONPG (4 mg/ml o-nitrophenyl-β-d-galactopyranoside in 0.1 M KPO4 pH 7.0) was added and the reaction was timed. When a medium-yellow colour developed, the reaction was stopped with 400 μl of 1 M Na2CO3. The OD420 nm of the supernatant was determined and the units were calculated with the equation: U = (OD420nm * 1000)/(OD660nm * time (in min) * volume of culture (in ml)). Experimental values represent the averages of four independent experiments and error was computed as standard deviation (SD).
Immunoblot analysis
Protein samples were separated by SDS-PAGE and blotted on PVDF (polyvinylidenfluoride) membranes (Merck Millipore). Membranes were blocked for 1 h with TBS, 0,1% Tween 20 and 5% dry milk and then incubated for overnight with the primary antibodies diluted in TBS, 0.1% Tween 20, 5% dry milk. The different antisera were used at the following dilutions: anti-SciP (1:5000) (17), anti-FljK (1:50 000) (Ardissone, unpublished), anti-CtrA (1:20 000) (Ardissone, unpublished), anti-PilA (1:10 000) (34), anti-HfsJ (1:10 000) (Eroglu, unpublished), anti-SpmX (1:50 000) (35) and anti-CCNA_00163 (1:10 000) (4) or anti-MreB (1:200 000) (36) as a loading control. The membranes were washed four times for 5 min in TBS, 0.1% Tween 20 and incubated for 1 h with the secondary antibody diluted in TBS, 0.1% Tween 20 and 5% dry milk. The membranes were finally washed again four times for 5 min in TBS, 0.1% Tween 20 and revealed with Immobilon Western Blotting Chemoluminescence HRP substrate (Merck Millipore) and Super RX-film (Fujifilm) or luminescent image analyzer (ChemidocTm MP, Biorad).
Microscopy
PYE cultivated cells in exponential or stationary growth phase were immobilized using a thin layer of 1% agarose. DIC and Phase microscopy images were taken with an Alpha Plan-Apochromatic 100×/1.46 DIC (UV) VIS-IR oil and Alpha Plan-Apochromatic 100×/1.46 Ph3 (UV) VIS-IR oil objectives respectively on an Axio Imager M2 microscope (Zeiss) with a 405 and 488 nm lasers (Visitron Systems GmbH, Puchheim, Germany) and a Photometrics Evolve camera (Photometrics) controlled through Metamorph V7.5 (Universal Imaging). Images were processed using Metamorph V7.5 and ImageJ.
Flow cytometry analysis
PYE cultivated cells to exponential (OD660nm = 0.3–0.6) or stationary growth phase were fixed into ice cold 77% ethanol solution. Fixed cells were resuspended in FACS Staining buffer pH 7.2 (10 mM Tris–HCl, 1 mM EDTA, 50 mM NaCitrate, 0.01% Triton X-100) and then treated with RNase A (Roche) at 0.1 mg/ml during 30 min at room temperature. Cells were stained in FACS Staining buffer containing 0.5 μM of SYTOX Green nucleic acid stain solution (Invitrogen) and then analysed using a BD Accuri C6 flow cytometer instrument (BD Biosciences, San Jose, CA, USA). Flow cytometry data were acquired using the CFlow Plus V1.0.264.15 software (Accuri Cytometers Inc.) and analysed using FlowJO software. Twenty thousand cells were analysed from each biological sample. The Green fluorescence (FL1-A) parameters were used to estimate cell chromosome contents. Relative chromosome number was directly estimated from the FL1-A value of WT cells treated with 30 μg/ml Rifampicin during 3 h at 30°C. Rifampicin treatment of cells blocks the initiation of chromosomal replication but allows ongoing rounds of replication to finish. Each experiment was repeated independently, and representative results are shown.
Motility assays and phage infectivity tests
Swarming properties were assessed by spotting 1.5 μl drops of overnight culture, adjusted to an OD660nm of 1, on PYE soft (0.3%) agar plates and grown for 48 hours. Phage susceptibility assays were conducted by mixing 300 μl of overnight culture in 6 ml soft PYE agar and overlaid on a PYE agar plate. Upon solidification of the soft (top) agar, 4 μl drops of serial dilution of phages (ϕCbK or ϕCr30) were spotted and scored for plaques after one day incubation at 4°C.
Viability test on plates
Viability of WT, ctrA401 and derivatives was assessed with 4 μl drops of serial dilutions of liquid culture (10−1 to 10−6), adjusted to OD660nm = 1, on PYE plates, then incubated for 2 days at 30°C or 37°C.
Chromatin ImmunoPrecipitation coupled to deep Sequencing (ChIP-Seq)
Exponential or stationary phase cells cultured in PYE were cross-linked in 10 mM sodium phosphate (pH 7.6) and 1% formaldehyde at room temperature for 10 min and on ice for 30 min thereafter and washed three times in phosphate-buffered saline (PBS). Cells were lysed in a Ready-Lyse lysozyme solution (Epicentre Technologies) according to manufacturer's instructions and lysates were sonicated in an ice-water bath for 15 cycles 30 s ON and 30 s OFF, to shear DNA fragments to an average length of 0.3–0.5 kbp and cleared by centrifugation at 14 000 g for 2 min at 4°C. Lysates were normalized by protein content, diluted to 1 ml using ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl (pH 8.1), 167 mM NaCl plus protease inhibitors (Roche, Switzerland) and pre-cleared with 80 μl of protein-A (rabbit antibodies) agarose (Roche) and 100 μg BSA. Ten percent of the supernatant was removed and used as total chromatin input DNA as described before (32).
Two microliters of rabbit polyclonal antibodies to CtrA were added to the remains of the supernatant, incubated overnight at 4°C with 80 μl of protein-A beads pre-saturated with BSA, washed once with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1) and 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1) and 500 mM NaCl) and LiCl buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA and 10 mM Tris–HCl (pH 8.1)), and twice with TE buffer (10 mM Tris–HCl (pH 8.1) and 1 mM EDTA). The protein DNA complexes were eluted in 500 μl freshly prepared elution buffer (1% SDS and 0.1 M NaHCO3), supplemented with NaCl to a final concentration of 300 mM and incubated overnight at 65°C to reverse the crosslinks. The samples were treated with 2 μg of Proteinase K for 2 h at 45°C in 40 mM EDTA and 40 mM Tris–HCl (pH 6.5). DNA was extracted using phenol:chloroform:isoamyl alcohol (25:24:1), ethanol precipitated using 20 μg of glycogen as carrier and resuspended in 50 μl of water.
Immunoprecipitated chromatin was used to prepare sample libraries used for deep-sequencing at Fasteris SA (Geneva, Switzerland). ChIP-Seq libraries were prepared using the DNA Sample Prep Kit (Illumina) following the manufacturer's instructions. Single-end run were performed on a Next-Generation DNA sequencing instrument (NGS, HiSeq3000/4000), 50 cycles were read and yielded several million reads. The single-end sequence reads stored in FastQ files were mapped against the genome of C. crescentus NA1000 (NC_011916.1) and converted to SAM using BWA and SAM tools respectively from the galaxy server (https://usegalaxy.org/). The resulting SAM was imported into SeqMonk (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/, version 1.45.1) to build sequence read profiles. The initial quantification of the sequencing data was done in SeqMonk: the genome was subdivided into 50 bp probes, and for every probe we calculated a value that represents a normalized read number per million (Supplementary Tables S2, S3 and S4).
Using the web-based analysis platform Galaxy (https://usegalaxy.org), CtrA ChIP-Seq peaks were called using MACS2 (37) relative to the total input DNA samples. The q-value (false discovery rate, FDR) cut-off for called peaks was 0.05. Peaks were rank-ordered according to fold-enrichment (Supplementary Table S5) and peaks with a fold-enrichment values >2 were retained for further analysis.
Supplementary Table S1 shows the datastore summary report.
The heatmaps in Supplementary Figure S1 were generated using SeqMonk and ChIP-Seq signals were plotted over 800 bp ranges at each C. crescentus Coding DNA Sequence (CDS) centered at each translation initiation codon. Sequence data have been deposited to the Gene Expression Omnibus (GEO) database (GSE134017 accession, samples GSM3933392–GSM3933401).
Molecular modelling of CtrA
Models of CtrA, CtrA(T170I) and CtrA(T168I/T170I) homo-dimers were generated based on the crystal structure of the DNA-binding transcriptional regulator BasR/PmrA from Klebsiella pneumoniae (PDB 4s05) (38), using the SWISS-MODEL server (39). Comparison with the solved structured of Brucella abortus receiver domain (PDB 4QPJ) (40) was performed with the built model to control the quality of the model. Pymol was used to visualize and map the location of the mutated amino acids within the structure.
RESULTS
CtrA401 uncouples the switch from late-S-phase to G1-phase promoters
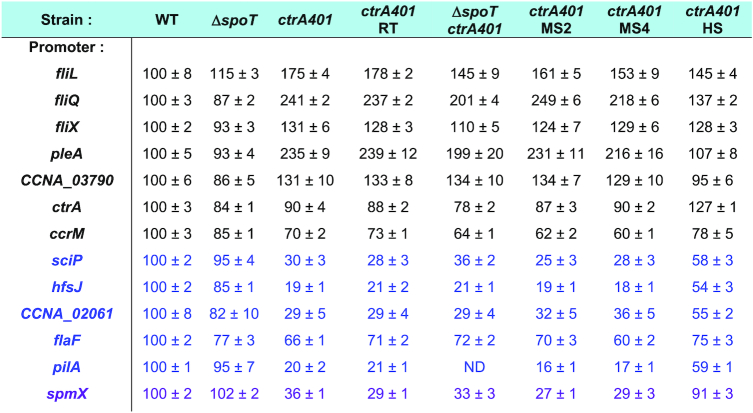
To determine if CtrA can indeed act negatively (directly or indirectly) on late S-phase promoters such as the CtrA-activated promoter PfliQ, we determined whether PfliQ activity is upregulated in ctrA401 cells at 30°C using a PfliQ-lacZ promoter probe plasmid in which PfliQ is transcriptionally fused to the promoterless lacZ gene. We observed a more than two-fold increase in LacZ activity in ctrA401 cells (241%, Table 2) versus WT cells growing exponentially at 30°C. To determine if other CtrA-activated promoters are upregulated in the same manner, we conducted LacZ-based promoter probe assays in WT and ctrA401 cells at 30°C (Table 2). Remarkably, we observed a similar behaviour with promoters that CtrA activates in late S-phase (e.g. PfliX,PfliL, PpleA, PcpaB and PCCNA_03790; Table 2), but not with promoters that CtrA activates in G1-phase (e.g. PpilA, PflaF,PhfsJ, PCCNA_02061 or PsciP; Table 2). In fact, promoters of the G1 class were poorly active in the ctrA401 background and immunoblotting revealed that proteins expressed from G1 phase promoters that depend directly (HfsJ, PilA, SciP; Figure 1B, Supplementary Figures S1B, S1D and S2B) or indirectly (SpmX; Figure 1B) on CtrA are not (or poorly) expressed in ctrA401 cells. These results suggest that ctrA401 cells are unable to activate these G1-phase promoters, but efficiently induce the late S-phase promoter class.
Table 2.
Activity of CtrA-activated promoters in C. crescentus WT & mutants
|
Promoter-probe assays of transcriptional reporters carrying a various lacZ- based CtrA dependent promoter in WT and ctrA401 cells and derivatives. Note that ctrA401 HS is isogenic to ctrA401 SS, encoding the CtrA (T168I/T170I) mutation. Black refers to late S-phase promoters and blue to the G1-phase promoters. spmX is represented in purple as it is not a direct target of CtrA. Values are expressed as percentages (activity in WT set at 100%). Data from four independent experiments, error bars are standard deviation. ND, Not determined.
To probe for commensurate changes in promoter occupancy of CtrA401 (T170I) versus WT CtrA, we conducted chromatin-immunoprecipitation followed by deep-sequencing (ChIP-Seq; Figure 1C, Supplementary Figure S1A) analyses of CtrA-bound promoters. Immunoprecipitations were done by incubating polyclonal antibodies to CtrA with chromatin extracted from WT and ctrA401 cells (grown exponentially at 30°C). Deep-sequencing revealed that the chromosomal occupancy of CtrA401 (T170I) is generally elevated compared to that of CtrA (WT), without noticeably affecting CtrA abundance inside cells (Figure 1C, inset). Thus, the T170I mutation does not compromise general binding to the CtrA box in vivo. As expected, the specific association of CtrA401 with late S-phase promoters such as PfliQ, PcpaB and PfliF is increased compared to that of CtrA (WT; Figure 1D), while at G1-phase promoters, such as PsciP, PhvyA, PhfsJ, PtacA and PpilA (Figure 1D), CtrA occupancy is clearly diminished. Moreover, a control promoter that CtrA does not directly target (the promoter of the fljL flagellin gene) showed poor occupancy of CtrA in vivo and little or no difference between WT and ctrA401 cells (Figure 1D, note change in scale).
We conclude that CtrA401 is impaired in the switch from late-S-phase to G1 phase promoter activation and binding. Since this defect also results in a failure to express the negative regulator of late S-phase promoters, SciP (Figure 4D, E, Supplementary Figure S1C, D), these promoters are no longer shut off and firing is maintained, likely resulting in elevated activity. Thus, the negative regulation of CtrA on late S-phase target promoters such as PfliQ is indirect, stemming from a defect in activating SciP expression in ctrA401 cells which explains why this allele of ctrA surfaced in the previous selection for loss of negative regulation at PfliQ (5). Importantly, and contrary to earlier interpretations of the phenotype of ctrA401 cells, our findings suggest that the phenotypes observed at the permissive temperature are not due to a general loss of CtrA function/binding at target promoters, but is instead due to a specific defect in G1-phase promoter firing and the resulting overactivation of late S-phase promoters.
Figure 4.

CtrA is substantially reprogrammed in stationary phase. (A) Promoter-probe assays of transcriptional reporters carrying a pleA and fliQ promoter in WT, ΔspoT, ctrA401 and derivatives. Transcription from PpleA-lacZ and PfliQ-lacZ in WT and ctrA401 is strongly induced in stationary phase in a SpoT-dependent manner. Values are expressed as percentages (activity in WT in exponential phase is set at 100%). (B) Genome-wide occupancies of CtrA on the Caulobacter WT genome in exponential phase compared to CtrA occupancies in WT, ΔspoT and ΔptsP genome in stationary phase as determined by ChIP-Seq using antibodies to CtrA. The x-axis represents the nucleotide position on the genome (bp), whereas the y-axis shows the normalized ChIP profiles in read per million (rpm). (C) ChIP-Seq traces of CtrA on different CtrA-binding promoter regions in WT cells in exponential phase, in WT and ΔspoT in stationary phase. Genes encoded are represented as boxes on the upper part of the graph, gene names and CCNA numbers gene annotation are indicated in the boxes or above. (D) Immunoblot showing steady-state levels of SciP in WT, ΔspoT, ctrA401, ΔspoT ctrA401, ΔsciP and Δlon, in exponential and stationary phase cells. MreB serves as a loading control. (E) Promoter-probe assays of sciP-translational reporters in WT, ΔspoT, ctrA401 and derivatives. Values are expressed as percentages (activity in WT set at 100%). Data from four independent experiments, error bars are standard deviation.
Capsule inhibits swarming motility of ctrA401 cells
Since the G1-phase is the principal phase of motility in C. crescentus, motility assays serve as proxy for perturbations in G1-phase duration (and gene expression). Consistent with the defect in G1-phase promoter activation by the ctrA401 mutation, these cells are poorly motile on swarm (0.3%) agar plates (Figure 2B) even though early flagellar promoter fire at an elevated level (Table 2; Figure 2A). Moreover, immunoblotting revealed that the FljK flagellin, a late flagellar component that is assembled into the flagellar filament, is also more abundant in ctrA401 cells than in WT cells (Figure 2B, Supplementary Figure S2A). Since a block in flagellar assembly prevents expression of FljK (32), these immunoblotting experiments indicate that ctrA401 cells do not suffer from a flagellar assembly defect and, thus, that swarming motility is impaired in ctrA401 cells for (an)other reason(s).
Figure 2.

Capsulation of the ctrA401 mutant affects the buoyancy switch, bacteriophage sensitivity and motility. (A) Promoter-probe assays of transcriptional reporters carrying a fljL, fliQ (class IV and class II genes, respectively) promoters in WT, ctrA401 and derivatives. Values are expressed as percentages (activity in WT set at 100%). (B) Motility plates (0,3% agar) inoculated with WT, ctrA401, ctrA401motility suppressors 1 and 2 (ctrA401-MS1 and ctrA401-MS2) strains and immunoblot showing the steady state levels of the alpha-flagellin FljK in WT, ΔspoT and ctrA401 cells. (C) Swarm (0.3%) agar inoculated with WT, ctrA401, ctrA401-MS, ΔMGE and Δ03998 (ΔCCNA_03998) cells. Note that motility is also improved when the ctrA401 mutation is transduced into ΔMGE cells (Supplementary Figure S3E), but not restored to the level seen for WT or ctrA401-MS, presumably because of yet unknown mechanism of motility inhibition in ctrA401 or because of unknown contributions conferred by the suppressor mutations on the MGE. (D) Schematic and pictures of cell buoyancy upon centrifugation on density gradient and sensitivity to bacteriophages ϕCr30 and ϕCbK for WT Caulobacter and ctrA401 mutants and derivatives. (E) Promoter-probe assays of hvyA-lacZ transcriptional (left graph) and translational reporters (right graph) in WT, ctrA401 and derivatives. Values are expressed as percentages (activity in WT set at 100%). Data from four independent experiments, error bars are standard deviation. (F) Swarming motility and buoyancy assays in cells overexpressing hvyA-tap fusion under control of Pvan on pMT335 plasmid restores the ‘heavy’ cell buoyancy to the ctrA401 cells.
To determine the basis of this motility defect, we isolated 10 motile suppressors (ctrA401-MS) emanating as flares on swarm agar from a background inoculum of (non-motile) ctrA401 cells (Figure 2B, C, Supplementary Figure S3E). Promoter probe assays with several early (class II) flagellar and other promoters in ctrA401-MS2 and ctrA401-MS4 cells (Table 2, Figure 2A, Supplementary Figure S3A) showed that CtrA-dependent regulation is not substantially altered in these mutants compared to ctrA401 parental cells. Thus, the suppressor mutation in ctrA401-MS cells lies outside of the CtrA regulatory hierarchy.
Surprisingly, ctrA401-MS cells rapidly sediment to the bottom of the cultivation tube, whereas ctrA401 parental or WT cultures remained in suspension during the same interval (Figure 2D). This sedimentation phenotype resembles that of capsule-less mutants that have a reduced cellular buoyancy (4). Indeed, buoyancy analyses by Percoll density gradient centrifugation revealed that ctrA401-MS2 cultures only contained ‘heavy’ cells (Figure 2D), whereas ctrA401 cells were only ‘light’. As WT cultures contain both ‘heavy’ (non-capsulated) G1-phase cells and ‘light’ (capsulated) S-phase cells (Figure 2D), we reasoned that ctrA401 cells remain ‘light’ because of a failure to activate a G1-phase promoter and that this block also perturbs swarming motility.
The CtrA-activated promoter of the hvyA gene, encoding a G1-specific capsulation inhibitor, fires in G1-phase (PhvyA) (15). Loss of HvyA leads to constitutive capsulation and thus only ‘light’ cells in the culture (4). After confirming that a transcriptional fusion to the PhvyA-lacZ and a PhvyA-hvyA::lacZ translational fusion is poorly active in ctrA401 cells (Figure 2E), we reasoned that the constitutive capsulation phenotype of ctrA401 cells is due to a failure in expressing HvyA. Indeed, constitutive expression of an epitope-tagged version of HvyA bearing a C-terminal Tandem Affinity Purification (TAP) (41) tag from a vanillate-inducible promoter on the pBBR-based plasmid pMT335 (42) not only reversed the buoyancy defect of ctrA401 cells (Figure 2F), but also improved swarming motility.
If ctrA401 cells are always capsulated and ‘light’, then the ‘heavy’ ctrA401 MS cells should lack capsule. Since capsule protects cells from infection by ϕCr30 by masking its receptor, the S-layer protein RsaA (43), we predicted that ctrA401 cells should be resistant to infection by ϕCr30, whereas ctrA401-MS2 should be hypersensitive to ϕCr30. The phage spot (lysis) tests shown in Figure 2D confirmed this prediction. As control for specificity we also conducted phage spot tests with caulophage ϕCbK which requires pili for infection (44). Both ctrA401 and ctrA401-MS2 mutants are resistant to ϕCbK because they do not express the pilA pilin gene encoding the structural subunit of the pilus filament that is expressed from the CtrA-activated PpilA promoter in G1-phase (Table 2, Supplementary Figures S1B, S2B). By contrast, WT cells are piliated and thus infected (and lysed) by ϕCbK.
Why might the ctrA401-MS mutants have lost the ability to form capsule? We suspected the acquisition of a mutation in a capsule biosynthesis gene in these mutants. A cluster of capsule biosynthetic proteins is encoded on a mobile genetic element (MGE; Supplementary Figure S3B) cells that can sporadically excise (45). PCR-based analyses of the ctrA401-MS mutants revealed that the MGE had been lost from ctrA401-MS4–7 cells as indicated by the increased abundance of the attB PCR product that is amplified when the attB junction is created by MGE excision (Supplementary Figure S3B, S3C), whereas the MGE is present in WT (NA1000) and ctrA401 cells. Further analysis by PCR revealed that ctrA401-MS4–7 cells lacked the capsule genes CCNA_03998 and CCNA_00477 encoded on the MGE. In the remaining mutants, CCNA_0477 is present, but CCNA_03998 was not detected in ctrA401-MS2 and a larger rearrangement of the CCNA_03998 had occurred in ctrA401-MS1 (Supplementary Figure S3C). Prompted by the observation that CCNA_03998 is rearranged or absent in the aforementioned ctrA401-MS mutants, we asked if the remaining four MS mutants (MS3, 8, 9, 10) had acquired point mutations in CCNA_03998 that would impair function. Indeed, PCR-sequencing revealed that ctrA401-MS3 has a nonsense mutation at codon 61 (changing the tryptophan codon to a stop codon), while ctrA401-MS8 and ctrA401-MS9 have an identical mutation at codon 331, changing a cysteine codon into a stop codon. The remaining mutant, ctrA401-MS10, has a missense mutation at codon 32, changing the proline codon to a histidine codon (Supplementary Figure S3D).
Taken together our analyses suggest that loss of the MGE and its functions (Figure 2C, Supplementary Figure S3E), particularly the loss of the capsule biosynthetic activity provided by the WbuB-like glycosyltransferase CCNA_03998 (4,15) (Figure 2C), promotes swarming motility of ctrA401 cells.
Intragenic suppressor mutations of ctrA401 alter DNA-binding
To investigate the G1-promoter block of ctrA401 cells further, we designed another suppressor screen to select for ameliorated G1-phase gene expression using the pilA::PpilA-nptII transcriptional reporter conferring resistance to kanamycin (13). For practical reasons, we chose ctrA401-MS2 as host for the reporter since this mutant showed the same regulatory defects on CtrA-dependent gene expression as ctrA401 parent cells (Table 2), with the added benefit of enabling convenient transfer of the pilA::PpilA-nptII reporter using ϕCr30-mediated generalized transduction, which is not possible for ctrA401 parental cells that do not support growth of ϕCr30 (see above). The pilA::PpilA-nptII reporter confers resistance to 40 μg/ml kanamycin (on plates) to WT cells (Supplementary Figure S4A). As expected, pilA::PpilA-nptII is poorly active in ctrA401-MS2 cells, precluding growth on kanamycin (40 μg/ml) plates (Supplementary Figure S4A). We found that transposon (Tn) mutagenesis of this reporter strain using a himar1 Tn conferring resistance to gentamicin, followed by selection for resistance to gentamicin and kanamycin, only revealed promoter-up Tn insertions at PpilA (Supplementary Figure S4B). Therefore, we sought intragenic suppressor mutations in ctrA401 that allow growth of ctrA401-MS2 pilA::PpilA-nptII reporter cells on plates containing kanamycin (40 μg/ml). To eliminate isolating mutants harboring promoter-up mutations in PpilA that permit growth on kanamycin plates, but that do not otherwise impact the CtrA regulon, we imposed a second visual screen for stalks in the kanamycin-resistant mutants, since the polar stalk is absent from ctrA401 cells (5) and it can be conveniently recognized by differential interference contrast (DIC) microscopy. As shown in Figure 3A, ctrA401 and ctrA401-MS2 pilA::PpilA-nptII parental cells are stalkless when grown in PYE, but stalks are produced in the ctrA401-SS (stalked suppressor) mutant. Moreover, DIC microscopy and fluorescence-activated cell sorting (FACS) revealed that the cell filamentation defect is also ameliorated in the SS mutant cells compared to ctrA401 and ctrA401-MS2 pilA::PpilA-nptII parental cells (Figure 3A).
Backcrossing experiments and DNA sequencing revealed that the ctrA401-SS mutant had acquired an additional mutation in ctrA, changing the threonine codon 168 into an isoleucine codon (T168I). The T168I mutation lies two residues ahead of the T170I mutation in CtrA401 (Figure 3B), within the DNA-binding domain of CtrA. The stretch from 167 to 191 in C. crescentus CtrA is highly conserved in the alpha-proteobacteria (Figure 3B), but strikingly T168 is also substituted by a hydrophobic residue (valine) at the corresponding position in the rickettsial lineage (for example in CtrA from Ehrlichia canis, Figure 3B), suggesting that this position in CtrA likely reflects a key residue in determining the DNA-binding activity of CtrA at G1-phase promoters in free-living alpha-proteobacteria and developmental (growth phase transition) control in the obligate intracellular lineage.
Modelling the promoter-bound structure of WT and mutant CtrA (Supplementary Figure S5), along with ChIP-Seq analysis of ctrA401-SS cells (obtained from backcrossing the ctrA401-SS allele into WT cells, Figure 1C) supported the key role of this residue in or near the DNA-binding domain (DBD) of CtrA. The CtrA401-SS (T168I/T170I) protein shows an increase in occupancy at G1-phase promoters and a reduction at late S-phase promoters, thus correcting the properties of the CtrA401 (T170I) mutant towards the WT properties (Figure 1C, D). Importantly, promoter probe assays confirmed that G1-phase promoter firing was also ameliorated in the ctrA401-SS mutant compared to ctrA401 cells (Table 2). Expectedly, the late S-phase promoters are down-regulated since PsciP activity is also elevated in ctrA401-SS compared to ctrA401 cells. Immunoblotting confirmed that the steady-state levels of HfsJ and SpmX that are either directly (HfsJ) or indirectly (SpmX) dependent on a CtrA-activated G1-phase promoter, are restored to near WT levels in ctrA401-SS cells compared to ctrA401 cells (Figure 1B).
In addition to stalk formation, other deficiencies of ctrA401 cells are also ameliorated by the ctrA401-SS mutation, including the resistance to ϕCbK (i.e. piliation, Figure 2D, Supplementary Figure S1B), as well as the resistance to ϕCr30 along with the exclusive ‘light’ buoyancy (i.e. constitutive capsulation, Figure 2D) phenotype. As expected, since the ctrA401-SS mutation allows the formation of non-capsulated G1 cells because of the improved expression of HvyA, motility is also improved compared to ctrA401 parental cells (Figure 3C). Importantly, ctrA401-SS cells have an elevated heat tolerance (plating efficiency at 37°C) compared to ctrA401 cells (Supplementary Figure S1C).
Thus, residues T170 and T168 define the binding capacity of CtrA at G1-phase promoters and confer opposing phenotypes depending on the side chain replacement. This notion is further reinforced by the previous finding of suppressor mutations in these residues (i.e. T168A and T170A) with the opposite effect, i.e. restoring motility to cells in which G1-promoters are de-repressed (15). Determining the transposon (Tn) insertions profile by Tn-Seq (transposon insertion sequencing) analysis of ctrA401 (T170I) and ctrA (T170A) cells also illustrated opposite mutant phenotypes (Figure 3D, Supplementary Figure S6). For example, for the divK gene that negatively regulates CtrA activity and induces a G1 arrest when disrupted (46), Tn insertions are 15-fold more frequent in ctrA401 (T170I) cells versus ctrA (T170A) cells (Figure 3D, Supplementary Table S7).
CtrA reprogramming in stationary phase is mediated by RpoB via (p)ppGpp
The fact that CtrA is implicated in the rickettsial growth phase (developmental) transition (24) along with the role of reside T168 described above, prompted us to investigate CtrA in stationary C. crescentus cells. We observed that the late S-phase promoter probe reporters PpleA-lacZ and PfliQ-lacZ are substantially upregulated in stationary phase compared to exponential phase ctrA401 cells or compared to stationary WT cells (Figure 4A). The stationary phase promoter induction of these two promoters by CtrA401 and (WT) CtrA is enhanced by (p)ppGpp, the alarmone that is produced by SpoT in stationary phase (47) as indicated by the fact the promoter activity is attenuated in ΔspoT cells. This finding prompted us to define the CtrA stationary phase target regulon by ChIP-Seq analysis in WT cells. We observed the ChIP-Seq profiles of CtrA to change substantially between exponential and stationary phase cells (Figure 4B, C). Moreover, SpoT enhances chromatin occupancy of CtrA in stationary phase (Figure 4B) and the activity of these CtrA-target promoters is indeed induced in stationary phase in a manner that depends on SpoT compared to exponential phase cells (Supplementary Figure S7).
As further confirmation for the role of (p)ppGpp in enhancing CtrA promoter occupancy in stationary phase, ChIP-Seq also revealed that CtrA was also less abundant on chromatin isolated from stationary cells lacking PtsP, the E1 component of an alternate phosphoenolpyruvate transfer system that activates the SpoT pathway upon nitrogen starvation (48–50) (Figure 4B). A prominent example of a stationary-phase induced CtrA-target promoter is the G1-phase promoter of the hfsJ gene (PhfsJ, see ChIP-Seq traces in Figure 4C). The strong SpoT-dependent increase in CtrA occupancy correlates well with the increase in HfsJ protein (51) steady-state levels in stationary phase by immunoblotting (Figure 1B). CtrA steady-state levels in stationary phase cells are reduced in the absence of SpoT (Figure 5A, right), suggesting that occupancy of CtrA is enhanced by (p)ppGpp because it promotes an increase in CtrA abundance.
Figure 5.

CtrA reprogramming in stationary phase is mediated by RpoB via (p)ppGpp. (A) Identification of the rpoBH559P mutation as motility suppressor of the ΔptsP ΔspoT mutant that restores CtrA steady state levels in stationary phase. Swarm (0.3%) agar plates inoculated with WT, ΔptsP ΔspoT, WT rpoB* and ΔptsP ΔspoT rpoB*. rpoBH559P mutation confers hypermotility to WT and ΔptsP ΔspoT cells. Immunoblot analysis (right) of the steady-state levels of CtrA in exponential and stationary phase of WT, ΔspoT, ΔptsP ΔspoT, WT rpoB* and ΔptsP ΔspoT rpoB* cells. CCNA_00163 serves as a loading control. (B) FACS analysis and DNA content (FL1-A) quantification in exponential and stationary phase of WT, ΔspoT, ctrA401, rpoB* and derivatives. Cells expressing the constitutive active form of E. coli RelA (RelA’) are also shown. rpoB* and ectopic induction of (p)ppGpp lead to G1 block in exponential and stationary phase. In stationary phase, ctrA401 cells are elongated and do not replicate while disruption of spoT improves this defect. (C) Genome-wide occupancies of CtrA on the Caulobacter WT, ΔptsP ΔspoT, WT rpoB* and ΔptsP ΔspoT rpoB* genome in exponential phase. The rpoB* mutation impacts the occupancy of CtrA on chromatin. Stars indicate two highly enriched peaks in rpoB* at the CCNA_03890 and CCNA_03426 loci. (D) ChIP-Seq traces of CtrA on the two CtrA-binding promoter regions highlighted with stars in (C) in WT and rpoB* cells in exponential phase and in WT and ΔspoT cells in stationary phase. Gene names are represented as boxes on the upper part of the graph and CCNA gene annotation are indicated in the boxes or above. (E) Promoter-probe assays of transcriptional reporters carrying the CCNA_03890 promoter in WT, rpoB*, ΔspoT rpoB* and ΔptsP ΔspoT rpoB*. Values are expressed as percentages (activity in WT set at 100%). (F) Immunoblots showing steady-state levels of SciP and CtrA in WT, ΔspoT, ctrA401, ΔspoT ctrA401, rpoB* and rpoB* ctrA401 in exponential and stationary phase.
Interestingly, FACS analysis revealed a pronounced cell cycle defect of ctrA401 cells in stationary phase compared to exponential phase or compared to stationary WT cells (Figure 5B). In fact, ctrA401 cells accumulate extra chromosomes and there are few S-phase cells compared to WT populations. The accumulation of extra chromosomes in stationary ctrA401 cells is mitigated by the ΔspoT mutation and the elevated (late S-phase) promoter activity in ctrA401 cells is also curbed when SpoT is inactivated (Table 2, Figure 4A). To test whether this contribution of SpoT/(p)ppGpp is mediated via allosteric control of RNA polymerase by (p)ppGpp, we sought suppressor mutations that render cells independent of (p)ppGpp for cell cycle control. To this end, we spotted ΔspoT ΔptsP double mutant cells on swarm agar and isolated spontaneous suppressor mutants that overcome the swarming defect of the parent. Genome sequencing of one such suppressor mutant and backcrossing of the mutation revealed a change of histidine codon to a tyrosine at position 559 of the rpoB gene (rpoB*), encoding the beta-subunit of RNA polymerase, to confer the increase in swarming motility (Figure 5A left). The rpoB* mutation also augments swarming motility when introduced into WT cells (Figure 5A left) and increases the fraction of the G1 population as determined by FACS, akin to WT cells engineered to ectopically produce (p)ppGpp with an inducible heterologous (p)ppGpp synthetase gene from E. coli (52) (relA’, lacking the C-terminal regulatory domain, Figure 5B) integrated at the xylX locus (30). Consistent with these results, the steady-state levels of CtrA are also upregulated in cells with the rpoB* mutation (Figure 5A, right) and the (ChIP-Seq) profile of CtrA-bound promoters precipitated from chromatin of exponentially growing cells revealed that the rpoB* mutation impacts the distribution of CtrA on its targets in vivo. Several promoters that CtrA prominently binds in exponential phase WT cells are further increased in rpoB* cells as confirmed by the ChIP-Seq traces presented in Figure 5D. The transcripts of CCNA_03890 and CCNA_03426, the former encoding a hypothetical protein conserved in the Caulobacterales order and the latter a MarR-like transcription factor, are strongly cell-cycle regulated like other CtrA-regulated transcripts (53), consistent with the notion that their promoters are directly activated by CtrA and enhanced in the presence of (p)ppGpp. In exponential phase cells, the rpoB* mutation does not fully recapitulate the CtrA occupancy profile of stationary stationary phase WT cells (Figures 4B, 5C, D), indicating that unknown determinants other than (p)ppGpp play a role in CtrA reprograming at certain promoters upon the transition into stationary phase. Together the reprogramming of CtrA and strong increase in SciP expression in stationary phase in a manner that depends on (p)ppGpp (Figure 4D, E) provides further support that cell cycle transcriptional regulators can also control growth phase transitions signaled by (p)ppGpp.
DISCUSSION
Coordination of G1-specific traits with CtrA
How CtrA can switch from activating late S-phase promoters to G1-phase promoters is fundamental to fully understand virulence gene expression underlying alpha-proteobacterial pathogenesis and symbiosis. In the human pathogen Brucella abortus G1-phase cells play an important role in driving the early stages of infection (20), while in the plant symbiont Sinorhizobium meliloti transcriptome studies in synchronized populations have documented the transcriptional switch from late S-phase to G1-phase (54). Upon studying the C. crescentus ctrA401 mutant we discovered a determinant uncoupling the two promoter classes activation by CtrA. Importantly, we observed that binding of CtrA401 at late S-phase promoters is not compromised in vivo. Instead, it is elevated compared to WT CtrA with a commensurate increase in promoter activity, while G1-phase promoters show the inverse behavior.
In C. crescentus, the promoter switch drives cell surface structure remodeling, including pili and capsule (1) that are known virulence determinants in bacterial pathogens. The acquisition of pili in G1 cells is triggered by expression of the PilA pilin (55), while capsule is lost from G1 cells upon expression of the negative regulator of capsulation, the HvyA transglutaminase homolog (4). These traits collectively promote the susceptibility of G1 cells to the bacteriophages ϕCr30 and ϕCbK that bind the S-layer and pili, respectively (43,44). The ctrA401 mutant is resistant to both phages because constitutive capsulation protects against infection by ϕCr30, while loss of piliation is due to the defect in PilA expression in ctrA401 cells and prevents infection by ϕCbK. Since cellular dispersal (G1-phase) functions are activated by the CtrA-dependent promoter switch, double stranded DNA phages such as ϕCr30 and ϕCbK deliberately infect G1 cells in order to introduce their genomes into the host before chromosome replication commences. Expression of HvyA not only reverses the constitutive capsulation phenotype of ctrA401 cells, but also ameliorates the swarming motility defect, indicating that capsule interferes with flagellar motility, explaining why HvyA expression has been incorporated into the regulatory program of Caulobacter G1 cells and its orthologs appear to underlie similar control. Thus, such coordination is likely critical for efficient dissemination and virulence properties of G1 cells in different alpha-proteobacteria.
The pivotal role of residues T168 and T170 in CtrA
CtrA is essential and its predicted essentiality formed the basis for the isolation of a lethal ‘loss of function’ (ctrA401) mutation that was isolated at the restrictive temperature of 37°C. The second selection criterion was impaired negative regulation of class II flagellar (late S-phase) activated by CtrA (5), despite the known role of CtrA being activating most target promoters (2). How the negative and positive regulation by CtrA on the same class of promoters could be instated was an unresolved question. The results described here provide a rational explanation for this effect, as we showed that only activation of the G1-promoter class is curbed by the CtrA mutation. In principle, the T170I mutation in CtrA401 can also be viewed as a ‘gain of function’ mutation endowing CtrA401 with an extended ChIP profile (Figure 1C), most likely reinforced by the reduction of sciP expression (Figure 1C, D; Supplementary Figure S1C, D) at 30°C.
By contrast, the activating function of CtrA (or stability) is lost at 37°C and thus cells suffer from insufficiency of essential cell division (and likely other) transcripts and/or de-regulation of DNA replication, ultimately causing cell death. Since the mutation in CtrA401 (T170I) lies in a highly conserved residue within the DBD (Supplementary Figure S5), our findings unearth a critical residue directing promoter, gene expression and thus deterministic phenotypic switches. The suppressors analyses revealed a compensatory mutation (T168I) in close proximity to the T170I mutation within the conserved DBD (Figure 3B). This mutation not only partially reinstates the promoter switch to CtrA401 (Table 2), but also ameliorates the temperature sensitivity of ctrA401 cells (Supplementary Figure S4C). Homology-based structural predictions of CtrA401 do not suggest a modification of the DNA-binding domain structure compared to WT CtrA (Supplementary Figure S5), but clearly the T168I mutation increases the occupancy of CtrA at G1-phase promoters, suggesting that DNA-affinity is increased or that this mutation promotes competition of CtrA against the repressor MucR at G1-phase promoters without directly affecting affinity for the CtrA-target box. While structural studies by NMR or X-ray crystallography might allow pinpointing the effect of the T168I mutation, modelling of CtrA onto the BasR/PmrA response regulator structures (45) predicts a head-to-tail binding of the two DBDs within a dimer, with T168 and T170 of CtrA being located close to important residues of the DBD. Both residues may act on each other, for example to promote cooperativity on DNA-binding within certain promoter sequences or local DNA architectures imposed by other DNA-binding regulators. Interestingly, residue T168 of CtrA was previously identified as a gain-of-function mutation to improve activation of late S-phase promoters to ΔmucR1/2 cells (15) (Figure 1E). The difference between having a small structural change between an alanine and isoleucine instead of T168 is not impossible to reconcile with changes in promoter activation and cooperativity upon DNA-binding depending on the forward genetic selection, but the exact consequences remain to be resolved at the atomic level using structural studies. Interestingly, CtrA in the rickettsial lineage naturally has a valine in place of threonine at position 168 of C. crescentus CtrA. With our genetic implications of the isoleucine or alanine (15) modification at this site in modulating the promoter switch of CtrA, depending on the context of a threonine or isoleucine at position 170, it will be revealing to investigate how rickettsial CtrA execute the suspected developmental switch, especially since this lineage does not encode homologs of the accessory negative regulators, SciP and MucR, in their genomes (1). Thus, we propose that functional Yin and Yang between T168 and T170 in C. crescentus CtrA fine-tunes the promoter switch by CtrA during the cell cycle and/or growth phase transitions.
(p)ppGpp is required for promoter reprogramming by CtrA during growth transition
Another major change in CtrA promoter occupancy occurs upon entry in stationary phase and such reprogramming may reflect an ancestral role of CtrA that is still exploited by the obligate intracellular rickettsial pathogens to direct the growth phase transition, for example in Ehrlichia chaffeensis from reticulate cells into dense-cored cells (24,26). While E. chaffeensis does not offer a powerful genetic system to dissect how growth-phase dependent reprogramming of CtrA occurs, we show that in C. crescentus alarmone-based nutritional signals acting through RNAP play an important role. Having shown previously that PctrA activity is increased in a (p)ppGpp dependent manner (50), we confirmed that CtrA levels are decreased in stationary ΔspoT cells and that a mutation in RpoB (H559Y) bypasses the need for (p)ppGpp. This mutation also augments the steady-levels of CtrA, consistent with the previous evidence that (p)ppGpp regulates CtrA stability (30). The role of (p)ppGpp in virulence and cell cycle control is well established for B. abortus (56,57) and S. meliloti (54,58,59), respectively, and even though members of the Ehrlichia genus do not encode a SpoT/RelA homologue in their genomes, many members of other rickettsial genera do. Moreover, mutations isolated in other alpha-proteobacteria that suppress (p)ppGpp null strain also mapped to rpoB and rpoC (59). Interestingly, the conserved H559 residue in C. crescentus RpoB corresponds to the H551 of E. coli that mimics (p)ppGpp binding (60). Thus, the role of the alarmone in transcription regulation is conserved in bacteria, and its action on CtrA and its targets to implement promoter switching provides an ideal and central entry point to modulate cell cycle and growth phase transitions.
DATA AVAILABILITY
Sequence data have been deposited to the Gene Expression Omnibus (GEO) database (GSE134017 accession, samples GSM3933392–GSM3933401).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mike Laub (Massachusetts Institute of Technology, USA), Lucy Shapiro (Stanford University, USA) and Justine Collier (University of Lausanne, CH) for materials, and Laurence Degeorges for excellent technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swiss National Science Foundation [31003A_182576]; University of Geneva. Funding for open access charge: Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung [31003A_182576].
Conflict of interest statement. None declared.
REFERENCES
- 1. Panis G., Murray S.R., Viollier P.H.. Versatility of global transcriptional regulators in alpha-Proteobacteria: from essential cell cycle control to ancillary functions. FEMS Microbiol. Rev. 2015; 39:120–133. [DOI] [PubMed] [Google Scholar]
- 2. Laub M.T., Shapiro L., McAdams H.H.. Systems biology of Caulobacter. Annu. Rev. Genet. 2007; 41:429–441. [DOI] [PubMed] [Google Scholar]
- 3. Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991; 204:372–384. [DOI] [PubMed] [Google Scholar]
- 4. Ardissone S., Fumeaux C., Bergé M., Beaussart A., Théraulaz L., Radhakrishnan S.K., Dufrene Y.F., Viollier P.H.. Cell cycle constraints on capsulation and bacteriophage susceptibility. eLife. 2014; 3:e03587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quon K.C., Marczynski G.T., Shapiro L.. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell. 1996; 84:83–93. [DOI] [PubMed] [Google Scholar]
- 6. Quon K.C., Yang B., Domian I.J., Shapiro L., Marczynski G.T.. Negative control of bacterial DNA replication by a cell cycle regulatory protein that binds at the chromosome origin. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bastedo D.P., Marczynski G.T.. CtrA response regulator binding to the Caulobacter chromosome replication origin is required during nutrient and antibiotic stress as well as during cell cycle progression. Mol. Microbiol. 2009; 72:139–154. [DOI] [PubMed] [Google Scholar]
- 8. Joshi K.K., Berge M., Radhakrishnan S.K., Viollier P.H., Chien P.. An adaptor hierarchy regulates proteolysis during a bacterial cell cycle. Cell. 2015; 163:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joshi K.K., Chien P.. Regulated proteolysis in Bacteria: Caulobacter. Annu. Rev. Genet. 2016; 50:423–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Domian I.J., Quon K.C., Shapiro L.. Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell. 1997; 90:415–424. [DOI] [PubMed] [Google Scholar]
- 11. Jacobs C., Domian I.J., Maddock J.R., Shapiro L.. Cell cycle-dependent polar localization of an essential bacterial histidine kinase that controls DNA replication and cell division. Cell. 1999; 97:111–120. [DOI] [PubMed] [Google Scholar]
- 12. Tsokos C.G., Perchuk B.S., Laub M.T.. A dynamic complex of signaling proteins uses polar localization to regulate cell-fate asymmetry in Caulobacter crescentus. Dev. Cell. 2011; 20:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iniesta A.A., McGrath P.T., Reisenauer A., McAdams H.H., Shapiro L.. A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:10935–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biondi E.G., Reisinger S.J., Skerker J.M., Arif M., Perchuk B.S., Ryan K.R., Laub M.T.. Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature. 2006; 444:899–904. [DOI] [PubMed] [Google Scholar]
- 15. Fumeaux C., Radhakrishnan S.K., Ardissone S., Theraulaz L., Frandi A., Martins D., Nesper J., Abel S., Jenal U., Viollier P.H.. Cell cycle transition from S-phase to G1 in Caulobacter is mediated by ancestral virulence regulators. Nat. Commun. 2014; 5:4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan M.H., Kozdon J.B., Shen X., Shapiro L., McAdams H.H.. An essential transcription factor, SciP, enhances robustness of Caulobacter cell cycle regulation. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:18985–18990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gora K.G., Tsokos C.G., Chen Y.E., Srinivasan B.S., Perchuk B.S., Laub M.T.. A cell-type-specific protein-protein interaction modulates transcriptional activity of a master regulator in Caulobacter crescentus. Mol. Cell. 2010; 39:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gora K.G., Cantin A., Wohlever M., Joshi K.K., Perchuk B.S., Chien P., Laub M.T.. Regulated proteolysis of a transcription factor complex is critical to cell cycle progression in Caulobacter crescentus. Mol. Microbiol. 2013; 87:1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ardissone S., Redder P., Russo G., Frandi A., Fumeaux C., Patrignani A., Schlapbach R., Falquet L., Viollier P.H.. Cell cycle constraints and environmental control of local DNA hypomethylation in alpha-Proteobacteria. PLos Genet. 2016; 12:e1006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deghelt M., Mullier C., Sternon J.F., Francis N., Laloux G., Dotreppe D., Van der Henst C., Jacobs-Wagner C., Letesson J.J., De Bolle X.. G1-arrested newborn cells are the predominant infectious form of the pathogen Brucella abortus. Nat. Commun. 2014; 5:4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mirabella A., Terwagne M., Zygmunt M.S., Cloeckaert A., De Bolle X., Letesson J.J.. Brucella melitensis MucR, an orthologue of Sinorhizobium meliloti MucR, is involved in resistance to oxidative, detergent, and saline stresses and cell envelope modifications. J. Bacteriol. 2013; 195:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caswell C.C., Elhassanny A.E., Planchin E.E., Roux C.M., Weeks-Gorospe J.N., Ficht T.A., Dunman P.M., Roop R.M. 2nd. The diverse genetic regulon of the virulence-associated transcriptional regulator MucR in Brucella abortus 2308. Infect. Immun. 2013; 81:1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pini F., De Nisco N.J., Ferri L., Penterman J., Fioravanti A., Brilli M., Mengoni A., Bazzicalupo M., Viollier P.H., Walker G.C. et al.. Cell cycle control by the master regulator CtrA in Sinorhizobium meliloti. PLos Genet. 2015; 11:e1005232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng Z., Miura K., Popov V.L., Kumagai Y., Rikihisa Y.. Insights into the CtrA regulon in development of stress resistance in obligatory intracellular pathogen Ehrlichia chaffeensis. Mol. Microbiol. 2011; 82:1217–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J.Z., Popov V.L., Gao S., Walker D.H., Yu X.J.. The developmental cycle of Ehrlichia chaffeensis in vertebrate cells. Cell Microbiol. 2007; 9:610–618. [DOI] [PubMed] [Google Scholar]
- 26. Rikihisa Y. Molecular pathogenesis of Ehrlichia chaffeensis infection. Annu. Rev. Microbiol. 2015; 69:283–304. [DOI] [PubMed] [Google Scholar]
- 27. Hauryliuk V., Atkinson G.C., Murakami K.S., Tenson T., Gerdes K.. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015; 13:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boutte C.C., Crosson S.. The complex logic of stringent response regulation in Caulobacter crescentus: starvation signalling in an oligotrophic environment. Mol. Microbiol. 2011; 80:695–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boutte C.C., Henry J.T., Crosson S.. ppGpp and polyphosphate modulate cell cycle progression in Caulobacter crescentus. J. Bacteriol. 2012; 194:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez D., Collier J.. Effects of (p)ppGpp on the progression of the cell cycle of Caulobacter crescentus. J. Bacteriol. 2014; 196:2514–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberati N.T., Urbach J.M., Miyata S., Lee D.G., Drenkard E., Wu G., Villanueva J., Wei T., Ausubel F.M.. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:2833–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murray S.M., Panis G., Fumeaux C., Viollier P.H., Howard M.. Computational and genetic reduction of a cell cycle to its simplest, primordial components. PLoS Biol. 2013; 11:e1001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Viollier P.H., Thanbichler M., McGrath P.T., West L., Meewan M., McAdams H.H., Shapiro L.. Rapid and sequential movement of individual chromosomal loci to specific subcellular locations during bacterial DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9257–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viollier P.H., Sternheim N., Shapiro L.. A dynamically localized histidine kinase controls the asymmetric distribution of polar pili proteins. EMBO J. 2002; 21:4420–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radhakrishnan S.K., Thanbichler M., Viollier P.H.. The dynamic interplay between a cell fate determinant and a lysozyme homolog drives the asymmetric division cycle of Caulobacter crescentus. Genes Dev. 2008; 22:212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Figge R.M., Divakaruni A.V., Gober J.W.. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol. Microbiol. 2004; 51:1321–1332. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lou Y.C., Weng T.H., Li Y.C., Kao Y.F., Lin W.F., Peng H.L., Chou S.H., Hsiao C.D., Chen C.. Structure and dynamics of polymyxin-resistance-associated response regulator PmrA in complex with promoter DNA. Nat. Commun. 2015; 6:8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al.. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Willett J.W., Herrou J., Briegel A., Rotskoff G., Crosson S.. Structural asymmetry in a conserved signaling system that regulates division, replication, and virulence of an intracellular pathogen. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B.. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001; 24:218–229. [DOI] [PubMed] [Google Scholar]
- 42. Thanbichler M., Iniesta A.A., Shapiro L.. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 2007; 35:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edwards P., Smit J.. A transducing bacteriophage for Caulobacter crescentus uses the paracrystalline surface layer protein as a receptor. J. Bacteriol. 1991; 173:5568–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guerrero-Ferreira R.C., Viollier P.H., Ely B., Poindexter J.S., Georgieva M., Jensen G.J., Wright E.R.. Alternative mechanism for bacteriophage adsorption to the motile bacterium Caulobacter crescentus. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9963–9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marks M.E., Castro-Rojas C.M., Teiling C., Du L., Kapatral V., Walunas T.L., Crosson S.. The genetic basis of laboratory adaptation in Caulobacter crescentus. J. Bacteriol. 2010; 192:3678–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hung D.Y., Shapiro L.. A signal transduction protein cues proteolytic events critical to Caulobacter cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13160–13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boutte C.C., Crosson S.. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013; 21:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hallez R., Delaby M., Sanselicio S., Viollier P.H.. Hit the right spots: cell cycle control by phosphorylated guanosines in alphaproteobacteria. Nat. Rev. Microbiol. 2017; 15:137–148. [DOI] [PubMed] [Google Scholar]
- 49. Ronneau S., Petit K., De Bolle X., Hallez R.. Phosphotransferase-dependent accumulation of (p)ppGpp in response to glutamine deprivation in Caulobacter crescentus. Nat. Commun. 2016; 7:11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanselicio S., Viollier P.H.. Convergence of alarmone and cell cycle signaling from trans-encoded sensory domains. mBio. 2015; 6:e01415-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fiebig A., Herrou J., Fumeaux C., Radhakrishnan S.K., Viollier P.H., Crosson S.. A cell cycle and nutritional checkpoint controlling bacterial surface adhesion. PLos Genet. 2014; 10:e1004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Turnbull K.J., Dzhygyr I., Lindemose S., Hauryliuk V., Roghanian M.. Intramolecular Interactions Dominate the Autoregulation of Escherichia coli Stringent Factor RelA. Front Microbiol. 2019; 10:1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schrader J.M., Li G.W., Childers W.S., Perez A.M., Weissman J.S., Shapiro L., McAdams H.H.. Dynamic translation regulation in Caulobacter cell cycle control. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6859–E6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Nisco N.J., Abo R.P., Wu C.M., Penterman J., Walker G.C.. Global analysis of cell cycle gene expression of the legume symbiont Sinorhizobium meliloti. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:3217–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skerker J.M., Shapiro L.. Identification and cell cycle control of a novel pilus system in Caulobacter crescentus. EMBO J. 2000; 19:3223–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim S., Watanabe K., Suzuki H., Watarai M.. Roles of Brucella abortus SpoT in morphological differentiation and intramacrophagic replication. Microbiology. 2005; 151:1607–1617. [DOI] [PubMed] [Google Scholar]
- 57. Dozot M., Boigegrain R.A., Delrue R.M., Hallez R., Ouahrani-Bettache S., Danese I., Letesson J.J., De Bolle X., Kohler S.. The stringent response mediator Rsh is required for Brucella melitensis and Brucella suis virulence, and for expression of the type IV secretion system virB. Cell Microbiol. 2006; 8:1791–1802. [DOI] [PubMed] [Google Scholar]
- 58. Wells D.H., Long S.R.. The Sinorhizobium meliloti stringent response affects multiple aspects of symbiosis. Mol. Microbiol. 2002; 43:1115–1127. [DOI] [PubMed] [Google Scholar]
- 59. Wells D.H., Long S.R.. Mutations in rpoBC suppress the defects of a Sinorhizobium meliloti relA mutant. J. Bacteriol. 2003; 185:5602–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trautinger B.W., Lloyd R.G.. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002; 21:6944–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data have been deposited to the Gene Expression Omnibus (GEO) database (GSE134017 accession, samples GSM3933392–GSM3933401).