


Abstract
Chromatin accessibility maps are important for the functional interpretation of the genome. Here, we systematically analysed assay specific differences between DNase I-seq, ATAC-seq and NOMe-seq in a side by side experimental and bioinformatic setup. We observe that most prominent nucleosome depleted regions (NDRs, e.g. in promoters) are roboustly called by all three or at least two assays. However, we also find a high proportion of assay specific NDRs that are often ‘called’ by only one of the assays. We show evidence that these assay specific NDRs are indeed genuine open chromatin sites and contribute important information for accurate gene expression prediction. While technically ATAC-seq and DNase I-seq provide a superb high NDR calling rate for relatively low sequencing costs in comparison to NOMe-seq, NOMe-seq singles out for its genome-wide coverage allowing to not only detect NDRs but also endogenous DNA methylation and as we show here genome wide segmentation into heterochromatic B domains and local phasing of nucleosomes outside of NDRs. In summary, our comparisons strongly suggest to consider assay specific differences for the experimental design and for generalized and comparative functional interpretations.
INTRODUCTION
The eukaryotic genome is largely organized in nucleosomes - the basic unit of chromatin. The precise mapping of nucleosome occupancy and the accessible DNA provides a widely used molecular approach to monitor chromatin organization in cells and the specific and local impact on gene expression. Three main experimental approaches , DNase I-seq, ATAC-seq and NOMe-seq, are mainly used to comprehensively analyze chromatin accessibility on a genome wide level. All these approaches have in the meantime been scaled down to single cell analyses making them the prime assays for functional studies (1–5). In some cases, these assays have even be applied to simultaneously measure gene expression (RNA-seq) and chromatin accessibility (NOMe-seq, ATAC-seq) from the same cell (5–7).
Several independent studies demonstrated that each of these methods come with advantages and disadvantages but so far very few systematic comparative analyses have been performed using standardized procedures. Our study fills this gap aiming to evaluate the comparability and complementarity of the three methods and at the same time showing their individual advantages or limitations to identify and interpret epigenetic and chromatin data.
The oldest and still widely used method to analyze nucleosome occupancy and positioning is DNase I-seq (8) (see Figure 1). DNase I cuts at freely accessible DNA to release small DNA fragments, which when coupled to next-generation sequencing (NGS) can be traced back to the genome to identify open chromatin regions. Extensive DNase I studies by ENCODE show that this approach boosts the identification of regulatory elements in different cells and showed that numerous genetic variants identified in genome-wide association studies (GWAS) locate to such DNase I hypersensitive regions (9). Still, the general use of DNase I-seq for open chromatin mapping can be quite difficult. First of all, DNase I experiments usually require a fair amount of pre-testing to identify the right incubation conditions, because, among other factors, under- or over-digestion of the chromatin will greatly influence the detection rate of open chromatin sites (10). For this the abundance of primary cell material can often be a problem. Usually, several experiments with different amounts of DNase I have to be performed to determine the optimum conditions. Once conditions are established the method can be scaled down to even single cells as showed by (2). Using paired end sequencing the prediction of nucleosome depleted regions (NDRs) from aligned DNase I-seq reads can be performed with conventional peak callers developed for ChIP-seq such as MACS2 (11).
Figure 1.

Chromatin accessibility assays. (A) NOMe-seq (left): Isolated nuclei are treated with M.CviPI, which methylates cytosines in GpC-dinucleotides. The DNA is purified, subjected to library preparation including bisulfite-conversion and sequenced. The resulting data give quantitative measures of GpC- and endogenous CpG-methylation at base-pair resolution across the whole genome. DNase I-seq (middle): After isolation of nuclei, chromatin is digested with DNase I. The DNA is purified and short double hit fragments are selected, which are used for library preparation and sequencing. The obtained data represent a relative enrichment of double-hit fragments in accessible regions. ATAC-seq (right): Isolated nuclei are treated with Tn5 transposase, which is loaded with sequencing adapters. After the tagmentation of chromatin (fragmentation and tagging by the transposome), the DNA is purified, barcoded by PCR and sequenced. The data give a relative read-out of tagmented fragments across accessible regions. (B) Snapshot of chromatin accessibility data relative to genomic coordinates for section of chr12. NOMe-seq (blue): Quantitative measure of GCH-methylation and NDRs called with gNOMeHMM are shown. The GCH coverage track indicates if number of reads is 5 (downward bars) or higher (upward bars). DNase I- (red) and ATAC-seq (orange): Read coverage and NDRs called with MACS2 are shown.
A second method with growing popularity is ATAC-seq (12) (see Figure 1), which stands for assay for transposase-accessible chromatin. ATAC-seq uses hyperactive Tn5 transposase-mediated cutting of genomic DNA combined with ligation-mediated insertion of DNA oligonucleotides which are pre-loaded (in vitro) to the enzyme. Following DNA isolation and PCR-amplification, libraries can directly be used for NGS. Because of its relative technical simplicity and high sensitivity this method is increasingly used (13–17). ATAC-seq requires very low amount (down to single cells) of (even frozen) input material to generate comprehensive maps (1000–50 000 cells). Since the ATAC reaction is an ‘end-point’ reaction it reduces the risk of chromatin over-digestion. Additionally, due to the relative simplicity of the protocol, ATAC-seq allows for high-throughput application to hundreds of bulk samples at relatively low cost making it applicable to larger clinical cohorts (18). A drawback of ATAC-seq is a frequently observed enrichment of mitochondrial (MT) sequence reads due to the unprotected nature of MT DNA which appears to be a preferred target for the Tn5 transposase in the cell. This can be avoided by depleting unwanted reads with a CRISPR/Cas9 based strategy (19,20). This addition however makes the method more laborious. ATAC-seq data can be processed using standard peak callers developed for ChIP-seq such as MACS2 (11).
The third method with growing popularity is a nuclease free method called NOMe-seq which stands for “nucleosome occupancy and methylation”. NOMe-seq utilizes the enzyme M.CviPI that specifically methylates cytosine dyads in a GpC sequence context originally used to identify local open chromatin regions (21) . Following the incubation of permeabilized cells or nuclei with M.CviPI, the extracted DNA is subjected to conventional bisulfite conversion followed by either regional (targeted) or genome-wide sequencing (WGBS). With this approach the DNA-methylation levels at GpC sites (NOMe reaction) and at endogenous CpG sites are determined simultaneously. The GpC methylation can be used to map DNA accessibility and determine nucleosome free regions. NOMe also has some interesting (partially unique) technical features: (i) NOMe-seq only requires low amounts of input material (down to single cells) and is applicable for many cell types. (ii) The in vitro methylation step is an endpoint reaction reducing the risk of over-exposure. (iii) NOMe-seq data provide a direct and single molecule quantitative readout, measuring the accessibility for each GpC in a single chromosome. (iv) NOMe can be seamlessly adopted for region-specific or whole genome-wide analysis of open chromatin. (v) NOMe-seq delivers the endogenous CpG methylation as a second readout enabling the simultaneous analysis of chromatin accessibility and DNA methylation on the same molecule in one experiment. The NOMe-Seq ‘readout’ of open chromatin sites is limited to GpC containing sequences and requires a high sequencing depth.
A recent NOMe-based assay, scNMT-seq, allows the combined sequencing of nucleosome occupancy together with transcriptome of the same cell (5). The current methods for NOMe-seq data interpretation leave room for improvement. (22) recently developed the findNDR tool, which computes enrichment scores of methylated reads for fixed size windows against a genome-wide average methylation rate. Here, we present a new algorithm for detecting NDRs in NOMe-seq data that allows for variable size length, performs a proper FDR correction, corrects for local, regional methylation backgrounds and corrects for abundance artifacts due to amplifications as commonly found in cancer genomes.
To perform a direct comparison of DNase I-, NOMe- and ATAC-seq data we decided to produce all data from a standard model cell line HepG2. We performed all experiments in our lab using the same stock of cells and the same cultivation conditions to minimize technical confounding variables. We deliberately used bulk cells to obtain deep and comprehensive genome wide overviews for each method. We observe that all three methods have a considerable overlap in detecting (strong) major open chromatin regions but also deviate to a large extend in other regions. We find that none of the methods calls the entire spectrum of open chromatin sites. This becomes most apparent when using these data for functional prediction of gene expression. We believe that our detailed comparison will contribute to better understand the technical and experimental limits of each of the three methods and at the same time allow to rationalize which approach should be used to address particular questions.
MATERIALS AND METHODS
Library preparation
Growing cells
HepG2 cell line was obtained from ATCC and cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal calf serum and 1% Pen/Strep mix under standard conditions (37°C, 5% CO2).
NOMe-seq
Optimal conditions for NOMe were first established by testing effects of several important technical parameters, such as nuclei extraction procedure, nuclei fixation, amount of M.CviPI enzyme and incubation time. As a read-out we used ultra-deep bisulfite amplicon sequencing for genomic regions with known chromatin accessibility states (as communicated with T. Kelly and/or published by (23)). The obtained results indicated that (i) after 3h accessible region is already fully methylated and ii) fixation of nuclei does not impact M.CviPI efficiency or nuclei integrity (Supplementary Figure S1). Another accessible region at the RPL27 gene with a known nuclei position (middle section of the amplicon) was used to evaluate the effect of different amounts of M.CviPI for 3 h in vitro methylation of 1 × 106 native HepG2 nuclei. From these tests, we concluded that incubation of HepG2 nuclei with 60 U of M.CviPI for 3 h results in a complete in vitro methylation of GpCs at accessible regions. For genome wide NOMe-seq, 1 million HepG2 nuclei were extracted using nuclei extraction buffer (60 mM KCl; 15 mM Tris–HCl, pH 8.0; 15 mM NaCl; 1 mM EDTA, pH 8.0; 0.5 mM EGTA, pH 8.0; 0.5 mM spermidine free base) supplemented with complete protease inhibitor cocktail (Roche, Basel, Switzerland) and 0.1% NP40 (Sigma-Aldrich, St. Louis, USA), and incubated on ice for 10 min. Nuclei were centrifuged (500 g, 4°C, 8 min), and the pellet was washed with nuclei extraction buffer. After another centrifugation step, the pellet was gently resuspended in 90 μl of 1× GpC buffer (NEB, Ipswich, USA) followed by addition of 70 μl of NOMe reaction mix 7 μl 10× GpC buffer (NEB), 1.5 μl of 32 mM SAM (NEB), 45 μl of 1 M sucrose and 60 U of M. CviPI (NEB). The reaction was incubated for 3 h at 37°C, and 0.5 μl of SAM was added after one and two hours. The reaction was stopped by adding 160 μl NOMe stop buffer (20 mM Tris–HCl, pH 8.0; 600 mM NaCl; 1% SDS, 10 mM EDTA) and 10 μl proteinase K (20 mg/ml, Sigma-Aldrich) and genomic DNA was extracted. Next, 100 ng was bisulfite-converted with the EZ DNA Methylation-Gold kit (Zymo, Irvine, USA) and then subjected to NGS library preparation using the TruSeq DNA Methylation Kit (Illumina, San Diego, USA) according to the manufacturer’s protocol. All libraries were checked for adapter dimers and fragment distribution on a Bioanalyzer HS chip (Agilent Technologies, USA). All samples were sequenced on an Illumina HiSeq2500 using V3 flowcells.
DNase I-seq
1 × 107 cells HepG2 cells have been subjected to nuclei isolation same as for NOMe and DNase I-seq was done as described previously (24).
ATAC-seq
ATAC-seq was performed on 50 000 HepG2 nuclei according to the protocol outlined by Buenrostro et al. (12).
Sequencing, pre-processing
Fastq files were trimmed for adapter sequence and low quality tails (Q < 20) with trim_galore (25), after which they were mapped to the human (hs37,1000G) genome (26). For WGBS and NOMe data, this was done with GSNAP (27) and for DNase I and ATAC data, GEM (28) was utilized (See Supplementary Table S1).
WGBS and NOMe
Unmapped reads were removed with samtools (29), before further processing with the Bis-SNP pipeline (30). Initially, reads were remapped in regions close to known insertions or deletions as supplied by Database of Single Nucleotide Polymorphisms (dbSNP); Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine (dbSNP Build ID: 138); available from: http://www.ncbi.nlm.nih.gov/SNP/. Duplicated reads were marked with Picard tools (http://broadinstitute.github.io/picard) and potentially overlapping sections between two paired reads were clipped with bamUtils (31). The quality was recalibrated in the context of dbSNP. Finally, methylation levels were called for all cytosines, and extracted with a modified version of the Bis-SNP vcf2bed.pl helper-script. For the NOMe samples, bed files were generated for cytosines in GCH and HCG context, where H is the IUPAC code for A, C or T. This corresponds to artificial and natural methylation, respectively. For WGBS samples, files for CG context were generated.
DNase I and ATAC
Duplicated reads were annotated with Picard tools. MACS2 (v2.1.0) (32) was used to call NDRs. In comparison to ChIP-seq, the cutting with DNase I and inclusion of adapters with the transposase in ATAC puts the focus on the start and end of a fragment, therefore MACS was executed with the following parameters: –shift -100, –extsize 200, –nomodel and –keep-dup all. Evaluation of other considered parameters can be found in Supplementary Text, section ‘On MACS2 peak calling for DNase I-seq and ATAC-seq’ and Supplementary Figure S20. All duplications were kept as MACS only takes one end of the fragment into account when designating duplicated reads compared to Picard tools that makes use of both.
External data
External data sources are listed in data file 1
Finding open chromatin regions with NOMe data
Given methylation values Mi, i = 1, …, m, for m cytosines in GCH context in the human genome, we use a Hidden Markov Model (HMM) to segment all cytosines in GCH context into two states (i) NDR and (ii) occupied region. As the methylation values are in range [0, 1] we use a binomial distribution in each HMM state. We use the Baum–Welch algorithm to fit the 2-state HMM using the HiddenMarkov R package (33). We stop the parameter optimization after either 1000 iterations or if the likelihood between two consecutive rounds drops <10−3. We fit an HMM for each chromosome and use parallelization with the SNOW package for fast computation. Each GC nucleotide is predicted to be open or closed based on posterior decoding using the fitted binomial HMM. Stretches of predicted NDRs, so called peaks, are further ranked by significance.
Initially, P-values were calculated with a one-sided Fisher’s exact test, contrasting the number of observations of methylated and unmethylated cytosines in GCH context within the region to a that of a background region. The background was selected as the closest 4 kb of closed chromatin up- and down-stream of the tested region.
To assign significance to the potential open chromatin regions found by the HMM, we computed empirical false discovery rates (FDRs) as well as the corresponding q-values. (34) The false discovery rate at significance threshold t(FDR(t)) is the expected value of the proportion of false discoveries significant at threshold t(f(t)) divided by the total number of discoveries that are significant at threshold t(s(t)):
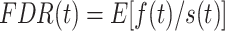 |
(1) |
This can be approximated by E[f(t)]/E[s(t)]. Using the HMM on our data set, we could directly get an estimate for E[f(t)] by counting regions with a P-value smaller than or equal to t. To estimate E[s(t)], we generated null data from the real data by shuffling methylation levels of the input data, leaving all other parts of the data intact. We then computed the segmentation for this data. This was performed to get the P-value distribution of regions falsely labeled as open.
Due to the coverage dependence of the chosen test, we implemented an automatic stratification step based on non-parametric mixture model clustering with the Mclust R-package (35). Assuming that regions with deviating copy numbers is the exception, we let the median represent the common coverage, and optimize the shrinkage parameter to minimize the number of clusters with mean coverage below the median. After each loci are assigned to a cluster, FDR values are estimated within each cluster. The gNOMeHMM package can be retrieved from https://github.com/karl616/gNOMePeaks.
Processing of RNA-seq data
TopHat 2.0.11 (36) and Bowtie 2.2.1 (37) were used to generate BAM files of RNA-seq reads, for NCBI build 37.1 in –library-type fr-firststrand and –b2-very-sensitive setting. Gene expression was quantified using Cufflinks 2.0.2 (38), the hg19 reference genome using the options frag-bias-correct, multi-read-correct, and compatible-hits-norm.
Determine characteristics of DNase I, ATAC and NOMe NDRs
To better understand the characteristics of the DNase I endonuclease, the Tn5 transposase, and the GpC methyltransferase M.CviPI, we generated sequence motifs, DNA shape predictions, as well as investigated DNA methylation at DNase I-seq and ATAC-seq cut-sites and at GpC sites for NOMe, respectively. We obtained the considered sequences using the bedtools getfasta (39) command on the 5′-cut-sites/GpC sites retrieved from the aligned reads. Note that all ATAC-seq sequences are shifted by 4 bp upstream, to consider the center of the Tn5 transposase (12). Per GpC sites, we sampled reads according to the methylation state of the respective GpC.
Generation of sequence motifs
Using 59 850 858 DNase I-sequences, 30 108 148 ATAC-seq sequences, and 126 202 679 NOMe-sequences, we generated sequence logos using the ggseqlogo R-package (40). The sequence motifs reported in literature are 6 bp for DNase I (41) and 20 bp for ATAC (12). To our knowledge, no bias motif has been reported in literature for NOMe-seq. To ensure these are captured and to harmonize with other figures, we used 31 bp centered on the enzyme activity sites of each assay.
Shape prediction
We use the DNAshapeR R-package (42) predictions for the minor groove width (MGW), Roll, propeller twist (ProT) and helix twist (HelT). Due to memory limitations, we randomly selected 2 million sequences per assay, constructed as described above, to be used for the shape computations. All spatial features are computed for each assay in a 31 bp window centered at the enzyme activity site.
DNA methylation
For each assay and sample, CpG methylation was extracted in 40bp windows centered on an enzyme event (bedtools (39)). Average methylation, weighted to coverage, was calculated for each relative position.
Logistic regression classifier for assay-specific NDRs
To identify characteristics of NDRs identified with only one distinct assay, we learn a multi-class logistic regression classifier using a variety of sequence based features, explained in the next section.
Feature definition
Within each assay specific NDR, we computed
A, T, C and G content,
CG content,
CpG and GpC count,
average CpG methylation,
NOMe-seq coverage,
and counts for all 5-mers.
We excluded GpC methylation as it would be an obviously strong feature for NOMe-seq NDRs, potentially overshadowing the remaining features. Additionally, we excluded NDR length as a feature, as this would mainly resemble peak caller specificities, as shown before for different peak callers on DNase I-seq data (11). The aforementioned features are computed for 12 415 DNase I NDRs, 19 323 ATAC-seq NDRs and 19 453 NOMe NDRs (excluding the Y-chromosome).
Logistic regression
We learn a multiclass logistic regression classifier using elastic net regularization, as implemented in the glmnet R-package (43). Elastic net regularization is producing sparse models and at the same times distributes the regression coefficients among correlated, yet predictive features, a property known as the grouping effect. This is achieved by combining two penalty terms, the lasso(L1) and the ridge(L2) penalty:
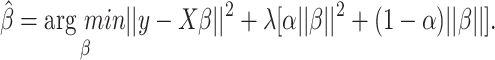 |
(2) |
Here, 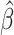 denotes the estimated model coefficients, β are the model coefficients, X is the feature matrix, which is scaled and centered, y is the response vector containing class assignments and α is a parameter regulating the trade-off between L1 and L2 penalty. We optimize the α parameter in a grid search from 0.0 to 1.0 with a step-size of 0.01. This is performed in scope of a 10-fold Monte-Carlo cross-validation procedure, in which the data is randomly split into
denotes the estimated model coefficients, β are the model coefficients, X is the feature matrix, which is scaled and centered, y is the response vector containing class assignments and α is a parameter regulating the trade-off between L1 and L2 penalty. We optimize the α parameter in a grid search from 0.0 to 1.0 with a step-size of 0.01. This is performed in scope of a 10-fold Monte-Carlo cross-validation procedure, in which the data is randomly split into 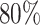 training and
training and 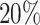 test data, assuring that both sets are balanced (see Supplementary Figure S3). Within each fold, we perform a nested-six fold inner cross validation to fit regression coefficients and determine the λ parameter controlling the total amount of regularization. We choose λ over the inner folds according to the minimum miss-classification error (λmin). Final model coefficients are determined using λmin on the entire training data set. Model performance is computed in terms of accuracy (ACC) on balanced hold-out test data using a 3 × 3 confusion matrix C:
test data, assuring that both sets are balanced (see Supplementary Figure S3). Within each fold, we perform a nested-six fold inner cross validation to fit regression coefficients and determine the λ parameter controlling the total amount of regularization. We choose λ over the inner folds according to the minimum miss-classification error (λmin). Final model coefficients are determined using λmin on the entire training data set. Model performance is computed in terms of accuracy (ACC) on balanced hold-out test data using a 3 × 3 confusion matrix C:
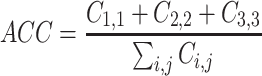 |
(3) |
Note that, since we perform a three-class classification, randomness is reflected by ACC ≤ 0.33.
Computation of nucleosome distances
We predicted nucleosome positions based on ChIP-seq data by running Nuchunter (44) with default parameters. The distance between neighboring predicted nucleosomes was calculated, the 220 315 gaps between 0 and 500 bp were selected and stratified into bins of 50 bp. Overlapping these gaps with NDRs and transcription factors allowed us to calculate enrichments in different size regimes. The enrichment was calculated as the difference between the observed and estimated number of overlapping features. For each feature and size bin, the estimated number of overlaps was calculated by distributing the observed overlaps over the bins in proportion to the number of expected overlaps. For plotting, the absolute values were log transformed, while keeping the sign of the original value
NDR clustering
For each set of NDRs, we calculated the average NOMe signal intensity in tiled 10 bp bins spanning 1kb up- and down-stream relative to the NDR summit. Subsequently, we identified all combinations of overlapping regions between the three NDR sets and the signal from these were combined. In the absence of a NDR the loci were defined by another assay, prioritized as NOMe, DNase I or ATAC. That is, a lacking NOMe NDR was approximated by DNase I and only if both NOMe and DNase I lacked NDR, the ATAC NDR was used as substitute. The resulting matrix consisted of rows with 200 bins from each assay. It was clustered into fifteen clusters with the k-means algorithm.
The resulting clusters were characterized with respect to the fraction of NDRs overlapping a ChromHMM segmentation of the DEEP HepG2 histone data and transcription factors with LOLA and an extended version of its core data base (45). The ChromHMM states are labeled in agreement to the 15-state core segmentation provided by Roadmap (46).
Linear regression predicting gene expression
To learn about the relationship between chromatin accessibility and gene expression, we fitted linear regression models predicting gene expression from predicted TFBSs.
Feature definition
We compute TFBS features using TEPIC (24,47) in all ATAC  , DNase I
, DNase I  and NOMe
and NOMe  NDR sets. Additionally, we consider the intersection
NDR sets. Additionally, we consider the intersection 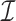 of the three sets, as well as their union
of the three sets, as well as their union 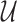 :
:
 |
(4) |
Furthermore, we consider three NDR sets extending 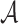 ,
, 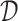 and
and 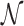 to match
to match 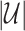 by randomly sampled regions that do not overlap with any of the already included open chromatin NDRs. We refer to these NDR sets with
by randomly sampled regions that do not overlap with any of the already included open chromatin NDRs. We refer to these NDR sets with 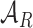 ,
, 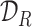 and
and 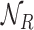 respectively. Thus, we consider in total eight different NDR sets
respectively. Thus, we consider in total eight different NDR sets 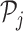 for j = 1, …, 8.
for j = 1, …, 8.
For each NDR 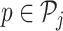 , we compute TF affinities ap, t for TF t using TRAP (48), normalized according to the length of the respective NDR |p|, for a set of 726 TFs. Position weight matrices are taken from the TEPIC 2.0 repository (47).
, we compute TF affinities ap, t for TF t using TRAP (48), normalized according to the length of the respective NDR |p|, for a set of 726 TFs. Position weight matrices are taken from the TEPIC 2.0 repository (47).
TF affinities ap, t are combined to TF-gene scores for gene g as suggested in (24),
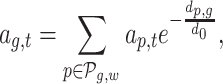 |
(5) |
using a window size w = 50 kb. In addition to TFBS features, we consider NDR length lg and NDR count cg as additional features per gene (as introduced in (49)):
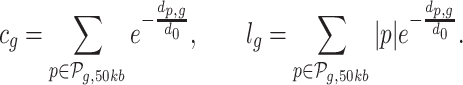 |
(6) |
Values for ag, t, cg and lg, are computed for all protein-coding genes that are associated to at least one DHS site. Where 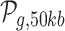 denotes all peaks in a 50 kb window around gene g.
denotes all peaks in a 50 kb window around gene g.
Linear regression
As for the logistic regression, we use elastic net regularization to learn a linear model of gene expression. Here, a row of the feature matrix X is composed of the TF gene scores ag, t as well as the values of cg and lg for a distinct gene g. The actual gene expression is denoted by the response vector y.
The learning strategy is identical to the one explained above, with the difference that the cross-validation error is measured as mean-squared error instead of miss-classification error. Final model performance is assessed using Spearman correlation computed between the actual gene expression y, and the predicted gene expression 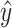 . Models are learned independently for each NDR set.
. Models are learned independently for each NDR set.
Hi-C data
Hi-C data of HepG2 from (50) was used (GSE113405). The interaction matrix and TADs were generated as described before (50). A/B compartment predictions were generated using HOMER tools (51) with the runHiCpca.pl command with the following parameters: -res 25000 -window 50 000. Positive values of pca1 correspond to A compartments while the negative values correspond to the B compartments. The compaction score Distal-to-Local [log2] Ratio (DLR) was calculated using analyzeHiC command with the following parameters: -res 5000 -window 15 000 -compactionStats auto. Figure 2A was generated using pyGenomicTracks (52).
Figure 2.

Genome wide chromatin landscape is reflected by NOMe signal. (A) zoomed out view (chr2:48,000,000-85,000,000) of open chromatin assay (NOMe-seq) with different epigenomic data tracks: from top to bottom; Hi-C contact matrix of HepG2 with called TADs (triangles), A/B compartments, ChromHMM segmentation using six histone marks (see Materials and Methods), two overlapping heterochromatic marks H3K9me3 and H3K27me3, GCH methylation signal filtered for NDRs, large-scale segmentation of NOMe signal (the horizontal line represents the value chosen by the classification model to define valleys) and UCSC genes. Valleys are identified from NOMe (see Methods) coincide with B-compartments, PMDs from (50), repressed polycomb (gray) and heterochromatic domains (Turquoise) and mostly pronounced at ZNF/repeats regions (Medium Aquamarine). (B) GCH methylation levels are low at heterochromatic and ZNF/repeats regions, while they are high in the repressive polycomb regions. (C) Compactness score derived from Hi-C data (DLR, see Methods) is higher in the heterochromatin and ZNF/Repeats regions in comparison to the repressive polycomb regions.
Large scale NOMe segmentation
GCH methylation values were first merged from both strands to calculate weighted average methylation per GpC and then smoothed using BSmooth (53) with h = 50 000 as a smoothing window. A set of genomic regions, after visual inspection, was manually selected and labeled accordingly as S1 (not valley) or S2 (valley). These regions were used to train a random forest classifier using the average GpC methylation in 30 kb tiles as variable vector and the aforementioned labels as response vector. Then the fitted model was used to predict the status of 30 kb tiles across the whole genome. All predicted consecutive S2 with gap length <30 kb were merged into one region. The final set of S2 regions were visualized in Figure 2A as valleys track. The training and prediction process were carried out using R caret package (54).
RESULTS
Genome wide comparison of open chromatin assay data
To monitor the comparability of the currently most widely used open chromatin assays we performed a direct data comparison between NOMe-seq, DNase I-seq and ATAC-seq, outlined in Figure 1, using data generated from the same batch of HepG2 cells grown in our lab. This approach reduces experimental confounding effects such as differences in cell batches and culture condition (growth, density). Data sets were generated following existing IHEC and BLUEPRINT protocols. The HepG2 cell line was selected as it is a major cell line used in ROADMAP and ENCODE analyses further allowing us to include external datasets for subsequent analyses.
A visual inspection of all three sequencing tracks indicated a fairly good agreement of local enrichment profiles at open chromatin sites (see Figure 1B) between ATAC- and DNase I peaks and NOMe signal enrichments. However, the NOMe-seq coverage is much more widespread as compared to the strong local enrichments of the two nuclease-based assays. To better compare the NOMe signal distribution with ATAC/DNase I ‘peak calling’ we calculated the genome wide GCH methylation levels of NOMe-seq data and related it to the FPKM values for DNase I-seq and ATAC-seq as numbers of 5′ read ends across the genome. We correlated these genome wide raw signal distributions aggregated in 500 bp bins (see Supplementary Figure S4). Following this approach we observe a moderate level of genome wide correlation between ATAC-seq and DNase I-seq read distributions (Spearman cor.: 0.41) while the correlation to NOMe-seq is far less only reaching 0.21 and 0.25 to ATAC and DNase I, respectively. To better understand the assay specific differences in the genome wide read distribution we started by investigating the sequence preferences of all three assays in more detail.
Sequence, structure and DNA-methylation influence DNase I, ATAC and NOMe
In line with (12) and (41) we observed that both endonucleases DNase I and the modified Tn5 (ATAC) show a slightly biased and distinct sequence preference at their cutting sites (see Figure 3A). This preference is found across multiple samples of different cell types (see Supplementary Figures S5 and S6). Also the ‘NOMe enzyme’ M.CviPI, which recognizes and methylates at the dinucleotide 5′GC3′ showed some minor sequence preferences at the flanking −2 and +2 position. However, this is only due to the (bioinformatic) exclusion of ambiguous GCG sites, which overlap with sites of endogenous CpG methylation and are therefore inconclusive. Despite the GCG effect, no other sequence biases were observed (see Supplementary Figure S7).
Figure 3.

Structural and sequence preferences for NOMe (left), DNase I (middle) and ATAC (right) are shown in columns. Cutsites (ATAC,DNase I) and sites of GC methylation (NOMe) are shown as position 0 (x-axis) with a window of 31 bp around it. For ATAC, a shift of 4 bp upstream is introduced to conserve symmetry. (A) Sequence logo for all assays, the larger a character is shown in a column the more often it occurs. (B) Analysis of structural and DNA methylation features. The top four panels contain structural features; in order, helix turn (HelT), minor groove width (MGW), propeller twist (ProT) and roll. These are displayed as median with a confidence band of median absolute deviation (MAD). The bottom panel contains the average CpG methylation with a MAD confidence band.
In an earlier line of work it was shown that CpG methylation in DNase I cut sites increases cleavage efficiency by altering the DNA structure (55) , which can also be predicted by looking at DNA shapes (56). To systematically compare the enzyme specific sequence preferences within each assay we predicted structural DNA shape features (see Methods) around the GpC methylation sites (NOMe) and the enzyme cutting sites (ATAC, DNase I) (Figure 3B and Supplementary Figure S8) across a number of available samples and datasets (see Supplementary Figure S8).
First, we investigated the structural features and observed that the sequences around the M.CviPI enzyme (NOMe) recognition site 5′GpC3′ show a pronounced signal for an increased Helix Twist (HelT) and Propeller Twist (ProT). Additionally, we find an increased Minor Groove Width (MGW) flanking the GC site. For DNase I, we observed an enlarged predicted MGW around the cut site, as reported before (55), and a slightly increased base roll. Both, M.CviPI and DNase I, act as monomers and show clear one sided effects around the recognition/cut site. The modified Tn5, on the other hand, acts as a dimer showing bidirectional oscillating changes in MGW, ProT, and Roll around the cut site. Together this analysis shows that the enzymes used in the three assays have sequence and structural preferences influencing their genome wide signal distribution.
Next, we examined if and how endogenous CpG methylation affects the three enzyme activities (see Figure 3B, bottom panel). DNase I indeed showed a slight but focused increase of CpG methylation around cutting sites—an observation confirming findings made by (55), while both M.CviPI and the modified Tn5 showed no position-specific effect of CpG methylation along the cutting site. However, we noticed that the average level of 5mC around DNase I and modified Tn5 (ATAC) cutting sites is strikingly lower as compared to M.CviPI (NOMe). This is a reflection of a much broader almost uniform (see Figure 1B) genome-wide coverage of NOMe reads.
Shared and specific features of nucleosome depleted regions recognized by NOMe, DNase I and ATAC assays
Next we focused our attention on comparing the performance of all three methods in open chromatin sites or Nucleosome Depleted Regions (NDRs). Such regions are widely used for functional genome annotations and interpretations. For NOMe we calculated NDRs with our own HMM based approach called gNOMeHMM (see Materials and Methods, (57)) which provides a robust genome wide NDR annotation (see Supplementary Figure S18 and Supplementary text, section ‘NOMe NDR prediction based on hidden Markov models’).
NDR detection for DNase I-seq and ATAC-seq data was performed with MACS2 (see Materials and Methods). Overall, we detected very similar numbers of NDRs for NOMe-seq (65 683), DNase I-seq (62 365) and ATAC-seq (67 675). Note that for all three assays libraries were sequenced at a depth to obtain sufficient coverage (NOMe = 10 × genome wide coverage of GpCs), DNase I 88 million reads, ATAC 84 million reads, see Materials and Methods and Supplementary Table S1). The total number of NDRs recognized by at least one of the three assays sums up to 105,081 of which most were found in intergenic (∼ 55%) or intronic (∼ 25%) regions. In total, 24% of NDRs were shared by all methods and additional 27% were supported by two methods (see Figure 4A).
Figure 4.

Comparison of accessibility measured with DNase I, ATAC and NOMe. (A) Comparison of NDR calls by all three methods, see text. (B) length distribution of NDRs. (C) NOMe signal at NDRs. (D) DNase I signal at NDRs. (E) ATAC signal at NDRs. (F) Results of classification of assay-unique NDRs. The confusion matrix shows actual (column) against predicted (row) class labels. (G–J) box plots of sequence characteristics selected by the classifier to have power in separation.
19 480 unique NDRs were predicted for NOMe, 12,854 for DNase I and 19,452 for ATAC. Most shared NDRs were found between ATAC and DNase I data, underpinning the commonalities in the enzymatic (cutting) reaction of DNase I and ATAC and also probably the commonalities in data processing and peak calling.
Common NDRs, i.e. NDRs shared by all three assays, tend to be longer, largely overlapping with previously known NDRs (see Figure 4B and Supplementary Figure S9B). They, in general, exhibit a strong above-average signal in all assays. NOMe-alone NDRs are an exception to this rule with slightly stronger signal as compared to common NDRs. Common NDRs are most strongly enriched for TSS, followed by CpG islands (CGIs) and LaminB1 sites and are heavily enriched for ENCODE mapped transcription factor binding sites (TFBSs) (see Supplementary Figure S9B). Assay-unique NDRs (ATAC-alone, DNase I-alone, NOMe-alone) have a relatively strong signal in only one of the data sets and the signal intensity drops or disappears in the other two assays (See Figure 4C–E). LOLA annotation of such assay-unique NDRs shows a specific enrichment of NOMe and ATAC unique NDRs for CTCF, Rad21 and SMC3 binding sites. This enrichment is even more pronounced in NOMe-unique NDRs. Rad21 and SMC3 are parts of the cohesin protein complex interacting with CTCF (58). Their co-localization suggests a widespread distribution of dynamic topological substructures (59) preferentially detected as NOMe and ATAC NDRs, respectively. Unique DNase I NDRs on the other hand are enriched for binding sites of the hepatocyte nuclear factors like FOXA1, FOXA2 and HNF4G indicating a higher sensitivity of DNase I to recognize networks of liver-specific NDRs. To resolve whether this is either a tissue driven or an assay driven effect, we analyzed 147 additional paired ATAC- and DNase I-seq samples from ENCODE (see Supplementary Figures S21–S23 and Supplementary Text, section ‘ENCODE ATAC-seq and DNase I-seq samples’). Although we commonly observed assay enrichment for unique ATAC and DNase I NDRs, there was no consistency among the enriched TFBS suggesting that the observed enrichments are at least partially depending on the analyzed tissue.
Sequence features and methylation enrichment around NDRs
To better characterize features separating the three sets of unique NDRs we applied a logistic regression classifier. The feature set included DNA methylation and sequence associated measures (see Methods). The accuracy of the classifier was 0.55 on a hold out test data set. Note that, as this is a three-class classification problem, a random classifier would achieve an accuracy of 0.33. When we additionally included the counts of 5-mers in the regions, the accuracy increased to 0.63. The most important features were A-content, C-content, GC count, and CG methylation (see Figure 4G-J). The former three separate NOMe from the enrichment assays, while CG methylation helps distinguish ATAC from NOMe and DNase I (list of top features shown in Supplementary Table S2). Additionally, some 5-mers were useful in separating the classes. For example, for classifying NOMe unique NDRs the 5-mer CGCGC was depleted, representing the GCG effect, while 5-mers enriched in DNase I unique NDRs resembled the observed DNase I sequence bias. Overall, the classifier was better at separating NOMe from ATAC and DNase I, and the most miss-classifications occurred between the ATAC and DNase I classes (see Figure 4F). The link between CG methylation and DNA accessibility is known (60). While only 10% of the Intersection NDRs have a CG methylation above 30%, there are 20% unique ATAC NDRs and about 30% unique NOMe and DNase I NDRs that fulfill this criteria (see Supplementary Figure S10A).
In addition, we looked to sequencing coverage and copy numbers (e.g. low complexity repeats) on NDR calling. Most NDRs are gathered around the median 16× read coverage, but there is a long tail toward high coverage. About 2-3% of unique NOMe NDRs had a coverage in the top 5%, while NDRs only detected with ATAC and/or DNase I had 8-9% (see Supplementary Figure S10B). This is concordant with a higher fraction of NDRs with at least 50% overlap to repeat masked regions among unique ATAC (38%) and DNase I (34%) NDRs as compared to unique NOMe NDRs (26%) and the intersection (15%) (see Supplementary Figure S10C). A feature singling out unique NOMe NDRs is the distance to the next NDR in the union set; 14% of unique NOMe NDRs are within 200 bp, while the next highest value is about 8% for other subsets containing NOMe NDRs in Figure 4A (see Supplementary Figure S10D).
Nucleosome phasing around NDRs
We analyzed the extent of nucleosome phasing around NDRs (see Supplementary Text, section ‘Considerations when observing phased nucleosomes’). While DNase I strongly uncovers the first nucleosomes 5′ and 3′ around the NDR, NOMe shows a nice ‘oscillating’ pattern of up to five nucleosomes around many NDRs (see Supplementary Figure S11). Also ATAC-seq has been shown to allow the detection of phased nucleosomes provided sufficient sequencing depth (12). In our setting ATAC-seq with 60 million reads nucleosome phasing around NDRs was only visible when fixing the summit of NDRs called by ATAC or at borders of NDRs called by NOMe (see Supplementary Figure S11). Considering that the actual sequencing coverage of NOMe-, DNase I- and ATAC-seq around NDRs is comparable, the detection of phased nucleosome patterns is much more pronounced for NOMe-seq. Reasons for this might be read information density and the genome wide coverage of NOMe-seq (see Supplementary text, section ‘Considerations when observing phased nucleosomes’).
NDRs can be grouped based on assays, size and nucleosome phasing
Next we performed K-means clustering of all NDRs in a 2 kb window centered around the NDR peak (see Figure 5A). We included all regions providing a NDR signal in at least one of the assays. The compilation of all NDRs illustrates that NDR size, signal strength and nucleosome phasing are prominent characteristics of both assay specific and assay independent patterns contributing to the formation of 15 NDR clusters. NDR clusters 3, 12, 13 and 15 are strongly enriched for TSS associated regions. They are marked by strong NDR signals mostly shared across all assays. These NDRs are characterized by an enrichment for general TFBS such as GABP, TAF1 and TBP (see Figure 5B). Clusters 10 and, to a lesser degree, 11 show a similar enrichment for TFBS but lack the strong TSS enrichment. Clusters 3 and 10 show an enrichment for TBP and bidirectional promoter activity. Cluster 1 and 6 have a moderate enrichment for active enhancers of class 1, also showing an enrichment for FOXA1, FOXA2 and to some degree HNF4G, i.e. a set of liver-specific transcription factors. NDR clusters 4 and 7 are most prominently called by NOMe-seq data. They were mildly (4) or strongly (7) enriched for CTCF TFBSs. Both clusters showed a regular distribution of open and closed chromatin signals indicating strong and extended nucleosome phasing around CTCF sites as reported by (22).
Figure 5.

Annotated clusters of NDRs with similar signal profiles. (A) NDRs signal profiles clustered into 15 clusters with K-means clustering. The left-most column shows the presence (yellow) of a called NDR for each method, respectively. For each assay, 2 kb centered on the NDR was split into 10 bp windows over which the signal was aggregated. With NOMe the raw methylation is displayed, while the DNase I and ATAC are represented by normalized log2-scaled read counts. (B) Annotation by LOLA of each cluster. The coloration of each tile corresponds to the log odds ratio of the enrichment test. These tests were only conducted against HepG2 tracks.
NDR clusters 4 and 7 also hold among the most narrow NDRs, which is in agreement with observations that nucleosome distance around NDRs with CTCF binding sites is smaller compared to, for instance, FOXA1 sites (see Figure 6A). Cluster 14 aggregates low signal NDRs, characterized by a lack of TFBS enrichment but an increased fraction of highly methylated and repeat masked loci. Together the clustering shows that parameters such as size, signal strength and nucleosome phasing are linked to functionally distinct NDR classes across the genome as also reported by (61). Particularly NOMe-seq and ATAC-seq show an enhanced sensitivity for detecting distinct groups of NDRs outside of annotated enhancers and TSS.
Figure 6.

Functional analysis of shared and assay-unique NDRs. (A) Analysis of nucleosome distances overlapping NDRs unqiue to any of the three assays as well as ChIP-seq peaks for the TF CTCF, RAD21, HNF4A, FOXA1 (rows). Intensity in the heatmap encodes enrichment compared to the background distribution in regions of the same length (column). (B) Comparison of gene expression prediction using TEPIC together with different NDR sets as basis for feature calculation (x-axis). The assay-specific NDR sets for ATAC, NOMe and DNase I are compared to the union or intersection NDR sets using all assays. In addition, each assay-specific NDR set was increased by adding random regions of the same size, as compared to the union set, denoted as ATAC R, NOMe R and DNase I R, respectively, to assess the effect on prediction performance with increased set sizes. Gene expression prediction performance is shown as a boxplot of Spearman correlation values (y-axis) between true and predicted expression values as part of 10-fold cross validation on genes not used for learning. ***t-test P < 0.01.
NDRs can be used to predict expression level of nearby genes
We recently developed TEPIC as an open chromatin based prediction model for gene expression using predicted TFBSs (24). With this approach we compared the prediction performance for NDRs individually called by ATAC, DNase I and NOMe, as well as the intersection and the union of those (see Figure 6B and Supplementary Figure S12). For each NDR subset, we computed TF binding predictions for 726 TFs and used these predictions in a gene-centric way as features to predict gene expression (see Materials and Methods). The worst performance was obtained with the NDRs obtained commonly by all three assays (intersection including 28 513 NDRs), which led to smaller performance Spearman correlation values although many of these NDRs share a strong signal in all assays. Note that these shared NDRs mostly cover TSS and active enhancers. For NDRs specific (not unique) to each assay, we observed that the results are comparable between assays with a slightly better performance of ATAC over NOMe and DNase I.
The best model performance was achieved by combining all three NDR sets (union). The union of NOMe and ATAC NDRs cannot be significantly distinguished from the full union although the trend is strongly in favor of the full union (see Supplementary Figure S12). To assure that this is not simply due to the increased number of NDRs, we extended each assay specific NDR set with randomly generated peaks to match the size of the union set. These models performed constantly worse than the actual assay specific sets and thus also worse than the union set. Put together this suggests that each assay fails to describe a certain part of the accessible chromatin landscape and thus, while the combination of assay specific NDRs allows to more comprehensively model the regulatory influence on gene expression.
Confirmation of assay specific NDRs
To verify the assignment of common and unique NOMe, DNase I and ATAC NDRs by an independent assay we selected 17 NDR regions showing distinct recognition patterns across the three assays (Supplementary Figure S13A). We analyzed these regions by targeted deep amplicon bisulfite sequencing following a NOMe treatment in HepG2-cells (see Supplementary Figure S13B for the experimental data of four examples). For NOMe-unqiue NDRs we observed a median GCH-methylation level of 35-70% (with the exception of the Fam35DP amplicon), while regions not called from NOMe-seq data (no NDR in any of the three methods or NDRs unique to DNase I and/or ATAC) show a reduced median GCH methylation of 9-26% (Supplementary Figure S13). Overall this confirms that NDRs are reliably called by gNOMeHMM showing a clear and specific GCH enrichment profile above background (except for FAM35DP with median GCH-methylation of 17%). Still, we also detect intriguing patterns of GCH-methylation in regions not called by gNOMeHMM, but only by the other methods. One such example is the DNase I- and ATAC-unique NDR NCK2 (Supplementary Figure S13B) and the other a non-NDR region within the UGGT1 (data not shown). Both show a clear GCH-methylation pattern in a subset of sequences indicating that a proportion of cells fulfill the criteria of a NOMe-specific NDR. This finding illustrates that some (partially) open chromatin regions might be missed by gNOMeHMM calling either due to low GCH-density or by low GCH-methylation levels indistinguishable from the neighboring ‘background signals’.
Genome wide information called by NOMe
In comparison to ATAC and DNase I, NOMe-seq data show an almost complete coverage across the genome. Outside of NDR peak regions this has mostly been treated as background noise. Here we had a deeper look into chromatin associated features linked to this genome wide ‘background’.
Nucleosome phasing around Intron-exon junctions
We first analyzed the nucleosome phasing in a 2 kb window around intron-exon junctions for all genes. In short we merged all NOMe data around intron-exon boundaries, excluding the first and the last two exons. To our surprise we detected a very pronounced phasing of up to 1 kb around the exon/intron junctions in genes with low expression when we applied K-means clustering (n = 5). This could not be observed for neither the unexpressed genes nor the moderate to highly expressed genes (see Supplementary Figure S14). Since expression is known to be related to histone elongation marks (62), we therefore analyzed the distribution of histone marks in the clusters in relation to phasing. Analogous to (63), we observe some reciprocal distribution of histone modifications at introns and exons (e.g. higher H3K36me3 in moderate to highly expressed exons) but no obvious direct link to the clusters showing phased or non-phased nucleosomes.
Genome wide distribution of NOMe signals in relation to chromatin states
While NOMe-seq has originally been used to assess chromatin accessibility, additional information content in the genome wide GpC methylation has largely been ignored. Visual inspection of GpC methylation in HepG2 cells indicated a minor variation of ‘background’ GpC signal across large domains (see Figure 2A and B), here called valleys. We therefore systematically analyzed the GpC methylation across 18 chromatin states for a series of cell types (HepG2, monocyte, machrophage, CD4 T central memory, CD4 T effector memory and CD4 T naive). We noticed that while the general level of genome wide GpC methylation changes with samples, the relative distribution over chromatin states remains constant (see Supplementary Figure S15). In fact, the lowest GpC signal was always in the Heterochromatin and ZNF genes & repeats states of the Roadmap 18-state ChromHMM segmentation.
In HepG2, we identified valleys using a classification model (see Materials and Methods). Surprisingly, the valleys are largely concurrent with partially methylated domains (PMDs), as defined by (50), and B-compartments retrieved from the Hi-C data of HepG2 (see Supplementary Figure S16). Moreover, the strongest segmental decrease of GpC methylation is co-localized with the most compacted heterochromatic domains, as measured by DLR score (distal-to-local ratio) based on Hi-C data (see Materials and Methods) (see Figure 2B and C).
Accurate genome wide CpG methylation calling
NOMe-seq has the great advantage to not only determine the genome wide distribution of closed and open chromatin but to also provide detailed information of the genome wide ‘endogenous’ CpG methylation status. To monitor this in detail we performed a direct comparison between NOMe-seq data and an independent WGBS-dataset generated from the same HepG2 cell batch. NOMe-seq called ‘endogenous’ CG methylation correlated excellent with the conventional WGBS data (Pearson correlation 0.95). This excellent correlation was observed on the level of single-CpG methylation as well as aggregated methylation levels at promoters, CpG islands and genome-wide tiled regions (see Supplementary Figure S17B and C). We noticed, however, that when confining our analysis to aggregated methylation levels in the regions identified as open chromatin by NOMe-seq (NDRs see below), correlation values slightly decreased as compared to genome-wide analysis, potentially due to the general depletion of CpG methylation in open chromatin (see Supplementary Figure S17B and D). We also observed a tendency of a decreased NOMe-seq read coverage at regions exhibiting the highest degree of differential methylation (see Supplementary Figure S17E). Interestingly, the highest ranking differentially methylated regions appear to have a higher CpG dinucleotide content whereas the GpC dinucleotide content is more uniformly distributed across all open chromatin regions (see Supplementary Figure S17E). We conclude that NOMe-seq data provide an overall excellent resource to determine endogenous CpG methylation largely independent from the GpC measured chromatin accessibility (see Supplementary Figure S17).
In summary, NOMe-seq provides not only openness/closeness information at NDRs such as TSS, enhancers or other ‘short’ regulatory regions, but also includes additional information about the genome wide chromatin compaction. Such additional features detectable in NOMe ‘bulk’ sequencing data sets may be useful for the interpretation of genome wide single cell NOMe-seq data.
DISCUSSION
There are many distinct technical approaches to map open chromatin such as MNase-seq (64,65), FAIRE-seq (66) and SONO-seq (67), but also using ChIP-seq data as reference (e.g. NucHunter (44)). Here, we focused on the three most commonly used methods ATAC, DNase I and NOMe, and thoroughly compared them in respect to their commonalities and differences as well as individual strengths and weaknesses.
Experimental differences and open chromatin calling
While all three methods require proper isolation of nuclei, the experimental challenges for the three assays differs. ATAC has the fewest working steps and nicely links labeling of accessible regions and NGS library preparation by the transposase-assisted incorporation of NGS adaptors into open chromatin. For DNase I it is important to avoid/minimize loss during the isolation of the small double-cut DNA fragments and the NGS library preparation. The experimental challenges for NOMe are comparable to DNase I although it is not an enrichment assay, but rather an extension of the commonly used bisulfite sequencing. This makes it possible to read out native CpG-methylation in addition to the chromosome accessibility and in scenarios where information from both epigenetic layers is desired, NOMe kills two birds with one stone. Since each NOMe sequencing read potentially reports on several linked events from a single cell, it provides an excellent opportunity to detect sub-population effects. With DNase I and ATAC, these events are commonly used to detect TFBS by genomic footprinting (68,69). Although, the coverage is relatively limited for genome-wide NOMe, such a procedure can be bolstered by multiple measurements from single cells as seen for CTCF (22). Our analysis also shows that irrespective of how NDRs are called NOMe allows (for most) a straight forward and very sensitive and quantitative way to deeply analysis the local chromatin accessibility via ultra deep sequencing of bisulfite amplicons.
Assay specific variation in NDR detection
A qualitative and quantitative interpretation of nucleosome depleted regions (NDRs) also called ‘open chromatin sites’ is the most valuable information deduced from chromatin accessibility data. Not surprisingly NDRs detected by all methods tested display strong signals. They are strongly enriched for TFBS and mainly cover active TSSs, common enhancers and other shared accessible/open loci (see Figure 5 and Supplementary Figure S9).
However, this core of strong and well-characterized NDRs is apparently less informative to predict gene expression, hinting towards the importance of weaker and more difficult to detect cell type specific NDRs outside of the intersection (see Figure 6B and Supplementary Figure S12). NDRs singularly or dually detected by the individual assays outperformed the intersection in expression prediction. Indeed the best predictions were made with the union of all or just ATAC and NOMe NDRs indicating that each set of unique NDRs carries important extra information not covered by the other assays.
To understand the differences in detection between the assays we investigated unique NDRs, i.e. NDRs called by one assay only, in more detail. To reach this point, we tested various NDR calling settings (see Supplementary Figures S19, S2 and Supplementary Text, section ‘On MACS2 peak calling for DNase I-seq and ATAC-seq’) and the results discussed here are representative throughout, but we would like to stress the importance in selecting optimal conditions when doing a more locus specific analysis. Although these NDRs were not called by one of the other two assays (note that we applied conventional sequencing depth for all assays), a deep NOMe-seq analysis suggests that seemingly unique NDRs can in principle also be detected by other assays (at least in one direction). Moreover, enrichment analysis of known NDRs (see Supplementary Figure S9), suggests that most of these (unique) NDRs are trustworthy. However, in HepG2, the unique sets of NDRs fall into two groups: DNase I NDRs are higher enriched for cell type (i.e. liver specific) enhancers overlapping with FOXA1, FOXA2 and HNF4G binding sites, while NOMe and ATAC NDRs often demarcate insulator regions associated with CTCF, Rad21 and SMC3 binding sites. Although the enrichment were different in other cell types (see Supplementary Figure S23), it remains clear that different assays detect different sub-populations of NDRs. Together these findings argue for a more careful assay specific and context dependent interpretation of open chromatin maps generated by only one assay and their limited use for trans-assay comparative analyses.
A potential reason for a preferential calling of insulator regions by ATAC and NOMe (see Figure 5) in our setting appears to be the variation in fragment length distribution between assays (see Figure 6A). DNase I-seq libraries are known to be dependent on enzyme concentration and insert size selection for library construction (10). NOMe and ATAC assays are used as ‘endpoint’ reactions and libraries are generated without size preselection. In our comparison they indeed cover a relatively smaller size range (see Figure 6A) such that the enrichment of relatively short insulator NRDs may be simply due to preparative differences.
Moreover, each assay comes with a set of inherent limitations affecting NDR detection. NOMe for example is only able to measure open chromatin in regions containing a sufficient 5′GpC3′ sequence context. Simultaneously, as shown by (12), (41) and confirmed here (see Figure 3, Supplementary Figures S5 and S6), the enzymes used for DNase I and ATAC assays come with a slight sequence preference. This could be handled by applying bias-correction approaches that adjust for sequence preferences, e.g. (69–71). We also show that the tertiary structure of the DNA has an apparent influence on enzyme activity (see Figure 3). Moreover there is an influence of endogenous 5′CpG3′ methylation surrounding the enzyme activity site. The increased methylation level at DNase I cut sites could be a sign of how at least one of these reside outside the NDR. In both NOMe and, especially, ATAC we observe a weak periodic pattern in the methylation bias that coincides with the minor groove width (see Supplementary Figure S8). It could be speculated whether this is due to how DNA is wrapped around the nucleosome as described by (60). In light of biases detected in previous and this study, we think that further research is needed to investigate effects on downstream bioinformatic analyses and biological interpretation.
With NOMe, we observed more NDRs close to other stronger NDRs (see Figure 5, cluster 5 and 8). These NDRs are probably not unique NDRs, but rather a result of phased nucleosomes and hence a sign of the strength and homogeneity of the stronger NDR. Our suggestion is to handle flanking NDRs with care and assign extra weight to the focal NDR at such a loci. For the enrichment assays—DNase I and ATAC—a potential sign of false positives would be high-ploidy or repeated regions artificially amplifying coverage and hence generating peaks. A common strategy to avoid this and other biases is the sequencing of a non-enriched library (cf. input sequencing for ChIP-seq). The most important factors, when applying machine learning to the classification problem of whether a NDR was unique to either assay, was A-, C-content, GC count and native CpG methylation.
Unique epigenomic information provided by NOMe
DNase I and ATAC-seq data are the most cost efficient and widely used techniques almost exclusively used for the local detection of open chromatin sites. In comparison to NOMe they only require a moderate level of sequencing depth and ongoing improvements in protocols are reducing that depth even further. However, what is often neglected is that the higher sequencing expenses for NOMe come with much deeper information content. First and most importantly in comparison to all other enrichment technologies (including MNase, FAIRE-seq and others), NOMe-Seq provides a single chromosome readout of multiple linked measurements allowing a direct localization and quantification of epigenetic changes. Recent developments in NOMe-Seq by (72) strongly argue for the use of NOMe for single molecule footprinting. Secondly, NOMe not only provides information of open chromatin sites (NDRs), it also allows to call endogenous DNA-methylation at the same time. These features have been appreciated by several authors making NOMe-seq a prime method for deep single cell epigenomics (4,5). However, so far most analyses concentrated on NDR detection and endogenous methylation calling, neglecting other genome wide informations we started to decipher in our analyses. Our extended study shows that GpC methylation profiles provide a measure for the extend and distribution of heterochromatin compartments and also the local nucleosome phasing outside of NDRs, e.g. around exon/intron junctions of genes across the genome. Surprisingly, this local phasing had some relation to gene expression strength. Additional work will be needed to understand the functional link between regular nucleosome spacing and low gene expression. We believe that such additional features are important add-ons that currently can comprehensively only be achieved by NOMe-seq.
In conclusion, our controlled side by side comparative approach of open chromatin assays revealed that all three most commonly used assays allow to call the most prominent NDRs covering a large fraction of the (mostly functionally annotated) highly accessible regions. However, we also find that many NDRs are less likely to be detected/called by individual assays and that these additional assay unique NDRs are extremely important to predict (and hence explain) the expression of genes. Our findings suggest that single assay approaches to detect open chromatin are less comprehensive than anticipated and incomplete for the calling of regulatory open chromatin information (at least under standard settings). Finally, we show that assays such as NOMe-seq that are more sequence consuming, but cover the genome in a comprehensive manner provide information on a series of useful additional epigenomic features important for functional interpretation.
DATA AVAILABILITY
All data sets used in this work are listed in supplementary file 1.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: K.N., F.S., G.G., F.M., P.E., M.S. and J.W. contributed to project conception. K.N., F.S., A.S., F.M., P.E., I.G.C., N.P. and M.S. contributed to the bioinformatic analysis. K.N., F.S., N.G., A.S., G.G., K.K., P.E. and J.W. contributed to the experimental analysis. N.G., G.G. and K.K. contributed to the primary data generation. K.N., F.S., N.G., A.S., G.G. and F.M. generated the figures. K.N. and J.W. drafted the article with the help of F.S., N.G., A.S., G.G., F.M., N.P. and M.S.
Notes
Present address: Fabian Müller, Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA.
Present address: Nico Pfeifer, Methods in Medical Informatics, Department of Computer Science, University of Tübingen, Tübingen, Germany.
Present address: Marcel H. Schulz, Institute for Cardiovascular Regeneration, Goethe University Frankfurt and German Center for Cardiovascular Research, Partner site Rhein-Main, Frankfurt am Main 60590, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Epigenome Programme (DEEP) by the Federal Ministry of Education and Research in Germany (BMBF 01KU1216); BMBF grant for de.NBI [031L0101D to K.N.]. Funding for open access charge: German Epigenome Programme (DEEP) by the Federal Ministry of Education and Research in Germany (BMBF 01KU1216); BMBF grant for de.NBI [031L0101D to K.N.].
Conflict of interest statement. The authors have no significant competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in this manuscript.
REFERENCES
- 1. Cusanovich D.A., Daza R., Adey A., Pliner H.A., Christiansen L., Gunderson K.L., Steemers F.J., Trapnell C., Shendure J.. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015; 348:910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jin W., Tang Q., Wan M., Cui K., Zhang Y., Ren G., Ni B., Sklar J., Przytycka T.M., Childs R. et al.. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015; 528:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife. 2017; 6:e23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo F., Li L., Li J., Wu X., Hu B., Zhu P., Wen L., Tang F.. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017; 27:967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clark S.J., Argelaguet R., Kapourani C.-A., Stubbs T.M., Lee H.J., Alda-Catalinas C., Krueger F., Sanguinetti G., Kelsey G., Marioni J.C. et al.. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 2018; 9:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao J., Cusanovich D.A., Ramani V., Aghamirzaie D., Pliner H.A., Hill A.J., Daza R.M., McFaline-Figueroa J.L., Packer J.S., Christiansen L. et al.. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018; 361:1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buenrostro J.D., Corces M.R., Lareau C.A., Wu B., Schep A.N., Aryee M.J., Majeti R., Chang H.Y., Greenleaf W.J.. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018; 173:1535–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. et al.. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M.. Linking disease associations with regulatory information in the human genome. Genome Res. 2012; 22:1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He H.H., Meyer C.A., Chen M.-W., Zang C., Liu Y., Rao P.K., Fei T., Xu H., Long H., Liu X.S. et al.. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods. 2014; 11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koohy H., Down T.A., Spivakov M., Hubbard T.. A comparison of peak callers used for DNase-Seq data. PLoS One. 2014; 9:e96303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu H., Yuan Z., Tan T., Wang J., Zhang J., Luo H.-J., Xia Y., Ji W., Gao F.. Improved tagmentation-based whole-genome bisulfite sequencing for input DNA from less than 100 mammalian cells. Epigenomics. 2015; 7:47–56. [DOI] [PubMed] [Google Scholar]
- 14. Picelli S., Björklund A.K., Reinius B., Sagasser S., Winberg G., Sandberg R.. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014; 24:2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Q., Gu L., Adey A., Radlwimmer B., Wang W., Hovestadt V., Bähr M., Wolf S., Shendure J., Eils R. et al.. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013; 8:2022–2032. [DOI] [PubMed] [Google Scholar]
- 16. Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J. et al.. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016; 48:1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W. et al.. The chromatin accessibility landscape of primary human cancers. Science. 2018; 362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montefiori L., Hernandez L., Zhang Z., Gilad Y., Ober C., Crawford G., Nobrega M., Sakabe N.J.. Reducing mitochondrial reads in ATAC-seq using CRISPR/Cas9. Scientific Rep. 2017; 7:2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu W., Crawford E.D., O’Donovan B., Wilson M.R., Chow E.D., Retallack H., DeRisi J.L.. Depletion of Abundant Sequences by Hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016; 17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kilgore J.A., Hoose S.A., Gustafson T.L., Porter W., Kladde M.P.. Single-molecule and population probing of chromatin structure using DNA methyltransferases. Methods. 2007; 41:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taberlay P.C., Statham A.L., Kelly T.K., Clark S.J., Jones P.A.. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer. Genome Res. 2014; 24:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly T.K., Liu Y., Lay F.D., Liang G., Berman B.P., Jones P.A.. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012; 22:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmidt F., Gasparoni N., Gasparoni G., Gianmoena K., Cadenas C., Polansky J.K., Ebert P., Nordstrom K., Barann M., Sinha A. et al.. Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res. 2017; 45:54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trim Galores software.
- 26. Consortium G.P. et al.. A global reference for human genetic variation. Nature. 2015; 526:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu T.D., Nacu S.. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010; 26:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marco-Sola S., Sammeth M., Guigó R., Ribeca P.. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012; 9:1185–1188. [DOI] [PubMed] [Google Scholar]
- 29. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y., Siegmund K.D., Laird P.W., Berman B.P.. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012; 13:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Breese M.R., Liu Y.. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. 2013; 29:494–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harte D. HiddenMarkov: Hidden Markov Models. R package version 1.8-4. 2015; Wellington: Statistics Research Associates. [Google Scholar]
- 34. Storey J.D., Tibshirani R.. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fraley C., Raftery A.. Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. 2002; 97: [Google Scholar]
- 36. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics. 2017; 33:3645–3647. [DOI] [PubMed] [Google Scholar]
- 41. Koohy H., Down T.A., Hubbard T.J.. Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS One. 2013; 8:e69853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiu T.P., Comoglio F., Zhou T., Yang L., Paro R., Rohs R.. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinformatics. 2016; 32:1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Friedman J., Hastie T., Tibshirani R.. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010; 33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 44. Mammana A., Vingron M., Chung H.-R.. Inferring nucleosome positions with their histone mark annotation from ChIP data. Bioinformatics. 2013; 29:2547–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheffield N.C., Bock C.. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016; 32:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmidt F., Kern F., Ebert P., Baumgarten N., Schulz M.H.. TEPIC 2—an extended framework for transcription factor binding prediction and integrative epigenomic analysis. Bioinformatics. 2018; 35:1608–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roider H.G., Kanhere A., Manke T., Vingron M.. Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics. 2007; 23:134–141. [DOI] [PubMed] [Google Scholar]
- 49. Schmidt F., Schulz M.H.. On the problem of confounders in modeling gene expression. Bioinformatics. 2018; 35:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salhab A., Nordström K., Gasparoni G., Kattler K., Ebert P., Ramirez F., Arrigoni L., Müller F., Polansky J.K., Cadenas C. et al.. A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol. 2018; 19:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramírez F., Bhardwaj V., Arrigoni L., Lam K.C., Grüning B.A., Villaveces J., Habermann B., Akhtar A., Manke T.. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 2018; 9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hansen K.D., Langmead B., Irizarry R.A.. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012; 13:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuhn M. Building predictive models in R using the caret package. J. Stat. Softw. 2008; 28:1–26.27774042 [Google Scholar]
- 55. Lazarovici A., Zhou T., Shafer A., Machado A.C.D., Riley T.R., Sandstrom R., Sabo P.J., Lu Y., Rohs R., Stamatoyannopoulos J.A. et al.. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rao S., Chiu T.-P., Kribelbauer J.F., Mann R.S., Bussemaker H.J., Rohs R.. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein–DNA binding. Epigenet. Chromatin. 2018; 11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Durek P., Nordström K., Gasparoni G., Salhab A., Kressler C., De Almeida M., Bassler K., Ulas T., Schmidt F., Xiong J. et al.. Epigenomic profiling of human CD4+ T cells supports a linear differentiation model and highlights molecular regulators of memory development. Immunity. 2016; 45:1148–1161. [DOI] [PubMed] [Google Scholar]
- 58. Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A.. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uusküla-Reimand L., Hou H., Samavarchi-Tehrani P., Rudan M.V., Liang M., Medina-Rivera A., Mohammed H., Schmidt D., Schwalie P., Young E.J. et al.. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016; 17:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Collings C.K., Anderson J.N.. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenet. Chromatin. 2017; 10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lai B., Gao W., Cui K., Xie W., Tang Q., Jin W., Hu G., Ni B., Zhao K.. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature. 2018; 562:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X.S., Ahringer J.. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009; 41:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huff J.T., Plocik A.M., Guthrie C., Yamamoto K.R.. Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 2010; 17:1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kent N.A., Adams S., Moorhouse A., Paszkiewicz K.. Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res. 2010; 39:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Henikoff J.G., Belsky J.A., Krassovsky K., MacAlpine D.M., Henikoff S.. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bianco S., Rodrigue S., Murphy B.D., Gévry N.. Global mapping of open chromatin regulatory elements by formaldehyde-assisted isolation of regulatory elements followed by sequencing (FAIRE-seq). DNA-Protein Interactions. 2015; Springer; 261–272. [DOI] [PubMed] [Google Scholar]
- 67. Auerbach R.K., Euskirchen G., Rozowsky J., Lamarre-Vincent N., Moqtaderi Z., Lefrançois P., Struhl K., Gerstein M., Snyder M.. Mapping accessible chromatin regions using Sono-Seq. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:14926–14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vierstra J., Stamatoyannopoulos J.A.. Genomic footprinting. Nat. Methods. 2016; 13:213. [DOI] [PubMed] [Google Scholar]
- 69. Li Z., Schulz M. H., Look T., Begemann M., Zenke M., Costa I. G, Z.. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019; 20:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martins A.L., Walavalkar N.M., Anderson W.D., Zang C., Guertin M.J.. Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res. 2017; 46:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Calviello A. K., Hirsekorn A., Wurmus R., Yusuf D., Ohler U.. Reproducible inference of transcription factor footprints in ATAC-seq and DNase-seq datasets using protocol-specific bias modeling. Genome Biol. 2019; 20:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krebs A.R., Imanci D., Hoerner L., Gaidatzis D., Burger L., Schübeler D.. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol. Cell. 2017; 67:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data sets used in this work are listed in supplementary file 1.