


Abstract
RNA polymerase-binding RNA aptamers (RAPs) are natural RNA elements that control transcription in cis by directly contacting RNA polymerase. Many RAPs inhibit transcription by inducing Rho-dependent termination in Escherichia coli. Here, we studied the role of inhibitory RAPs (iRAPs) in modulation of antisense transcription (AT) using in silico and in vivo approaches. We revisited the antisense transcriptome in cells with impaired AT regulators (Rho, H-NS and RNaseIII) and searched for the presence of RAPs within antisense RNAs. Many of these RAPs were found at key genomic positions where they terminate AT. By exploring the activity of several RAPs both in a reporter system and in their natural genomic context, we confirmed their significant role in AT regulation. RAPs coordinate Rho activity at the antisense strand and terminate antisense transcripts. In some cases, they stimulated sense expression by alleviating ongoing transcriptional interference. Essentially, our data postulate RAPs as key determinants of Rho-mediated AT regulation in E. coli.
INTRODUCTION
The pervasive activity of the Escherichia coli RNA polymerase (RNAP) leads to transcription not only of defined genes but also of as-yet-unannotated RNAs encoded across the genome (1,2). In particular, the antisense strand—the one opposite to genes—can be pervasively transcribed into thousands of different antisense RNAs (3,4). Transcription from the antisense strand might have a strong impact on the expression levels of the corresponding sense transcriptional unit (5). RNAPs transcribing the sense strand can be transcriptionally hindered by antisense transcription (AT) in a process termed transcriptional interference (TI), which influences the rate of transcription from the sense promoter (6). Furthermore, AT regulates gene expression by several mechanisms involving sense-antisense RNA interactions such as transcription attenuation, translation inhibition/activation or modulation of RNA stability (7). The capacity of AT to regulate gene expression has been demonstrated in diverse bacterial species (8–10).
Transcription of the antisense strand is tightly controlled. In E. coli, three main layers of AT regulation have been proposed. Transcription initiation at the antisense strand can be repressed by the DNA-binding protein H-NS via promoter occlusion (11). Actively transcribing antisense RNAPs can be stopped by the RNA helicase Rho, which promotes premature transcription termination (12,13). Finally, if an antisense RNA is transcribed, it can be processed by specialized RNases (7). Specifically, the double-strand RNA (dsRNA) endonuclease RNaseIII degrades antisense RNAs bound to their sense cognate transcripts (10). Apart from being involved in AT regulation, H-NS, Rho and RNaseIII are key regulators of global gene expression (14–16). Rho is a multi-functional factor that, together with NusG, is responsible for transcription termination across the genome (17–21). Although Rho has been suggested to play an essential role in regulation of ubiquitous AT (13), the specific sequence determinants that allow this activity have not been defined to date.
Recently, our group described a novel class of widespread RNA regulatory elements, termed RAPs (RNAP-binding RNA aptamers) that modulate transcription in E. coli (22) and Saccharomyces cerevisiae (23). Initially selected by genomic SELEX using E. coli RNAP holoenzyme as a bait (24), the bacterial RAPs have been further tested for their transcription modulating potential in cis. We defined an abundant subset of these RNA elements, named inhibitory RAPs (iRAPs), that inhibit transcription in cis by causing Rho-dependent transcription termination. They are widespread across the bacterial genome with the majority encoded on the antisense strand to annotated genes (22). We therefore hypothesized that RAPs are actively involved in AT regulation by triggering Rho termination.
Here, we focus on iRAPs frequently located on the antisense strand opposite to coding genes, as well as some intergenic regions of E. coli genome. By extensively characterizing these RAPs, we demonstrated how they mitigate TI between convergent genes and increase sense gene expression by repressing AT. We also show that iRAPs regulate the expression of antisense transcripts from their endogenous genomic loci in cis by promoting Rho-dependent termination.
MATERIALS AND METHODS
Bacterial strains, plasmids, oligonucleotides and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table S1. Oligonucleotides used in this study are listed in Supplementary Table S2.
Bacterial strain E. coli: str. K-12 substr. MG1655 Δhns was constructed by transducing the hns::kan allele from E. coli: str. BW25113 Δhns using generalized transduction with bacteriophage T4 (25).
The bacterial vector pRAP#-TI (SAH141.1) is an mCherry/BFP fluorescence reporter which allows the detection of RAP-mediated regulation of TI. It was derived from pTI (SAH141, (26)) by AgeI/HindIII restriction and addition of a cloning site containing KpnI and XhoI restriction sites.
The bacterial cells were cultured in solid LB-agar media for cloning and maintaining strains. The media was supplemented with kanamycin (50 μg/ml), spectinomycin (100 μg/ml) or tetracycline (35 μg/ml) when needed. Liquid cultures of the strains in LB were used for fluorescence detection assays and RNA extraction. The media was supplemented with the antibiotics listed above and bicyclomycin (BCM, 8–20 μg/ml) when needed. The cells were cultivated with constant aeration, shaking at 200 rpm.
Total RNA library preparation for NGS
Escherichia coli: str. K-12 substr. MG1655 was grown in 100 ml flasks filled with 10 ml of rich media (LB), with rigorous shaking at 37°C until they reached exponential phase with OD600 = 0.4–0.5. Cells were then split into smaller aliquots (300 μl of culture in 2 ml tubes) and further cultivated under similar conditions, but in the absence or presence of BCM (added to a final concentration of 50 μg/ml) for another 15 min. At this point samples were treated with 40 μl of Stop Solution (95% ethanol, 5% phenol), cells collected with centrifugation and subjected to RNA extraction with hot phenol (see ‘RNA isolation’ step below).
DNA-free total RNA was first depleted of ribosomal RNA (using Ribo Minus™ Transcriptome Isolation Kit, bacteria, from Thermo Fisher) according to the manufacturer's instructions.
Strand-specific libraries for NGS were prepared from 30 to 60 ng of ribosomal RNA-depleted total RNA using Illumina TruSeq Stranded mRNA according to the manufacturer's preparation guide, with 15 cycles of final amplification. Libraries were indexed to allow further multiplexing. Quality control at the major steps of libraries preparation, as well as the final libraries, was performed using Agilent 2100 Bioanalyzer and Fragment Analyzer (performed by Vienna Biocenter Core Facilities, VBCF, http://ngs.vbcf.ac.at/).
Sequencing RNA libraries
The libraries were sequenced on an Illumina NextSeq 500 platform using HighOutput 75 cycles kit. A total of 633M paired end reads passing the filter was obtained, which resulted in a minimum of 25M paired end reads per sample after demultiplexing (reaching total of 25–95M reads per sample).
Computational analysis for antisense regions
Sequenced reads were quality controlled using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and subsequently adaptor clipped and quality trimmed with Trimmomatic (27). Processed reads were mapped against the E. coli reference genome (NC_000913.3) using bowtie2 (28). To make the paired end libraries compatible with downstream tools, the strand assignment of first-in-pair reads were flipped. The presence of systemic strand bias was tested as described previously (29), and deemed to be absent (<0.1%). To determine regions with modulated transcriptional activity, the genome was segmented into windows of length 100 nt with a step size of 60 nt (so that two consecutive windows overlap by 40 nt). Read counts per window were deduced with bedtools multicov (30). Differential abundance was tested using DESeq2 (31), with default settings beside the count normalization. Since BCM treatment leads to vast changes in the antisense and intergenic gene expression, only read counts of windows sense to annotated genes were used to deduce the sample size factors, which then were applied to all read counts. Finally, all upregulated windows with an adjusted P-value < 0.01 were merged using bedtools if they exhibited an overlap.
H-NS samples (ENA study accession number: PRJEB14391) were treated in the same way, except that processing was adapted for the fact that those samples were sequenced in single-end read mode.
Flow cytometry
Overnight cultures of E. coli: DH5 alpha cells carrying the plasmids of interest were harvested by centrifugation for 1 min at 13 400 rpm and resuspended in 1× PBS. Washed cells were diluted 1/100 in fresh 1× PBS. An aliquot of diluted cells was stored for further processing (unsorted) and another was sorted using FACS Aria Ilu (BD Biosciences). Low-fluorescent cells were separated from the population and collected by applying a stringent gating based on the lowest fluorescence values from control cells carrying pRAP#-GFP (no RAP). Unsorted and sorted cells were cultured in fresh LB supplemented with the appropriate antibiotic overnight. FlowJo X software (Tree Star) was used for data analysis and graphical representation.
DNA preparation for large scale identification of active RAPs
Two plasmid populations (from unsorted and sorted bacterial cells) were used as PCR templates to obtain the DNA for sequencing. The plasmids were extracted using PureYield™ Plasmid Miniprep System (Promega) and amplified using modified designed Illumina TruSeq primers (Forward 5′- AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNGAATTCGGAGCGGGGCAGC-3′; Reverse 5′- CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCGGATCCTCGGGGCTGGGA -3′) by 20 cycles of PCR using Q5 High Fidelity DNA polymerase (New England Biolabs). The PCR products were purified using QIAquick Gel Extraction Kit (Qiagen) and analyzed on an Agilent 2100 Bioanalyzer. The DNA libraries were sequenced on individual MiSeq lanes (150 bp, paired end) using a standard Illumina Multiplexing Index Read Sequencing primer (5′-GATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3′) at the VBCF NGS unit.
Deep-sequencing analyses for large scale identification of inhibitory RAPs
Reads obtained from FACS-sorted and -unsorted amplicon-seq libraries were adaptor clipped and quality trimmed with cutadapt (32) and Trimmomatic (27). The processed first in pair reads were then mapped in single-end mode against the E. coli reference genome (NC_000913.3) augmented by the employed expression vectors (pRAP#-GFP) with the read aligner STAR (33) with non-default parameters -alignIntronMin 14 -chimSeqmentMin 14 -alignEndsType Local -twopassMode Basic. Read count per annotated RAP was deduced with htseq-count (34) with non-default parameters -mode intersect-nonempty -nonunique all -secondary-alignments score -supplementary-alignments score. Subsequently, reads per million was calculated from the raw read counts.
A stringent analysis of the data was performed to avoid false positives. Only RAPs with 10 or more reads detected in the deep-sequencing analysis of unsorted cells were considered for further analysis. The number of reads from each RAP relative to the total number of reads from each sample (sorted and unsorted cells) was taken for further calculations. The logarithm of the ratio between the relative reads detected from a RAP from the sorted cells and the relative reads detected from a RAP from the unsorted cells was calculated. All RAPs with log-ratio values larger than zero were considered to be enriched in the FACS-sorted cell population with reduced GFP expression; hence, they were classified as inhibiting RAPs (iRAPs).
Motif prediction for iRAPs
For motif finding, MEME (35) was used in classic mode, on all RAP sequences identified as iRAPs in the large scale identification assay. Beside the set ‘zero or one occurrence’ and the ‘search given strand only’, the default parameters were kept.
Regional association enrichment test
To evaluate if pre-defined genomic regions (e.g. Rho_as_regions) harbor a RAP more often than expected, intersections of regions and RAPs are produced by bedtools intersect (30) and counted. The procedure is iterated 10 000 times with randomized RAP regions, conserving the number and lengths of the RAPs, randomizing only their position along the genome. To evaluate if RAPs are closer than expected to particular positions in the genome (e.g. 3′ ends of Rho_as_regions), the distance distribution was deduced applying bedtools (30) closest for the native RAP annotation and 100 times for a randomized RAP annotation. The obtained distributions were tested for significantly different median values applying a one-sided Mann-Whitney U test implemented in the R function wilcox.test.
Fluorescence detection assays
GFP fluorescence
Escherichia coli strains carrying pGFP and derivatives were grown in 5 ml LB media supplemented with 50 μg/ml kanamycin up to OD600 = 1.2–1.4. The cultures were diluted with fresh media to OD600 = 0.05 and 150 μl of each diluted culture was transferred to single wells of a sterile 96-well black plate with flat clear bottom and incubated with a clear lid. The cultures were grown at 37°C for 9–10 h with constant orbital shaking (1 mm amplitude) in Tecan Infinite F500. Measurements of OD612 and GFP fluorescence (485 nm and 535 nm, gain 30) were taken from below the plate every 10 min. Relative GFP fluorescence was calculated normalizing the fluorescence intensity measured from cells at stationary phase (OD612 = ∼0.9) to their cell density. Then, the values were plotted relative to the control GFP intensity (p#RAP-GFP with no RAP cloned). Each value is processed from measurements taken from three technical replicates for each biological triplicate.
mCherry fluorescence
Measurements of mCherry fluorescence were performed as described previously (26). Relative mCherry fluorescence values were calculated as for the relative GFP fluorescence, described above.
RNA isolation
Total RNA isolation was performed as described previously (10) following the hot phenol method.
Northern blot analysis
For northern blotting RNA transcripts, 10 μg of DNAseI-treated (Roche) total RNA was separated by gel electrophoresis in 1% formaldehyde/MOPS denaturing agarose gels in 1× MOPS. RNA was denatured in 2X RNA load dye (ThermoFisher Scientific) with incubation for 15 min at 70°C before loading on a gel. RNA was transferred to HybondXL membranes by capillary action using 20X SSC buffer for 10–12 h. The membranes were UV cross-linked (150 mJ/cm2) and probed with 5′end γ32P-labeled DNA oligonucleotide probes using ULTRAhyb-Oligo Hybridization Buffer (Ambion) following the manufacturer's instructions. DNA probe labeling was performed using T4 PNK (New England Biolabs) according to the manufacturer's instructions. The northern blot output was visualized in Amersham Typhoon Biomolecular Imager (GE Healthcare). All northern blots were performed at least twice in independent experiments showing the same results.
Quantification and statistical analyses
Statistical parameters for each experiment are reported in the corresponding figure legends. The reported data were obtained from at least three biological replicates unless otherwise stated. The significance was tested using Student's t test. Statistical analysis was performed using GraphPad Quick Calcs (https://www.graphpad.com/quickcalcs/ttest1.cfm).
RESULTS
Evidence for ubiquitous RAP-mediated transcription regulation on the antisense strand
To elucidate the involvement of RAPs in regulating transcription on the antisense strand, we investigated the E. coli transcriptome after inhibition or deletion of Rho, H-NS or RNaseIII and studied the co-localization of RAPs within these antisense transcripts (Figure 1A). First, we obtained and analyzed high-resolution total RNA sequencing data from E. coli grown to exponential phase in rich media in the absence and presence of the Rho inhibitor bicyclomycin (BCM) (36) (see Materials and Methods). Additionally, we merged our RAP data with a previously published dataset for an H-NS mutant strain of E. coli (37). We performed differential expression analyses to detect which genomic regions antisense to annotated genes were overexpressed upon the inhibition/deletion of the regulatory factors (Figure 1B). We detected 2968 and 487 upregulated antisense regions upon inhibition of Rho and mutation of H-NS, respectively, termed ‘Rho_as_regions’ and ‘H-NS_as_regions’ (Figure 1C, Supplementary Table S3). Figure 1D shows their length distribution, with an average length of 1173 and 421 nt for ‘Rho_as_regions’ and ‘H-NS_as_regions’, respectively, and their cumulative length.
Figure 1.

Evidence for ubiquitous RAP-mediated transcription regulation on the antisense strand. (A) Workflow schematics of the computational analysis to define: (i) the antisense genomic regions upregulated upon inactivation of AT regulators (Rho, H-NS and RNaseIII) (Supplementary Table S3); (ii) RAPs that can terminate AT (Supplementary Table S4); (iii) RAPs enrichment within the overexpressed antisense regions (tests shown in (F). (B) Genome schematics representing Rho, H-NS, RIII_as_regions, and RAPs with the potential of terminating AT (i.e. locating within Rho, H-NS and RIII_as_regions and/or close to termination points: 5′ ends of Rho_as_regions and 3′ ends of H-NS/RIII_as_regions). Green arrows represent annotated genes encoded on the + strand, curved arrowed lines represent antisense transcripts, arrow-boxes represent RAPs encoded antisense to genes. WT, ‘WT’, ‘WT+BCM’, ‘hns-’ and ‘rnc-’ indicate the genotype/growth conditions under which the represented antisense RNAs were transcribed. ‘WT’ wild type E. coli K-12 substr. MG1655; ‘hns-’ E. coli str. K-12 substr. MG1655 H-NS mutant lacking the repressor activity, ‘rnc-’ RNaseIII mutant lacking enzymatic activity; ‘+BCM’ (50 μg/ml). (C) Bar graph showing the number of overexpressed antisense regions detected upon deletion/inhibition of an AT regulator based on our RNA sequencing analysis (Rho and H-NS) and previously published data (RNaseIII). The x-axis represents the percentage (darker color) of these regions that contains at least one RAP and the percentage that does not (lighter color). (D) Length distribution and total cumulative length of overexpressed antisense regions from (C). (E) Venn diagram showing how many RAPs are found within antisense overexpressed regions (Rho, H-NS and RIII_as_regions). (F) Regional association enrichment tests of RAPs and Rho, H-NS and RIII_as_regions. Expected values were obtained using randomized RAP genomic coordinates. Observed values were obtained using the original RAP genomic coordinates (Supplementary Table S4). ***P < 0.0001; **P < 0.001; *P < 0.01 (one-sided P-value). (G) Distribution of RAPs closest to the 5′ ends of Rho_as_regions. The plot shows the distribution of the RAP subset (red) compared to a randomized RAP subset (blue). Dotted lines indicate the median value from each dataset, also indicated in the top corners. The values were statistically significant (one-sided Mann-Whitney U test: P-value 7.6e–09).
To define antisense RNAs controlled by RNaseIII, we used a dsRNA dataset obtained in our group using an E. coli RNaseIII mutant (10). This work reports 316 dsRNAs detected in the absence of RNaseIII activity, 238 of which are formed by RNAs antisense to annotated genes and their cognate sense transcripts. We used these data to define 238 genomic regions where antisense RNAs are transcribed and further processed by RNaseIII termed here ‘RIII_as_regions’ (Figure 1C, Supplementary Table S3). Their cumulative length and length distribution is shown in Figure 1D, with an average length of 177 nt.
Once the antisense regions have been defined, we combined these data with genomic coordinates of all RAPs and performed regional association enrichment tests (see Materials and Methods). The co-localization of RAPs within these antisense regions suggested their potential to terminate antisense transcripts. Indeed, 67.5% of Rho_as_regions encoded at least one RAP (Figure 1C), significantly more than expected by chance (Figure 1F) Furthermore, a significant percentage (42.9%) of RIII_as_regions encoded at least one RAP (Figure 1C and F). The lack of apparent enrichment of H-NS_as_regions with RAPs (Figure 1F) does not rule out RAP-mediated regulation here. As discussed below, a close look at RAP distribution within H-NS_as_regions revealed their regulatory potential.
The location of RAPs suggests they terminate AT
To define the RAPs that can regulate AT, we analyzed their abundance along the upregulated antisense regions discussed above. We found 45.7% of all RAPs to locate within these regions (Figure E), significantly more than expected by chance (Figure 1F, Supplementary Table S4). Many RAPs can cause transcriptional termination (22). We hypothesized that if RAPs terminate transcription of antisense RNAs, they should be encoded near their 3′ ends (Figure 1B). Indeed, we observed a significant tendency for RAPs to be clustered towards the 3′ ends of H-NS_as_regions, with 44.8% of the RAPs found in H-NS_as_regions being located within the last 100 bp (Figure 1F and Supplementary Table S4). Although not statistically significant, a large percentage of the RAPs (64.2%) within RIII_as_regions were also found close to 3′ ends (Figure 1F and Supplementary Table S4). As a large subset of RAPs is implicated in Rho termination, the inhibition of Rho by BCM should reveal the transcripts terminated by these RAPs (i.e. enhanced signals downstream of RAPs). Therefore, if a RAP were found close to the 5′ end of a Rho_as_region (Figure 1B, RAP#Y), it would indicate that this element promoted termination of the antisense transcript in the absence of BCM. We analyzed the distance distribution of the RAPs closest to 5′ ends of Rho_as_regions compared to randomized RAPs and observed a significant trend for RAPs being found directly upstream of the start of Rho_as_regions (Figure 1G). This result indicates that they are responsible for terminating transcription at those locations. Several examples of the cases discussed above (where RAPs are found in key locations within Rho, H-NS and RIII_as_regions) are presented as screenshots including RNA sequencing data in Supplementary Figure S1.
RAPs have potential to alleviate transcriptional interference (TI)
TI is a direct consequence of AT and can affect sense expression (6). Therefore, RAPs that are active on the antisense strand can modulate expression of the genes encoded opposite to them. To explore which RAPs regulate TI, we first defined genes that could be downregulated by AT. For this, we used our total RNA-sequencing data from BCM-treated cells. On inhibiting Rho and therefore impairing the activity of many RAPs that cause Rho-dependent termination, RNAPs continue transcribing over genes and sense promoters. We determined how many of the antisense regions upregulated upon the BCM treatment (i.e. Rho_as_regions) would overlap with a downregulated sense gene annotated on the opposite strand. Since there are different mechanistic models for TI (38,39), we considered two scenarios to define downregulated genes by the action of antisense RNAPs: (i) the polymerase should transcribe over a defined promoter of a gene or operon (promoter database obtained from (40)) or (ii) the RNAP should transcribe completely over the coding sequence (CDS) of a gene. The results of this analysis (Supplementary Table S5) showed that in all the cases where a Rho_as_region overlapped the CDS of a certain gene/operon it also overlapped its promoter. However, there were many cases where only the promoter region of a downregulated gene overlapped with a Rho_as_region. We then identified a subset of 302 genes potentially downregulated by TI caused by BCM-upregulated AT (Supplementary Table S5). Approximately 76% of the Rho_as_regions that overlapped the downregulated genes contained at least one RAP (Supplementary Figure S2A), significantly more than expected by chance (Supplementary Figure S2B). This situation of RAPs modulating TI is exemplified with the cases of yacL and mrr (Supplementary Figure S2C and D). From these data we are not able to rule out the downstream effects of inhibiting Rho (for instance, misregulation of certain transcription factors or readthrough of Rho-dependent terminators), but we argue that TI could be directly responsible for downregulating these genomic loci. Thus, the 551 RAPs (Supplementary Table S4) regulating AT along these loci would indirectly modulate sense gene expression by preventing TI.
Taken together these results argue that a large subset of RAPs has the potential to regulate AT events across the genome.
A fifth of RAPs encoded antisense to genes are iRAPs
Based on extrapolation, approximately a quarter of all RAPs had been classified as iRAPs due to their transcription inhibitory potential (22). To provide a better estimate, we developed a high-throughput cell sorting-based assay to test the in cis activity of many RAPs in parallel. To do so, we first cloned the original SELEX RAP pool with all the RAP sequences in the form of DNA (24) into a GFP-based reporter plasmid (pGFP, (22), Supplementary Figure S3). We further sorted the bacterial populations with RAP-containing plasmids (pSELEX_RAPs-GFP) to obtain a fraction of cells with reduced GFP expression (i.e. the cells containing an iRAP upstream of the GFP, Supplementary Figure S3A). Based on preliminary tests with individual iRAPs and a promoter-less pGFP (Supplementary Figure S4A), we applied stringent gating resulting in a collected fraction of ∼9% of all cells (Supplementary Figure S3B). We deep-sequenced the reporter plasmids from this subgroup and analyzed the output to identify RAPs that cause Rho-dependent termination (Supplementary Table S4). To confirm that the reduction in GFP in the cell population containing pGFP-SELEX cDNA pool was due to iRAP activity, we applied BCM and examined GFP fluorescence (Supplementary Figure S4B–D). As a control, we cloned random DNA sequences into pGFP with the same length range as RAPs and also tested its GFP fluorescence with or without BCM (Supplementary Figure S4B-D). From almost 2000 different RAPs cloned, ∼19% demonstrated inhibiting properties. Interestingly, 75% of the cloned RAPs were located antisense to annotated genes with one fifth of them demonstrating inhibitory properties. Extrapolating these values to the total number of RAPs found antisense to genes, ∼2,000 RAPs can be expected to inhibit transcription across the antisense strand.
Approximately 54% of the 375 iRAPs (Supplementary Table S4) identified in the screen were encoded within Rho, H-NS or RIII_as_regions (Supplementary Figure S5A), significantly more than expected by chance (Supplementary Figure S5C). iRAPs tended to cluster upstream of Rho_as_regions 5′ ends (Supplementary Figure S5D) and were significantly enriched at the 3′ ends of H-NS_as_regions compared to randomly positioned RAPs (Supplementary Figure S5E). These observations suggest that iRAPs terminate transcription at these locations. Moreover, 20 iRAPs were encoded antisense to some of the genes potentially downregulated by TI discussed in the previous section (Supplementary Table S5). Thus, these elements could relieve TI by terminating AT.
The absence of a consensus motif among the discovered RAPs suggested that there might be different types of RAP activities or RNAP-binding sites (22). We repeated the motif analysis using a subset of 375 iRAPs described above. Of these iRAPs, 28.5% shared a common CA-rich motif (Supplementary Figure S5B).
Taking into account the data discussed in these four sections we defined a small set of RAPs (Supplementary Table S4, highlighted rows) to evaluate our hypothesis of RAP-mediated regulation of AT. These elements are located close to the 3′ and 5′ ends of H-NS/RIII_as_regions and Rho_as_regions, respectively, indicating their potential role in terminating those transcripts. Indeed, some of these RAPs showed inhibitory properties in our screening (Supplementary Figure S3B), so we further tested them in different systems and in their natural context.
Inhibitory RAPs regulate transcriptional interference between convergent genes
To explore the in cis regulatory effect of the 22 RAPs of interest (Supplementary Table S4, highlighted rows), we first used pGFP with RAPs encoded within the 5′ UTR of the reporter gene (Supplementary Figure S6A). All 22 RAPs downregulated the reporter expression (Supplementary Figure S6B) and this effect was inhibited by BCM, implying Rho-dependent termination in accordance with the iRAP model of activity. Considering natural gene organization in bacteria, we further modified our test strategy by using a reporter system that models closely spaced convergent genes. This transcriptional interference reporter (pTI (26)) harbors two convergent fluorescent genes, mCherry in sense and blue fluorescent protein (BFP) in antisense, constitutively expressed by a weak and a strong promoter, respectively (Figure 2A). The strong BFP promoter causes TI, preventing mCherry transcription (26). This TI was alleviated by cloning iRAPs within the BFP strand. The presence of iRAPs in the BFP strand caused premature transcription termination and an up to 5-fold increase in mCherry expression with the most active iRAP (Figure 2B). This effect can be also triggered in the presence of the canonical long Rho utilization site sequence (rut). Upon the addition of BCM, the transcriptional termination triggered by iRAPs and rut was compromised and the mCherry expression repressed again by the TI effect (Figure 2B).
Figure 2.
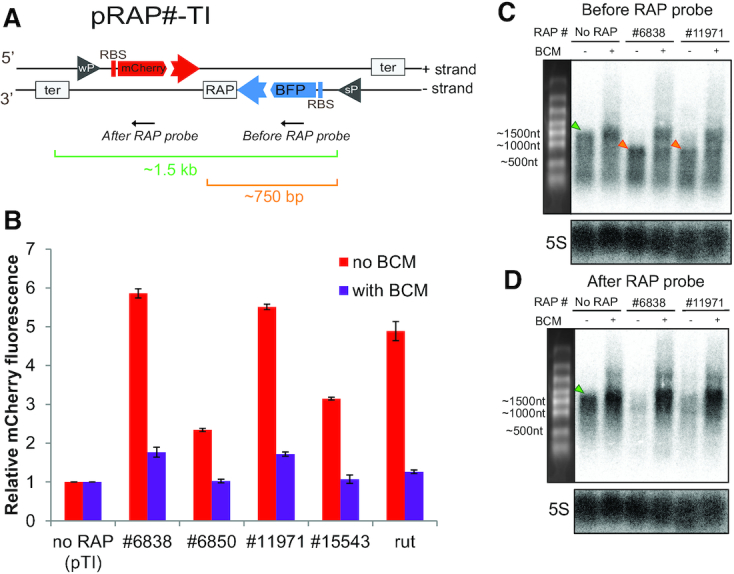
RAPs mitigate TI between convergent genes. (A) Schematic of pRAP#-TI reporter system. ‘wP’ and ‘sP’ inside a grey triangle indicate the location of a weak and a strong promoter, respectively. ‘RBS’ indicates the ribosome-binding site. The ‘RAP’ box indicates the site where RAPs can be cloned. The grey boxes ‘ter’ indicate the terminators of mCherry and BFP transcriptional units. The black arrows indicate the annealing location of the DNA probes used in the northern blot analysis shown in (C) and (D). (B) mCherry fluorescence levels of pRAP#-TI containing different RAPs, indicated here as ‘# and the RAP number’. The levels are relative to the reporter without a RAP sequence cloned after BFP (no RAP). Red bars indicate the mCherry levels when no BCM is added to the culture. Violet bars indicate the mCherry levels when 8 μg/ml BCM is added. All values represented in bars show the mean of measurements from three biological replicates relative to the ‘no RAP’ control. Error bars indicate the standard deviation. BCM treatment caused a statistically significant (P < 0.0001, t test) decrease of mCherry expression in all cases. (C) Northern blot analysis of the transcripts produced from the - strand represented in (A) using the ‘Before RAP probe’. The upper labels indicate the samples used for the analysis. ‘no RAP’ indicates that the RNA comes from pRAP#-TI without any RAP cloned after BFP. ‘#6838′ and #11971 represent RNA coming from pRAP#6838-TI and pRAP#11971-TI, respectively. The ‘BCM’ label indicates which lane contains RNA extracted from cells grown in the presence of 20 μg/ml BCM. The left panel shows the RNA marker bands with their approximate length. The green triangles indicate the full length ∼1500 nt RNA isoform. The orange triangles indicate the ∼750 nt terminated version of the BFP transcript. 5S ribosomal RNA served as a loading control. (D) Northern blot analysis of the transcripts produced from the - strand represented in (A) using the ‘After RAP probe’. The analysis is depicted as in (C).
To further explore the termination effect of iRAPs in the TI system, we performed a northern blot analysis of the BFP transcript in the presence or absence of iRAPs. When no RAP was cloned after the BFP coding sequence, we detected a full-length ∼1500-nt transcript (Figure 2C). However, when iRAPs #6838 and #11971 were located after BFP, we detected a ∼750 nt long transcript. Upon the addition of BCM, this transcription inhibition was relieved and we again detected the full-length transcript. The shorter isoform of the BFP mRNA produced in the presence of iRAP could not be detected by a probe annealing after the RAP (Figure 2D). Only a weak band corresponding to the full-length transcript was observed, presumably produced by a few RNAPs reading through the iRAPs. We have also reproduced these results cloning the model iRAP#15 in the TI system (Supplementary Figure S7). These observations are in agreement with the detected mCherry levels and demonstrate that transcription termination is facilitated by iRAPs via Rho-dependent termination. Using this system to explore the activity of 22 experimentally confirmed iRAPs (Supplementary Figure S6B), we found that all of them alleviated TI (Figure 2 and Supplementary Figure S8).
Inhibitory RAPs terminate antisense transcription in their natural context
The capacity of many RAPs to efficiently terminate transcription and their abundance on the antisense strand suggest that they control AT events across the bacterial genome. To confirm this hypothesis, we studied the transcriptional impact of several iRAPs found within transcribed antisense regions of the genome in their natural context. iRAP#6850 is found at the end of a transcript antisense to yoeH, the transcription start site of which is located ∼200 bp upstream of the RAP (41) (Figure 3A). This transcript is degraded by RNaseIII and can only be detected in the background of the endonuclease mutation (rnc-) (10). Since iRAP#6850 triggers Rho-dependent termination (Figure 2B and Supplementary Figure S6B), it may be responsible for stopping transcription of the antisense RNA. Indeed, we detected by northern blotting a ∼150–200-nt long antisense RNA using a probe annealing before the RAP location (Figure 3B, lane 3) but not if the probe annealed after it (Figure 3C, lane 3). In the latter case, we only observed a weak smear corresponding to longer RNA isoforms that might be due to the RNAP reading through the iRAP. Adding BCM blocked the termination effect of iRAP#6850, allowing the RNAP to continue transcribing over the regulatory element. The action of BCM allowed the detection of a much longer antisense transcript with probes annealing both before and after the RAP (Figures 3B and C, lane 4). However, as expected, the latter probe only detected the RNA isoforms longer than ∼200 nt. Furthermore, no RNA was detected using a probe annealing upstream of the original antisense RNA. The last two observations clearly demonstrate that the transcription of longer RNA isoforms downstream of the iRAP is the result of BCM action and not misregulation of upstream transcriptional units. An additional example of an RNaseIII-regulated antisense transcript terminated by an iRAP is shown in Supplementary Figure S9.
Figure 3.

RAPs terminate AT in their natural context. (A) Schematic of RAP#6850 genomic locus. The sense yoeH transcript is depicted as a green line. The transcripts antisense to yoeH are shown as violet lines with their approximate maximum length indicated. ‘-BCM’ and ‘+BCM’ represent the growth conditions in which this transcript is produced, without or with BCM (20 μg/ml), respectively. The red arrows symbolize different DNA probes and their annealing locations used in the northern blot analysis shown in (B) and (C). (B and C) Northern blot analysis of antisense transcripts produced at the genomic locus described in (A). The upper titles describe the probe used to detect RNA. The numbering on the right shows the approximate length of the RNA marker. The description of each lane is shown at the bottom: ‘WT’ wild type E. coli strain; ‘rnc-’ RNaseIII mutant lacking enzymatic activity; ‘+BCM’ growth conditions, with BCM (20 μg/ml) added to the media. 5S ribosomal RNA served as a loading control. The blue triangle indicates the ∼200 nt terminated isoform of the antisense RNA. The pink triangles indicate the longest antisense RNA isoforms (∼700 nt) detected upon addition of BCM. (D) Schematic of RAP#2751 genomic locus. The transcripts antisense to ybhN are shown as violet lines with their approximate maximum length indicated. ‘-BCM’ and ‘+BCM’ represent the growth conditions in which this transcript is produced, without or with BCM (30 μg/ml), respectively. The red arrows symbolize different DNA probes and their annealing locations used in the northern blot analysis shown in (E) and (F). (E and F) Northern blot analysis of antisense transcripts produced at the genomic locus described in (D). The upper titles describe the probe used to detect RNA. The numbering on the right shows the approximate length of the RNA marker. The description of each lane is shown at the bottom: ‘WT’ wild type E. coli str. K-12 substr. MG1655; ‘hns-’ E. coli str. K-12 substr. MG1655 H-NS mutant lacking the repressor activity; ‘+BCM’ growth conditions, with BCM (30 μg/ml) added to the media. 5S ribosomal RNA served as a loading control. The blue triangle indicates the ∼500 nt terminated isoform of the antisense RNA. The pink triangles indicate the longest antisense RNA isoforms (∼1500 nt) detected upon addition of BCM. (G) Schematic of RAP#10218 genomic locus. The transcripts antisense to yghE are shown as violet lines with their approximate maximum length indicated. ‘-BCM’ and ‘+BCM’ represent the growth conditions in which this transcript is produced, without or with BCM (30 μg/ml), respectively. The red arrows symbolize different DNA probes and their annealing locations used in the northern blot analysis shown in (H) and (I). (H and I) Northern blot analysis of antisense transcripts produced at the genomic locus described in (G). The upper titles describe the probe used to detect RNA. The numbering on the right shows the approximate length of the RNA marker. The description of each lane is shown at the bottom: ‘WT’ wild type E. coli str. K-12 substr. MG1655; ‘+BCM’ growth conditions, with BCM (30 μg/ml) added to the media. 5S ribosomal RNA served as a loading control. The blue triangle indicates the ∼350–400 nt terminated isoform of the antisense RNA. The pink triangles indicate the longest antisense RNA isoforms (∼1,500 nt) detected upon addition of BCM.
We observed a similar trend when studying the genomic context of iRAP#2751 by northern blotting. This empirically confirmed inhibitory element (Supplementary Figures S6 and S8) is encoded at the 3′ end of an H-NS_as_region (Figure 3D), suggesting its involvement in the termination of this transcript. This ∼500 nt antisense RNA was detected in the absence of H-NS (hns-) (Figure 3E) using a probe annealing before the iRAP location (Figure 3E, lane 3), but not a probe annealing after it (Figure 3F, lane 3). However, similar to the previous case, upon the addition of BCM, the length of the antisense transcript increased to ∼1500 nt. The fact that the longer transcript version can be detected by probes annealing before and after iRAP#2751 suggests that this aptamer is responsible of terminating AT at the ybhN locus.
A similar case of iRAP-mediated regulation of genomic AT is presented in Figure 3G–I. We detected a ∼350–400 nt long transcript antisense to yghE transcribed from an antisense promoter (41) by northern blot in wild-type E. coli cells (Figure 3H, lane 1). This transcript contains iRAP#10218 at its 3′ end, which promotes termination. Upon the addition of BCM, the iRAP effect was inhibited and this transcript became longer (Figure 3H, lane 4). Only the 1200 nt long RNA isoform was detected using a probe downstream of the iRAP position. Moreover, no RNA was detected using a probe annealing upstream of the transcript antisense to yghE.
Taken together, these results suggest that iRAP#6850, iRAP#2751, iRAP#10218 and iRAP#4740 terminate transcription of RNAs antisense to yoeH, ybhN, yghE and tpx respectively. They serve as proof of principle that RAPs modulate AT in E. coli by causing Rho-dependent termination.
DISCUSSION
The functionality of antisense transcription (AT) in bacteria has been extensively debated over the last decade (2). Recent advances in NGS techniques have allowed an in-depth exploration of RNAs produced from both strands, clearly pointing to the impact of AT on total transcriptome composition and its significant regulatory role (42). To ensure proper gene expression, transcription of the antisense strand must be suppressed to prevent potential interference with sense transcription (5). E. coli RNA helicase Rho has been suggested to play a major role in keeping the levels of antisense RNAs low (13).
Recently, we discovered that iRAPs, a novel class of RNA elements distinct from canonical rut sites, can promote Rho-dependent termination (22). As the majority of iRAPs are encoded opposite to the coding strand, we combined in silico and in vivo approaches to further explore the idea that antisense-encoded iRAPs serve as widespread potentiators of Rho termination of AT.
Integrative in silico analysis suggests RAP involvement in AT regulation
Our computational analysis provides strong evidence for RAP-mediated regulation of AT across the E. coli genome. We revisited the antisense transcriptome of E. coli upon inactivation of three main AT regulators (Rho, H-NS and RNaseIII) and identified the antisense genomic areas that became overexpressed. Our high-resolution RNA deep sequencing data (with and without BCM) establishes Rho as the main AT suppressor. More antisense genomic material is transcribed upon inhibition of Rho than that of H-NS or RNaseIII (Figure 1C and D). The coverage of Rho_as_regions is larger than previously reported (13), substantially expanding the number of antisense genomic regions controlled by Rho. By merging these transcriptomic data with the genomic location of 15 724 RAPs, we explored their enrichment within antisense transcripts (Figure 1F), revealing their potential for terminating AT over the whole genome. We identified hundreds of RAPs located close to the start of Rho_as_regions, with a significant tendency to be positioned directly upstream of their 5′ ends (Figure 1F). These observations suggest their involvement in controlling AT via Rho-dependent termination. Hundreds of RAPs were located within the 3′ ends of antisense transcripts overexpressed in H-NS and RNaseIII mutants, also indicating their potential as terminator elements (Figure 1F). We showed that RAPs are significantly enriched at the 3′ of H-NS but not in RIII_as_regions. Although RAP enrichment at the 3′ ends of RIII_as_regions was not statistically significant, this does not rule out that RAPs terminate these transcripts. RIII_as_regions are on average short (Figure 1D) and, overall, they are enriched with RAPs (Figure 1F). Thus, any RAP occurring within RIII_as_regions might be responsible for their termination.
High-throughput in vivo screening assay for iRAP activity
We developed and applied a high-throughput screening for iRAPs (Supplementary Figures S3 and S5A) to identify and focus only on RAPs that trigger Rho-dependent termination. A combination of FACS and NGS for massive iRAP characterization represents a powerful framework to study transcription regulation in bacteria. By doing so, we demonstrated a significant overrepresentation of iRAPs within Rho, H-NS and RIII_as_regions (Supplementary Figure S5C), pointing to their potential for controlling the expression of these genomic regions. The tendency of iRAPs to cluster right upstream of 5′ ends of Rho_as_regions and significantly at the 3′ ends of H-NS_as_regions (Supplementary Figures S5D and E) strongly indicates that iRAPs are terminating transcription of the antisense RNAs. Due to the iRAP screening limitations, we explored only a few iRAPs encoded at the 3′ end of RIII_as_regions (Supplementary Figure S5A). Future experimental studies will test iRAPs from the 3′ end of H-NS_as_regions and RIII_as_regions for their inhibitory properties. Additional iRAP elements identified in the new screening procedure allowed us to find a CA-rich motif (Supplementary Figure S5B) shared by almost a third of iRAPs, similar to the one we already described for this sub-group of RAPs (22).
iRAP-mediated prevention of transcription interference
AT has been suggested to decrease sense expression by TI (4). Thus, RAPs involved in AT regulation might increase sense transcription by terminating antisense RNAPs. We searched for such RAPs within genomic regions where TI decreases sense transcription. In contrast to the whole-genome method previously applied (13), our approach identifies 302 genes potentially downregulated due to the increased antisense transcription upon Rho inhibition (Supplementary Figure S2). This finding suggests a novel regulatory role for Rho: it preserves sense transcription by curbing the expression of the antisense strand. Almost 76% of those genes harbor RAPs encoded antisense to them (Supplementary Figure S2A) that likely facilitate Rho-mediated suppression of antisense RNAPs. This is consistent with the fact that these genes contain significantly more RAPs within their antisense strand than expected by change (Supplementary Figure S2B), suggesting that the presence of those RAPs at these loci is biologically significant.
In this work we identified and focused on examples of closely spaced convergent genes where the BCM treatment led to readthrough of a terminator, causing AT and potentially downregulating the opposite gene (Supplementary Figure S2D). Remarkably, the presence of multiple iRAPs right after the Rho-dependent terminator additionally suggests a protective role for RAPs serving as multiple barriers to ensure transcription termination. These observations can be linked to the idea of additional RNA elements triggering the action of Rho after terminators (43), giving this RNA helicase the role of termination quality control.
We emulated this situation of convergent transcription with a reporter TI system resembling natural gene organization in bacteria (Figure 2A) and demonstrated the significant regulatory potential of iRAPs in TI regulation. The antisense RNAPs transcribing BFP in the reporter plasmid were terminated by any downstream RAP with inhibitory activity in cis (Figure 2C, D, Supplementary Figures S6 and S7). This increased the expression of the convergent mCherry gene (both at the RNA and protein levels) by reducing the load of transcribing RNAPs on the opposite strand (Figure 2B–D and Supplementary Figure S8). The effect on TI regulation was eliminated by Rho-inhibiting BCM, consistent with the proposed mechanism for iRAPs (22). Therefore, we suggest how the transcriptional pressure caused by expression from convergent promoters is alleviated by the action of iRAPs.
Using the endogenous iRAPs #6850, #2571, #10218 as examples, we demonstrated how iRAPs can terminate endogenous AT in their natural context. Based on prediction of their regulatory potential from transcriptomics data (RNAseIII, H-NS and Rho_as_regions, respectively), we further inspected the in vivo effect of these iRAPs at each of the corresponding loci by northern blotting. The transcript produced by the pseudogene yoeH (44) base pairs with an antisense RNA that is only detectable in an RNAseIII mutant ((10) and Figure 3B). We demonstrated that iRAP#6850, located within this antisense RNA (Figure 3A), is responsible for its termination via Rho action. This data is consistent with the fact that there is a putative antisense promoter (41) ∼200 bp upstream of the iRAP, corresponding to the length of the detectable transcript in Figure 3B, lane 3. The data on this iRAP together with iRAP#4740 (Supplementary Figure S9) show how important RAPs are in promoting termination of antisense transcripts. As the function of these antisense RNAs is to modulate the levels of sense transcripts via direct base pairing (10), RAPs assure that they are terminated and not to overlap with downstream genes.
Similarly, we found that iRAP #2571 is responsible for terminating transcription of an H-NS-regulated antisense transcript (Figure 3E). This RNA is antisense to ybhN, a gene encoding a predicted inner membrane protein (45). The antisense transcript was detected in our computational analysis and serves as an example of how iRAPs found close to the 3′ end of H-NS_as_regions are responsible for terminating AT. Finally, in wild-type E. coli cells, a naturally occurring transcript antisense to yghE detected in our deep sequencing data was terminated by iRAP#10218 (Figure 3G). In this case, the RAP action is essential to stop transcription starting from an antisense promoter (41) located within the pseudogene encoding a putative type II secretion system protein (46). Interestingly, this antisense RNA was recently reported to be translated (47). Therefore, iRAP#10218 functions as the terminator element of this antisense mRNA. The same study identified seven more antisense RNAs that are translated into small proteins. Among them, three harbor a RAP at the end of their ORF (Supplementary Table S4). As iRAPs are capable of promoting Rho-dependent termination within coding regions (22), these RAPs could terminate transcription of these translated antisense RNAs. The data shown in Figure 3 demonstrate that the studied iRAPs terminate the transcription of RNAs antisense to yoeH, ybhN and yghE. These cases of RAP-mediated AT regulation exemplify the interplay between H-NS, RNaseIII and Rho. Even though a particular antisense transcript is not detected because it is degraded by RNaseIII or it is low abundant due to the H-NS repression, it is still transcribed and it has to be terminated. That is where Rho would play its role at the antisense strand and iRAPs would coordinate its action.
As we observed previously (22), iRAPs stimulate Rho termination within ribosome-free RNA stretches. As only few antisense RNAs are known to be translated in E. coli, antisense iRAPs should act similarly to iRAPs located within UTRs. Our previous data suggested that UTR iRAPs may either help Rho loading onto RNA and/or promoting RNAP pausing (22). It is still unclear whether these aptamers bind first RNAP and then Rho, they bind both simultaneously, and/or by interacting with RNAP they render it termination-prone. Further mechanistic studies will address these questions.
Taken together, this work expands the biological role of RAPs as widespread transcriptional regulators, not only of annotated genes but also of antisense RNAs. Here we confirm it experimentally for a few of these regulatory elements. However, our in silico analysis suggests that RAPs regulate numerous antisense transcriptional units across the genome. This analysis could be expanded by applying RNA deep sequencing techniques to H-NS and RNaseIII E. coli mutants growing in the presence of BCM. With this approach one could find hundreds of antisense RNAs with RAPs at their 3′ ends that are not terminated when Rho is inhibited. Further exploration of cryptic antisense transcripts may reveal novel mechanisms for regulating the ‘dark matter’ of the transcriptome.
DATA AVAILABILITY
Raw sequencing read data produced in the course of this study are available from the European Nucleotide Archive under the study accession number PRJEB31692. Sample accession numbers ERS3223839 to ERS3223844 correspond to the transcriptome data of wild type and BCM-treated E. coli, respectively. Sample accession numbers ERS3223845 and ERS3223846 correspond to the Amplicon-seq data used to characterize RAPs as iRAPs.
Differential expression analyses and annotation of upregulated antisense regions and RAPs can be visualized in http://genome-euro.ucsc.edu/cgi-bin/hgHubConnect by copying the following URL in ‘My Hubs’: http://nibiru.tbi.univie.ac.at/publicNGS/Assembly_Hubs/EcoliRAP_hub/hub.txt.
Original northern blot pictures were deposited in Mendeley data and can be accessed via the following link: https://data.mendeley.com/datasets/x8gkh63mp8/draft?a=d8210201-bf4d-4286-b58c-768593e1613e.
Flow cytometry data were deposited in Flow Repository under the Repository ID FR-FCM-Z239 and can be accessed via the following link: http://flowrepository.org/id/RvFrRjEeIn4djSNl0eqewEWCZESxEwstPfPUj6jAOs71fXcxik3OXc8SlMg3b2gW.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Joseph T. Wade and Boris Görke for helpful discussions and ideas and for sharing plasmids and strains. We thank Katja M. Arndt for sharing plasmids. We also thank the Vienna BioCenter Core Facilities (VBCF) for NGS of FACS-selected constructs and the BioOptics-FACS facility at the MFPL for cell sorting. We thank Life Science Editors for language editing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Doctoral program RNA Biology [W1207-B09 to A.M.]; Austrian Science Fund (Fonds zur Förderung der wissenschaftlichen Forschung) [I538-B12, F4301, F4308 to R.S]; National Institutes of Health [R01 GM126891 to E.N.]; Blavatnik Family Foundation [to E.N.]; Howard Hughes Medical Institute [to E.N.]. Funding for open access charge: Austrian Science Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Dornenburg J.E., Devita A.M., Palumbo M.J., Wade J.T.. Widespread antisense transcription in Escherichia coli. MBio. 2010; 1:e00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lybecker M., Bilusic I., Raghavan R.. Pervasive transcription: detecting functional RNAs in bacteria. Transcription. 2014; 5:e944039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Georg J., Hess W.R.. cis -Antisense RNA, Another Level of Gene Regulation in Bacteria. Microbiol. Mol. Biol. Rev. 2011; 75:286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wade J.T., Grainger D.C.. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014; 12:647–653. [DOI] [PubMed] [Google Scholar]
- 5. Wade J.T., Grainger D.C.. Spurious transcription and its impact on cell function. Transcription. 2018; 9:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Courtney C.M., Chatterjee A.. cis-Antisense RNA and transcriptional interference: coupled layers of gene regulation. J. Gene Ther. 2014; 2:1–9. [Google Scholar]
- 7. Thomason M.K., Storz G.. Bacterial antisense RNAs: how many are there, and what are they doing. Annu. Rev. Genet. 2010; 44:167–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lasa I., Toledo-Arana A., Dobin A., Villanueva M., de los Mozos I.R., Vergara-Irigaray M., Segura V., Fagegaltier D., Penadés J.R., Valle J. et al.. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20172–20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mellin J.R., Tiensuu T., Bécavin C., Gouin E., Johansson J.. A riboswitch-regulated antisense RNA in Listeria monocytogenes. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lybecker M., Zimmermann B., Bilusic I., Tukhtubaeva N., Schroeder R.. The double-stranded transcriptome of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:3134–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S.S., Singh N., Bonocora R.P., Fitzgerald D.M., Wade J.T., Grainger D.C.. Widespread suppression of intragenic transcription initiation by H-NS. Genes Dev. 2014; 28:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cardinale C.J., Washburn R.S., Tadigotla V.R., Brown L.M., Gottesman M.E., Nudler E.. Termination factor rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008; 320:935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peters J.M., Mooney R.A., Grass J.A., Jessen E.D., Tran F., Landick R.. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 2012; 26:2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grainger D.C. Structure and function of bacterial H-NS protein. Biochem. Soc. Trans. 2016; 44:1561–1569. [DOI] [PubMed] [Google Scholar]
- 15. Mitra P., Ghosh G., Hafeezunnisa M., Sen R.. Rho protein: roles and mechanisms. Annu. Rev. Microbiol. 2017; 71:687–709. [DOI] [PubMed] [Google Scholar]
- 16. Arraiano C.M., Andrade J.M., Domingues S., Guinote I.B., Malecki M., Matos R.G., Moreira R.N., Pobre V., Reis F.P., Saramago M. et al.. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010; 34:883–923. [DOI] [PubMed] [Google Scholar]
- 17. Nudler E., Gottesman M.E.. Transcription termination and anti-termination in E. coli. Genes Cells. 2002; 7:755–768. [DOI] [PubMed] [Google Scholar]
- 18. Epshtein V., Dutta D., Wade J., Nudler E.. An allosteric mechanism of Rho-dependent transcription termination. Nature. 2010; 463:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boudvillain M., Figueroa-Bossi N., Bossi L.. Terminator still moving forward: Expanding roles for Rho factor. Curr. Opin. Microbiol. 2013; 16:118–124. [DOI] [PubMed] [Google Scholar]
- 20. Sedlyarova N., Shamovsky I., Bharati B.K., Epshtein V., Chen J., Gottesman S., Schroeder R., Nudler E.. sRNA-Mediated Control of Transcription Termination in E. coli. Cell. 2016; 167:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts J.W. Mechanisms of bacterial transcription termination. J. Mol. Biol. 2019; doi:10.1016/j.jmb.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 22. Sedlyarova N., Rescheneder P., Magán A., Popitsch N., Rziha N., Bilusic I., Epshtein V., Zimmermann B., Lybecker M., Sedlyarov V. et al.. Natural RNA polymerase aptamers regulate transcription in E. coli. Mol. Cell. 2017; 67:30–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klopf E., Moes M., Amman F., Zimmermann B., Von Pelchrzim F., Wagner C., Schroeder R.. Nascent RNA signaling to yeast RNA Pol II during transcription elongation. PLoS One. 2018; 13:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Windbichler N., Pelchrzim F. Von, Mayer O., Csaszar E., Schroeder R.. Isolation of small RNA-binding proteins from E. coli: Evidence for frequent interaction of RNAs with RNA polymerase. RNA Biol. 2008; 5:30–40. [DOI] [PubMed] [Google Scholar]
- 25. Wilson G.G., Young K.K.Y., Edlin G.J., Konigsberg W.. High-frequency generalised transduction by bacteriophage T4. Nature. 1979; 280:80–82. [DOI] [PubMed] [Google Scholar]
- 26. Hoffmann S.A., Kruse S.M., Arndt K.M.. Long-range transcriptional interference in E. coli used to construct a dual positive selection system for genetic switches. Nucleic Acids Res. 2016; 44:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bolger A.M., Lohse M., Usadel B.. Genome analysis Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2 Ben. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amman F., Halluin A.D., Antoine R., Huot L., Bibova I., Keidel K.. Primary transcriptome analysis reveals importance of IS elements for the shaping of the transcriptional landscape of Bordetella pertussis. RNA Biol. 2018; 15:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal. 2011; 17:10–12. [Google Scholar]
- 33. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anders S., Pyl P.T., Huber W.. Genome analysis HTSeq — a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bailey T.L., Williams N., Misleh C., Li W.W.. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006; 34:W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zwiefka A., Kohn H., Widger W.R.. Transcription termination factor rho: the site of bicyclomycin inhibition in Escherichia coli. Biochemistry. 1993; 32:3564–3570. [DOI] [PubMed] [Google Scholar]
- 37. Lamberte L.E., Baniulyte G., Singh S.S., Stringer A.M., Bonocora R.P., Stracy M., Kapanidis A.N., Wade J.T., Grainger D.C.. Horizontally acquired AT-rich genes in Escherichia coli cause toxicity by sequestering RNA polymerase. Nat. Microbiol. 2017; 2:16249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shearwin K.E., Callen B.P., Egan J.B.. Transcriptional interference—a crash course. Trends Genet. 2005; 21:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mazo A., Hodgson J.W., Petruk S., Sedkov Y., Brock H.W.. Transcriptional interference: an unexpected layer of complexity in gene regulation. J. Cell Sci. 2007; 120:2755–2761. [DOI] [PubMed] [Google Scholar]
- 40. Cohen O., Doron S., Wurtzel O., Dar D., Edelheit S., Karunker I., Mick E., Sorek R.. Comparative transcriptomics across the prokaryotic tree of life. Nucleic Acids Res. 2016; 44:W46–W53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thomason M.K., Bischler T., Eisenbart S.K., Förstner K.U., Zhang A., Herbig A., Nieselt K., Sharma C.M., Storza G.. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 2015; 197:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Georg J., Hess W.R.. Widespread antisense transcription in prokaryotes. Microbiol. Spectr. 2018; 6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dar D., Sorek R.. High-resolution RNA 3-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res. 2018; 46:6797–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blattner F.R., Plunkett G. III, Bloch C.A., Perna N.T., Valerie B., Riley M., Collado-Vides J., Glasner J.D., Rode Christopher K., Mayhew G.F. et al.. The Complete Genome Sequence of Escherichia coli K-12. Science. 1997; 277:1453–1462. [DOI] [PubMed] [Google Scholar]
- 45. Daley D.O., Rapp M., Granseth E., Melén K., Drew D., von Heijne G.. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 2005; 308:1321–1323. [DOI] [PubMed] [Google Scholar]
- 46. Yang J., Baldi D.L., Tauschek M., Strugnell R.A., Robins-Browne R.M.. Transcriptional regulation of the yghJ-pppA-yghG-gspCDEFGHIJKLM cluster encoding the Type II secretion pathway in enterotoxigenic Escherichia coli. J. Bacteriol. 2007; 189:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weaver J., Mohammad F., Buskirk A.R., Storz G.. Identifying small proteins by ribosome profiling with stalled initiation complexes. MBio. 2019; 10:e02819-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing read data produced in the course of this study are available from the European Nucleotide Archive under the study accession number PRJEB31692. Sample accession numbers ERS3223839 to ERS3223844 correspond to the transcriptome data of wild type and BCM-treated E. coli, respectively. Sample accession numbers ERS3223845 and ERS3223846 correspond to the Amplicon-seq data used to characterize RAPs as iRAPs.
Differential expression analyses and annotation of upregulated antisense regions and RAPs can be visualized in http://genome-euro.ucsc.edu/cgi-bin/hgHubConnect by copying the following URL in ‘My Hubs’: http://nibiru.tbi.univie.ac.at/publicNGS/Assembly_Hubs/EcoliRAP_hub/hub.txt.
Original northern blot pictures were deposited in Mendeley data and can be accessed via the following link: https://data.mendeley.com/datasets/x8gkh63mp8/draft?a=d8210201-bf4d-4286-b58c-768593e1613e.
Flow cytometry data were deposited in Flow Repository under the Repository ID FR-FCM-Z239 and can be accessed via the following link: http://flowrepository.org/id/RvFrRjEeIn4djSNl0eqewEWCZESxEwstPfPUj6jAOs71fXcxik3OXc8SlMg3b2gW.