

Abstract
Identification of key regulators and regulatory pathways is an important step in the discovery of genes involved in cancer. Here, we propose a method to identify key regulators in prostate cancer (PCa) from a network constructed from gene expression datasets of PCa patients. Overexpressed genes were identified using BioXpress, having a mutational status according to COSMIC, followed by the construction of PCa Interactome network using the curated genes. The topological parameters of the network exhibited power law nature indicating hierarchical scale-free properties and five levels of organization. Highest degree hubs (k ≥ 65) were selected from the PCa network, traced, and 19 of them was identified as novel key regulators, as they participated at all network levels serving as backbone. Of the 19 hubs, some have been reported in literature to be associated with PCa and other cancers. Based on participation coefficient values most of these are connector or kinless hubs suggesting significant roles in modular linkage. The observation of non-monotonicity in the rich club formation suggested the importance of intermediate hubs in network integration, and they may play crucial roles in network stabilization. The network was self-organized as evident from fractal nature in topological parameters of it and lacked a central control mechanism.
Subject terms: Regulatory networks, Oncology
Introduction
Prostate is a gland of the male reproductive system which secretes seminal fluid in human adult1. According to World Cancer Report 2014, the cancer of prostate or Prostate cancer (PCa) in man is second most common cancer, after lung cancer, and is responsible for a fifth of cancer deaths in males worldwide2. PCa, based on the type of origin in prostate, can be classified into five types: (i) acinar adenocarcinoma, (ii) ductal adenocarcinoma, (iii) transitional cell (or urothelial) cancer, (iv) squamous cell cancer and (v) small cell prostate cancer, with adenocarcinomas being the most common, even though metastasis is much quicker in other types3,4.
In recent years, gene expression studies using high-throughput techniques namely next generation sequencing, microarray and proteomics have led to the identification of new genes and pathways in PCa. The identification of novel key regulators is important as the current therapeutic modalities against PCa, including the use of antiandrogens and blocking androgen synthetic pathway5 and using Luteinizing hormone-releasing hormone (LHRH) agonists and antagonists along with cytotoxic anticancer drugs, cause notable side effects6,7. Moreover, PCa diagnosis, which is largely dependent on the Prostate specific antigen (PSA) and Digital rectal examination (DRE), has its own limitations8,9. PSA is also elevated in benign prostatic hyperplasia and other noncancerous conditions9. This necessitates the discovery of more reliable biomarkers for better and early diagnosis, as well as identification of new targets other than the genes involved in androgen metabolism for the discovery and development of new and more potent drugs which have less toxicity and lesser side effects.
Genes are regulated in a coordinated way and the expression of one gene usually depends on the presence or absence of another gene (gene interaction). Network theory, which studies the relations between discrete objects through graphs as their representations, can be used to study complex gene regulatory networks which can have different types (random, scale-free, small world and hierarchical networks). The development of algorithms to study of these networks can provide an important tool to find/identify disease-associated genes in complex diseases such as cancer. Earlier, the network theory-based methods have been used to predict disease genes from networks generated using curated list of genes reported to be associated with the disease and mapping them to the human gene interaction network (HPRD database)10. In such approach, the studies have been limited to the curated gene list forming the network not completely representing the system and patient-specific information is not considered. Moreover, current studies on complex networks in human disease models to discover key disease genes rely mostly on clustering and identifying the high degree hubs or/and motif discovery from the networks11,12. Therefore, the application of network theoretical methods to the protein–protein interaction (PPI) networks of cancer associated genes constructed from the corresponding genes by analyzing high-throughput gene expression datasets of human cancer patients may be used for better sensitivity and forecast in understanding the key regulating genes of the corresponding disease. The clinical impact of using patients’ gene expression data over gene expression data from cancer cell lines will also give a systematic insight in predicting key regulator genes expressed in cancer and understanding their roles in disease manifestation and progression. In this study, we have used the gene expression data (RNAseq) of PCa patients to construct complex PPI network and analyze it. The study gives equal importance to the hubs, motifs and modules of the network to identify the key regulators and regulatory pathways not restricting only to overrepresented motifs or hubs identification, establishing a relationship between them in gene-disease association studies using network theory. The method used in this study is new and takes a holistic approach for predicting key disease genes and their pathways within network theoretical framework using datasets of PCa patients.
Materials and Methods
Identification and selection of PCa-associated genes
BioXpress v3.0 (https://hive.biochemistry.gwu.edu/bioxpress), which uses TCGA (https://tcga-data.nci.nih.gov/) RNA sequencing datasets derived from the human cancer patients13, was used to differentiate the deregulated genes in cancer. The cancer browser tool of COSMIC (https://cancer.sanger.ac.uk/cosmic)14 was used for the mutational status and accordingly, non-redundant genes overexpressed in human PCa were identified. Systematic flow chart of methodology is given in Fig. 1.
Figure 1.

Flowchart of the methodology.
Construction of protein-protein interaction (PPI) network
After excluding the redundancy and redundant copies, out of 4,890 genes found to be significantly overexpressed (FC > 1, adjusted p < 0.05) in PCa patients from BioXpress, 3,871 genes, which had mutational status in PCa according to COSMIC, were used to construct an interactome network using GeneMANIA app15 in Cytoscape 3.6.016. From the network, only the physical interaction network, which represented the protein-protein interaction network of PCa-associated genes, was extracted. After curation of the network (removal of isolated node/nodes), a protein-protein interaction network of 2,960 nodes and 20,372 edges was finally constructed as primary network representing a graph denoted by G(N, E), where, N is the set of nodes with N = {ni}; i = 1, 2, …, N and E the set of edges with E = {eij}; i, j = 1, 2, 3, …., N.
Method for detection of levels of organization
Considering the size of the network and its sensitivity, Louvain method of modularity (Q) maximization was used for community detection17. The first level of organization was established by the interaction of communities constructed from primary PPI network. The sub-communities constructed from all communities in the first level of organization constituted second level of organization. In the same way, successive levels were constructed until the level of motifs, thereby each smaller community had a minimum of one triangular motif defined by sub-graph G(3,3). Since the triangular motifs are overrepresented in PPI network and serve as controlling unit in a network18, we used motif G(3,3) as qualifying criteria for a community/subcommunity as a constituting member at a certain level of organization. Further, each community or smaller community landed up to different level of organization.
Topological analyses of the networks
Cytoscape plugins, NetworkAnalyzer19 and CytoNCA20 were used to analyse the topological properties of the network for centralities, degree distribution, clustering coefficients and neighbourhood connectivity. The highest degree nodes were identified as hubs of the PCa network. Top 103 hub proteins having degree k ≥ 65 were considered for tracing the key regulators of the network. Other topological parameters, viz., Rich club coefficients (Φ), Participation coefficients (Pi) and Within-module degree (Zi score) were calculated using Igraph package “brainGraph” (https://github.com/cwatson/brainGraph) in R. Another parameter subgraph centrality was also calculated using Igraph functions.
Degree (k)
In the analysis of network, degree k indicates the total number of links established by a node in a network and is used to measure the local significance of a node in regulating the network. In a graph represented by G = (N, E), where N denotes nodes and E the edges, the degree of ith node (ki) is expressed as , where Aij denotes the adjacency matrix elements of the graph.
Probability of degree distribution (P(k))
It is the probability of a random node to have a degree k out of the total number of nodes in the network and is represented as fraction of nodes having degree (k), as shown in Eq. (1), where Nk is the total number of nodes with degree k and N, total nodes in the network.
| 1 |
P(k) of random and small-world networks follow Poison distribution in degree distribution against degree, but most real-world networks, scale-free and hierarchical networks follow power law distribution P(k) ~k-γ, where, 4 ≥ γ ≥ 2. In hierarchical networks, γ ~2.26 (mean-field value) indicating a modular organization at different topological levels21. Therefore, P(k) pattern defines the characteristic topology of a network.
Clustering coefficients C(k)
The strength of internal connectivity among the nodes neighbourhoods which quantifies the inherent clustering tendency of the nodes in the network is characterised by the Clustering coefficient C(k), which is the ratio between the number of triangular motifs formed by a node with its nearest neighbours and the maximum possible number of triangular motifs in the network. For any node i having degree ki in an undirected graph, C(k) can be expressed as Eq. (2), where mi is the total number of edges among its nearest neighbours. In scale-free networks C(k) ~ constant, but it exhibits power law in hierarchical network against degree, C(k) ~ k−α, with α ~ 121.
| 2 |
Neighbourhood connectivity CN(k)
The node neighbourhood connectivity is the average connectivity established by the nearest-neighbours of a node with degree k, represented by CN(k) can be expressed as shown in Eq. (3), where, P(q|k) is conditional probability of the links of a node with k connections to another node having q connections.
| 3 |
In hierarchical network topology, CN(k) exhibit power law against degree k, that is, CN(k) ~ kβ, where, β ~ 0.522. Further, the positivity or negativity of the exponent β can be defined as, respectively, the assortivity or disassortivity nature of a network topology23.
Centrality measures
A node’s global functional significance in regulating a network through information processing is estimated by the basic Centrality measures - Closeness centrality CC, Betweenness centrality CB and Eigenvector centrality CE24. Another centrality measure, Subgraph centrality CS is also used to describe the participation of nodes in other subgraphs in the network25. These centrality measures collectively determine the cost effectiveness and efficiency of information processing in a network.
The closeness centrality CC represents the total geodesic distance from a given node to all its other connected nodes. It represents the speed of spreading of information in a network from a node to other connected nodes26. CC of a node i in a network is calculated by the division of total number of nodes in the network, n by sum of geodesic path lengths between nodes i and j which is represented by dij in Eq. (4).
| 4 |
Betweenness Centrality CB is the measure of a node which is the share of all shortest-path traffic from all possible routes through nodes i to j. Thus, it characterizes a node’s ability to benefit extraction from the information flow in the network27 and its controlling ability of signal processing over other nodes in the network28. If dij(v) denotes the number of geodesic paths from node i to node j passing through node v, then CB(v) of node v can be obtained by Eq. (5).
| 5 |
If M denotes the number of node pairs, excluding v, then normalized betweenness centrality is given by the Eq. (6).
| 6 |
Eigenvector centrality CE is proportional to the sum of the centrality of all neighbours of a node and it reflects the intensity of these most prominent nodes influencing the signal processing in the network29. If nearest neighbours of node i in the network is denoted by nn(i) with eigenvalue λ and eigenvector vi of eigen-value equations, Avi = vi(v) where, A is the network adjacency matrix, CE can be shown by the Eq. (7),
| 7 |
CE score corresponds to maximum positive eigenvalue, λmax, of the principal eigenvector of A29. Since a node’s CE function depends on the centralities of its neighbours, it varies across different networks association of high CE nodes; within closely connected locality of such nodes reduces the chances of isolation of nodes29. Thus, CE becomes a powerful indicator of information transmission power of a node in the network.
The subgraph centrality CS of a node calculates the number of subgraphs the node participates in a network. It can be calculated using eigenvalues and eigenvectors of adjacency matrix of the graph, as shown in Eq. (8), where λj is the jth eigenvalue and vj(i), the ith element of the associated eigenvector. The weightages are higher for smaller graphs. Higher subgraph centrality of a node corresponds to better efficiency of information transmission and increase in essentiality of the node in the network25.
| 8 |
Within-module degree and Participation coefficients of the hubs
In complex networks the characterization of hubs as high degree nodes with higher centrality values is incomplete without exploring the role of nodes at the modular levels30. The role of nodes at the modular level is determined through the participation of nodes in establishing links between the nodes within the module as well as outside the module and calculating the modular degree of the nodes. Within-module degree or Z-score, Zi, signifies the connections of a node i in the modules and categorizes a node as modular hub-node with high (Zi ≥ 2.5) signifying more intra-module connectivity of the node than inter-module, whereas, lower Z values, Zi < 2.5, categorizes as non-hub nodes with less intra-module connectivity30. The Z- score can be calculated as shown in Eq. (9), where ki represents the number of links of node i to other nodes in its modules Si and , the average of degree (k) over all nodes in Si; , is the standard deviation of k in Si.
| 9 |
The participation coefficient, Pi determines the participation of the node i in linking the nodes inside and outside its module30. Pi values lie in the range of 0−1 with higher values corresponding to the participation of nodes in establishing links outside the modules with homogeneous distribution of its links among all modules, and if kis is taken to represent the number of links of node i to nodes in modules s and ki, the total degree of node i, Pi can be calculated as in Eq. (10), where, NM is the number of modules in the network.
| 10 |
Rich-club analysis
Identification of hubs in a network generally is done through general centrality measures, especially higher degree nodes are commonly considered as hubs and existence of high degree nodes in a network correlate with the local regulatory roles of these high degree hubs in the network31. This phenomenon of formation of rich club connection between high degree hubs exhibit the robustness of the network and the resilience when the hubs are targeted32. The existence of rich club phenomenon among hubs is investigated by calculating the Rich-club coefficients Φ(k) across the degree range32. Φ(k) is equivalent to the clustering coefficient among a subgroup of nodes with degrees ≥k. In order to remove the random interconnection probability factor, normalization of the rich club coefficients can be done by the Eq. (11), where Φrand(k) is the rich-club coefficient of random networks with similar size and degree sequence and Φnorm(k) > 1 indicating rich-club formation. This rich club phenomenon is associated with the assortivity nature of the networks and is important to understand the roles played by these hubs’ roles in the network integration and efficient transmission of signals33.
| 11 |
Tracking the key regulators in the networks
The most influential genes in the PCa network were identified first through calculating the centrality measures. Since, higher degree nodes have higher centrality values, top 103 highest degree nodes (Degree k ≥ 65) were considered among the hub nodes of the network for tracing the key regulators which may play important role in regulating the network. Then tracing of nodes from the primary network up to motif level G(3, 3) was done on the basis of representation of the respective nodes (proteins) across the sub modules obtained from Louvain method of community detection/clustering17. Finally, the hub-nodes (proteins) which were represented at the modules at every hierarchical level were considered as key regulators of the PCa network.
Functional association analysis of modules
The modules at all levels of hierarchy were analysed for their functional annotations with DAVID functional annotation tool34. The functions and pathways with corrected p < 0.05 were considered statistically significant.
Results
PPI network in PCa follows hierarchical scale-free topology composed of modules at five levels of hierarchy
From the interactome network of 3,871 PCa genes, the physical interacting PPI network of 2,960 proteins with 2,960 nodes and 20,372 edges was constructed as the primary network (Fig. 1). Analysis of this primary PCa network showed that the network followed power law distributions for probability of node degree distribution, P(k), clustering coefficient C(k) and neighbourhood connectivity distribution CN(k) against degree (k) with negative exponents22 (Eq. 12) (Fig. 2). This power law feature indicates that the network exhibited hierarchical-scale free behaviour with systems level organization of modules/communities. Further, community finding using Louvain modularity optimization method17 led to the detection of communities and sub-communities at various levels of organization (Fig. 3A). Thus, a total of 436 communities and smaller communities were detected, out of which 38 reached up to level V, the level of motif G(3,3).
Figure 2.

Topological properties of PCa and the modules/communities at the first hierarchical level. Degree distribution probability (P(k)), clustering coefficient (C(k)), neighbourhood connectivity (CN(K)) as function of degree (k) and centrality measurement closeness (CC(k)), betweenness centrality (CB(k)), eigenvector centrality ((CE(k)), subgraph centrality (CS) as a function of degree.
Figure 3.

(A) Communities/modules of PCa PPI network. (B) Interacting partners of the 19 key regulators at motif level. (C) Protein Protein Interaction of key regulators with AR through TP53, CTNNB1 and AKT1 constructed from GeneMANIA.
Communities at the first hierarchical level also showed power law distribution for P(k), C(k) and CN(k) against degree distribution with negative exponents indicating further systems level organization of modules (Eq. 12) except in case of communities C8, C10 and C15 where the CN(k) exhibit power law against degree k with positive exponents (β ~ 0.05, 0.13, 0.14 respectively) (Fig. 2). This indicates assortivity nature in the modules indicating the possibility of rich-club formation in these modules, where, hubs play significant role in maintaining network properties and stability22.
| 12 |
Nineteen (19) novel regulators served as backbone of the network
Centrality measures are used to assess the importance of the nodes in information processing in a network. Betweenness centrality CB, Closeness centrality CC, Eigenvector centrality CE and Subgraph centrality CS are various topological properties which can determine the efficiency of signal transmission in a network25. In PCa network and modules at the first hierarchical level, these parameters also exhibited power law as a function of degree (k) with positive exponents where the centralities tend to increase with higher degree nodes (Eq. 13) (Fig. 2). This behaviour revealed the increase in efficiency of signal processing with higher degree nodes in the network showing the importance of these nodes in controlling the flow of information, thereby regulating and stabilizing the network. Hence, hub proteins had a significant influence in regulating the network and might be playing an important role in PCa. In order to identify the most influential key regulator proteins in the network, top 103 hub-proteins having degree (k) ≥ 65 were considered for identification of the key regulators through their representation at every topological level (Supplementary Table 1). After tracing hubs at every topological level, 19 (RPL11, RPL15, RPL19, RPL23A, RPL3, RPL5, RPL6, RPLP0, RPS11, RPS8, RPSA, HSPA5, NOP2, RANBP2, SNU13, CUL7, CCT4, ASHA1 and EIF3A) (Tables 1, 2) were found to be the backbone of the network. These key regulators along with their partners forming the motifs (Fig. 3B), might be playing the most important roles in regulating and maintaining the stability (network integrity, optimization of signal processing, dynamics etc.) of the network.
| 13 |
Table 1.
Key regulators and their topological properties.
| Sl. No. | ID | Gene | Function | Degree (k) |
Closeness centrality (CC) |
Betweenness centrality (CB) |
Eigenvector centrality (CE) |
Subgraph centrality (CS) |
|---|---|---|---|---|---|---|---|---|
| 1 | AHSA1 | Activator of Hsp90 ATPase activity 1 | Positive regulation of ATPase | 75 | 0.379262 | 0.00331 | 0.054588 | 2.47E + 23 |
| 2 | CCT4 | Chaperonin containing TCP1 subunit 4 | Protein folding | 66 | 0.384435 | 0.003334 | 0.033523 | 9.29E + 22 |
| 3 | CUL7 | Cullin 7 | Ubiquitin-dependent protein catabolism | 270 | 0.435916 | 0.034463 | 0.151248 | 1.89E + 24 |
| 4 | EIF3A | Eukaryotic translation initiation factor 3 subunit A | Translation pre-initiation complex formation | 85 | 0.37006 | 0.002209 | 0.079073 | 5.18E + 23 |
| 5 | HSPA5 | Heat shock protein family A (Hsp70) member 5 | Activation of signaling protein activity involved in unfolded protein response | 111 | 0.408758 | 0.012349 | 0.062591 | 3.24E + 23 |
| 6 | NOP2 | NOP2 nucleolar protein | rRNA base methylation, | 68 | 0.375317 | 0.001319 | 0.076741 | 4.88E + 23 |
| 7 | RANBP2 | RAN binding protein 2 | Protein sumoylation | 72 | 0.386242 | 0.004881 | 0.031476 | 8.19E + 22 |
| 8 | RPL11 | Ribosomal protein L11 | Ribosomal large subunit assembly | 83 | 0.383638 | 0.001239 | 0.114333 | 1.08E + 24 |
| 9 | RPL15 | Ribosomal protein L15 | Nuclear-transcribed mRNA catabolic process | 79 | 0.382201 | 0.001289 | 0.109354 | 9.91E + 23 |
| 10 | RPL19 | Ribosomal protein L19 | Nuclear-transcribed mRNA catabolic process | 78 | 0.381856 | 0.000469 | 0.113259 | 1.06E + 24 |
| 11 | RPL23A | Ribosomal protein L23a | Ribosomal large subunit assembly | 84 | 0.379602 | 0.001182 | 0.109817 | 9.99E + 23 |
| 12 | RPL3 | Ribosomal protein L3 | Ribosomal large subunit assembly | 67 | 0.385588 | 0.000788 | 0.100707 | 8.41E + 23 |
| 13 | RPL5 | Ribosomal protein L5 | Ribosomal large subunit assembly | 92 | 0.383141 | 0.001438 | 0.114815 | 1.09E + 24 |
| 14 | RPL6 | Ribosomal protein L6 | Ribosomal large subunit assembly | 113 | 0.39809 | 0.003601 | 0.129049 | 1.38E + 24 |
| 15 | RPLP0 | Ribosomal protein lateral stalk subunit P0 | Nuclear-transcribed mRNA catabolic process | 88 | 0.394955 | 0.002486 | 0.110764 | 1.02E + 24 |
| 16 | RPS11 | Ribosomal protein S11 | Nuclear-transcribed mRNA catabolic process | 74 | 0.376943 | 0.000751 | 0.102129 | 8.64E + 23 |
| 17 | RPS8 | Ribosomal protein S8 | Nuclear-transcribed mRNA catabolic process | 120 | 0.394218 | 0.004481 | 0.126136 | 1.32E + 24 |
| 18 | RPSA | Ribosomal protein SA | Ribosomal small subunit assembly | 79 | 0.378002 | 0.002864 | 0.094957 | 7.47E + 23 |
| 19 | SNU13 | SNU13 homolog, small nuclear ribonucleoprotein (U4/U6, U5) | mRNA splicing, via spliceosome | 87 | 0.370245 | 0.003088 | 0.072649 | 4.37E + 23 |
Table 2.
The key regulator identified in this study and their key functions in disease conditions.
| Genes | Function/mechanism | Condition/Disease | Reference |
|---|---|---|---|
| Ribosomal genes | |||
|
RPL5, RPL6, RPL11 |
Stabilizes p53/TAp73 (by binding to the MDM2/MDMX/HDM2) and inhibits ubiquitination of p53/TAp73 | Arrests cell growth arrest, promote apoptosis | 66 |
| RPL23A | Stabilizes p53 and inhibits RAS-mediated tumorigenesis | Arrests cell growth arrest, promote apoptosis | 67 |
| RPL11 | Either prevents the binding of co-activator TRRAP to MYC promoter, or act via miR-24/miRISC silencing complex | Inactivates c-MYC transcription, or promote its degradation | 68 |
| RPL6 | Upregulates Cyclin E, promoting cell growth and cell cycle progression; inhibition is reported to downregulate Cyclin E, arresting cell cycle at G1 | Gastric cancer and Multi drug resistance gastric cancer | 69 |
| RPS8 | Overexpression | Pancreatic ductal carcinoma, gastric, colorectal, breast and oral cancers | 70– 74 |
| RPLP0 | The gene product RPLP0 interacts with Cathepsin X; knockdown arrests cell cycle at G1, increasing apoptosis | Gastric, ovarian and endometrial cancers | 75, 76 |
| RPSA | A cell surface receptor (binding to lamin), facilitates cell adhesion and activation of signal transduction pathways; overexpression linked to tumor aggression and metastasis | Colorectal cancer | 77, 78 |
| RPS11, RPL19, RPL15, RPL3 | Overexpression. RPL19 overexpression serve as a Prognostic marker in PCa | Glioblastoma, colorectal, gastric, lung, and prostate cancers | 79– 83 |
| Non-ribosomal regulators | |||
| SNU13 | Interacts with several RPs | Strengthens the role of RPs | 41 |
| CCT4 | Mutated | Hereditary sensory neuropathy | 42 |
| AHSA1 | Wnt/β-catenin signaling pathway | Cell growth, apoptosis, migration and invasion | 46 |
| CUL7 | ERK-SNAI2 signalling, affecting cell adhesion inhibition of p53; Cyclin A overexpression and affecting microtubule dynamics by increasing α-tubulin accumulation | Epithelial-mesenchymal transition in metastasis, inhibits apoptosis, cycle progression, cell proliferation and migration lung, breast cancer etc. | 47– 49 |
| EIF3A | Translation initiation and regulation of mTOR pathway | Translation of genes involved in cell proliferation, cell differentiation, apoptosis | 51 |
| HSPA5 | Unfolded protein response in ER stress | Escaping cell death in cancers | 52, 53 |
| NOP2 | Regulates cell cycle progression from G1 to S phase | Biomarker for cell transformation | 55, 56 |
| RANBP2 | Involved in SUMOylation of topioisomerase II-α and the p150/importin β/RANBP2 pathway | Lung cancer and myelomocytic leukemia | 57 |
Modules of the network were associated with specific functions
Community detection of the network using Louvain modularity optimization method leads to clustering of the primary PCa network up to the level of motifs (Fig. 3A). This clustering showed that Modularity (Q) of the networks exhibited an increasing pattern with topological levels with highest average Modularity (Q = 0.5527) seen at the first hierarchical level, and lowest (Q = 0.0013) at the level V, the motif level35,36.
In complex PPI networks the modules have biological meanings relating to functions and gene ontology analyses have revealed enrichment of certain known functions and pathways in the modules37. Our primary PCa-network was composed of 14 modules deduced from the community detection and their mean clustering coefficients C(k) ~ 0.094−0.392 (Table 3). Among these, modules C12 and C13 which were the largest and had the highest mean clustering coefficients C(k) = 0.392 and 0.218 respectively, showing a functional homogeneity in the modules. These modules were analysed for their functional annotations with DAVID functional annotation tool34 to reveal association with different functions (Table 3).
Table 3.
Average Clustering coefficients of the PCa modules at first hierarchical level.
| Sl. No. | Modules | Most enriched function | Avg. Clustering coefficient | Corrected p-value |
|---|---|---|---|---|
| 1. | C2 | RNA mediated gene silencing | 0.114 | 2.07E − 07 |
| 2. | C3 | Unfolded protein folding | 0.209 | 4.12E − 16 |
| 3. | C4 | ATP/nucleotide binding | 0.171 | 2.64E − 14 |
| 4. | C5 | MRNA transport | 0.165 | 2.25E − 15 |
| 5. | C6 | Transcription initiation | 0.374 | 2.68E − 12 |
| 6. | C7 | Endoplasmic reticulum membrane proteins | 0.094 | 0.019 |
| 7. | C8 | Endocytosis | 0.14 | 2.67E − 09 |
| 8. | C9 | Mitocondrial proteins | 0.124 | 6.75E − 33 |
| 9. | C10 | Proteosome | 0.182 | 1.02E − 16 |
| 10. | C11 | Ubiquitin protein ligase activity | 0.107 | 1.13E − 10 |
| 11. | C12 | Ribonucleoprotein | 0.392 | 7.50E − 104 |
| 12. | C13 | Transcription regulation | 0.218 | 6.11E − 74 |
| 13. | C14 | Transmembrane helix | 0.096 | 4.07E − 04 |
| 14. | C15 | DNA repair | 0.31 | 6.61E − 08 |
Hubs in the PCa network coordinate the modules acting as modular hubs
In complex hierarchical networks, the modularity of sub-communities and the roles played by the nodes in the modules is defined with the nodes Within-module Z score, Zi along with their Participation coefficients Pi30. Zi gives the degree of the nodes within their modules, and Pi describes the influence of a node inside the module, as well outside it, in terms of signal processing as well as maintaining network stabilization. Hence, Zi and Pi were calculated for each node in the modules using Eqs (9), (10), respectively. Accordingly, within-module Z score, the nodes are classified as follows:
Modular non-hub nodes Zi < 2.5: (R1) Ultraperipheral nodes: The nodes linking all other nodes within their modules, Pi ≤ 0.05(R2) Peripheral nodes: nodes linking most other nodes in their modules, 0.05 < Pi ≤ 0.62; (R3) non-hub connector nodes: nodes linking many nodes in other modules, 0.62 < Pi ≤ 0.80; and (R4) Non-hub kinless nodes: nodes linking all other modules, Pi > 0.80.
Modular hubs Zi ≥ 2.5: (R5) Provincial hubs; hub nodes linking vast majority nodes within their modules, Pi ≤ 0.30; (R6) Connector hubs; hubs linking most the other modules, 0.30 < Pi ≤ 0.75; and (R7) Kinless hubs; hubs linking among all modules, Pi > 0.75.
In the PCa PPI network™study, many hub-proteins were acting as modular hubs, helping in establishing connection between the modules at different hierarchical levels. For example, CUL7 and RANBP2 were among important key regulator protein hubs in PCa which also acted as modular kinless and connector hubs of module C3 and C5 at the first hierarchical level (Fig. 4A,B). P53, E2F1 and c−MYC acted as kinless global hubs of module C9 connecting with all the modules and other proteins in the network. NOP56, FBL, RNF2 and NPM1 also acted as connector modular hubs of C12 module connecting other modules at the same level (Fig. 4A,C).
Figure 4.

Identification of modular hubs. (A) In the primary PCa network and the modules at first Hierarchical level with within module Z score Zi, and their participation coefficients Pi. (B) Identification of modular hubs among 19 key regulators. (C) Participation coefficient Pi vs degree k in PCa primary network and the modules at first hierarchical level.
PCa network exhibited non-monotonicity in rich-club formation across the hierarchy
Identification of rich club nodes is another common feature to study the influence of hubs in the network forming a strong connection among them which is done by calculating normalized rich club coefficient Φnorm across the degree range k (Eq. 11). Normalized rich-club coefficient Φnorm > 1 indicates the existence of rich club among the nodes which play key role in network integration, increasing its stability and improving the efficiency of transmission of information among hub proteins. Since, PCa network is hierarchical and shows disassortativity in nature with node neighbourhood connectivity CN(k) following power law distribution against degree (k) with negative value of exponent β (Eq. 13), rich club formation among the hub proteins is quite unlikely32,38. Although rich club formation is not exhibited among high degree hub proteins, the moderate intermediate degree protein with degree 19 ≤ k ≤ 107 showed higher rich club coefficients than the hubs in PCa network (Fig. 5). In the PCa network across the hierarchy, different patterns of rich club coefficients were exhibited among the modules (Fig. 5), showing the phenomenon of non-monotonic behaviour at different hierarchical levels. With respect to modules C12 and C13 at first hierarchical level, they exhibit rich club formation between the high degree nodes but the pattern changes moving at the lower levels. However, in the modules C8, C10 and C15, the topological properties of these modules exhibit assortativity nature due to (i) the node neighbourhood connectivity CN(k) in these modules follow power law with positive β exponents, (ii) Φ increases monotonically with degree k, and (iii) Φnorm approximately increases with degree k with values of Φnorm > 1 (Fig. 6), indicating the possibility of rich club formation among the high degree nodes (Fig. 6A). Considering the nodes with degrees whose Φnorm is larger than one, the approximate range of degrees of nodes forming rich-club in these three modules are 61≥k≥14 (C8), 52≥k≥6 (C10), 37≥k≥6 (C15), and clearly show rich-club formations in the respective network modules (red coloured nodes in the respective modules in Fig. 6).
Figure 5.
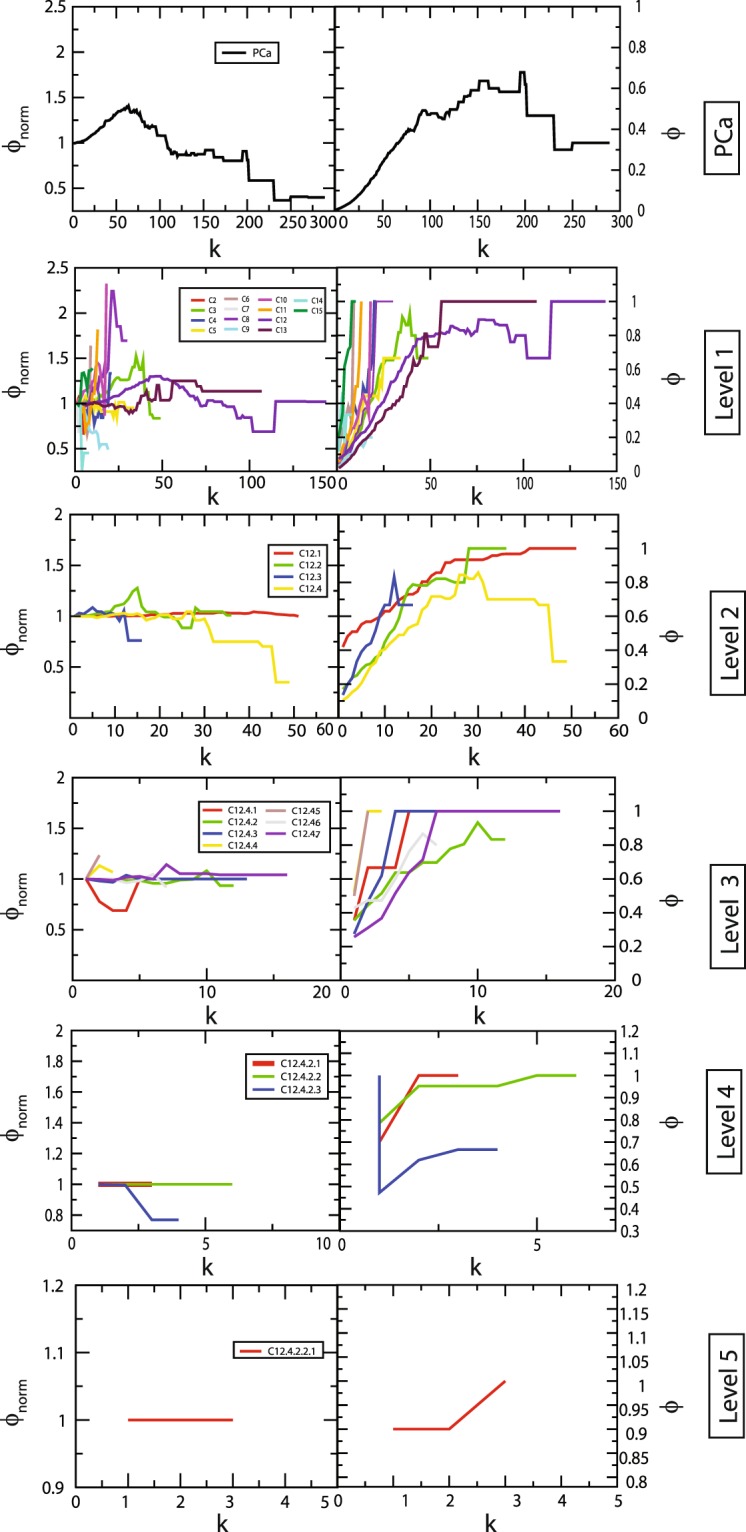
Rich club analysis of PCa PPI network and the communities upto the motif level.
Figure 6.

(A) Rich formation in C8, C10 and C15 in first hierarchal level of PCa. (B) Degree maximum and minimum degrees of rich club forming hubs.
Discussion
The real-world complex networks generally have hierarchically organized community structure, which is evident from fractal studies and scaling behaviour of these networks21. Even though there is no specific definition of communities or modules in a network, each community/module is established by densely interconnected nodes forming clusters around the hub nodes which generally have their own local properties and organization35. The hubs have highest interactions in the network due to their high degree, constitute both intra and inter communities’ interactions in the network in a hierarchical manner, and thus play a central role in information processing in the network31. The primary PPI PCa network constructed in this study for tracking the hubs up to the level of motifs led to the identification of 19 key regulators (hubs) from 3,871 genes found to be significantly overexpressed in human prostate adenocarcinomas. There have been limited community finding methods in complex networks, among which the Newman and Girvan leading eigenvector algorithm35,36, is commonly used. However, in comparatively large complex networks, Louvain method, which is based on modularity, Q maximization/optimization17, is the most suitable, sensitive and comparatively faster. In our study, considering the size of the network and its sensitivity, we used Louvain method for community detection and while giving equal importance to the hubs, motifs and modules of the network, we identified the novel key regulators. 11 key regulators (RPL11, RPL15, RPL19, RPL23A, RPL3, RPL5, RPL6, RPLP0, RPS11, RPS8 and RPSA) belong to the family of ribosomal proteins (RPs) which are involved in ribosomal biosynthesis and other eight predicted regulators (HSPA5, NOP2, RANBP2, SNU13, CUL7, CCT4, ASHA1 and EIF3A) have important functions reported to be associated with various other cancers. Moreover, at the level of motifs these key regulators interact with other proteins which may also be playing important roles in PCa and establishing themselves to be the candidate disease-genes along with key PCa regulators (Fig. 3B).
The emergence of 11 RPs as key regulators in PCa is an important finding in this study. It could be due to the crucial role of RPs in cell growth and proliferation propagated through protein synthesis. In cancers, ribosomal biosynthesis increases to meet the requirement of rapidly growing/proliferating cells39. Some RPs take part in extra-ribosomal functions involved in tumorigenesis, immune cell signalling, and development and regulating diseases through translocation across the nuclear pore complex40. RPs have been associated with tumorigenesis either as oncoproteins or tumour suppressors, with differential roles being reported in different cancers. During ribosomal or nucleolar stress such as hypoxia, lack of nutrient, starvation, deregulation of genes etc., RPs modulate the p53-mediated apoptosis. The association of RPs with cancers as discussed in Table 2 suggests a potential unexplored function of these proteins in PCa, both as therapeutic target and predictive biomarker. An understanding of the functions and the pathways of key RPs, for example their role in stabilizing p53 during ribosomal stress and role in cell growth/proliferation in PCa patients is of immense significance as it provides new insights into the control and prevention of PCa.
Besides, other non-ribosomal predicted key regulators identified in this study, SNU13, CCT4, AHSA1, CUL7, EIF3A, HSPA5, NOP2 and RANBP2, are also vital in cell physiology and are equally important for their involvement in cell growth and proliferation in one way or another. The NHP2−likeprotein1(SNU13) identified in this study as another key regulator, is a component of the spliceosome complex41 which interacts with several RPs and strengthens the role of RPs in cancers. CCT4, Chaperonin containing TCP1 subunit 4, is a chaperone which when mutated is associated with hereditary sensory neuropathy42.
AHSA1, theActivatorofHSP90ATPaseActivity1, is a positive regulator of the heat shock protein 90 (HSP90) and when activated HSP90 forms a complex with HSP70 which helps in either binding of the tumour suppressor p53 to DNA, or its degradation by ubiquitination43. In cancers, activated HSP90 stabilizes the mutated p53 which decreases its DNA binding activity and degradation through binding with its inhibitor MDM2, thus promoting tumour progression44. The activation and transportation of steroid hormones (androgen receptor, AR and oestrogen receptor, ER) to the nucleus is also mediated by HSP9045; thus, AHSA1 activation of HSP90 may influence the androgen metabolism in PCa. Moreover, AHSA1 is a regulator of the cell growth, apoptosis, migration and invasion through Wnt/β−catenin signaling pathway46, which suggests its role as a candidate-disease gene in PCa.
CUL7, Culin7, is a component of an E3ubiquitin−proteinligase complex and interacts with p53, CUL9 and FBXW8, and is reported to be an antiapoptotic oncogene47. CUL7 has been associated with various cancer types, but its promotion of epithelial-mesenchymal transition in metastasis and its regulation of ERK−SNAI2 signalling affecting the expression of cell adhesion proteins, E−cadherins, fibronectin, N−cadherin and vimentin in cancer is well studied48. CUL7 inhibits apoptosis in lung cancer through inhibition of p53 which regulates c−MYC cell cycle progression47. CUL7 regulates cell cycle progression through CyclinA overexpression and affects the cell migration, which is a hallmark of cancer, influencing microtubule dynamics in breast cancer49. Therefore, the targeted knockdown and silencing of CUL7 has led to a decrease in cell proliferation, weaker −tubulin accumulation in microtubules, promoting their stability and decreasing cell migration (in breast, liver and lung carcinoma cells) and has been suggested as a potential therapeutic target in various cancers47–49.
The Eukaryotic translation initiationfactor 3 subunit A(EIF3A) forms 43SPre−initiation complex(43SPIC) with other initiation factors and 40Sribosome and initiates the protein synthesis process. This translates mainly genes involved in cell proliferation, cell differentiation, apoptosis etc. and exerts transcriptional activation/repression through forming different forms of stem loop binding with the mRNAs50. Dysregulation of translation initiation and the role of EIF3 has been studied in cancers and involvement of EIF3 complex in regulation of mTOR pathway51, makes it an interesting protein to study for its regulatory role in PCa.
The Heat shock protein family A (HSP70) member 5(HSPA5) or glucose−regulated protein 78kDa (GRP78), is a chaperone localized in endoplasmic reticulum (ER) and involved in folding and assembly of proteins and plays an active role in unfolded protein response in ER stress, promoting cell survival which is a common process of escaping cell death in cancers52,53. Due to this activity, HSPA5 is an emerging therapeutic drug target for cancer.
NOP2(p120) is a putative RNA methyl transferase protein and its expression is detectable in proliferating normal and tumour cells, but undetectable in non-proliferating normal cells54. Its role in regulating cell cycle progression from G1 to S phase and transformation of normal fibroblast cells55,56 makes NOP2 an interesting protein which can be used as biomarker for cell transformation. Ran binding protein 2 (RANBP2) is another key regulator identified in this study which is involved in the SUMOylation of TopioisomeraseII− before the onset of anaphase, helping in separation of chromatids from the centromere and its under-expression, mutation or deficiency has been observed in various cancers specially lung cancer and myelomocytic leukemia acting as tumor suppressor genes57. Since SUMOylation plays an important role in tumour progression58, the p150/importin/RANBP2 pathway may also play a significant role in PCa progression.
In PCa, p53 and AR are the most mutated genes reported according to COSMIC14. The protein-protein interactome of GeneMANIA15 showed that out of the 19 key regulators identified in this study, 12 (CUL7, HSPA5, CCT4, RPL19, RPL11, RPL3, RPL6, RPLP0, RANBP2, RPS8, RPL23A and RPL15) interact directly with p53 and other key regulators through them (Fig. 3C). Association of mutation in the Androgen Receptor gene (AR) which causes the mutated receptor to remain in activated state and continue to maintain androgen receptor mediated downstream signalling even in lower level of circulating androgens leading to discovery of androgen independency in prostate cancer59. A recent report suggests several mutations in the AR gene in different metastatic castration-resistance (CRPC) patients in prostate cancer suggesting AR mutants as a good biomarker candidate60. β−catenin (CTNNB1) and GSK−3β are other co-regulators of Androgen receptor and phosphorylation of AR by GSK−3β which inhibit AR driven transcription, but in prostate cancer, the increase in the activity of AKT suppression of GSK−3β due to phosphorylation helps in PCa progression61. In the PCa, loss of tumour suppressor PTEN gene also releases the inhibitory effect on AR increasing its trans localization to nucleus and transcriptional activity62. Therefore, the interaction of the key regulators on AR acted indirectly through p53 and β-catenin(CTNNB1) (Fig. 3C), where the 12 key regulators interact with p53 which regulates GSK−3 and PTEN which are the upstream regulators of AR. In addition, key regulators, RPSA and HSPA5 interact with AR indirectly through β−catenin (CTNNB1) and AKT1 suggesting an important role of the reported key regulators in regulating the functions mediated through p53 and AR in PCa. The findings reiterate the putative roles of these hubs in PCa manifestation and progression. This study may prove fundamental in characterizing the potential therapeutic targets and biomarkers for sensitive intervention and diagnosis of PCa.
It is to be noted that in this study the PCa PPI network followed hierarchical scale free topology. Along with the conventional centrality measures, CB, CC, CE and CS, probability degree distribution P(k), clustering coefficient C(k) and node neighbourhood connectivity distribution CN(k) are used to characterize a network whether one is scale-free, random, small-network or hierarchical network21. PCa PPI network followed power law distributions for probability of node degree distribution, P(k), clustering coefficient, C(k), and neighbourhood connectivity distribution against degree k with negative exponents21 (Eq. 12) (Fig. 2), indicating the network falls in hierarchical-scale free behaviour which can exhibit systems level organization of modules/communities.
Since, node neighbourhood connectivity distribution CN(k) as a function of degree k obeyed power law with negative exponent β, it showed its disassortative nature indicating that there is no signature of rich club formation among high degree nodes in the network32. Degree centrality is the most commonly used centrality measure used to define the hubs which are the high degree nodes in the network. This disassortivity may be due to the sparse distribution of the hubs among the modules playing key roles in coordinating specific function within each module as well as establishing the connections among the modules32. Furthermore, we used Louvain modularity optimization method17 to detect, find communities and sub-communities and their organization at various levels of organization (Fig. 3A). The communities/sub-communities at various hierarchically organized levels also exhibited hierarchical scale-free topology, as was the case in the primary PCa network (Fig. 2). This hierarchical organization shows the systematic coordinating role of the emerged modules/communities and hubs in regulating and maintaining the properties of the network10. In such type of networks, the centrality-lethality rule31 is not obeyed which indicates that disturbing the hub/hubs in the network will not cause the whole network collapse.
Another important feature we found in PCa network is the observation of the non-monotonic behaviour in the rich club formation in the PCa PPI network and across its hierarchy (Fig. 5). The intermediate degree nodes (19 ≤ k ≤ 107) in PCa network showed normalised rich club coefficients (Φnorm > 1) greater than the highest degree hubs, indicating an important role of these intermediate degree nodes (even AR also falls in this category) in regulating the network organization and maintaining stability through establishing key links between the low degree nodes and high degree hubs. Hence, this category of nodes could perform key roles specially in integrating various types of nodes in the network to optimize topological properties of the network. Formation of rich club among the high degree nodes in the communities C8, C10 and C15 (Fig. 6A) indicating an increase in sensitivity of these hubs on being targeted hence take significant roles in regulating their respective modular functions, i.e., endocytosis, proteosome and DNA repair mechanisms (Table 3). These high degree hubs in these modules fall among the intermediate degree nodes in the primary PCa PPI network (Fig. 6B). Thus the varying pattern of rich club signatures across the hierarchy may possibly relate to the change in popularity of the proteins at different levels of organization, and hence hub-proteins preserve their level-dependent influence across the hierarchy10. Such behaviour in the PPI networks can be correlated to their weaker resilience and instability at sub-system/modular level which may be critical for certain functional modules due to malfunctions in the key regulator hub-proteins.
The Centrality measures are used to assess the importance of the nodes in information processing in the network. Betweenness centrality CB, closeness centrality CC, eigenvector centrality CE and subgraph centrality CS are the topological properties which can determine efficiency of signal transmission in a network25. The behaviour of these parameters exhibiting power law as a function of degree k with positive exponents, where the centralities tend to increase with higher degree nodes (Eq. 13) (Fig. 2), reveals the increase in efficiency of signal processing with higher degree nodes in PCa network, showing the importance of hubs in controlling the flow of information, thereby regulating and stabilizing the network organization. Therefore, hub-proteins have a significant influence in regulating the network although they do not control the whole network completely, thereby increasing the risk of being targeted in the network. Hence, the certain hubs might be acting as key regulators in PCa and the 19 predicted key regulators might serve as a backbone of the network.
Community detection of the network using Louvain modularity optimization method led to clustering of the primary PCa network up to the level of motifs (Fig. 3A). This clustering showed that modularity, Q, of the networks exhibit an increasing pattern with the topological levels with highest average modularity (Q = 0.5527) seen at the first hierarchical level of PCa network and lowest (Q = 0.0013) at level V, that is, at the motif level35,36. In complex PPI network the modules have biological meanings and gene ontology analyses have revealed enrichment of certain known functions and pathways in the modules37. The functional homogeneity in the modules of PCa network has been correlated to their mean clustering coefficients as modules with higher mean clustering coefficients have better chance to be associated with specific functions63,64. Moreover, in disease interactome, the disease modules which are unique modules representing the interaction between disease genes and their neighbourhood, overlaps with the topological modules derived from the network and functional modules associated with functions and are interrelated65. Primary PCa network is composed of 14 modules deduced from the community detection method with their mean clustering coefficients C(k) ~ 0.094−0.392 (Table 3). Among them modules C12 and C13 which were the largest had the highest mean clustering coefficients, C(k) = 0.392 & 0.218, respectively, showing a functional homogeneity in these modules. These modules have been analysed for their functional annotations with DAVID functional annotation tool34 which revealed association with different functions (Table 3). Modules C12 and C13 are represented with ribosomal biosynthesis and transcriptional regulation, respectively. This suggests a bigger role of RPs in PCa which is also evident from the representation of various RPs (RPL3, 5, 6, 11, 15, 19 etc.) as key regulators in PCa network. Transcriptional regulation is the most important level of gene regulation which is accomplished mainly through interaction of transcription factors along with their cofactors to the promoter regions of many genes. The tumour suppressor transcription factor (TF) p53 gene—the most mutated among all PCa—is one of the hub proteins represented in this community. Another important TF, c−MYC—an oncogene acting as a regulator of the cell cycle progression and cell division—is also represented in this community. Moreover, reports on regulations of p53 with the key ribosomal proteins (RPL5, RPL6, RPL11 etc.) and c−MYC key regulator CUL7 through p53 in several cancers suggest a critical association of transcriptional regulation in PCa.
Since the study of complex hierarchical networks is incomplete without understanding the modularity of sub-communities and the roles played by the nodes in the modules, our study applied the approach to characterize the nodes in PCa network through defining their within-module Z score Zi with their participation coefficients Pi30. In the PCa network many hub proteins act as modular kinless hubs or connector modular hubs maintaining the links within the modules as well as connecting other modules at the same level (Fig. 4A–C). This shows the importance of the hub-proteins in the hierarchical organization of the network exhibiting their involvement in establishing links among the nodes in each module as well as among the modules in the network which are associated with specific functions.
Conclusions
This paper introduces a new method for finding key regulators in prostate adenocarcinomas using biological networks constructed from high throughput datasets of Prostate cancer patients. The network theoretical approach used here placed equal emphasis on the hubs, motifs and modules of the network to identify key regulators/regulatory pathways, not restricting only to overrepresented motifs or hubs. It established a relationship between hubs, modules and motifs. The network used all genes associated with the disease, rather than using manually curated datasets. Highest degree hubs (k ≥ 65) were identified, out of which 19 were novel key regulators. The network, as evident from fractal nature in topological parameters, was a self-organized network and lacked a central control mechanism. Identification of novel key regulators in prostate cancer, particularly ribosomal proteins add new dimension to the understanding of PCa and its treatment and predicting key disease genes/pathways within network theoretical framework. This method can be used to any networks constructed from patients’ datasets which follow hierarchical topology.
Supplementary information
Acknowledgements
I.R.M. acknowledges Deshbandhu College, University of Delhi for the study leave to pursue doctoral research. M.Z.M. acknowledges financial assistance from Department of Health Research, Ministry of Health and Family Welfare, Government of India under Young Scientist scheme (Sanction File No. R.12014/01/2018-HR, FTS No. 3146887). S.A. acknowledges the Department of Biotechnology, Ministry of Science and Technology, Government of India for the bioinformatics facility at Jamia Hamdard under BTISNet, the Biotechnology Information System Network (Sanction no. BT/BI/25/062/2012(BIF). S.A. and O.K. acknowledge Indian Council of Medical Research for International fellowship to SA to visit Emory Winship Cancer Institute, Atlanta. R.K.B.S. acknowledges Jawaharlal Nehru University and UGC for UPE-II (Sanction no. 101) for financial assistance.
Author contributions
R.K.B.S., S.A., I.R.M. and M.Z.M. conceived the model and conducted numerical experiments. I.R.M. and M.Z.M. prepared figures of the numerical results. I.R.M., M.Z.M., O.K., S.A. and R.K.B.S. analysed and interpreted the simulation results and wrote the manuscript. R.K.B.S. and S.A. jointly supervised the study and approved the final draft.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Irengbam Rocky Mangangcha and Md Zubbair Malik.
Contributor Information
Shakir Ali, Email: sali@jamiahamdard.ac.in.
R. K. Brojen Singh, Email: brojen@jnu.ac.in.
Supplementary information
is available for this paper at 10.1038/s41598-019-52896-x.
References
- 1.Aaron LT, Franco OE, Hayward SW. Review of Prostate Anatomy and Embryology and the etiology of Benign Prostatic Hyperplasia. Urologic Clinics. 2016;43(3):279–288. doi: 10.1016/j.ucl.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Cancer Report 2014. World Health Organization. pp. Chapter 5.11. ISBN 9283204298.
- 3.Tobias, J. & Hochhauser, D. Cancer and its management (7th edition). (Wiley-Blackwell, West Sussex, UK 2015).
- 4.Edge, S. B. et al. American Joint Committee on Cancer (AJCC). Cancer Staging Manual. 7th ed. Springer, New York, USA (2009).
- 5.Mateo. J, Smith A, Ong M, de Bono JS. Novel drugs targeting the androgen receptor pathway in prostate cancer. Cancer metastasis reviews. 2014;33:567–579. doi: 10.1007/s10555-013-9472-2. [DOI] [PubMed] [Google Scholar]
- 6.Ritch CR, Cookson MS. Advances in the management of castration resistant prostate cancer. BMJ. 2016;355:i4405. doi: 10.1136/bmj.i4405. [DOI] [PubMed] [Google Scholar]
- 7.Erdogan B, Kostek O, Bekirhacioglu M. Enzalutamide in Prostate Cancer, A Review on Enzalutamide and cancer. EJMO. 2018;2(3):121–129. [Google Scholar]
- 8.Saini S. PSA and beyond: alternative prostate cancer biomarkers. Cellular Oncology. 2016;39(2):97–106. doi: 10.1007/s13402-016-0268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naji L, et al. Digital Rectal Examination for Prostate Cancer Screening in Primary Care: A Systematic Review and Meta-Analysis. Ann. Fam. Med. 2018;16(2):149–154. doi: 10.1370/afm.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali S, et al. Exploring novel key regulators in breast cancer network. Plos One. 2018;13(6):e0198525. doi: 10.1371/journal.pone.0198525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milo R, et al. Network Motifs: Simple Building Blocks of Complex Networks. Science. 2002;298(5594):824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 12.Alon U. Network motifs: theory and experimental approaches. Nature Reviews, Genetics. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 13.Dingerdissen HM, et al. BioMuta and BioXpress: mutation and expression knowledgebases for cancer biomarker discovery. Nucleic Acids Research. 2018;46(Database issue):D1128–D1136. doi: 10.1093/nar/gkx907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tate JG, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warde-Farley D, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment. 2008;10:P10008. doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 18.Yeger-Lotem E, et al. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. PNAS. 2004;101(16):5934–5939. doi: 10.1073/pnas.0306752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assenov Y, Ramírez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y, Li M, Wang J, Pan Y, Wu F-X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi: 10.1016/j.biosystems.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Ravasz, E., & Barabási, A.-L. Hierarchical organization in complex networks. Physical Review E, 67(2) (2003). [DOI] [PubMed]
- 22.Pastor-Satorras R, Vzquez A, Vespignani A. Dynamical and correlation properties of the Internet. Physical review letters. 2001;87(25):258701. doi: 10.1103/PhysRevLett.87.258701. [DOI] [PubMed] [Google Scholar]
- 23.Barrat A, Barthelemy M, Pastor-Satorras R, Vespignani A. The architecture of complex weighted networks. PNAS, USA. 2004;101(11):3747–3752. doi: 10.1073/pnas.0400087101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman MEJ, Girvan M. Finding and evaluating community structure in networks. Physical Review E. 2004;69(2):026113. doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 25.Estrada E, Rodríguez-Velázquez JA. Subgraph centrality in complex networks. Physical Review E. 2005;71(5):056103–1-9. doi: 10.1103/PhysRevE.71.056103. [DOI] [PubMed] [Google Scholar]
- 26.Canright G, Engo-Monsen K. Roles in networks. Science of Computer Programming. 2004;53(2):195–214. doi: 10.1016/j.scico.2003.12.008. [DOI] [Google Scholar]
- 27.Borgatti SP, Everett MG. A graph-theoretic perspective on centrality. Social networks. 2006;28(4):466–484. doi: 10.1016/j.socnet.2005.11.005. [DOI] [Google Scholar]
- 28.Brandes U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001;25:163–177. doi: 10.1080/0022250X.2001.9990249. [DOI] [Google Scholar]
- 29.Canright GS, Engo-Monsen K. Spreading on networks: a topographic view. Complexus. 2006;3(1–3):131–146. doi: 10.1159/000094195. [DOI] [Google Scholar]
- 30.Guimerà R, Nunes Amaral LA. Functional cartography of complex metabolic networks. Nature. 2005;433(7028):895–900. doi: 10.1038/nature03288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 32.Colizza V, Flammini A, Serrano MA, Vespignani A. Detecting rich-club ordering in complex networks. Nature Physics. 2006;2(2):110–115. doi: 10.1038/nphys209. [DOI] [Google Scholar]
- 33.Rubinov M, Sporns O. Complex network measures of brain connectivity: Uses and interpretations. NeuroImage. 2010;52(3):1059–1069. doi: 10.1016/j.neuroimage.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 35.Newman M. E., J. Modularity and community structure in networks. PNAS. 2006;103(23):8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman MEJ. The Structure and Function of Complex Networks. SIAM Review. 2003;45(2):167–256. doi: 10.1137/S003614450342480. [DOI] [Google Scholar]
- 37.Dong J, Horvath S. Understanding network concepts in modules. BMC Systems Biology. 2007;1(1):24. doi: 10.1186/1752-0509-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou S, Mondragon RJ. The Rich-Club Phenomenon in the Internet Topology. IEEE Communications Letters. 2004;8(3):180–182. doi: 10.1109/LCOMM.2004.823426. [DOI] [Google Scholar]
- 39.Dolezal JM, Dash AP, Prochownik EV. Diagnostic and prognostic implications of ribosomal protein transcript expression patterns in human cancers. BMC Cancer. 2018;18:275. doi: 10.1186/s12885-018-4178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou X, Liao W-J, Liao J-M, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. Journal of Molecular Cell Biology. 2015;7(2):92–104. doi: 10.1093/jmcb/mjv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertram K, et al. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell. 2017;170(4):701–713.e11. doi: 10.1016/j.cell.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823(3):624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Müller L, Schaupp A, Walerych D, Wegele H, Buchner J. Hsp90 Regulates the Activity of Wild Type p53 under Physiological and Elevated Temperatures. Journal of Biological Chemistry. 2004;279(47):48846–48854. doi: 10.1074/jbc.M407687200. [DOI] [PubMed] [Google Scholar]
- 44.Peng Y, Chen L, Li C, Lu W, Chen J. Inhibition of MDM2 by hsp90 Contributes to Mutant p53 Stabilization. Journal of Biological Chemistry. 2001;276(44):40583–40590. doi: 10.1074/jbc.M102817200. [DOI] [PubMed] [Google Scholar]
- 45.Ratajczak T, Cluning C, Ward BK. Steroid Receptor-Associated Immunophilins: A Gateway to Steroid Signalling. Clin. Biochem. Rev. 2015;36(2):31–52. [PMC free article] [PubMed] [Google Scholar]
- 46.Shao J, Wang L, Zhong C, Qi R, Li Y. AHSA1 regulates proliferation, apoptosis, migration, and invasion of osteosarcoma. Biomedicine & Pharmacotherapy. 2016;77:45–51. doi: 10.1016/j.biopha.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 47.Kim SS, et al. CUL7 Is a Novel Antiapoptotic Oncogene. Cancer Research. 2007;67(20):9616–9622. doi: 10.1158/0008-5472.CAN-07-0644. [DOI] [PubMed] [Google Scholar]
- 48.Tian P, Liu D, Sun L, Sun H. Cullin7 promotes epithelial-mesenchymal transition of esophageal carcinoma via the ERK-SNAI2 signaling pathway. Molecular Medicine Reports. 2018;17(4):5362–5367. doi: 10.3892/mmr.2018.8503. [DOI] [PubMed] [Google Scholar]
- 49.Qiu N, et al. Cullin7 is a predictor of poor prognosis in breast cancer patients and is involved in the proliferation and invasion of breast cancer cells by regulating the cell cycle and microtubule stability. Oncology Reports. 2017;39:603–610. doi: 10.3892/or.2017.6106. [DOI] [PubMed] [Google Scholar]
- 50.Lee ASY, Kranzusch PJ, Doudna JA, Cate JHD. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature. 2016;536(7614):96–99. doi: 10.1038/nature18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schipany K, Rosner M, Ionce L, Hengstschläger M, Kovacic B. eIF3 controls cell size independently of S6K1-activity. Oncotarget. 2015;6:24361–24375. doi: 10.18632/oncotarget.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxidants & Redox Signaling. 2009;11(9):2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cerezo M, Rocchi S. New anti-cancer molecules targeting HSPA5/BIP to induce endoplasmic reticulum stress, autophagy and apoptosis. Autophagy. 2016;13(1):216–217. doi: 10.1080/15548627.2016.1246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Beus E, Brockenbrough JS, Hong B, Aris JP. Yeast NOP2 encodes an essential nucleolar protein with homology to a human proliferation marker. J. Cell Biol. 1994;127:1799–1813. doi: 10.1083/jcb.127.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perlaky L, et al. Increased growth of NIH/3T3 cells by transfection with human p120 complementary DNA and inhibition by a p120 antisense construct. Cancer Research. 1992;52(2):428–436. [PubMed] [Google Scholar]
- 56.Fonagy A, et al. Cell cycle regulated expression of nucleolar antigen P120 in normal and transformed human fibroblasts. Journal of Cellular Physiology. 1993;154(1):16–27. doi: 10.1002/jcp.1041540104. [DOI] [PubMed] [Google Scholar]
- 57.Lim JH, et al. RANBP2-ALK fusion combined with monosomy 7 in acute myelomonocytic leukemia. Cancer. Genetics. 2014;207(1–2):40–45. doi: 10.1016/j.cancergen.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Eifler K, Vertegaal ACO. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends in Biochemical Sciences. 2015;40(12):779–793. doi: 10.1016/j.tibs.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Marzo AM, et al. Pathological and molecular mechanisms of prostate carcinogenesis: Implications for diagnosis, detection, prevention, and treatment. Journal of Cellular Biochemistry. 2004;91(3):459–477. doi: 10.1002/jcb.10747. [DOI] [PubMed] [Google Scholar]
- 60.Lallous, N. et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biology, 17(1) (2016). [DOI] [PMC free article] [PubMed]
- 61.Salas TR, et al. Glycogen Synthase Kinase-3β Is Involved in the Phosphorylation and Suppression of Androgen Receptor Activity. Journal of Biological Chemistry. 2004;279(18):19191–19200. doi: 10.1074/jbc.M309560200. [DOI] [PubMed] [Google Scholar]
- 62.Lin H-K, Hu Y-C, Lee DK, Chang C. Regulation of Androgen Receptor Signaling by PTEN (Phosphatase and Tensin Homolog Deleted on Chromosome 10) Tumor Suppressor through Distinct Mechanisms in Prostate Cancer Cells. Molecular Endocrinology. 2004;18(10):2409–2423. doi: 10.1210/me.2004-0117. [DOI] [PubMed] [Google Scholar]
- 63.Colizza V, Flammini A, Maritan A, Vespignani A. Characterization and modeling of protein–protein interaction networks. Physica A: Statistical Mechanics and Its Applications. 2005;352(1):1–27. doi: 10.1016/j.physa.2004.12.030. [DOI] [Google Scholar]
- 64.Lewis AC, Jones NS, Porter MA, Charlotte DM. The function of communities in protein interaction networks at multiple scales. BMC Systems Biology. 2010;4(1):100. doi: 10.1186/1752-0509-4-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nature Reviews Genetics. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014;588:1–9. doi: 10.1016/j.febslet.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 67.Meng X, et al. RPL23 Links Oncogenic RAS Signaling to p53-Mediated Tumor Suppression. Cancer Research. 2016;76(17):5030–5039. doi: 10.1158/0008-5472.CAN-15-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dai M-S, Arnold H, Sun X-X, Sears R, Lu H. Inhibition of c-Myc activity by ribosomal protein L11. The EMBO Journal. 2007;26(14):3332–3345. doi: 10.1038/sj.emboj.7601776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gou Y, et al. Ribosomal protein L6 promotes growth and cell cycle progression through upregulating cyclin E in gastric cancer cells. Biochemical and Biophysical Research Communications. 2010;393:788–793. doi: 10.1016/j.bbrc.2010.02.083. [DOI] [PubMed] [Google Scholar]
- 70.Chen R, et al. Proteins associated with pancreatic cancer survival in patients with resectable pancreatic ductal adenocarcinoma. Laboratory Investigation. 2014;95(1):43–55. doi: 10.1038/labinvest.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang YZ, et al. Discovery and validation of prognostic markers in gastric cancer by genome-wide expression profiling. World J Gastroenterol. 2011;17:1710–1717. doi: 10.3748/wjg.v17.i13.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mao-De L, Jing X. Ribosomal Proteins and Colorectal Cancer. Current Genomics. 2007;8:43–49. doi: 10.2174/138920207780076938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Callari M, et al. Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res Treat. 2011;127:601–10. doi: 10.1007/s10549-010-1015-8. [DOI] [PubMed] [Google Scholar]
- 74.Kato Y, et al. Gene expression pattern in oral cancer cervical lymph node metastasis. Oncology Reports. 2006;16:1009–1014. [PubMed] [Google Scholar]
- 75.Teller A, et al. Dysregulation of apoptotic signaling pathways by interaction of RPLP0 and cathepsin X/Z in gastric cancer. Pathology - Research and Practice. 2015;211(1):62–70. doi: 10.1016/j.prp.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 76.Artero-Castro A, et al. Expression of the ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors. Human Pathology. 2011;42(2):194–203. doi: 10.1016/j.humpath.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 77.Zhang S-C, et al. RPSA Gene Mutants Associated with Risk of Colorectal Cancer among the Chinese Population. Asian Pac J Cancer Prev. 2013;14(12):7127–7131. doi: 10.7314/APJCP.2013.14.12.7127. [DOI] [PubMed] [Google Scholar]
- 78.Jiang G, et al. A novel biomarker C6orf106 promotes the malignant progression of breast cancer. Tumor Biology. 2015;36(10):7881–7889. doi: 10.1007/s13277-015-3500-5. [DOI] [PubMed] [Google Scholar]
- 79.Yong WH, et al. Ribosomal Proteins RPS11 and RPS20, Two Stress-Response Markers of Glioblastoma Stem Cells, Are Novel Predictors of Poor Prognosis in Glioblastoma Patients. PLOS ONE. 2015;10(10):e0141334. doi: 10.1371/journal.pone.0141334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sethi MK, et al. Quantitative proteomic analysis of paired colorectal cancer and non-tumorigenic tissues reveals signature proteins and perturbed pathways involved in CRC progression and metastasis. Journal of Proteomics. 2015;126:54–67. doi: 10.1016/j.jprot.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 81.Bee A, et al. Ribosomal Protein L19 Is a Prognostic Marker for Human Prostate Cancer. Clinical Cancer Research. 2006;12(7):2061–2065. doi: 10.1158/1078-0432.CCR-05-2445. [DOI] [PubMed] [Google Scholar]
- 82.Russo A, et al. Regulatory role of rpL3 in cell response to nucleolar stress induced by Act D in tumor cells lacking functional p53. Cell Cycle. 2015;15(1):41–51. doi: 10.1080/15384101.2015.1120926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bee A, et al. siRNA Knockdown of Ribosomal Protein Gene RPL19 Abrogates the Aggressive Phenotype of Human Prostate Cancer. Plos One. 2011;6(7):e22672. doi: 10.1371/journal.pone.0022672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.