

Abstract
Cardiovascular disease remains the leading cause of death for both men and women. The observation that premenopausal women are protected from cardiovascular disease relative to age-matched men, and that this protection is lost with menopause, has led to extensive study of the role of sex steroid hormones in the pathogenesis of cardiovascular disease. However, the molecular basis for sex differences in cardiovascular disease is still not fully understood, limiting the ability to tailor therapies to male and female patients. Therefore, there is a growing need to investigate molecular pathways outside of traditional sex hormone signaling to fully understand sex differences in cardiovascular disease. Emerging evidence points to the mineralocorticoid receptor (MR), a steroid hormone receptor activated by the adrenal hormone aldosterone, as one such mediator of cardiovascular disease risk, potentially serving as a sex-dependent link between cardiovascular risk factors and disease. Enhanced activation of the MR by aldosterone is associated with increased risk of cardiovascular disease. Emerging evidence implicates the MR specifically within the endothelial cells lining the blood vessels in mediating some of the sex differences observed in cardiovascular pathology. This review summarizes the available clinical and preclinical literature concerning the role of the MR in the pathophysiology of endothelial dysfunction, hypertension, atherosclerosis, and heart failure, with a special emphasis on sex differences in the role of endothelial-specific MR in these pathologies. The available data regarding the molecular mechanisms by which endothelial-specific MR may contribute to sex differences in cardiovascular disease is also summarized. A paradigm emerges from synthesis of the literature in which endothelial-specific MR regulates vascular function in a sex-dependent manner in response to cardiovascular risk factors to contribute to disease. Limitations in this field include the relative paucity of women in clinical trials and, until recently, the nearly exclusive use of male animals in preclinical investigations. Enhanced understanding of the sex-specific roles of endothelial MR could lead to novel mechanistic insights underlying sex differences in cardiovascular disease incidence and outcomes and could identify additional therapeutic targets to effectively treat cardiovascular disease in men and women.
Keywords: endothelial cell, mineralocorticoid receptor, cardiovascular disease, sex differences, aldosterone, estrogen
I. INTRODUCTION
1. Gaps in Knowledge of the Mechanisms Underlying Sex Differences in Cardiovascular Disease
Cardiovascular pathologies such as hypertension, atherosclerosis, and heart failure lead to substantial morbidity, and heart disease remains the leading cause of death in both men and women (Xu et al. 2018). While premenopausal women are protected from cardiovascular disease relative to age-matched men, this protection is lost with menopause, implicating sex hormones in the pathogenesis of cardiovascular disease. As such, the role of sex hormones in the cardiovascular system, particularly signaling through estrogen receptor (ER) isoforms a and β, has been extensively studied (Arnold et al. 2017). However, due to the complex nature of sex steroid signaling pathways, the molecular basis for sex differences in cardiovascular disease is still not fully understood, limiting the ability to tailor therapies to male and female patients.
Additionally, common cardiovascular risk factors such as metabolic syndrome and obesity abolish the protection from cardiovascular disease in women even prior to menopause (Barrett-Connor et al. 1991; Sowers 1998; Wilson et al. 2002), highlighting the need to investigate molecular pathways outside of traditional sex hormone signaling to fully understand sex differences in cardiovascular disease. However, this area is currently understudied in both the clinical and preclinical literature. Generally, the patient cohorts in cardiovascular disease clinical trials are heavily weighted towards male patients, with women constituting only a minority of study participants. Further, most preclinical studies in the cardiovascular field focus on male animals, with very few directly comparing the sexes.
2. The Mineralocorticoid Receptor: Regulator of Blood Pressure, Mediator of Cardiovascular Disease
The mineralocorticoid receptor (MR) was first described to contribute to blood pressure control by regulating the transcription and expression of sodium transport proteins in the distal nephron (Arriza et al. 1987). Emerging evidence now points to the MR as a broader mediator of cardiovascular disease risk, potentially serving as a sex-dependent link between cardiovascular risk factors and disease (Davel et al. 2018a). The MR is a transcription factor that can be activated either by glucocorticoids such as cortisol (corticosterone in rodents), which circulate at high levels, or by its more specific but less abundant ligand aldosterone (Aldo) (Funder 2010). Individual tissues maintain specificity of the MR for Aldo by expression of the 11β-hydroxysteroid dehydrogenase 2 (11βHSD2) enzyme, which converts MR-binding glucocorticoids to metabolites that cannot bind to the MR and thus affords Aldo specificity to the MR (Naray-Fejes-Toth et al. 1998).
Independently of the relationship between the MR and blood pressure, elevated serum Aldo levels are associated with a substantially increased risk of stroke, myocardial infarction (MI), and sudden cardiac death (Ivanes et al. 2012; Milliez et al. 2005). Conversely, inhibition of the MR in large randomized clinical trials such as the RALES, EPHESUS, and EMPHASIS-HF results in significant reductions in mortality in heart failure patients. This decrease in mortality is associated with only modest changes in blood pressure along with trends towards decreased MI risk when secondary endpoints are examined (Zannad et al. 2011; Pitt et al. 1999; Pitt et al. 2003b). As such, substantial investigation in the preclinical literature has focused on understanding the role of MR signaling in non-renal cells in the development of cardiovascular disease, which has the potential to nominate additional therapeutic targets related to MR signaling.
3. Vascular Cell-Specific Mineralocorticoid Receptors Contribute to Cardiovascular Disease
The vascular wall is made up of three parts: an inner layer of endothelial cells (ECs) that forms the interface between circulating blood and underlying tissues; a medial layer of smooth muscle cells (SMCs) which contract or relax to control vessel diameter thereby regulating blood flow to downstream organs; and an outer layer of adventitial fibroblasts and adipose cells that provide structural support and regulatory mediators to the inner two layers. The inner EC layer contributes to vasodilation by activating ion channels and releasing paracrine factors to stimulate dilation of the underlying SMCs, including the anti-inflammatory, antioxidant gas nitric oxide (NO) (Vanhoutte et al. 2016). The endothelium also regulates inflammatory cell recruitment by modulating expression of endothelial-leukocyte adhesion molecules and by the generation of reactive oxygen species (ROS) to produce oxidative stress.
The MR is expressed in vascular SMCs and ECs. In its genomic role as a transcription factor, the MR within ECs (EC-MR) regulates genes that contribute to critical EC functions, including expression of inflammatory mediators and regulators of endothelial sodium handling and junctional integrity (Kusche-Vihrog et al. 2010; Moss and Jaffe 2015; Kirsch et al. 2013). EC-MR also contributes to NO bioavailability and oxidative stress via rapid, “non-genomic” signaling outside of its traditional, gene-transcription role (Wehling 2018). Multiple studies have demonstrated that ECs express 11βHSD2 that is capable of inactivating cortisol (Christy et al. 2003; Caprio et al. 2008; Liu et al. 2009), thus it is likely that Aldo is the relevant ligand for EC-MR. However, some studies show low or variable 11βHSD2 expression in ECs that may depend on cell conditions (Gong et al. 2008), raising the possibility that glucocorticoids may activate EC-MR under certain conditions. Regardless of the ligand, however, studies in mice with EC-specific MR deletion reveal that EC-MR contributes to the cardiovascular pathology that develops in the setting of risk factors such as obesity, diabetes, and hyperlipidemia (Davel et al. 2017).
In addition to ECs, functional MR is expressed in human vascular SMCs (Jaffe and Mendelsohn 2005), where it has been shown to contribute to vasoconstriction and blood pressure regulation (McCurley et al. 2012; DuPont et al. 2016; Galmiche et al. 2014; Amador et al. 2016) and to vascular remodeling in response to injury, aging, and hypertension (Pruthi et al. 2014; Galmiche et al. 2014; Kim et al. 2018) in vivo in males. In vitro, SMC-MR may also contribute to SMC calcification (Jaffe et al. 2007) and cytokine production (McGraw et al. 2013), although it was recently shown not to contribute to the pathogenesis of atherosclerosis in male mice (Moss et al. 2018). The MR also contributes to inflammatory phenotypes in a number of leukocyte cell types, such as T cells, neutrophils, and monocytes (Bene et al. 2014). In vitro, macrophage MR contributes to the production of ROS and inflammatory cytokines and promotes pro-inflammatory “M1-like” macrophage polarization (Usher et al. 2010; Bene et al. 2014) and contributes to plaque development in atherosclerosis models (Shen et al. 2017). Recent in vivo studies further implicate T cell MR in the pathogenesis of hypertension (Sun et al. 2017) and pressure overload-induced cardiac dysfunction (Li et al. 2017a). Although this review focuses on the role of the MR specifically within ECs in cardiovascular disease, additional investigations of the role of the MR in other cell types will certainly provide substantial insight into the mechanisms driving cardiovascular disease.
4. Endothelial Cell Mineralocorticoid Receptors in Cardiovascular Disease: Is There Effect Modification by Sex?
Substantial recent exploration reveals a role for EC-specific MR in endothelial dysfunction, hypertension, atherosclerosis, and heart failure. However, the vast majority of preclinical investigations into the function of EC-MR have been conducted only in male animals, and those that do use female animals do not typically compare them to male counterparts to examine sex differences. However, rare publications in the existing literature that do directly compare the role of EC-MR between males and females reveal striking sex differences in the role of this receptor in the vascular endothelium. Further, critical analysis of studies performed in each sex separately may yield insight into potential sex-specific mechanisms of EC-MR function in the cardiovascular system.
Here we review the recent literature exploring the role of the MR in mediating sex differences in 1) endothelial dysfunction, 2) hypertension, 3) atherosclerosis, and 4) heart failure, with a focus on the MR in the vascular endothelium. The first part of the review focuses on the clinical literature supporting a sex-specific role for the MR in each cardiovascular disorder. The second part examines the preclinical literature specifically assessing the role of EC-MR in animal models of each disease, commenting on effect modification by sex where there are available data. Finally, the third part of this review summarizes the data regarding the molecular mechanisms that may mediate a sex-specific role for EC-MR in cardiovascular disease. The available data supports that EC-MR may be a key player in determining sex differences in cardiovascular disease and reveals many areas warranting further study.
II. CLINICAL DATA: CONTRIBUTION OF THE MR TO CARDIOVASCULAR DISEASE IN MEN AND WOMEN
Activation of the MR in the setting of cardiovascular stress or risk factors appears to contribute to the development of cardiovascular diseases. However, whether there is a difference in this role by sex that might contribute to sex differences in cardiovascular disease risk and outcomes is just beginning to be elucidated. In this section, we review the existing clinical literature on the contribution of the MR to 1) endothelial dysfunction, 2) hypertension, 3) atherosclerosis, and 4) heart failure, with a focus on differentiating the role of the MR between men and women. A summary of the clinical studies using MR antagonists cited in this section can be found in Table 1.
Table 1:
Clinical Trials of MR Antagonism in Cardiovascular Pathology Cited in This Review
| Study Population | MRA Used | MRA Dosing (Duration) | Study Type | N: Control/MRA | Sex: Men/Women | Outcome | MRA Improved, Worsened, or No Effect on Outcome | Reference |
|---|---|---|---|---|---|---|---|---|
| ENDOTHELIAL DYSFUNCTION | ||||||||
| Healthy older adults | Epl | 100 mg/day (2 days) | RCT, double-blind, crossover | 22/22 | 8/14 | Endothelial function | Worsened | Hwang et al. 2016 |
| Obesity | Epl | 100 mg/day (1 month) | RCT, double-blind, crossover | 22/22 | 10/12 | Endothelial function | Improved† | Hwang et al. 2013b |
| Obesity | Spiro | 50 mg/day (6 weeks) | RCT, double-blind | 16/16 | 10/22 | Endothelial function | No Effect | Garg et al. 2014 |
| Metabolic Syndrome | Epl | 100 mg/day (1 month) | RCT, double-blind, crossover | 8/8 | 4/4 | Endothelial function | No Effect | Hwang et al. 2015 |
| Type 2 Diabetes | Spiro | 25 mg/day (6 months) | RCT, double-blind | 23/17 | 27/13 | Endothelial function | Improved | Garg et al. 2015 |
| Hypertension | Epl | 50 mg/day (11 months) | Randomized, double-blind, pre-post | 40/20* | 45/15 | Endothelial function | Improved | Fujimura et al. 2012 |
| HFrEF | Spiro | 50 mg/day (1 month) | RCT, double-blind, crossover | 10/10 | 10/0 | Endothelial function | Improved | Farquharson and Struthers 2000 |
| HFrEF | Spiro | 12.5-50 mg/day (3 months) | RCT, double-blind, crossover | 43/43 | 35/8 | Endothelial function | Improved | Macdonald et al. 2004 |
| Polycystic Ovary Syndrome | Spiro | 100 mg/day# (6 months) | Pre-post | 0/30 | 0/30 | Endothelial function | Improved | Studen et al. 2011 |
| Rheumatoid Arthritis | Spiro | 2 mg/kg/day (12 weeks) | Pre-post | 0/24 | 2/22 | Endothelial function | Improved | Syngle et al. 2009 |
| HYPERTENSION | ||||||||
| Class I and II Hypertension | Epl | 50-200 mg/day (12 months) | Randomized, double-blind, pre-post | 246/253** | 276/223 | Blood pressure | Improved | Williams et al. 2004 |
| Hypertension and LV Hypertrophy | Epl | 200 mg/day (9 months) | Randomized, double-blind, pre-post | 54/50** | 95/58 | Blood pressure | Improved | Pitt et al. 2003a (4E) |
| Resistant Hypertension | Spiro or Epl | 25 mg/day Spiro or 50 mg/day Epl (3 months) | Retrospective cohort | 0/46 | 30/16 | Blood pressure | Improved‡ | Khosla et al. 2009 |
| ATHEROSCLEROSIS | ||||||||
| Hemodialysis | Spiro | 50 mg 3x/week (2 years) | RCT, double-blind | 23/30 | 34/19 | Intima-media thickness | Improved | Vukusich et al. 2010 |
| Primary Aldosteronism | Epl | 50 mg/day (12 months) | Pre-post | 0/14 | 2/12 | Intima-media thickness | Improved | Matsuda et al. 2016 |
| HEART FAILURE | ||||||||
| LV dysfunction after MI | Epl | 25-50 mg/day (2 years) | RCT, double-blind | 3319/3313 | 4714/1918 | Mortality, hospitalization | Improved | Pitt et al. 2003b (EPHESUS) |
| HFrEF | Spiro | 25-50 mg/day (2 years) | RCT, double-blind | 841/822 | 1217/446 | Mortality, hospitalization | Improved | Pitt et al. 1999 (RALES) |
| HFrEF | Epl | 25-50 mg/day (2 years) | RCT, double-blind | 1373/1364 | 2127/610 | Mortality, hospitalization | Improved | Zannad et al. 2011 (EMPHASISHF) |
| HFpEF | Spiro | 25 mg/day (4 months) | Open-label, uncontrolled | 0/11 | 0/11 | Exercise capacity | Improved | Daniel et al. 2009 |
| HFpEF | Spiro | 25 mg/day (12 months) | RCT, double-blind | 209/213 | 201/221 | LV structure and function | Improved | Edelmann et al. 2013 (Aldo-DHF) |
| HFpEF | Spiro | 15-45 mg/day (3 years) | RCT, double-blind | 1723/1722 | 1670/1775 | Hospitalization | Improved | Pitt et al. 2014 (TOPCAT) |
| HFpEF | Spiro | 15-45 mg/day (3 years) | RCT, double-blind | 1723/1722 | 1670/1775 | Mortality | No Effect | Pitt et al. 2014 (TOPCAT) |
| HFpEF | Spiro | 25 mg/day (6 months) | RCT, double-blind | 67/64 | 21/110 | Exercise capacity | Improved | Kosmala et al. 2016 |
| HFpEF | Spiro | 25 mg/day (9 months) | RCT, double-blind | 38/42 | 16/64 | Exercise capacity, quality of life | No Effect | Upadhya et al. 2017a |
| VASCULAR STIFFNESS | ||||||||
| Healthy subjects aged 55-79 | Epl | 100 mg/day (1 month) | RCT, double-blind, crossover | 23/23 | 10/13 | Aortic stiffness | Improved | Hwang et al. 2013a |
| Dilated Cardiomyopathy | Spiro | 25-100 mg/day (6 months) | Randomized, open-label, blinded endpoint | 51/51 | 28/74 | Aortic stiffness | Improved | Vizzardi et al. 2015 |
| HFpEF | Spiro | 25 mg/day (9 months) | RCT, double-blind | 38/42 | 16/64 | Aortic stiffness | No Effect | Upadhya et al. 2017a |
Controls were 40 mg/day nifedipine (n=20) or 100 mg/day losartan (n=20).
Control was 10-40 mg/day enalapril.
Treatment paradigm: 21 days on, 7 days off.
Effect in obese>lean, improvement also correlated with fasting glucose levels.
Effect in women>men, obese>lean.
MR=mineralocorticoid receptor; MRA=MR antagonist; Spiro=Spironolactone; Epl=Eplerenone; RCT=randomized controlled trial; HFrEF=heart failure with reduced ejection fraction; HFpEF=heart failure with preserved ejection fraction; LV=left ventricle; MI=myocardial infarction.
1. Endothelial Dysfunction
A. Epidemiology
Endothelial dysfunction is marked by impaired endothelium-dependent vasodilation, reduced NO biosynthesis, and increased vascular inflammation and is the earliest measurable defect in the pathogenesis of vascular diseases. A sub-analysis of the offspring of Framingham Heart Study participants found that female sex significantly correlated with defects in endothelial-dependent dilation, while male sex did not (Hamburg et al. 2011). These epidemiologic data suggest sex-specific mechanisms of endothelial dysfunction and support a potential a role for EC-MR, as mineralocorticoid signaling in the endothelium appears to play a substantial role in the development of endothelial dysfunction in the setting of cardiometabolic risk factors (Davel et al. 2017).
B. The MR May Contribute to Endothelial Dysfunction in the Setting of Cardiovascular Risk Factors
Data supports that under baseline conditions without cardiovascular risk factors, the MR does not play a substantial role in vascular dysfunction (reviewed in Biwer et al. 2019). Indeed, chronic MR antagonism had no beneficial effect on endothelial function in a group of younger (age 40’s) obese subjects without associated diabetes or other cardiac risk factors (Garg et al. 2014), in older (age 60’s) otherwise healthy adults acutely administered eplerenone (Hwang et al. 2016), or in a study of 8 older adults with metabolic syndrome (Hwang et al. 2015).
However, several clinical studies do support a role for the MR in the development of endothelial dysfunction when multiple or severe cardiovascular risk factors are present. This is illustrated by one study which found that MR inhibition had no effect on endothelial function in lean older adults but improved endothelial function in older adults with obesity and/or impaired glucose tolerance (Hwang et al. 2013b). Spironolactone improved NO bioactivity and brachial artery endothelial function in two studies of patients with heart failure (Farquharson and Struthers 2000; Macdonald et al. 2004). MR inhibition likewise improved coronary flow reserve, a measure of coronary vessel endothelial function, in type 2 diabetics (Garg et al. 2015) and improved brachial artery endothelial function in patients with hypertension (Fujimura et al. 2012). In another study, acute Aldo administration triggered microvascular endothelial dysfunction in normotensive African Americans; conversely, MR inhibition with spironolactone improved resistance vessel endothelial function in ex vivo vessels from hypertensive African Americans regardless of gender (Mohandas et al. 2015).
While it seems that premenopausal women are protected from a wide variety of cardiovascular pathologies relative to age-matched men (Benjamin et al. 2018), studies point to a role for the MR in endothelial dysfunction even prior to menopause in women with enhanced cardiovascular risk. For example, young women with polycystic ovarian syndrome, which is characterized by increased androgen synthesis along with other cardiometabolic risk factors such as obesity, diabetes, and hypertension (Marciniak et al. 2016), have an increased risk of cardiovascular disease. These women also develop endothelial dysfunction (Paradisi et al. 2001), to which the MR may contribute. Aldo levels are elevated in women with polycystic ovarian syndrome compared to weight-matched controls (Cascella et al. 2006), and prolonged treatment with spironolactone was shown to improve endothelial function in a cohort of polycystic ovarian syndrome patients (Studen et al. 2011). It is important to note that spironolactone also inhibits the androgen receptor (AR) (Yang and Young 2016), thus it is difficult to distinguish whether the protective effects of spironolactone in this latter study are due to is anti-MR or its anti-androgen effects. However, in the context of rheumatoid arthritis, an autoimmune condition that also confers greater cardiovascular disease risk to women even prior to menopause, spironolactone treatment also significantly improved endothelial function and reduced inflammatory indicators in this predominantly-female cohort (Syngle et al. 2009). These data suggest that the MR may contribute to endothelial dysfunction even in premenopausal women if additional cardiovascular risk factors are present (see Table 1).
2. Hypertension
A. Epidemiology
High blood pressure affects 30% of American adults (Fryar et al. 2017). Hypertension increases the risk of MI and stroke, and prolonged exposure to hypertension can lead to heart and kidney failure. Many pharmacotherapies exist to combat hypertension. This includes MR inhibitors, which have been demonstrated to be effective antihypertensive medications in clinical trials (Pitt et al. 2003a; Williams et al. 2004). Despite this, nearly half of hypertensive patients are inadequately controlled with current antihypertensive drugs (Fryar et al. 2017). Emerging data support that therapy-resistant hypertension is more likely to be dependent on MR signaling than therapy-responsive subtypes (Yugar-Toledo et al. 2017; Dudenbostel and Calhoun 2017). Indeed, MR antagonism with the competitive inhibitors spironolactone or eplerenone effectively reduces blood pressure in patients with therapy-resistant hypertension (Glicklich and Frishman 2015; Fernet et al. 2018; Rossignol et al. 2018).
The prevalence of hypertension in premenopausal women is lower than that of age-matched men, although hypertension still affects nearly 28% of American women. However, after menopause this is reversed, with women 60 years of age and older experiencing significantly higher rates of hypertension than age-matched men. Further, an increase in therapy-resistant hypertension in postmenopausal women (Fryar et al. 2017) suggests that the mechanisms driving hypertension in women may change with age and estrogen status, potentially becoming more dependent on MR signaling after menopause.
B. The MR Contributes to Hypertension in Women
The MR has long been known to regulate blood pressure via its role in controlling renal sodium balance, and data from human studies supports a role for the MR in blood pressure regulation specifically in women. In a recent study of over 1,500 individuals from the HyperPATH consortium, women had a significantly greater rise in blood pressure compared to men in response to stimuli that increase Aldo secretion including salt restriction (Jurgens and Graudal 2004) and angiotensin-ll infusion (Shukri et al. 2018). Similarly, in a study of obese patients with chronic kidney disease, female subjects experienced a greater decrease in blood pressure with MR antagonism than males (Khosla et al. 2009). A gene variant of ERβ was also shown to associate with salt sensitivity of blood pressure specifically in premenopausal women (Manosroi et al. 2017), suggesting the potential for cross-regulation of blood pressure by MR and estrogen signaling.
3. Atherosclerosis
A. Epidemiology
Atherosclerosis is increasingly common, and downstream consequences of atherosclerosis, including MI and ischemic stroke, account for a majority of deaths worldwide (Barquera et al. 2015) Atherosclerosis is a diffuse vascular pathology in which inflamed, lipid-laden plaques accumulate in the vascular wall. Under conditions of excess inflammation, these plaques can rupture and thrombose, occluding the vessel and preventing blood flow to downstream tissues. The clinical consequence of this occlusion is ischemia resulting in damage to the brain in stroke, to the heart in MI, and to the skeletal muscle in critical limb ischemia.
It is quite clear that sex differences exist in the incidence of cardiovascular ischemic events in humans, with premenopausal women experiencing significantly fewer of these events than age-matched men (Benjamin et al. 2018). The actions of both estrogen and testosterone have been demonstrated to be beneficial in atherosclerosis in clinical and preclinical models (Boese et al. 2017). However, it is not clear whether women’s protection from cardiovascular ischemic events prior to menopause is due to less plaque burden or fewer plaque rupture events. One study found carotid intima-media thickness, a clinical index of plaque size, to be only slightly higher in young men than women, while coronary calcium score, which correlates with plaque inflammation and susceptibility to rupture, is substantially higher in men than in women (Benjamin et al. 2018).
B. MR Activation Contributes to Atherosclerosis in Men and Women
Clinical studies of the role of the MR in atherosclerosis in human patients tend to combine men and women. However, the available clinical data suggests that in both men and women, Aldo and MR activation contribute to atherosclerosis progression and complications. de Rita et al. (2012) reported that elevated plasma Aldo concentration significantly correlated with plaque progression while sex and age did not. One study of end-stage renal disease patients, a population at high risk for atherosclerotic ischemic events, analyzed plaques in men and women separately and showed that spironolactone treatment prevented increases in intima-media thickness in both sexes (Vukusich et al. 2010). Finally, Matsuda et al. (2016) demonstrated that eplerenone reduced intima-media thickness in a small cohort of 12 primary aldosteronism patients, 10 of whom were women.
While atherosclerotic plaque progression can contribute to chronic angina and symptoms that reduce quality of life, plaque rupture is more dependent on inflammation and contributes most to the morbidity and mortality associated with atherosclerosis (Libby et al. 2013). In atherosclerosis patients, Aldo levels correlated with serum inflammatory factors regardless of gender, suggesting a pro-inflammatory role of Aldo in both men and women (Tomaschitz et al. 2011). Further, multiple studies correlate MR activation with increased risk of ischemic events downstream of plaque rupture (Milliez et al. 2005; Ivanes et al. 2012). These studies include both men and women but do not separate the data by sex. Thus, the available data in human observational studies and clinical trials suggests that MR inhibitors could be a useful tool to reduce atherosclerotic plaque progression and complications in both men and women and warrant further clinical study and exploration of the mechanisms by which the MR contributes to vascular disease in both sexes.
4. Heart Failure
A. Epidemiology
When the cardiac systolic pump function becomes impaired, this is known as heart failure with reduced ejection fraction (HFrEF). When the heart is unable to fully relax during diastole to allow blood to fill the ventricles, but is still able to contract, this is known as heart failure with preserved ejection fraction (HFpEF). HFrEF and HFpEF each account for about half of the total burden of heart failure (Dunlay et al. 2017). Men are somewhat more likely to develop HFrEF, likely due to higher rates of hypertension and MI in younger men, both of which are common causes of HFrEF (Dunlay et al. 2017; Benjamin et al. 2018). By contrast, HFpEF is more common in women and is the most common type of heart failure in the growing population over 65 years of age (Upadhya et al. 2017b). Risk factors for HFpEF include advanced age and obesity, both of which are increasingly common in women (Owan et al. 2006; Flegal et al. 2016; Tsujimoto and Kajio 2017). Although women with HFpEF generally have improved survival compared to men, this protection is lost in women with diabetes (Martinez-Selles et al. 2012) and women with HFpEF reported reduced quality of life relative to men in a recent study (Faxen et al. 2018). Thus, HFpEF is a growing clinical problem, especially in the rapidly growing elderly population and in women with cardiovascular risk factors.
B. Clear Benefits of MR Antagonism in Heart Failure with Reduced Ejection Fraction
Inhibition of the MR is well known to prevent mortality and improve outcomes in patients with HFrEF (Pitt et al. 1999; Pitt et al. 2003b; Zannad et al. 2011). The benefits of MR antagonism on HFrEF may apply to both men and women, but the data supporting this is scarce (see Tables 1 and 2). A sub-analysis of the Framingham Heart Study showed that serum Aldo levels correlate with cardiac remodeling in women but not men, suggesting that there may indeed be differences between the sexes in the way the MR signaling pathway contributes to heart failure (Vasan et al. 2004). Despite these data, clinical trials investigating the role of the MR in HFrEF continue to recruit predominantly male subjects. For example, the landmark RALES, EPHESUS, and EMPHASIS-HF trials that showed a clear mortality benefit of MR antagonism in HFrEF patients were heavily weighted towards male participants, with women making up only 27% of study subjects. While this resulted in the individual studies being under-powered to assess sex differences, combination of the data in a recent meta-analysis did enable sub-analysis of the data by sex. In this combined data, the male sub-group retained the significant mortality benefit of MR antagonism, while the female sub-group also tended towards a decline in sudden cardiac death with MR antagonism that was not statistically significant (Rossello et al. 2019). Thus, it is unclear whether MR antagonism is beneficial in female HFrEF patients, as has been shown definitively for men. It will be critical to include more women in future trials of MR antagonists in heart failure in order to fully understand the role of this receptor in HFrEF in women.
Table 2:
Clinical and Preclinical Evidence for a Sex-Specific Role of the MR and EC-MR in Cardiovascular Disease
| Model | Sex | Intervention | Results | References |
|---|---|---|---|---|
| ENDOTHELIAL DYSFUNCTION | ||||
| MR in humans | Men | Spiro, Epl | MR antagonists improve endothelial function in heart failure, diabetes, hypertension (mostly male cohorts). | Farquharson and Struthers 2000; Macdonald et al. 2004; Fujimura et al. 2012; Garg et al. 2015 |
| Women | Spiro | MR antagonists improve endothelial function in women with polycystic ovarian syndrome. | Studen et al. 2011 | |
| Spiro | MR antagonists improve endothelial function in women with Rheumatoid Arthritis. | Syngle et al. 2009 | ||
| Both | Spiro | MR antagonists improve endothelial function in hypertensive African American men and women (analyzed separately). | Mohandas et al. 2015 | |
| MR in animals | Male | Epl, Finerenone | MR antagonists improve endothelial function. | Rajagopalan et al. 2002; Gonzalez-Blazquez et al. 2018 |
| Female | Spiro | MR antagonism improves endothelial function in leptin-sensitized obese females. | Huby et al. 2016 | |
| EC-MR-KO mice | Male | Tie2 Cre | EC-MR-KO improves endothelial function of the aorta in obesity. | Schafer et al. 2013 |
| VE-Cadherin Cre | EC-MR-KO improves endothelial function of the mesenteric arterioles in hypertension. | Mueller et al. 2015 | ||
| VE-Cadherin Cre | EC-MR-KO does not alter endothelial function of the mesenteric arterioles in obesity or hyperlipidemia. | Davel et al. 2018b | ||
| Female | VE-Cadherin Cre | EC-MR-KO corrects the endothelial dysfunction with obesity and hyperlipidemia. | Davel et al. 2018b | |
| HYPERTENSION | ||||
| MR in humans | Men | Epl | MR antagonism reduces blood pressure (mostly male cohort). | Pitt et al. 2003a |
| Women | Salt restriction, angiotensin-II | RAAS stimulation increases blood pressure more in women than in men. | Jurgens and Graudal 2004; Shukri et al. 2018 | |
| Spiro, Epl | Women with Resistant Hypertension experienced greater blood pressure decrease with MR antagonism than men. | Khosla et al. 2009 | ||
| Both | Epl | MR antagonism reduces blood pressure (roughly equal male/female cohort). | Williams et al. 2004 | |
| MR in animals | Male | Spiro, Epl | MR antagonism reduces blood pressure in gonad-intact and castrated males. | Michaelis et al. 2012; reviewed in DuPont and Jaffe 2017 |
| Female | Spiro | MR antagonism reduces blood pressure in obese females. | Huby et al. 2016 | |
| Spiro | MR antagonism does not reduce blood pressure in ovariectomized females. | Michaelis et al. 2012 | ||
| Epl | MR antagonism prevents endothelial tight junction remodeling in the cerebral arteries of hypertensive females. | Tada et al. 2010 | ||
| EC-MR-KO mice | Male | VE-Cadherin tetOFF overexpression | EC-MR overexpression increases blood pressure. | Nguyen Dinh Cat et al. 2010 |
| Tie2 Cre, VE-Cadherin Cre | EC-MR-KO does not affect blood pressure at baseline or in disease models. | Rickard et al. 2014; Mueller et al. 2015; Dinh et al. 2016; Lother et al. 2016; Salvador et al. 2017; Laursen et al. 2018 | ||
| Tie2 Cre, VE-Cadherin Cre | EC-MR-KO attenuates the pathologic remodeling that occurs with hypertension. | Rickard et al. 2014; Lother et al. 2016; Diaz-Otero et al. 2017; Diaz-Otero et al. 2018 | ||
| Female | VE-Cadherin Cre | EC-MR-KO likely does not affect blood pressure at baseline or in disease models. | Jia et al. 2015b; Davel et al. 2018b | |
| ATHEROSCLEROSIS | ||||
| MR in humans | Men | Aldo (observational) | Aldo levels correlate with cardiovascular ischemia (mostly male cohorts). | Milliez et al. 2005; Ivanes et al. 2012 |
| Women | Epl | MR antagonism reduces IMT in primary hyperaldosteronism (mostly female cohort). | Matsuda et al. 2015 | |
| Both | Aldo (observational) | Aldo levels correlate with larger plaques independently of sex. | de Rita et al. 2013 | |
| Spiro | MR antagonism reduces plaque volume in men and women with end-stage renal disease. | Vukusich et al. 2010 | ||
| Aldo (observational) | Aldo levels correlate with soluble inflammatory markers independently of sex. | Tomaschitz et al. 2011 | ||
| MR in animals | Male | Aldo, 11βHSD2-KO | MR activation increases plaque size, inflammation. | Deuchar et al. 2011; McGraw et al. 2013; Marzolla et al. 2017 |
| Spiro, Epl | MR antagonism reduce plaque size, inflammation. | Rajagopalan et al. 2002; Keidar et al. 2003; Suzuki et al. 2006; Raz-Pasteur et al. 2012; Raz-Pasteur et al. 2014; Kratz et al. 2016; Li et al. 2017; Moss et al. 2019 | ||
| Female | Spiro | MR antagonism does not reduce plaque inflammation in female mice | Moss et al. 2019 | |
| EC-MR-KO mice | Male | VE-Cadherin Cre | EC-MR-KO attenuates atherosclerotic plaque inflammation and inflammatory cell recruitment without changing plaque size. | Moss et al. 2019 |
| Female | VE-Cadherin Cre | Females have less plaque inflammation and inflammatory cell recruitment than males, and EC-MR-KO does not confer further protection in females. | Moss et al. 2019 | |
| HFrEF | ||||
| MR in humans | Men | Epl | MR antagonists reduce mortality (mostly male cohorts). Significant effect remains in male sub-group of meta-analysis. | Pitt et al. 1999; Pitt et al. 2003b; Zannad et al. 2011; Rossello et al. 2019 |
| Women | Epl | MR antagonism may reduce mortality in women (trend but not significant in meta-analysis). | Rossello et al. 2019 | |
| Aldo (observational) | Aldo levels correlate with cardiac remodeling in women but not men. | Vasan et al. 2004 | ||
| MR in animals | Male | Epl, Aldosterone synthase inhibitor | MR antagonists improve systolic function and reduce mortality. | Fraccarollo et al. 2003; Wang et al. 2004; Munoz-Pacheco et al. 2013; Furuzono et al. 2017 |
| pH1tet-inducible anti-MR shRNA expression | Inducible genetic MR knockdown improves systolic function and reduces mortality. | Montes-Cobos et al. 2015 | ||
| Female | Epl | Female rats had larger improvements in systolic function and cardiac remodeling than males. | Kanashiro-Takeuchi et al. 2009 | |
| EC-MR-KO mice | Male | Tie2 Cre, VE-Cadherin Cre | EC-MR-KO improves systolic function and prevents cardiac remodeling. | Rickard et al. 2014; Lother et al. 2016; Salvador et al. 2016; Salvador et al. 2017 |
| Female | --- | Not studied | --- | |
| HFpEF | ||||
| MR in humans | Men | Spiro | MR antagonism does not improve mortality in men (no trend or significant effect in TOPCAT meta-analysis). | Merrill et al. 2019 |
| Women | Spiro | MR antagonism improves diastolic dysfunction (mostly-female cohorts). | Pandey et al. 2015; Fukuta et al. 2018 | |
| Spiro | MR antagonism may or may not improve exercise capacity (mostly-female cohorts). | Daniel et al. 2009; Upadhya et al. 2017a | ||
| Spiro | MR antagonism improves mortality in women (significant effect in TOPCAT meta-analysis). | Merrill et al. 2019 | ||
| Both | Spiro | MR antagonism improves left ventricular function and structure and reduces hospitalization rates, with no overall effect on mortality (roughly equal male/female cohorts). | Edelmann et al. 2013; Pitt et al. 2014 | |
| MR in animals | Male | --- | Male mice do not develop diastolic dysfunction with Western diet. | Manrique et al. 2013 |
| Female | Spiro | MR antagonism prevents diastolic dysfunction and cardiac inflammation with Western diet. | Bostick et al. 2015 | |
| EC-MR-KO mice | Male | --- | Not studied | --- |
| Female | VE-Cadherin Cre | EC-MR-KO prevents diastolic dysfunction and cardiac inflammation with Western diet. | Jia et al. 2015b | |
MR=mineralocorticoid receptor; EC-MR=endothelial-specific MR; KO=knockout; Aldo=aldosterone; Spiro=spironolactone; Epl=eplerenone; RAAS=renin-angiotensin-aldosterone system; 11βHSD2=11β-hydroxysteroid dehydrogenase 2; shRNA=short hairpin RNA
C. MR Antagonism May Specifically Benefit Women with Heart Failure with Preserved Ejection Fraction
In contrast to the clear benefit observed with MR antagonism in HFrEF, investigations into the role of the MR in HFpEF in humans have produced variable results. However, the growing body of literature does suggest a role for the MR in this disease. In an open-label trial in which 11 women with HFpEF were administered spironolactone, the authors observed an improvement in peak exercise capacity from baseline and a reduction in the median heart failure score (Daniel et al. 2009). Subsequent studies and meta-analyses in mostly-female cohorts have largely found that spironolactone improves diastolic function in HFpEF patients (Pandey et al. 2015; Fukuta et al. 2018). Results vary as to whether spironolactone improves exercise tolerance in HFpEF patients, with some studies showing increased exercise capacity with MR antagonism (Daniel et al. 2009; Kosmala et al. 2016) and others showing no benefit (Upadhya et al. 2017a; Pandey et al. 2015; Fukuta et al. 2018).
Larger randomized trials of MR antagonism in HFpEF have produced extensive controversy in recent years. The Aldo-DHF trial randomized over 400 patients and demonstrated improved left ventricular functional and structural parameters in HFpEF patients randomized to spironolactone (Edelmann et al. 2013). Subsequently, however, the TOPCAT trial randomized over 3,000 HFpEF patients in 6 countries to either placebo or spironolactone, and the results revealed that MR antagonism reduced the rate of hospitalization for heart failure but did not significantly affect mortality (Pitt et al. 2014). Subsequent sub-analysis of this otherwise negative trial revealed heterogeneity in the data that may have masked the beneficial effects of MR antagonism in certain patient subgroups. For example, subjects who qualified for the study based on natriuretic peptide levels (the majority of patients enrolled in the Americas) had a significant mortality benefit with spironolactone, while patients who qualified based solely on clinical criteria (the majority of patients enrolled in Russia and Georgia) did not (Pfeffer et al. 2015; Bristow et al. 2016) Another sub-analysis, which included 1,767 of the randomized patients and was equally comprised of men and women, demonstrated that women with HFpEF had a significant reduction in cardiovascular and all-cause mortality with spironolactone, while men did not (Merrill et al. 2019). While such post-hoc analyses are hypothesis-generating, the results may help to contextualize the sex differences observed in mortality in HFpEF patients and provide opportunities for further study to identify sex-specific therapies for this disease, for which there are currently no available pharmacotherapeutic options.
5. Summary of the Clinical Data
In summary, observational studies and clinical trials support that the MR contributes to the pathogenesis endothelial dysfunction, hypertension, atherosclerosis, and heart failure, as MR antagonist therapy has been shown to improve outcomes in patients with these conditions (see Tables 1 and 2). In some cases, there appear to be differences between men and women in the role of the MR in disease, as evidenced by the female predominance of salt-sensitivity of blood pressure and sex differences in the efficacy of MR therapy in heart failure. Importantly, many clinical trials use spironolactone as an MR antagonist, while others use the less potent but more selective eplerenone. Thus, it is possible that off-target effects of spironolactone on the progesterone or androgen receptors could contribute to sex differences observed in the effect of MR inhibition in various cardiovascular pathologies. Further careful study will be needed to fully understand the sex-specific contributions of the MR to cardiovascular disease in humans, with a greater focus on the inclusion of female patients in clinical trials and the use of selective MR inhibitors.
III. PRECLINICAL DATA: SEX-SPECIFIC ROLES FOR THE ENDOTHELIAL MINERALOCORTICOID RECEPTOR IN CARDIOVASCULAR DISEASE
The clinical literature points to a role for the MR in cardiovascular disease, in some cases with sex-specific effects. However, studies in humans by necessity rely on the use of systemic MR inhibitors, thus precluding examination of the role of the MR in particular cell types. By contrast, genetic animal models of cell-specific MR deletion have enabled investigations into the contribution of the MR specifically within the vascular endothelium to cardiovascular disease. In this section, the preclinical literature implicating EC-MR in 1) endothelial dysfunction, 2) hypertension, 3) atherosclerosis, and 4) heart failure is discussed, particularly in light of new evidence supporting a sex-specific role for EC-MR in cardiovascular disease. See Table 2 for a summary of the studies in this section describing the sex-specific role of the MR in cardiovascular disease and potential correlations to human clinical data.
1. A Sex-Specific Role for EC-MR in Endothelial Dysfunction
A. MR Inhibition in Animals Improves Endothelial Function, Particularly in the Context of Obesity
As has been demonstrated in human studies (Table 1 ), MR inhibition in animal models improves indices of endothelial function, particularly in the context of cardiovascular risk factors. Specifically, MR antagonist treatment improved aortic endothelial function and peak relaxations and reduced ROS generation in male hyperlipidemic rabbits (Rajagopalan et al. 2002) and in rats with chronic kidney disease (Gonzalez-Blazquez et al. 2018).
Obesity in particular may represent a state of enhanced MR activation, as higher body mass index correlates with higher Aldo levels in patients administered a high-salt diet (Bentley-Lewis et al. 2007). This is likely due to adipocyte-derived factors that increase Aldo release either directly from the fat (Briones et al. 2012) or from the adrenal gland (Huby et al. 2016). Specifically, the adipokine leptin, which circulates at significantly higher levels in obese females than males (Deng and Scherer 2010), may mediate the role of the MR in endothelial dysfunction in obesity in females. Leptin was shown in preclinical models to increase Aldo secretion from the adrenal gland and to induce endothelial dysfunction in females in an MR-dependent manner (Huby et al. 2016; Faulkner and Belin de Chantemele 2019). This may be especially important for the pathogenesis of endothelial dysfunction in females, as leptin levels and rates of obesity are both higher in women than in men (Flegal et al. 2016; Deng and Scherer 2010).
B. EC-MR Contributes to Endothelial Dysfunction, with Sex- and Vascular Bed-Dependent Effects
Preclinical animal studies reveal that in the setting of cardiovascular risk factors, EC-MR is a mediator of endothelial dysfunction and its specific deletion from ECs has a positive impact on vascular function (Davel et al. 2017). Either global MR inhibition with eplerenone or EC-specific MR deletion improved dilation of aortic rings in obese male mice and in lean mice with Aldo infusion (Schafer et al. 2013). Similarly, EC-MR deletion improved resistance vessel endothelial function in a model of male mice exposed to angiotensin-II-induced hypertension (Mueller et al. 2015). In female mice, EC-MR deletion also prevented Western diet-induced aortic endothelial dysfunction (Jia et al. 2016).
One recent study directly compared the role of EC-MR in mesenteric microvessel dysfunction in the setting of obesity and hyperlipidemia in male and female littermates. In this study, obese male mice were able to compensate for endothelial dysfunction, with no role for EC-MR in endothelial dysfunction of the mesenteric arteries. By contrast, diet-induced obesity did result in endothelial dysfunction in female mice, and genetic deletion of EC-MR restored endothelial-dependent microvessel relaxation by increasing NO bioavailability. Notably, there was no role for EC-MR in endothelial function in healthy male or female mice; the EC-MR-dependent endothelial dysfunction was observed in females only with the addition of obesity and/or hyperlipidemia (Davel et al. 2018b).
The study by Davel et al. (2018b) was the first to directly compare the role of the MR in endothelial function between males and females, revealing significant sex differences in the role of EC-MR in vasodilatory pathways and microvascular endothelial dysfunction in response to cardiometabolic risk factors. Comparison to the prior literature in males suggest that the role of EC-MR in modulating endothelial function depends on the vascular bed and cardiovascular risk factor interrogated. Whereas Davel et al. (2018b) found no role for EC-MR in mesenteric microvessel dysfunction that occurs with obesity in males, Schafer et al. (2013) previously demonstrated a role for EC-MR in aortic endothelial dysfunction in obese males. Further, Mueller et al. (2015) showed improvement in microvessel function with EC-MR deletion in male mice subjected to angiotensin-II hypertension.
Thus, the role of EC-MR in endothelial function may depend on sex, vascular bed, and clinical context. Further studies comparing the role of the MR in endothelial dysfunction between men and women and in the setting of a variety of cardiovascular risk factors could translate these preclinical results into actionable sex-specific therapies to reverse endothelial dysfunction and prevent further cardiovascular disease.
2. A Nuanced Role for EC-MR in Hypertension
A. The MR May Regulate Blood Pressure in Females with Cardiometabolic Risk Factors, As Has Been Shown for Males
As in humans, ample data demonstrates that the MR influences blood pressure in male animal models (reviewed in DuPont and Jaffe 2017). As most mechanistic studies exploring the role of the MR in blood pressure changes have used only male animals, less is known regarding females. One notable exception is a study by Huby et al. (2016) in which MR inhibition with spironolactone substantially reduced blood pressure in female agouti yellow obese mice. Another study directly compared the blood pressure-lowering effect of spironolactone between gonadectomized male and female rats, revealing that while high salt diet increased blood pressure in both sexes, MR inhibition with spironolactone reduced blood pressure only in males (Michaelis et al. 2012). These studies highlight the need for further detailed exploration into MR-mediated mechanisms of hypertension in females and for direct comparisons between the sexes.
B. EC-MR May Not Directly Control Blood Pressure but Modulates the Response to Hypertension
A focus on the specific role of endothelial MR reveals that genetic deletion of EC-MR in male mice does not affect blood pressure at baseline (Salvador et al. 2017; Mueller et al. 2015) or in models of experimentally-induced hypertension (Rickard et al. 2014; Dinh et al. 2016; Lother et al. 2016; Laursen et al. 2018; Mueller et al. 2015). By contrast, male mice overexpressing the MR specifically in ECs have higher systolic blood pressure and exaggerated vasoconstrictor responses, suggesting that under conditions where EC-MR is upregulated, it may contribute to elevated blood pressure potentially via crosstalk between vascular ECs and SMCs (Nguyen Dinh Cat et al. 2010).
Data concerning the contribution of EC-MR to blood pressure regulation or hypertension in females is scarce but suggests that EC-MR may not play a role in blood pressure regulation in females, as has been shown rigorously for males. In their recent study of sex differences in endothelial function, Davel et al. (2018b) measured blood pressure by tail cuff plethysmography in a subset of animals and reported no effect of EC-MR deletion in either sex under any of the dietary conditions studied. Likewise, measurement of blood pressure in anesthetized female animals after Western diet feeding revealed no difference in blood pressure with EC-MR deletion (Jia et al. 2015b). However, studies directly comparing males and females and using sensitive blood pressure measurement techniques such as radiotelemetry in conscious mice are needed to confirm this lack of a role for EC-MR in blood pressure regulation in females.
Although EC-MR may not contribute to blood pressure regulation per se, studies indicate that it may be critical for the pathologic arterial and myocardial remodeling observed as a consequence of hypertension. In a study of male mice with angiotensin-II-induced hypertension, EC-MR deletion completely prevented the decreases in cerebral vessel outer diameter, lumen diameter, and cross-sectional area observed in MR-intact littermates with the same degree of hypertension. This indicates that EC-MR is necessary for the pathologic cerebral arterial remodeling observed in hypertension (Diaz-Otero et al. 2017), and further studies suggest a role for the MR in cognitive dysfunction induced by hypertension (Diaz-Otero et al. 2018). EC-MR deletion also prevented pathologic cardiac remodeling without modulating blood pressure in two studies of male mice in models of experimental hypertension-induced cardiac dysfunction (Rickard et al. 2014; Lother et al. 2016).
Thus, the role of EC-MR in hypertension and its downstream consequences is well characterized in male animal models, but less is known regarding its role in females. As hypertension affects over a quarter of all women in America, and two-thirds of women over the age of 60, understanding the sex-specific mechanisms and downstream consequences of this pathology in females will be critical to crafting appropriate sex-specific therapies and preventative strategies.
3. EC-MR Differentially Contributes to Atherosclerosis in Males and Females
A. The MR Promotes Atherosclerosis in Male Animals
Despite human data suggesting a role for the MR in atherosclerosis in both men and women, preclinical investigations exploring mechanisms in animal models have almost exclusively focused on the pathology in males. In the apolipoprotein-E-knockout atherogenic mouse model, Aldo administration along with high fat diet increases plaque size and inflammation in males in as little as 4 weeks (McGraw et al. 2013; Marzolla et al. 2017). Similarly, deletion of 11βHSD2, which leads to constitutive activation of the MR by corticosterone, accelerates plaque formation and inflammation in male apolipoprotein-E-knockout mice (Deuchar et al. 2011). Conversely, MR inhibition with eplerenone or spironolactone has repeatedly been shown to decrease plaque size and inflammation in male mice (Raz-Pasteur et al. 2014; Raz-Pasteur et al. 2012; Keidar et al. 2003; Suzuki et al. 2006; Kratz et al. 2016; Moss et al. 2019), rabbits (Rajagopalan et al. 2002), and pigs (Li et al. 2017b).
B. EC-MR Contributes to Atherosclerotic Plaque Inflammation, with Sex-Specific Effects
Preclinical studies indicate that EC-MR plays a critical role in the inflammation of the atherosclerotic plaque. In male apolipoprotein-E-knockout mice, intracellular adhesion molecule (ICAM)-1, a surface protein expressed on endothelial cells that mediates leukocyte-endothelial interactions, was found to be necessary for Aldo to increase plaque formation and inflammation (Marzolla et al. 2017). In male mice, activation of the MR by genetic 11βHSD2 ablation also increased endothelial expression of vascular cell adhesion molecule (VCAM)-1, another mediator of leukocyte-endothelial adhesion (Deuchar et al. 2011).
A recent study further explored the possibility that EC-MR regulates inflammation of the atherosclerotic plaque, this time directly comparing male and female mice. In this study, EC-MR deletion in males significantly reduced atherosclerotic plaque inflammation and leukocyte rolling and adhesion to the vasculature in vivo. By contrast, gonad-intact female littermates exhibited less atherosclerotic plaque inflammation and fewer leukocyte-endothelial interactions, even with intact MR. Moreover, in females, EC-MR deletion did not provide additional protection against atherosclerotic vascular inflammation, in contrast to the observed benefit of EC-MR deletion in males (Moss et al. 2019). These data reveal a significant sex difference not only in atherosclerotic vascular inflammation overall, but in the role of EC-MR in regulating inflammation in the context of atherosclerosis. The results of this study suggest new mechanisms for the contribution of Aldo and the MR to cardiovascular ischemia in humans and for the protection from atherosclerotic plaque rupture observed in premenopausal women. Further investigations into the relationship between female sex and EC-MR function, such as studies using ovariectomized versus gonad-intact females with intact MR or EC-MR deletion, could identify mechanisms by which MR and female sex hormone signaling interact in vivo in the context of atherosclerosis.
4. EC-MR Contributes to the Pathogenesis of Heart Failure with Both Reduced and Preserved Ejection Fraction
A. MR Inhibitors Improve Function and Survival in Animals with Heart Failure
Inhibition of the MR has been demonstrated to improve clinically relevant features of HFrEF in animal models, especially when combined with other standard heart failure therapies. Addition of eplerenone to a standard regimen consisting of an angiotensin-converting enzyme inhibitor, a thiazide diuretic, and a β-adrenergic blocker prevented left ventricular hypertrophy and echocardiographic anomalies in male spontaneously hypertensive heart failure rats beyond the effect of standard therapy alone (Munoz-Pacheco et al. 2013). Combination of MR inhibition with angiotensin-converting enzyme blockade appears to be especially effective at attenuating cardiac contraction defects and fibrosis in male rats (Fraccarollo et al. 2003) and mice (Wang et al. 2004) subjected to MI to induce heart failure. In the transverse aortic constriction model of pressure overload-induced heart failure, MR inhibition either by inducible whole-body genetic knockdown (Montes-Cobos et al. 2015) or by inhibition of the Aldo synthase enzyme (Furuzono et al. 2017) reduced mortality and improved cardiac function in male mice, even without additional therapies.
Only one study has compared male and female animals side-by-side to assess sex-specific roles of MR signaling in experimental heart failure. Kanashiro-Takeuchi et al. (2009) found that after MI, female rats benefited more from eplerenone therapy than males. Specifically, ejection fraction, infarct size, cardiac fibrosis, and contraction anomalies were all improved in female rats, while males experienced smaller changes in these parameters that did not reach statistical significance in this study.
B. In Animal Models, EC-MR Contributes to HFrEF in Males and to HFpEF in Females, but Sex Differences Have Not Been Studied
EC-MR has also been shown to contribute to the pathophysiology of HFrEF, at least in male animals. EC-MR deletion reduced ventricle weight and prevented an increase in cardiac fibrosis in male hypertensive mice (Rickard et al. 2014; Lother et al. 2016) and improved ejection fraction in male mice in the transverse aortic constriction model (Salvador et al. 2017), independent of effects on inflammation (Salvador et al. 2016). As no study has investigated the role of EC-MR in HFrEF in female animals, further studies are needed to understand whether EC-MR contributes to this pathology in females.
Much of the preclinical literature in HFpEF focuses on female animal models, opposite of the trends in the other cardiovascular outcomes described in this Review. This is largely due to the activities of the Sowers research group, which uses a model of female mice fed a Western diet (containing high fat and high sucrose) resulting in obesity-induced cardiac diastolic dysfunction. This group found that only female mice, not male mice, develop diastolic dysfunction in this treatment paradigm, suggesting sex differences in the mechanisms driving HFpEF (Manrique et al. 2013). In this model, MR antagonism improved diastolic function and reduced cardiac fibrosis, inflammation, and other markers of adverse myocardial remodeling (Bostick et al. 2015). This correlates with clinical studies in human HFpEF patients demonstrating beneficial effects of MR antagonists on diastolic function (Pandey et al. 2015; Fukuta et al. 2018) (see Table 2). EC-MR deletion in female mice recapitulates most of these benefits of pharmacologic MR blockade, implying that the MR within ECs plays a critical role in the development of diastolic dysfunction in this model (Jia et al. 2015b). Future work is needed to explore potential sex differences in the role of EC-MR in HFpEF in different model systems in which both sexes develop dysfunction. Such investigations could shed light on the mechanisms driving the sex differences in outcomes and quality of life in patients with HFpEF (Faxen et al. 2018; Martinez-Selles et al. 2012) and would support future clinical trials of MR inhibition in HFpEF, particularly in the context of obesity.
5. Summary of the Data from Animal Models
In animal models, the MR specifically within the vascular endothelium promotes endothelial dysfunction, mediates inflammation in atherosclerosis, and contributes to cardiac remodeling in heart failure. In many cases, these preclinical data are consistent with the effects of MR inhibition that have been observed in human clinical cohorts (Table 2). While sex differences in these diseases have been directly investigated in a few cases, for the most part our understanding of the mechanisms driving cardiovascular disease comes from studies in male model systems or comparisons of males and females studied separately. Notable exceptions, such as studies of the role of the MR in endothelial-dependent relaxation (Davel et al. 2018b), atherosclerotic inflammation (Moss et al. 2019), and diastolic dysfunction in diet-induced obesity (Manrique et al. 2013; Jia et al. 2015b) point to intriguing sex differences in the function of the MR in the vascular endothelium. Additional studies directly comparing male and female animals are needed to provide critical insight into the mechanisms mediating sex differences in cardiovascular disease in humans.
IV. MOLECULAR MECHANISMS FOR THE SEX-SPECIFIC ROLES OF ENDOTHELIAL MINERALOCORTICOID RECEPTORS IN CARDIOVASCULAR DISEASE
In this section, we review the literature describing the contribution of EC-MR to: 1) inflammation; 2) vascular stiffness; and 3) oxidative stress as potential mechanisms for the sex-dependent role of EC-MR in various cardiovascular pathologies. We further discuss the relevant mechanistic insight gleaned from studies exploring 4) crosstalk between the MR and sex hormone signaling. Figure 1 provides a model summarizing these data.
Figure 1. Molecular Mechanisms for the Sex-Specific Contributions of EC-MR to Cardiovascular Disease.
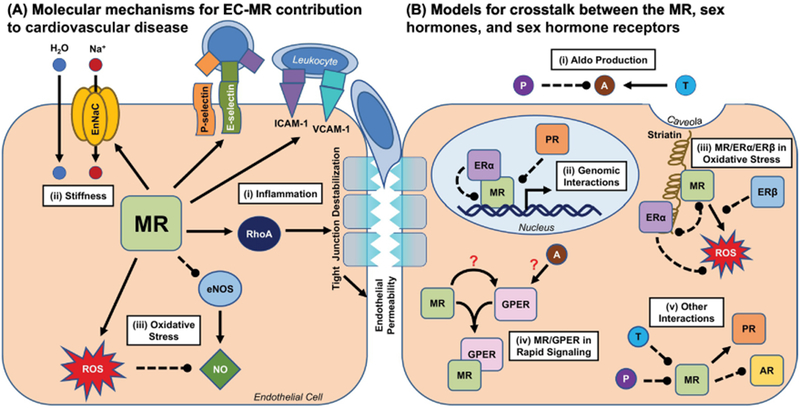
(A) The MR participates in a number of processes in ECs that may contribute to cardiovascular disease in a sex-specific manner. (i) EC-MR promotes the expression of endothelial adhesion molecules such as P- and E-selectin and ICAM-1, and this differs by sex for E-selectin and ICAM-1. This results in differential leukocyte recruitment to the vasculature in males and females. EC-MR also promotes endothelial permeability by activating RhoA, which leads to tight junction destabilization and may facilitate leukocyte trans-endothelial migration. (ii) The MR is well known to promote the expression of sodium transport proteins such as EnNaC, which in the endothelium can promote vascular stiffness. Whether this differs by sex is unclear, as all studies of EC-MR in vascular stiffness have been performed in female mice. (iii) EC-MR promotes oxidative stress in both males and females, though the mechanism for this effect may differ by sex. The ROS produced by this effect inactivate NO, thus preventing effective endothelium-dependent dilation of the underlying smooth muscle cells. This effect appears to vary by sex, arterial bed, and disease model. (B) There are several potential nodes for crosstalk between the MR and sex hormone receptors, many of which have yet to be fully explored. (i) Sex hormones may modulate production of the MR ligand Aldo at the level of the adrenal gland: testosterone may increase Aldo production, while progesterone may inhibit it. (ii) Activated ERα can bind to and inhibit the transcriptional function of the MR, which requires nuclear translocation but does not require ERα itself to bind DNA. The PR has also been demonstrated to inhibit MR transcriptional activities. (iii) The MR and ERα may compete for occupancy of striatin at the caveolar membrane, where they mediate non-genomic effects on eNOS and other rapid signaling cascades. (iv) Possible interactions between Aldo, the MR, and GPER are particularly controversial. Activation of either the MR or GPER can activate similar rapid signaling pathways, and many of these effects can be blocked by either MR inhibition or GPER inhibition. Possible models for this crosstalk include activation of GPER by MR, direct binding of Aldo to GPER, and complex formation between the MR and GPER. (v) Progesterone has been shown to bind to and inhibit the MR, and testosterone has been hypothesized to do the same. AR/MR interactions are not well characterized but may include inhibition of the AR by MR. ERβ has also been demonstrated to attenuate Aldo-induced ROS production, through unclear mechanisms. The MR may also promote PR activity. Solid arrow=positive regulation, dotted line=negative regulation; A=Aldo; AR=androgen receptor; EnNaC=endothelial epithelial sodium channel; eNOS=endothelial nitric oxide synthase; ER=estrogen receptor; GPER=G protein-coupled estrogen receptor; NO=nitric oxide; P=progesterone; PR=progesterone receptor; ROS=reactive oxygen species; T=testosterone.
1. Inflammation
Inflammation plays a critical role in the pathophysiology of a number of cardiovascular diseases, including hypertension, atherosclerosis, and heart failure (Ruparelia et al. 2017). EC-MR has been demonstrated to contribute to a number of inflammatory processes, in some cases sex-dependently. This may represent a molecular mechanism by which EC-MR contributes to a wide variety of cardiovascular diseases.
A. EC-MR Regulates Inflammatory Endothelial Adhesion Molecules
The first description of the MR gene regulatory function within ECs was in human coronary artery ECs, where EC-MR was shown to transcriptionally regulate ICAM-1, a key endothelial mediator of leukocyte adhesion (Caprio et al. 2008). Later studies in apolipoprotein-E-knockout male mice demonstrated that ICAM-1 is necessary for Aldo to enhance atherosclerosis (Marzolla et al. 2017), and MR inhibition with eplerenone in Dahl salt-sensitive rats decreased renal ICAM-1 expression (Kobayashi et al. 2005), further implicating MR regulation of ICAM-1 in tissue inflammation (Figure 1 A-i). Additional in vitro studies using human ECs demonstrated that estrogen, via ERα, inhibits MR transcription of ICAM-1, suggesting that estrogen signaling diminishes the role of MR-induced ICAM-1 in inflammation (Barrett Mueller et al. 2014). This is in line with a study in which the effect of estrogen on atherosclerosis in females was found to be independent of ICAM-1 (Gourdy et al. 2003), while in male mice ICAM-1 deletion has been shown to reduce lesion size (Bourdillon et al. 2000). Taken together, these data suggest a model in which EC-MR regulates ICAM-1 in males to promote inflammation, while in females ERα blocks this function of EC-MR (Figure 1B-iv).
The MR has also been linked to regulation of VCAM-1, another endothelial molecule involved in leukocyte adhesion to the vasculature. Deletion of 11βHSD2, which leads to overactivation of the MR by corticosterone, increased endothelial VCAM-1 expression in the aortic roots of male apolipoprotein-E-knockout mice (Deuchar et al. 2011). In another study, VCAM-1 expression was inhibited by eplerenone in the renal tissue of Dahl salt-sensitive rats (Kobayashi et al. 2005). Conversely, VCAM-1 may be negatively regulated by estrogen: in a study of ovariectomized female atherosclerotic mice, addition of estrogen decreased VCAM-1 relative to placebo (Gourdy et al. 2003). Scant data studying VCAM-1 regulation in EC-MR deficient mice points to potential endothelial-specific regulation of this molecule in males that may vary by the model used. In one model of male mice subjected to mineralocorticoid/high-salt hypertension, EC-MR deletion prevented VCAM-1 upregulation in cardiac ECs (Lother et al. 2016). By contrast, EC-MR deletion did not alter whole-heart VCAM-1 expression in males subjected to pressure-overload cardiac hypertrophy (Salvador et al. 2017). No study has yet explored the role of EC-MR in regulating VCAM-1 in females.
B. EC-MR Sex-Dependently Regulates the Selectins, Endothelial Molecules Critical for Leukocyte Recruitment
The selectins are a family of molecules expressed on the EC surface that mediate leukocyte rolling interactions with the endothelium, the necessary first step for tissue inflammation. P-selectin is involved in leukocyte capture and fast rolling, while E-selectin is necessary for leukocyte slow-rolling interactions, which precede firm adhesion and trans-endothelial migration (Sundd et al. 2011). EC-MR was recently found to regulate E-selectin in vivo in males. When compared directly to female littermates, TNFα-induced mesenteric venous expression of E-selectin was lower than that of males and not further affected by the deletion of EC-MR. This pattern of E-selectin expression correlated with sex-dependent effects on leukocyte slow rolling in the vasculature in the setting of an acute inflammatory stimulus and with the accumulation of inflammatory cells in aortic plaques in a model of hyperlipidemia-induced atherosclerosis (Moss et al. 2019). This recent study is consistent with prior in vitro investigations suggesting E-selectin regulation by the MR (Seeger et al. 2009; Hashikabe et al. 2006) and demonstrating that patients with high Aldo levels have higher circulating levels of soluble E-selectin (Tomaschitz et al. 2011). Further, E-selectin has been demonstrated to be negatively regulated by estrogen signaling (Tyree et al. 2002), consistent with the reduction in E-selectin expression observed in female mice compared to males (Moss et al. 2019).
P-selectin may also be regulated by EC-MR, though the data supporting this is less certain than that for E-selectin. P-selectin expression in whole-kidney lysates was increased in Dahl salt-sensitive rats relative to normotensive rats, and this expression was reduced by eplerenone (Kobayashi et al. 2005). In vitro, the Aldo-induced increase in leukocyte adhesion to ECs in static culture could be prevented by P-selectin inhibition, implicating P-selectin in this effect of EC-MR (Jeong et al. 2009). P-selectin is critical for leukocyte fast rolling interactions with the endothelium, and Moss et al. (2019) found that leukocyte fast rolling tended to be reduced by EC-MR deletion in males and females, however this was not statistically significant and P-selectin expression was not assessed in that study. That this tendency was the same in both sexes is consistent with data indicating that P-selectin is not involved in the protective effect of estrogen on atherosclerosis, suggesting that it is not an estrogen target and therefore may not be differentially regulated between the sexes (Gourdy et al. 2003).
C. A Role for EC-MR in Endothelial Permeability
The integrity of the endothelial tight junction also contributes to inflammation, as endothelial permeability to proteins, lipids, and leukocytes facilitates inflammation of underlying tissues. MR activation by Aldo treatment disrupted the membrane localization of tight junction proteins in human cultured ECs, resulting in permeability of the endothelial monolayer to labeled dextrans (Kirsch et al. 2013). Conversely, in female rats, eplerenone blocked degradation of tight junction proteins in response to hemodynamic instability, thereby preventing cerebral aneurysm formation (Tada et al. 2010). Thus, EC-MR may contribute to endothelial permeability, at least in females. This may be via its regulation of the RhoA signaling pathway, which among other activities promotes EC-EC junction stability via actions on the cytoskeleton (Shimokawa et al. 2016). Aldo has been found to activate RhoA in various cardiovascular cell types (Kirsch et al. 2013; Lavall et al. 2014; Nguyen Dinh Cat et al. 2018), leading to F-actin stress fiber formation. In cultured human ECs, this promotes disruption of endothelial junction proteins and permeability of the endothelial monolayer (Kirsch et al. 2013). Genomic ER signaling may also activate RhoA in ECs (Oviedo et al. 2011; Simoncini et al. 2006), although non-genomic estrogen signaling may counteract this effect (Li et al. 2016). Thus, in the case of endothelial junction integrity, estrogen signaling may not block MR effects on endothelial junction integrity and may instead work in parallel to promote endothelial permeability.
D. EC-MR-Mediated Inflammation May Contribute to Cardiovascular Fibrosis
Fibrosis is often a consequence of inflammation. While no studies have directly compared the role of EC-MR in cardiovascular fibrosis between males and females, analysis of the existing literature reveals the possibility of sex differences. One study found that EC-MR deletion did not alter cardiac inflammation or fibrosis in male mice subjected to transverse aortic constriction, a model of pressure overload-induced cardiac remodeling (Salvador et al. 2017). By contrast, EC-MR deletion in females attenuated cardiac (Jia et al. 2015b) and aortic (Jia et al. 2016) fibrosis in a Western diet-fed model, which corresponded to reductions in inflammatory markers in these mice. Western diet-fed females also develop renal artery dysfunction, inflammation, and fibrosis, which was recently also shown to be prevented by EC-MR deletion (Aroor et al. 2019). Thus, the limited data so far could be interpreted to suggest that EC-MR may specifically contribute to cardiovascular fibrosis only in females via effects on inflammation. However, since each study was performed in only one sex and in different models of cardiovascular fibrosis, it is not possible to distinguish true sex differences from differences in the models or methods used by different investigators. Direct comparison of males and females in the same model system is needed to definitively interrogate these potential sex differences in the role of EC-MR in fibrosis.
In summary, EC-MR appears to contribute to inflammation by regulating EC adhesion molecule expression and endothelial permeability (Figure 1A-i). In some circumstances, these processes are differentially regulated in males and females and appear to be subject to opposite regulation by estrogen signaling. Further work, especially studies comparing inflammation in male and female animal models, will be instrumental in elucidating the sex-specific mechanisms by which EC-MR contributes to inflammation to induce cardiovascular pathology.
2. Vascular Stiffness
Vascular stiffening occurs with aging and in response to chronic cardiometabolic risk factors and precedes and predicts the development of cardiovascular diseases including hypertension and atherosclerosis (Huveneers et al. 2015). The phenomenon of vascular stiffening involves dysfunction of all parts of the vessel wall, including the vascular SMCs, ECs, and extracellular matrix (Jia et al. 2015a). Mineralocorticoid signaling contributes to stiffening of the vascular wall, particularly in the presence of cardiovascular disease or risk factors, as MR blockade reduced aortic stiffness in human subjects with dilated cardiomyopathy (Vizzardi et al. 2015) and attenuated the aortic stiffening observed in female mice fed a Western diet (DeMarco et al. 2015). This role for the MR in vascular stiffness may only emerge in the presence of cardiovascular risk factors, as one study found that in older but otherwise healthy individuals, MR blockade did not change indices of arterial stiffness (Hwang et al. 2013a).
The MR in vascular SMCs contributes to arterial stiffening, as specific deletion of smooth muscle cell MR was recently shown to attenuate aortic stiffness in aging male mice (Kim et al. 2018). However, the MR specifically within the vascular endothelium has also been found to contribute to arterial stiffness via regulation of endothelial ion channels. ECs stiffness is modulated by changes in intracellular ion concentrations thereby altering intracellular water content. The resulting mechano-signals are then transmitted to neighboring ECs and the SMCs of the vessel wall. In female mice, Aldo administration induced aortic stiffness via endothelial expression of the epithelial sodium channel (EnNaC, Figure 1A-ii), a well-known gene target of the MR in the renal epithelium (Jia et al. 2018b; Kusche-Vihrog et al. 2010). Conversely, EC-MR deletion attenuated aortic stiffness observed in female mice administered Aldo or fed a Western diet, also via regulation of EnNaC (Jia et al. 2016). Downstream of EC-MR, EnNaC activity in the endothelium also promotes endothelial permeability and inflammation, thus increasing susceptibility to further cardiovascular dysfunction (Jia et al. 2018a).
In addition to EnNaC, the MR also regulates other ion channels, though many of these investigations have been performed in non-ECs (reviewed in DuPont et al. 2014). Notably, in breast cancer-derived ECs, Aldo has been shown to upregulate expression of the sodium/hydrogen exchanger via a mechanism that involves both the MR and rapid estrogen signaling (Rigiracciolo et al. 2016). Further study is required to determine whether EC-MR may regulate homeostasis of ion channels beyond EnNaC or the sodium/hydrogen exchanger to promote endothelial and vascular stiffness and thus contribute to the pathogenesis of cardiovascular disease.
3. Oxidative Stress
The role of the MR in oxidative stress and its contribution to endothelial dysfunction has been reviewed elsewhere (Queisser and Schupp 2012; Davel et al. 2017). The activity of the MR in the endothelium appears to be critical for these activities in both sexes. In male mice, EC-MR deletion prevented Aldo-induced increases in superoxide formation in the cerebral arteries (Dinh et al. 2016). In females fed a Western diet, EC-MR deletion increased eNOS activation and reduced nitrogen free radicals in the aorta (Jia et al. 2016). Thus, it appears that in both male and female animal models, EC-MR contributes to vascular oxidative stress. However, the mechanism by which EC-MR exerts these effects may differ between males and females. Hyperlipidemic male mice were recently found to developed endothelial dysfunction characterized by impaired endothelium-mediated vasodilation that was not ameliorated with genetic EC-MR deletion. Female hyperlipidemic littermates also developed endothelial dysfunction, but in females, EC-MR deletion resulted in a compensatory increase in NO production and NO-mediated dilation (Davel et al. 2018b) (Figure 1A-iii). This enhanced role for EC-MR in females may have been possible due to higher Aldo levels or potentially also low estrogen levels in these female mice with cardiometabolic risk factors (Davel et al. 2018b). Indeed, data from human studies suggests that Aldo is increased (Bentley-Lewis et al. 2007) and sex hormones may be dysregulated in the context of obesity (Poddar et al. 2017), which may activate EC-MR to promote vascular oxidative stress in obese females. This is supported by a separate study of ovariectomized female spontaneously hypertensive rats, in which estrogen replacement attenuated oxidative stress in the coronary arteries, while the addition of drospirenone, a progestin with anti-MR activity, had no additional effect in estrogen-replete females (Borgo et al. 2016).
4. Crosstalk Between the Mineralocorticoid Receptor and Sex Hormones
A. Sex-Dependent Regulation of Aldo Production
Women tend to have higher levels of circulating Aldo than men, both at baseline (Sequeira et al. 1986) and in pathogenic states (Szymanski et al. 2011; Shukri et al. 2018; Bentley-Lewis et al. 2007), a finding that has been recapitulated in rodent models (Tang 1985; Davel et al. 2018b; Faulkner and Belin de Chantemele 2018). Sex-specific effects on Aldo levels may be related to sex differences in adrenal Aldo production in the zona glomerulosa (ZG). Female rat ZG cells produced more Aldo at baseline than cells from male rats (Huang et al. 2018), and Aldo synthase expression was increased in female mice exposed to either leptin sensitization or obesity, resulting in higher circulating Aldo levels and blood pressure (Huby et al. 2016). By contrast, female rats exhibit a higher Aldo clearance rate than males (Morris et al. 1975), suggesting sex-specific control of Aldo balance at both the production and excretion levels.
The mechanism for the sex dependence of Aldo homeostasis may also be related to sex hormones other than estrogen. Women in the luteal phase of the menstrual cycle, when progesterone levels are highest, have higher Aldo levels than in the follicular phase, when progesterone levels drop (Szmuilowicz et al. 2006). This study also observed an increase in Aldo production in the ZG cells of female rats when treated with progesterone. It is unclear whether serum levels of glucocorticoids, which can also activate the MR, follow the same pattern, as two small studies show discrepant results: one study of 5 women found that both Aldo and corticosterone were higher in the luteal phase than the follicular (Schwartz and Abraham 1975), while another study found no difference in cortisol levels between the phases in 4 women (Stewart et al. 1993). The progesterone-related effect on Aldo production may be due to increased secretion of the hormone, rather than synthesis, as a separate study found that progesterone inhibited the Aldo synthase enzyme in transfected cells (Vecchiola et al. 2013).
By contrast, data support that estrogen likely does not to influence Aldo production. Estrogen did not correlate with Aldo levels in the above study of menstrual cycle variation, nor did estrogen alter Aldo production in rat ZG cells (Szmuilowicz et al. 2006) or in a separate study of human adrenocortical cells (Yanes and Romero 2009). Estrogen also did not affect the activity of the Aldo synthase enzyme in transfected cells (Vecchiola et al. 2013). Consistent with these results, a study of human adrenocortical cells found that estrogen increased Aldo production only when ERβ was inhibited, indicating that ERβ may prevent Aldo secretion that may otherwise occur with estrogen exposure (Caroccia et al. 2014).
Testosterone and AR signaling also appear to influence Aldo production, with opposite effects depending on the timing of exposure. AR signaling during prenatal development promotes Aldo production in male offspring (Martinez-Arguelles et al. 2011), while in adult male animals AR signaling inhibits Aldo production (Kau et al. 1999; Hofmann et al. 2012; Carsia et al. 2018). It is unclear whether AR signaling may regulate Aldo production in females, as one study demonstrated decreased Aldo production in testosterone-treated female geckos (Carsia et al. 2018) while another study observed no effect on Aldo production in ovariectomized female rats treated with an AR inhibitor (Hofmann et al. 2012). Thus, Aldo levels in males and females appear to be regulated by sex steroid hormone signaling. The female sex hormone progesterone generally upregulates Aldo production in women and in rat adrenal cells, while testosterone inhibits Aldo production in adult male animals (Figure 1B-i).
B. Interactions Between the MR and Estrogen Receptors
In addition to sex differences in Aldo levels that may result in differential MR activation in males and females, there is also evidence that the MR can interact with sex hormone receptors directly in the effector cells, providing another mechanistic link between MR signaling and sex differences in cardiovascular disease. In particular, the α and β isoforms of the ER have been shown to modulate MR function. In a study of human ECs in vitro, ERα activation triggered the formation of a complex containing ERα and the MR that inhibited MR transcriptional function. This repression of the MR required ERα to be able to translocate to the nucleus but did not involve the DNA-binding domain or the rapid non-genomic signaling functions of ERα (Figure 1B-ii). In functional assays, Aldo treatment of human ECs induced ICAM-1 expression and leukocyte adhesion, effects that were blocked by co-administration of estrogen (Barrett Mueller et al. 2014). Recently, EC-MR was shown to regulate ICAM-1 and E-selectin only in males in vivo or in the absence of estrogen in vitro, further implicating MR-estrogen crosstalk in the regulation of endothelial inflammatory mediators (Moss et al. 2019). Other data also suggests that the MR can interact with ERs in vascular SMCs: in these cells, both ERα and ERβ attenuated Aldo-induced oxidative stress (Muehlfelder et al. 2012), suggesting that ERβ, like ERα, may antagonize MR-mediated processes in the vasculature, through mechanisms that have not yet been elucidated (Figure 1B-iii). Thus, interactions between the MR and ERs are likely not limited to ECs but may occur in many cell types throughout the body. Gene expression profiling has been performed in vascular tissue to describe the gene sets activated by estrogen (Schnoes et al. 2008), ERα and ERβ (O’Lone et al. 2007), and Aldo (Newfell et al. 2011). Independent pathway analyses from these studies supports that the MR activates genes in the vasculature related to oxidative stress and inflammation, while estrogen signaling appears to inhibit similar pathways. Direct comparison of the data sets described in these three publications and further studies on the impact of estrogen signaling on MR-mediated vascular gene expression could provide exciting insight into potential genomic crosstalk between the MR and ERs.
Current evidence suggests additional non-genomic interactions between estrogen- and Aldo-mediated pathways via the scaffolding protein striatin (Figure 1B-iii). In a cultured human EC line (EAhy.926), the scaffolding protein striatin recruits ERα to the caveolar membrane, thus facilitating its activation by estradiol and rapid downstream phosphorylation and activation of eNOS (Lu et al. 2004). Striatin protein expression is upregulated by the MR in EAhy.926 cells (Pojoga et al. 2012), and striatin facilitates the non-genomic phosphorylation of ERK1/2 and induction of ROS observed upon MR activation (Coutinho et al. 2014; Grossmann et al. 2005). The striatin pathway appears to confer protection from salt sensitivity of blood pressure, at least in male rodents (Garza et al. 2015). Thus, the binding of both the MR and ERα to the striatin scaffold could be an additional mechanism for interactions between the non-genomic functions of these two receptors. However, as this link is currently only circumstantial, a direct investigation into this mechanism is certainly warranted.
An additional emerging mechanism for nongenomic crosstalk between MR and estrogen signaling is via the G-protein coupled ER (GPER), which was first described as a mediator of rapid estrogen effects (Filardo et al. 2000). Since then, various pharmacologic and genetic perturbations of this receptor have shown it to be involved in a number of disease processes from obesity and metabolic syndrome to inflammation, often in a sex-specific manner (Sharma and Prossnitz 2017). Further studies implicate GPER as a potential mediator of nongenomic Aldo signaling as well, though whether this occurs via direct binding of Aldo to GPER or downstream of traditional binding of Aldo to the MR is still controversial (Figure 1B-iv). GPER was first suggested as an Aldo-binding receptor by Gros et al. (2011), wherein the authors demonstrated that Aldo can induce ERK1/2 phosphorylation and apoptosis in rat vascular SMCs infected with MR or GPER overexpression vectors, effects which could be inhibited by the GPER antagonist G15. Subsequently, Aldo stimulation of ERK1/2 phosphorylation was shown to be blocked by either G15 or short hairpin RNA-mediated knockdown of GPER in rat vascular ECs (Gros et al. 2013). By contrast, Ferreira et al. (2015) showed that GPER was involved in Aldo-induced vasoconstriction but not vasodilation in the mesenteric resistance arteries of female mice, suggesting GPER effects on SMCs but not ECs. Finally, Aldo induces vasoconstriction of the afferent renal arteriole, an effect that can be blocked by GPER inhibition (Ren et al. 2016). While these data implicate GPER in the rapid effects of Aldo on vascular cells, it is not clear that GPER is the sole mediator of nongenomic Aldo signaling. In all of the above-mentioned studies, the addition of MR antagonists eplerenone and spironolactone blocked the rapid effects attributed to GPER, as did siRNA knockdown of the MR in a study of breast cancer cell lines (Rigiracciolo et al. 2016). This indicates that the canonical MR is also involved in the rapid, nongenomic effects of Aldo. Further, data from Cheng et al. (2014) suggested that Aldo may not directly bind to GPER, furthering the controversy over whether GPER may be a novel Aldo receptor.
Despite the controversy, growing evidence supports that GPER and the MR mediate rapid Aldo-induced signaling in concert (Figure 1B-iv). Indeed, GPER and the MR have been shown to colocalize in the presence of Aldo in breast cancer cell lines (Rigiracciolo et al. 2016). Further studies to enhance our understanding of the potential interactions between the MR, Aldo, estrogen, and GPER could provide additional insight into the mechanisms of sex differences observed in Aldo- and MR-mediated cardiovascular disease.
C. Interactions Between the MR and the Progesterone Receptor
Progesterone and the progesterone receptor (PR) may also interact with the MR, providing yet another link between MR signaling and female sex hormones. Whereas progesterone levels positively correlate with Aldo secretion as described above, progesterone itself can bind to and inhibit the MR in mammalian cells (Rupprecht et al. 1993; Mooij et al. 2015) (Figure 1B-v). While progesterone negatively regulates the wild-type MR, a point mutation in the ligand-binding domain of the MR has been identified that instead leads to activation of the MR by progesterone, resulting in early-onset hypertension and severe pregnancy-associated hypertension (Geller et al. 2000). PR has also been shown to inhibit MR transcriptional activity (McDonnell et al. 1994), while the MR may in turn activate the PR. Aldo promoted cell spreading and F-actin stress fiber formation in PR-positive breast cancer cells, an effect that was largely absent in PR-negative cells (Leo et al. 2004) (Figure 1B-v). However, this paradigm of mutual signaling is tentative at best, and specific study of the interactions between the MR and PR will be critical to understanding what role, if any, progestin signaling may have in MR-mediated cardiovascular disease.
D. Interactions Between the MR and the Androgen Receptor
The majority of data concerning potential interactions between MR and AR signaling comes from the prostate cancer literature. Prostate cancer cell lines have been shown to express 11βHSD2, thereby confering Aldo specificity to the MR by modifying cortisol to the MR-inactive cortisone (Page et al. 1994). In a recent study, Aldo treatment sensitized prostate cancer cells to the AR inhibitor enzalutamide and MR knockdown increased AR expression (Shiota et al. 2018). By contrast, testosterone and its active metabolite dihydrotestosterone have been shown to bind to and inhibit the MR (Takeda et al. 2007). In addition, the AR coactivator XRCC6 can also bind to the MR, inhibiting its transcription of target genes in the H9c2 embryonic cardiac myocyte cell line (Yang et al. 2014). Of note, this latter result is the only evidence to date linking the MR to the AR in a cardiovascular-relevant cell type and this paradigm has not been studied in ECs. Taken together, these data suggest a feedback model in which MR activation suppresses AR expression and activity, and androgens themselves may in turn inhibit the MR (Figure 1B-v). Much more study is needed to confirm this hypothesis and the explore possible physiologic ramifications of MR/AR crosstalk in the cardiovascular system.
5. Summary of the Molecular Mechanistic Data
The existing data indicates that the MR within the vascular endothelium contributes to cardiovascular disease via several molecular mechanisms. EC-MR regulates the expression of inflammatory adhesion molecules and promotes endothelial barrier permeability (Figure 1A-i), which together promote tissue inflammation in cardiovascular disease models such as atherosclerosis and heart failure. By regulating expression of the epithelial sodium channel and other ion channels in ECs, EC-MR promotes vascular stiffness, a precursor to hypertension and risk factor for atherosclerosis and cardiac dysfunction (Figure 1A-ii). By promoting oxidative stress and inhibiting NO availability, EC-MR contributes to impaired endothelial function in the setting of risk factors including hypertension, obesity and hyperlipidemia (Figure 1A-iii).
Where it has been studied, the role of EC-MR in promoting tissue inflammation, vascular stiffness, and endothelial dysfunction has often been found to be sex-specific. This is particularly evident in animal models of cardiovascular risk factors such as obesity and hyperlipidemia. This may be due to a variety of interactions between the MR and sex hormone receptors that either promote or inhibit MR activity. Progesterone and testosterone have been shown to regulate adrenal production of Aldo (Figure 1B-i); estrogen and progesterone receptors inhibit the genomic activity of the MR (Figure 1B-ii); the MR and various estrogen receptors may cooperate or inhibit one another in the context of rapid, non-genomic signaling (Figure 1B-iii–iv); and progesterone and testosterone themselves have been shown to directly inhibit the MR (Figure 1B-v). The MR may in turn regulate the activity of the androgen and progesterone receptors (Figure 1B-v). In our review of the literature, we found no evidence to suggest that sex hormones can directly activate the wild-type MR.
The current understanding of this complex ecosystem of ligands, receptors, and subcellular process regulation is limited by a scarcity of literature rigorously comparing differences in these mechanisms between the sexes. However, the available data suggests that in general, sex hormones tend to inhibit the harmful effects of the MR in the cardiovascular system. This paradigm could help to explain increases in cardiovascular disease risk in postmenopausal women, when ovarian hormone levels are low (Benjamin et al. 2018), and in men with low testosterone levels (Channer and Jones 2003; Rovira-Llopis et al. 2017). This may also be consistent with activated MR promoting cardiovascular disease even in premenopausal women who exhibit additional cardiovascular risk factors such as obesity, where hormone production and MR activity are often dysregulated (reviewed in Poddar et al. 2017). Certainly, further study is warranted to fully understand the nature of the interactions between the MR and sex hormones in order to adequately design therapies to combat cardiovascular disease in both sexes.
V. CONCLUSIONS AND PERSPECTIVES
In this review, we have explored the evidence for a sex-specific role for EC-MR in cardiovascular disease. In humans, the MR is involved in the pathophysiology of endothelial dysfunction, hypertension, atherosclerosis, and heart failure, and inhibition of the MR has been demonstrated to be beneficial in each of these conditions. In male animal models EC-MR has been shown to contribute to endothelial dysfunction in response to cardiovascular risk factors, to tissue inflammation, and to the adverse cardiac remodeling that occurs in models of heart failure and hypertension, without contributing to the blood pressure regulation itself. In women the role of the MR becomes evident after menopause, when preclinical data suggests that the MR may be more active due to low levels of MR-inhibiting sex hormones and increased Aldo levels in obese females. However, a role for the MR in cardiovascular disease can be observed even in premenopausal women if additional cardiovascular risk factors are present that may diminish the beneficial effects of female sex hormones. The specific role of EC-MR in cardiovascular pathology in females is just beginning to be understood, with data supporting a role for this receptor in endothelial dysfunction and cardiac diastolic dysfunction but not inflammation or hypertension. However, substantial further investigation is needed to fully appreciate the nuances of potential sex differences and sex hormone effects in many different models of cardiovascular disease.
An important limitation in this field is the paucity of women in clinical trials and, until recently, the nearly exclusive use of male animals in preclinical investigations. With the recent requirement by the National Institutes of Health that biological sex be addressed as an important variable in basic science and clinical research (McCullough et al. 2014), data is beginning to surface demonstrating sex differences in the role of the MR in cardiovascular disease, with mechanistic insights likely to expand. Further, tens of thousands of men and women have been randomized to MR antagonist therapy in clinical trials. Sub-analysis of this existing wealth of data by sex and equitable inclusion of women in future studies would provide excellent opportunities to understand sex differences in the renin-angiotensin-aldosterone system in human subjects.
It is critical to understand the differences between men and women in the etiology, natural history, and downstream consequences of cardiovascular pathology. Endothelial-specific MR may be a tantalizing factor mediating sex differences in endothelial dysfunction, atherosclerosis, and heart failure. By contrast, EC-MR may not contribute to sex differences in hypertension incidence, instead mediating the adverse consequences of elevated blood pressure. Additionally, the MR has been suggested to contribute to the pathophysiology of MI (Beygui et al. 2006), cardiac arrhythmia (Neefs et al. 2017), and certain pathologies of the heart valves (Liu et al. 2018), but whether there is an effect of sex or a role for EC-MR has not yet been explored. Such an understanding could profoundly impact the clinical management of male and female patients, with current and emerging new MR antagonists as versatile tools in the treatment of cardiovascular disease. Furthermore, understanding the molecular mechanisms driving sex differences in the role of EC-MR in cardiovascular disease has the potential to nominate additional therapeutic targets downstream of EC-MR that could allow for tailored treatment of cardiovascular disease to improve outcomes in both men and women.
Acknowledgments
Funding Sources
This work was funded by grants from the National Institutes of Health (NHLBI HL095590 [to IZJ] and F30HL137255 [to MEM]) and from the American Heart Association (EIA18290005 [to IZJ]).
List of Abbreviations
- 11βHSD2
11β-hydroxysteroid dehydrogenase 2
- AAA
abdominal aortic aneurysm
- Aldo
aldosterone
- AR
androgen receptor
- AF
atrial fibrillation
- EC
endothelial cell
- EnNaC
endothelial epithelial sodium channel
- eNOS
endothelial nitric oxide synthase
- ER
estrogen receptor
- ERK
extracellular signal-related kinase
- GPER
G protein-coupled estrogen receptor
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- ICAM-1
intracellular adhesion molecule-1
- MR
mineralocorticoid receptor
- MI
myocardial infarction
- NO
nitric oxide
- PR
progesterone receptor
- ROS
reactive oxygen species
- SMCs
smooth muscle cells
- VCAM-1
vascular cell adhesion molecule-1
- ZG
zona glomerulosa
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no conflicts of interest.
REFERENCES CITED
- Amador CA, Bertocchio JP, Andre-Gregoire G, Placier S, Duong Van Huyen JP, El Moghrabi S, Berger S, Warnock DG, Chatziantoniou C, Jaffe IZ, Rieu P, and Jaisser F 2016. ‘Deletion of mineralocorticoid receptors in smooth muscle cells blunts renal vascular resistance following acute cyclosporine administration’, Kidney Int, 89: 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold AP, Cassis LA, Eghbali M, Reue K, and Sandberg K 2017. ‘Sex Hormones and Sex Chromosomes Cause Sex Differences in the Development of Cardiovascular Diseases’, Arterioscler Thromb Vasc Biol, 37: 746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroor AR, Habibi J, Nistala R, Ramirez-Perez FI, Martinez-Lemus LA, Jaffe IZ, Sowers JR, Jia G, and Whaley-Connell A 2019. ‘Diet-Induced Obesity Promotes Kidney Endothelial Stiffening and Fibrosis Dependent on the Endothelial Mineralocorticoid Receptor’, Hypertension, 73: 849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, and Evans RM 1987. ‘Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor’, Science, 237: 268–75. [DOI] [PubMed] [Google Scholar]
- Barquera S, Pedroza-Tobias A, Medina C, Hernandez-Barrera L, Bibbins-Domingo K, Lozano R, and Moran AE 2015. ‘Global Overview of the Epidemiology of Atherosclerotic Cardiovascular Disease’, Arch Med Res, 46: 328–38. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor EL, Cohn BA, Wingard DL, and Edelstein SL 1991. ‘Why is diabetes mellitus a stronger risk factor for fatal ischemic heart disease in women than in men? The Rancho Bernardo Study’, JAMA, 265: 627–31. [PubMed] [Google Scholar]
- Barrett Mueller K, Lu Q, Mohammad NN, Luu V, McCurley A, Williams GH, Adler GK, Karas RH, and Jaffe IZ 2014. ‘Estrogen receptor inhibits mineralocorticoid receptor transcriptional regulatory function’, Endocrinology, 155: 4461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bene NC, Alcaide P, Wortis HH, and Jaffe IZ 2014. ‘Mineralocorticoid receptors in immune cells: emerging role in cardiovascular disease’, Steroids, 91: 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Council on, Epidemiology, Prevention Statistics, Committee, and Stroke Statistics, Subcommittee. 2018. ‘Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association’, Circulation, 137: e67–e492. [DOI] [PubMed] [Google Scholar]
- Bentley-Lewis R, Adler GK, Perlstein T, Seely EW, Hopkins PN, Williams GH, and Garg R 2007. ‘Body mass index predicts aldosterone production in normotensive adults on a high-salt diet’, J Clin Endocrinol Metab, 92: 4472–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beygui F, Collet JP, Benoliel JJ, Vignolles N, Dumaine R, Barthelemy O, and Montalescot G 2006. ‘High plasma aldosterone levels on admission are associated with death in patients presenting with acute ST-elevation myocardial infarction’, Circulation, 114: 2604–10. [DOI] [PubMed] [Google Scholar]
- Biwer LA, Wallingford MC, and Jaffe IZ 2019. ‘Vascular Mineralocorticoid Receptor: Evolutionary Mediator of Wound Healing Turned Harmful by Our Modern Lifestyle’, Am J Hypertens, 32: 123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boese AC, Kim SC, Yin KJ, Lee JP, and Hamblin MH 2017. ‘Sex differences in vascular physiology and pathophysiology: estrogen and androgen signaling in health and disease’, Am. J. Physiol Heart Circ. Physiol, 313: H524–H45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgo MV, Claudio ER, Silva FB, Romero WG, Gouvea SA, Moyses MR, Santos RL, Almeida SA, Podratz PL, Graceli JB, and Abreu GR 2016. ‘Hormonal therapy with estradiol and drospirenone improves endothelium-dependent vasodilation in the coronary bed of ovariectomized spontaneously hypertensive rats’, Braz J Med Biol Res, 49: e4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Habibi J, DeMarco VG, Jia G, Domeier TL, Lambert MD, Aroor AR, Nistala R, Bender SB, Garro M, Hayden MR, Ma L, Manrique C, and Sowers JR 2015. ‘Mineralocorticoid receptor blockade prevents Western diet-induced diastolic dysfunction in female mice’, Am J Physiol Heart Circ Physiol, 308: H1126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdillon MC, Poston RN, Covacho C, Chignier E, Bricca G, and McGregor JL 2000. ‘ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(−/−)/ICAM-1(−/−)) fed a fat or a chow diet’, Arterioscler Thromb Vasc Biol, 20: 2630–5. [DOI] [PubMed] [Google Scholar]
- Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Correa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, Sorisky A, Ooi TC, Ruzicka M, Burns KD, and Touyz RM 2012. ‘Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction’, Hypertension, 59: 1069–78. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Enciso JS, Gersh BJ, Grady C, Rice MM, Singh S, Sopko G, Boineau R, Rosenberg Y, and Greenberg BH 2016. ‘Detection and Management of Geographic Disparities in the TOPCAT Trial: Lessons Learned and Derivative Recommendations’, JACC Basic Transl Sci, 1: 180–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, and Jaffe IZ 2008. ‘Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion’, Circ. Res, 102: 1359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroccia B, Seccia TM, Campos AG, Gioco F, Kuppusamy M, Ceolotto G, Guerzoni E, Simonato F, Mareso S, Lenzini L, Fassina A, and Rossi GP 2014. ‘GPER-1 and estrogen receptor-beta ligands modulate aldosterone synthesis’, Endocrinology, 155: 4296–304. [DOI] [PubMed] [Google Scholar]
- Carsia RV, McIlroy PJ, and John-Alder HB 2018. ‘Modulation of adrenal steroidogenesis by testosterone in the lizard, Coleonyx elegans’, Gen Comp Endocrinol, 259: 93–103. [DOI] [PubMed] [Google Scholar]
- Cascella T, Palomba S, Tauchmanova L, Manguso F, Di Biase S, Labella D, Giallauria F, Vigorito C, Colao A, Lombardi G, and Orio F 2006. ‘Serum aldosterone concentration and cardiovascular risk in women with polycystic ovarian syndrome’, J Clin Endocrinol Metab, 91: 4395–400. [DOI] [PubMed] [Google Scholar]
- Channer KS, and Jones TH 2003. ‘Cardiovascular effects of testosterone: implications of the “male menopause”?’, Heart, 89: 121–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SB, Dong J, Pang Y, LaRocca J, Hixon M, Thomas P, and Filardo EJ 2014. ‘Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone’, Mol Cell Endocrinol, 382: 950–9. [DOI] [PubMed] [Google Scholar]
- Christy C, Hadoke PW, Paterson JM, Mullins JJ, Seckl JR, and Walker BR 2003. ‘11beta-hydroxysteroid dehydrogenase type 2 in mouse aorta: localization and influence on response to glucocorticoids’, Hypertension, 42: 580–7. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Vega C, Pojoga LH, Rivera A, Prado GN, Yao TM, Adler G, Torres-Grajales M, Maldonado ER, Ramos-Rivera A, Williams JS, Williams G, and Romero JR 2014. ‘Aldosterone’s rapid, nongenomic effects are mediated by striatin: a modulator of aldosterone’s effect on estrogen action’, Endocrinology, 155: 2233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel KR, Wells G, Stewart K, Moore B, and Kitzman DW 2009. ‘Effect of aldosterone antagonism on exercise tolerance, Doppler diastolic function, and quality of life in older women with diastolic heart failure’, Congest Heart Fail, 15: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davel AP, Anwar IJ, and Jaffe IZ 2017. ‘The endothelial mineralocorticoid receptor: mediator of the switch from vascular health to disease’, Curr Opin Nephrol Hypertens, 26: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davel AP, Jaffe IZ, Tostes RC, Jaisser F, and Belin de Chantemele EJ 2018a. ‘New roles of aldosterone and mineralocorticoid receptors in cardiovascular disease: translational and sex-specific effects’, Am J Physiol Heart Circ Physiol, 315: H989–H99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davel AP, Lu Q, Moss ME, Rao S, Anwar IJ, DuPont JJ, and Jaffe IZ 2018b. ‘Sex-Specific Mechanisms of Resistance Vessel Endothelial Dysfunction Induced by Cardiometabolic Risk Factors’, J Am Heart Assoc, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rita O, Hackam DG, and Spence JD 2012. ‘Effects of aldosterone on human atherosclerosis: plasma aldosterone and progression of carotid plaque’, Can. J. Cardiol, 28: 706–11. [DOI] [PubMed] [Google Scholar]
- DeMarco VG, Habibi J, Jia G, Aroor AR, Ramirez-Perez FI, Martinez-Lemus LA, Bender SB, Garro M, Hayden MR, Sun Z, Meininger GA, Manrique C, Whaley-Connell A, and Sowers JR 2015. ‘Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice’, Hypertension, 66: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, and Scherer PE 2010. ‘Adipokines as novel biomarkers and regulators of the metabolic syndrome’, Ann N Y Acad Sci, 1212: E1–E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuchar GA, McLean D, Hadoke PW, Brownstein DG, Webb DJ, Mullins JJ, Chapman K, Seckl JR, and Kotelevtsev YV 2011. ‘11beta-hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe−/− mice’, Endocrinology, 152: 236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Otero JM, Fisher C, Downs K, Moss ME, Jaffe IZ, Jackson WF, and Dorrance AM 2017. ‘Endothelial Mineralocorticoid Receptor Mediates Parenchymal Arteriole and Posterior Cerebral Artery Remodeling During Angiotensin II-Induced Hypertension’, Hypertension, 70: 1113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Otero JM, Yen TC, Fisher C, Bota D, Jackson WF, and Dorrance AM 2018. ‘Mineralocorticoid receptor antagonism improves parenchymal arteriole dilation via a TRPV4-dependent mechanism and prevents cognitive dysfunction in hypertension’, Am J Physiol Heart Circ Physiol, 315: H1304–H15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh QN, Young MJ, Evans MA, Drummond GR, Sobey CG, and Chrissobolis S 2016. ‘Aldosterone-induced oxidative stress and inflammation in the brain are mediated by the endothelial cell mineralocorticoid receptor’, Brain Res, 1637: 146–53. [DOI] [PubMed] [Google Scholar]
- Dudenbostel T, and Calhoun DA 2017. ‘Use of Aldosterone Antagonists for Treatment of Uncontrolled Resistant Hypertension’, Am J Hypertens, 30: 103–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlay SM, Roger VL, and Redfield MM 2017. ‘Epidemiology of heart failure with preserved ejection fraction’, Nat Rev Cardiol, 14: 591–602. [DOI] [PubMed] [Google Scholar]
- DuPont JJ, Hill MA, Bender SB, Jaisser F, and Jaffe IZ 2014. ‘Aldosterone and vascular mineralocorticoid receptors: regulators of ion channels beyond the kidney’, Hypertension, 63: 632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont JJ, and Jaffe IZ 2017. ‘30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The role of the mineralocorticoid receptor in the vasculature’, J Endocrinol, 234: T67–T82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA, and Jaffe IZ 2016. ‘Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging’, JCI. Insight, 1: e88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann F, Wachter R, Schmidt AG, Kraigher-Krainer E, Colantonio C, Kamke W, Duvinage A, Stahrenberg R, Durstewitz K, Loffler M, Dungen HD, Tschope C, Herrmann-Lingen C, Halle M, Hasenfuss G, Gelbrich G, Pieske B, and Aldo DHF Investigators 2013. ‘Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial’, JAMA, 309: 781–91. [DOI] [PubMed] [Google Scholar]
- Farquharson CA, and Struthers AD 2000. ‘Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure’, Circulation, 101: 594–7. [DOI] [PubMed] [Google Scholar]
- Faulkner JL, and Belin de Chantemele EJ 2018. ‘Sex Differences in Mechanisms of Hypertension Associated With Obesity’, Hypertension, 71: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JL, and Belin de Chantemele EJ 2019. ‘Leptin and Aldosterone’, Vitam Horm, 109: 265–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faxen UL, Hage C, Donal E, Daubert JC, Linde C, and Lund LH 2018. ‘Patient reported outcome in HFpEF: Sex-specific differences in quality of life and association with outcome’, Int J Cardiol, 267: 128–32. [DOI] [PubMed] [Google Scholar]
- Fernet M, Beckerman B, Abreu P, Lins K, Vincent J, and Burgess E 2018. ‘Antihypertensive effect of the mineralocorticoid receptor antagonist eplerenone: a pooled analysis of patient-level data from comparative trials using regulatory-approved doses’, Vasc Health Risk Manag, 14: 233–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira NS, Cau SB, Silva MA, Manzato CP, Mestriner FL, Matsumoto T, Carneiro FS, and Tostes RC 2015. ‘Diabetes impairs the vascular effects of aldosterone mediated by G protein-coupled estrogen receptor activation’, Front Pharmacol, 6: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, and Frackelton AR Jr. 2000. ‘Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF’, Mol Endocrinol, 14: 1649–60. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, and Ogden CL 2016. ‘Trends in Obesity Among Adults in the United States, 2005 to 2014’, JAMA, 315: 2284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraccarollo D, Galuppo P, Hildemann S, Christ M, Ertl G, and Bauersachs J 2003. ‘Additive improvement of left ventricular remodeling and neurohormonal activation by aldosterone receptor blockade with eplerenone and ACE inhibition in rats with myocardial infarction’, J Am Coll Cardiol, 42: 1666–73. [DOI] [PubMed] [Google Scholar]
- Fryar CD, Ostchega Y, Hales CM, Zhang G, and Kruszon-Moran D 2017. ‘Hypertension Prevalence and Control Among Adults: United States, 2015–2016’, NCHS Data Brief. 1–8. [PubMed] [Google Scholar]
- Fujimura N, Noma K, Hata T, Soga J, Hidaka T, Idei N, Fujii Y, Mikami S, Maruhashi T, Iwamoto Y, Kihara Y, Chayama K, Kato H, Liao JK, Higashi Y, and Group, Rock Study. 2012. ‘Mineralocorticoid receptor blocker eplerenone improves endothelial function and inhibits Rho-associated kinase activity in patients with hypertension’, Clin Pharmacol Ther, 91: 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuta H, Goto T, Wakami K, Kamiya T, and Ohte N 2018. ‘Effects of mineralocorticoid receptor antagonists on left ventricular diastolic function, exercise capacity, and quality of life in heart failure with preserved ejection fraction: a meta-analysis of randomized controlled trials’, Heart Vessels. [DOI] [PubMed] [Google Scholar]
- Funder JW 2010. ‘Minireview: Aldosterone and mineralocorticoid receptors: past, present, and future’, Endocrinology, 151: 5098–102. [DOI] [PubMed] [Google Scholar]
- Furuzono S, Meguro M, Miyauchi S, Inoue S, Homma T, Yamada K, Tagawa YI, Nara F, and Nagayama T 2017. ‘A novel aldosterone synthase inhibitor ameliorates mortality in pressure-overload mice with heart failure’, Eur J Pharmacol, 795: 58–65. [DOI] [PubMed] [Google Scholar]
- Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, and Jaisser F 2014. ‘Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness’, Hypertension, 63: 520–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Kneen L, Williams GH, and Adler GK 2014. ‘Effect of mineralocorticoid receptor antagonist on insulin resistance and endothelial function in obese subjects’, Diabetes Obes Metab, 16: 268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Rao AD, Baimas-George M, Hurwitz S, Foster C, Shah RV, Jerosch-Herold M, Kwong RY, Di Carli MF, and Adler GK 2015. ‘Mineralocorticoid receptor blockade improves coronary microvascular function in individuals with type 2 diabetes’, Diabetes, 64: 236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza AE, Rariy CM, Sun B, Williams J, Lasky-Su J, Baudrand R, Yao T, Moize B, Hafiz WM, Romero JR, Adler GK, Ferri C, Hopkins PN, Pojoga LH, and Williams GH 2015. ‘Variants in striatin gene are associated with salt-sensitive blood pressure in mice and humans’, Hypertension, 65: 211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB, and Lifton RP 2000. ‘Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy’, Science, 289: 119–23. [DOI] [PubMed] [Google Scholar]
- Glicklich D, and Frishman WH 2015. ‘Drug therapy of apparent treatment-resistant hypertension: focus on mineralocorticoid receptor antagonists’, Drugs, 75: 473–85. [DOI] [PubMed] [Google Scholar]
- Gong R, Morris DJ, and Brem AS 2008. ‘Variable expression of 11beta Hydroxysteroid dehydrogenase (11beta-HSD) isoforms in vascular endothelial cells’, Steroids, 73: 1187–96. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Blazquez R, Somoza B, Gil-Ortega M, Martin Ramos M, Ramiro-Cortijo D, Vega-Martin E, Schulz A, Ruilope LM, Kolkhof P, Kreutz R, and Fernandez-Alfonso MS 2018. ‘Finerenone Attenuates Endothelial Dysfunction and Albuminuria in a Chronic Kidney Disease Model by a Reduction in Oxidative Stress’, Front Pharmacol, 9: 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdy P, Mallat Z, Castano C, Garmy-Susini B, Mac Gregor JL, Tedgui A, Arnal JF, and Bayard F 2003. ‘The atheroprotective effect of 17 beta-estradiol is not altered in P-selectin- or ICAM-1-deficient hypercholesterolemic mice’, Atherosclerosis, 166: 41–8. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Liu B, Chorazyczewski J, and Feldman RD 2013. ‘Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation’, Am J Physiol Cell Physiol, 304: C532–40. [DOI] [PubMed] [Google Scholar]
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, and Feldman RD 2011. ‘GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone’, Hypertension, 57: 442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann C, Benesic A, Krug AW, Freudinger R, Mildenberger S, Gassner B, and Gekle M 2005. ‘Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions’, Mol Endocrinol, 19: 1697–710. [DOI] [PubMed] [Google Scholar]
- Hamburg NM, Palmisano J, Larson MG, Sullivan LM, Lehman BT, Vasan RS, Levy D, Mitchell GF, Vita JA, and Benjamin EJ 2011. ‘Relation of brachial and digital measures of vascular function in the community: the Framingham heart study’, Hypertension, 57: 390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashikabe Y, Suzuki K, Jojima T, Uchida K, and Hattori Y 2006. ‘Aldosterone impairs vascular endothelial cell function’, J Cardiovasc Pharmacol, 47: 609–13. [DOI] [PubMed] [Google Scholar]
- Hofmann PJ, Michaelis M, Gotz F, Bartel C, Kienitz T, and Quinkler M 2012. ‘Flutamide increases aldosterone levels in gonadectomized male but not female Wistar rats’, Am J Hypertens, 25: 697–703. [DOI] [PubMed] [Google Scholar]
- Huang Y, Ting PY, Yao TM, Homma T, Brooks D, Katayama Rangel IA, Adler GK, Romero JR, Williams JS, Pojoga LH, and Williams GH 2018. ‘Histone demethylase LSD1 and biological sex: impact on blood pressure and aldosterone production’, J Endocrinol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huby AC, Otvos L Jr., and Belin de Chantemele EJ 2016. ‘Leptin Induces Hypertension and Endothelial Dysfunction via Aldosterone-Dependent Mechanisms in Obese Female Mice’, Hypertension, 67: 1020–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, Daemen MJ, and Hordijk PL 2015. ‘Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease’, Circ Res, 116: 895–908. [DOI] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Nichols WW, and Christou DD 2013a. ‘Role of mineralocorticoid receptors in arterial stiffness in human aging’, Exp Gerontol, 48: 701–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Segal MS, and Christou DD 2013b. ‘Mineralocorticoid receptors modulate vascular endothelial function in human obesity’, Clin Sci (Lond), 125: 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Talcott S, Jaffe IZ, and Christou DD 2016. ‘Acute effect of mineralocorticoid receptor antagonism on vascular function in healthy older adults’, Exp Gerontol, 73: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Meade TH, English M, and Christou DD 2015. ‘Effect of Selective Mineralocorticoid Receptor Blockade on Flow-Mediated Dilation and Insulin Resistance in Older Adults with Metabolic Syndrome’, Metab Syndr Relat Disord, 13: 356–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanes F, Susen S, Mouquet F, Pigny P, Cuilleret F, Sautiere K, Collet JP, Beygui F, Hennache B, Ennezat PV, Juthier F, Richard F, Dallongeville J, Hillaert MA, Doevendans PA, Jude B, Bertrand M, Montalescot G, and van, Belle E 2012. ‘Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure’, Eur. Heart J, 33: 191–202. [DOI] [PubMed] [Google Scholar]
- Jaffe IZ, and Mendelsohn ME 2005. ‘Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells’, Circ Res, 96: 643–50. [DOI] [PubMed] [Google Scholar]
- Jaffe IZ, Tintut Y, Newfell BG, Demer LL, and Mendelsohn ME 2007. ‘Mineralocorticoid receptor activation promotes vascular cell calcification’, Arterioscler Thromb Vasc Biol, 27: 799–805. [DOI] [PubMed] [Google Scholar]
- Jeong Y, Chaupin DF, Matsushita K, Yamakuchi M, Cameron SJ, Morrell CN, and Lowenstein CJ 2009. ‘Aldosterone activates endothelial exocytosis’, Proc Natl Acad Sci U S A, 106: 3782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Aroor AR, DeMarco VG, Martinez-Lemus LA, Meininger GA, and Sowers JR 2015a. ‘Vascular stiffness in insulin resistance and obesity’, Front Physiol, 6: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Habibi J, Aroor AR, Hill MA, DeMarco VG, Lee LE, Ma L, Barron BJ, Whaley-Connell A, and Sowers JR 2018a. ‘Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice’, Metabolism, 78: 69–79. [DOI] [PubMed] [Google Scholar]
- Jia G, Habibi J, Aroor AR, Hill MA, Yang Y, Whaley-Connell A, Jaisser F, and Sowers JR 2018b. ‘Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction’, Hypertension, 72: 731–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, and Sowers JR 2016. ‘Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females’, Circ Res, 118: 935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, Aroor AR, Domeier TL, Zhu Y, Meininger GA, Barrett, Mueller K, Jaffe IZ, and Sowers JR 2015b. ‘Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females’, Hypertension, 66: 1159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens G, and Graudal NA 2004. ‘Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterols, and triglyceride’, Cochrane Database Syst Rev. CD004022. [DOI] [PubMed] [Google Scholar]
- Kanashiro-Takeuchi RM, Heidecker B, Lamirault G, Dharamsi JW, and Hare JM 2009. ‘Sex-specific impact of aldosterone receptor antagonism on ventricular remodeling and gene expression after myocardial infarction’, Clin Transl Sci, 2: 134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau MM, Lo MJ, Wang SW, Tsai SC, Chen JJ, Chiao YC, Yeh JY, Lin H, Shum AY, Fang VS, Ho LT, and Wang PS 1999. ‘Inhibition of aldosterone production by testosterone in male rats’, Metabolism, 48: 1108–14. [DOI] [PubMed] [Google Scholar]
- Keidar S, Hayek T, Kaplan M, Pavlotzky E, Hamoud S, Coleman R, and Aviram M 2003. ‘Effect of eplerenone, a selective aldosterone blocker, on blood pressure, serum and macrophage oxidative stress, and atherosclerosis in apolipoprotein E-deficient mice’, J. Cardiovasc. Pharmacol, 41: 955–63. [DOI] [PubMed] [Google Scholar]
- Khosla N, Kalaitzidis R, and Bakris GL 2009. ‘Predictors of hyperkalemia risk following hypertension control with aldosterone blockade’, Am J Nephrol, 30: 418–24. [DOI] [PubMed] [Google Scholar]
- Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, Karumanchi SA, Christou DD, and Jaffe IZ 2018. ‘Smooth Muscle Cell-Mineralocorticoid Receptor as a Mediator of Cardiovascular Stiffness With Aging’, Hypertension, 71: 609–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch T, Beese M, Wyss K, Klinge U, Haller H, Haubitz M, and Fiebeler A 2013. ‘Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton’, Hypertension, 61: 501–08. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Hara K, Tojo A, Onozato ML, Honda T, Yoshida K, Mita S, Nakano S, Tsubokou Y, and Matsuoka H 2005. ‘Eplerenone shows renoprotective effect by reducing LOX-1-mediated adhesion molecule, PKCepsilon-MAPK-p90RSK, and Rho-kinase pathway’, Hypertension, 45: 538–44. [DOI] [PubMed] [Google Scholar]
- Kosmala W, Rojek A, Przewlocka-Kosmala M, Wright L, Mysiak A, and Marwick TH 2016. ‘Effect of Aldosterone Antagonism on Exercise Tolerance in Heart Failure With Preserved Ejection Fraction’, J Am Coll Cardiol, 68: 1823–34. [DOI] [PubMed] [Google Scholar]
- Kratz MT, Schirmer SH, Baumhakel M, and Bohm M 2016. ‘Improvement of endothelial function in a murine model of mild cholesterol-induced atherosclerosis by mineralocorticoid antagonism’, Atherosclerosis, 251: 291–98. [DOI] [PubMed] [Google Scholar]
- Kusche-Vihrog K, Callies C, Fels J, and Oberleithner H 2010. ‘The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium?’, Steroids, 75: 544–9. [DOI] [PubMed] [Google Scholar]
- Laursen SB, Finsen S, Marcussen N, Quaggin SE, Hansen PBL, and Dimke H 2018. ‘Endothelial mineralocorticoid receptor ablation does not alter blood pressure, kidney function or renal vessel contractility’, PLoS One, 13: e0193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavall D, Selzer C, Schuster P, Lenski M, Adam O, Schafers HJ, Bohm M, and Laufs U 2014. ‘The mineralocorticoid receptor promotes fibrotic remodeling in atrial fibrillation’, J. Biol. Chem, 289: 6656–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo JC, Guo C, Woon CT, Aw SE, and Lin VC 2004. ‘Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells’, Endocrinology, 145: 1314–21. [DOI] [PubMed] [Google Scholar]
- Li C, Sun XN, Zeng MR, Zheng XJ, Zhang YY, Wan Q, Zhang WC, Shi C, Du LJ, Ai TJ, Liu Y, Liu Y, Du LL, Yi Y, Yu Y, and Duan SZ 2017a. ‘Mineralocorticoid Receptor Deficiency in T Cells Attenuates Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction Through Modulating T-Cell Activation’, Hypertension, 70: 137–47. [DOI] [PubMed] [Google Scholar]
- Li W, Chen X, Riley AM, Hiett SC, Temm CJ, Beli E, Long X, Chakraborty S, Alloosh M, White FA, Grant MB, Sturek M, and Obukhov AG 2017b. ‘Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs’, Basic Res Cardiol, 112: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cheng L, Liang H, Duan W, Hu J, Zhi W, Yang J, Liu Z, Zhao M, and Liu J 2016. ‘GPER inhibits diabetes-mediated RhoA activation to prevent vascular endothelial dysfunction’, Eur J Cell Biol, 95: 100–13. [DOI] [PubMed] [Google Scholar]
- Libby P, Lichtman AH, and Hansson GK 2013. ‘Immune effector mechanisms implicated in atherosclerosis: from mice to humans’, Immunity, 38: 1092–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WH, Fang YN, Wu CC, Chen MC, Chang JP, Lin YS, Pan KL, Ho WC, Chang TH, Huang YK, Fang CY, Chen CJ, and Lee WC 2018. ‘Differential Gene Expression Profile of Renin-Angiotensin System in the Left Atrium in Mitral Regurgitation Patients’, Dis Markers, 2018: 6924608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Mladinov D, Pietrusz JL, Usa K, and Liang M 2009. ‘Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression’, Cardiovasc Res, 81: 140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lother A, Furst D, Bergemann S, Gilsbach R, Grahammer F, Huber TB, Hilgendorf I, Bode C, Moser M, and Hein L 2016. ‘Deoxycorticosterone Acetate/Salt-Induced Cardiac But Not Renal Injury Is Mediated By Endothelial Mineralocorticoid Receptors Independently From Blood Pressure’, Hypertension, 67: 130–8. [DOI] [PubMed] [Google Scholar]
- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, and Karas RH 2004. ‘Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha’, Proc Natl Acad Sci U S A, 101: 17126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald JE, Kennedy N, and Struthers AD 2004. ‘Effects of spironolactone on endothelial function, vascular angiotensin converting enzyme activity, and other prognostic markers in patients with mild heart failure already taking optimal treatment’, Heart, 90: 765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manosroi W, Tan JW, Rariy CM, Sun B, Goodarzi MO, Saxena AR, Williams JS, Pojoga LH, Lasky-Su J, Cui J, Guo X, Taylor KD, Chen YI, Xiang AH, Hsueh WA, Raffel LJ, Buchanan TA, Rotter JI, Williams GH, and Seely EW 2017. ‘The Association of Estrogen Receptor-beta Gene Variation With Salt-Sensitive Blood Pressure’, J Clin Endocrinol Metab, 102: 4124–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique C, DeMarco VG, Aroor AR, Mugerfeld I, Garro M, Habibi J, Hayden MR, and Sowers JR 2013. ‘Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet’, Endocrinology, 154: 3632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak A, Nawrocka Rutkowska J, Brodowska A, Wisniewska B, and Starczewski A 2016. ‘Cardiovascular system diseases in patients with polycystic ovary syndrome - the role of inflammation process in this pathology and possibility of early diagnosis and prevention’, Ann Agric Environ Med, 23: 537–41. [DOI] [PubMed] [Google Scholar]
- Martinez-Arguelles DB, Guichard T, Culty M, Zirkin BR, and Papadopoulos V 2011. ‘In utero exposure to the antiandrogen di-(2-ethylhexyl) phthalate decreases adrenal aldosterone production in the adult rat’, Biol Reprod, 85: 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Selles M, Doughty RN, Poppe K, Whalley GA, Earle N, Tribouilloy C, McMurray JJ, Swedberg K, Kober L, Berry C, Squire I, and Meta-Analysis Global Group In Chronic Heart, Failure. 2012. ‘Gender and survival in patients with heart failure: interactions with diabetes and aetiology. Results from the MAGGIC individual patient meta-analysis’, Eur J Heart Fail, 14: 473–9. [DOI] [PubMed] [Google Scholar]
- Marzolla V, Armani A, Mammi C, Moss ME, Pagliarini V, Pontecorvo L, Antelmi A, Fabbri A, Rosano G, Jaffe IZ, and Caprio M 2017. ‘Essential role of ICAM-1 in aldosterone-induced atherosclerosis’, Int. J. Cardiol, 232: 233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Kawate H, Matsuzaki C, Sakamoto R, Shibue K, Ohnaka K, Anzai K, Nomura M, and Takayanagi R 2016. ‘Eplerenone improves carotid intima-media thickness (IMT) in patients with primary aldosteronism’, Endocr. J, 63: 249–55. [DOI] [PubMed] [Google Scholar]
- McCullough LD, de Vries GJ, Miller VM, Becker JB, Sandberg K, and McCarthy MM 2014. ‘NIH initiative to balance sex of animals in preclinical studies: generative questions to guide policy, implementation, and metrics’, Biol Sex Differ, 5: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, and Jaffe IZ 2012. ‘Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors’, Nat. Med, 18: 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, Shahbaz MM, Vegeto E, and Goldman ME 1994. ‘The human progesterone receptor A-form functions as a transcriptional modulator of mineralocorticoid receptor transcriptional activity’, J Steroid Biochem Mol Biol, 48: 425–32. [DOI] [PubMed] [Google Scholar]
- McGraw AP, Bagley J, Chen WS, Galayda C, Nickerson H, Armani A, Caprio M, Carmeliet P, and Jaffe IZ 2013. ‘Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism’, J. Am. Heart Assoc, 2: e000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill M, Sweitzer NK, Lindenfeld J, and Kao DP 2019. ‘Sex Differences in Outcomes and Responses to Spironolactone in Heart Failure With Preserved Ejection Fraction: A Secondary Analysis of TOPCAT Trial’, JACC Heart Fail, 7: 228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M, Hofmann PJ, Gotz F, Bartel C, Kienitz T, and Quinkler M 2012. ‘Sex-specific effects of spironolactone on blood pressure in gonadectomized male and female Wistar rats’, Horm Metab Res, 44: 291–5. [DOI] [PubMed] [Google Scholar]
- Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, and Mourad JJ 2005. ‘Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism’, J. Am. Coll. Cardiol, 45: 1243–48. [DOI] [PubMed] [Google Scholar]
- Mohandas A, Suboc TB, Wang J, Ying R, Tarima S, Dharmashankar K, Malik M, and Widlansky ME 2015. ‘Mineralocorticoid exposure and receptor activity modulate microvascular endothelial function in African Americans with and without hypertension’, Vasc. Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes-Cobos E, Li X, Fischer HJ, Sasse A, Kugler S, Didie M, Toischer K, Fassnacht M, Dressel R, and Reichardt HM 2015. ‘Inducible Knock-Down of the Mineralocorticoid Receptor in Mice Disturbs Regulation of the Renin-Angiotensin-Aldosterone System and Attenuates Heart Failure Induced by Pressure Overload’, PLoS One, 10: e0143954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooij CF, Parajes S, Pijnenburg-Kleizen KJ, Arlt W, Krone N, and Claahsen-van der Grinten HL 2015. ‘Influence of 17-Hydroxyprogesterone, Progesterone and Sex Steroids on Mineralocorticoid Receptor Transactivation in Congenital Adrenal Hyperplasia’, Horm Res Paediatr. [DOI] [PubMed] [Google Scholar]
- Morris DJ, Caron PC, Graham WC, Silverman JA, and DeConti GA 1975. ‘Sex dependence of clearance rates of aldosterone and its metabolites from plasma of intact rats’, Steroids, 25: 763–71. [DOI] [PubMed] [Google Scholar]
- Moss ME, DuPont JJ, Iyer SL, McGraw AP, and Jaffe IZ 2018. ‘No Significant Role for Smooth Muscle Cell Mineralocorticoid Receptors in Atherosclerosis in the Apolipoprotein-E Knockout Mouse Model’, Front Cardiovasc Med, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ME, Lu Q, Iyer SL, Engelbertsen D, Marzolla V, Caprio M, Lichtman AH, and Jaffe IZ 2019. ‘Endothelial Mineralocorticoid Receptors Contribute to Vascular Inflammation in Atherosclerosis in a Sex-Specific Manner’, Arterioscler Thromb Vasc Biol, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ME, and Jaffe IZ 2015. ‘Mineralocorticoid Receptors in the Pathophysiology of Vascular Inflammation and Atherosclerosis’, Front Endocrinol. (Lausanne), 6: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muehlfelder M, Arias-Loza PA, Fritzemeier KH, and Pelzer T 2012. ‘Both estrogen receptor subtypes, ERalpha and ERbeta, prevent aldosterone-induced oxidative stress in VSMC via increased NADPH bioavailability’, Biochem Biophys Res Commun, 423: 850–6. [DOI] [PubMed] [Google Scholar]
- Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, and Jaffe IZ 2015. ‘Endothelial Mineralocorticoid Receptors Differentially Contribute to Coronary and Mesenteric Vascular Function Without Modulating Blood Pressure’, Hypertension, 66: 988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Pacheco P, Ortega-Hernandez A, Caro-Vadillo A, Casanueva-Eliceiry S, Aragoncillo P, Egido J, Fernandez-Cruz A, and Gomez-Garre D 2013. ‘Eplerenone enhances cardioprotective effects of standard heart failure therapy through matricellular proteins in hypertensive heart failure’, J Hypertens, 31: 2309–18; discussion 19. [DOI] [PubMed] [Google Scholar]
- Naray-Fejes-Toth A, Colombowala IK, and Fejes-Toth G 1998. ‘The role of 11beta-hydroxysteroid dehydrogenase in steroid hormone specificity’, J Steroid Biochem Mol Biol, 65: 311–6. [DOI] [PubMed] [Google Scholar]
- Neefs J, van den Berg NW, Limpens J, Berger WR, Boekholdt SM, Sanders P, and de Groot JR 2017. ‘Aldosterone Pathway Blockade to Prevent Atrial Fibrillation: A Systematic Review and Meta-Analysis’, Int J Cardiol, 231: 155–61. [DOI] [PubMed] [Google Scholar]
- Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME, and Jaffe IZ 2011. ‘Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways’, Arterioscler. Thromb. Vasc. Biol, 31: 1871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Dinh Cat A, Callera GE, Friederich-Persson M, Sanchez A, Dulak-Lis MG, Tsiropoulou S, Montezano AC, He Y, Briones AM, Jaisser F, and Touyz RM 2018. ‘Vascular dysfunction in obese diabetic db/db mice involves the interplay between aldosterone/mineralocorticoid receptor and Rho kinase signaling’, Sci Rep, 8: 2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Dinh Cat A, Griol-Charhbili V, Loufrani L, Labat C, Benjamin L, Farman N, Lacolley P, Henrion D, and Jaisser F 2010. ‘The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure’, FASEB J, 24: 2454–63. [DOI] [PubMed] [Google Scholar]
- O’Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, and Hansen U 2007. ‘Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species’, Mol Endocrinol, 21: 1281–96. [DOI] [PubMed] [Google Scholar]
- Oviedo PJ, Sobrino A, Laguna-Fernandez A, Novella S, Tarin JJ, Garcia-Perez MA, Sanchis J, Cano A, and Hermenegildo C 2011. ‘Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway’, Mol Cell Endocrinol, 335: 96–103. [DOI] [PubMed] [Google Scholar]
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, and Redfield MM 2006. ‘Trends in prevalence and outcome of heart failure with preserved ejection fraction’, N Engl J Med, 355: 251–9. [DOI] [PubMed] [Google Scholar]
- Page N, Warriar N, and Govindan MV 1994. ‘11 beta-Hydroxysteroid dehydrogenase and tissue specificity of androgen action in human prostate cancer cell LNCaP’, J Steroid Biochem Mol Biol, 49: 173–81. [DOI] [PubMed] [Google Scholar]
- Pandey A, Garg S, Matulevicius SA, Shah AM, Garg J, Drazner MH, Amin A, Berry JD, Marwick TH, Marso SP, de Lemos JA, and Kumbhani DJ 2015. ‘Effect of Mineralocorticoid Receptor Antagonists on Cardiac Structure and Function in Patients With Diastolic Dysfunction and Heart Failure With Preserved Ejection Fraction: A Meta-Analysis and Systematic Review’, J Am Heart Assoc, 4: e002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradisi G, Steinberg HO, Hempfling A, Cronin J, Hook G, Shepard MK, and Baron AD 2001. ‘Polycystic ovary syndrome is associated with endothelial dysfunction’, Circulation, 103: 1410–5. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Claggett B, Assmann SF, Boineau R, Anand IS, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Heitner JF, Lewis EF, O’Meara E, Rouleau JL, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, McKinlay SM, and Pitt B 2015. ‘Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial’, Circulation, 131: 34–42. [DOI] [PubMed] [Google Scholar]
- Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O’Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, McKinlay SM, and Investigators, Topcat. 2014. ‘Spironolactone for heart failure with preserved ejection fraction’, N Engl J Med, 370: 1383–92. [DOI] [PubMed] [Google Scholar]
- Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, Roniker B, Kleiman J, Krause S, Burns D, and Williams GH 2003a. ‘Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study’, Circulation, 108: 1831–8. [DOI] [PubMed] [Google Scholar]
- Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleiman J, Gatlin M, Eplerenone Post-Acute Myocardial Infarction Heart Failure, Efficacy, and Survival Study, Investigators. 2003b. ‘Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction’, N Engl J Med, 348: 1309–21. [DOI] [PubMed] [Google Scholar]
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, and Wittes J 1999. ‘The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators’, N. Engl. J. Med, 341: 709–17. [DOI] [PubMed] [Google Scholar]
- Poddar M, Chetty Y, and Chetty VT 2017. ‘How does obesity affect the endocrine system? A narrative review’, Clin Obes, 7: 136–44. [DOI] [PubMed] [Google Scholar]
- Pojoga LH, Coutinho P, Rivera A, Yao TM, Maldonado ER, Youte R, Adler GK, Williams J, Turchin A, Williams GH, and Romero JR 2012. ‘Activation of the mineralocorticoid receptor increases striatin levels’, Am J Hypertens, 25: 243–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, and Jaffe IZ 2014. ‘Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors’, Arterioscler. Thromb. Vasc. Biol, 34: 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queisser N, and Schupp N 2012. ‘Aldosterone, oxidative stress, and NF-kappaB activation in hypertension-related cardiovascular and renal diseases’, Free Radic Biol Med, 53: 314–27. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Duquaine D, King S, Pitt B, and Patel P 2002. ‘Mineralocorticoid receptor antagonism in experimental atherosclerosis’, Circulation, 105: 2212–16. [DOI] [PubMed] [Google Scholar]
- Raz-Pasteur A, Gamliel-Lazarovich A, Coleman R, and Keidar S 2012. ‘Eplerenone reduced lesion size in early but not advanced atherosclerosis in apolipoprotein E-deficient mice’, J. Cardiovasc. Pharmacol, 60: 508–12. [DOI] [PubMed] [Google Scholar]
- Raz-Pasteur A, Gamliel-Lazarovich A, Gantman A, Coleman R, and Keidar S 2014. ‘Mineralocorticoid receptor blockade inhibits accelerated atherosclerosis induced by a low sodium diet in apolipoprotein E-deficient mice’, J. Renin. Angiotensin. Aldosterone. Syst, 15: 228–35. [DOI] [PubMed] [Google Scholar]
- Ren Y, Janic B, Kutskill K, Peterson EL, and Carretero OA 2016. ‘Mechanisms of connecting tubule glomerular feedback enhancement by aldosterone’, Am J Physiol Renal Physiol, 311: F1182–F88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, and Young MJ 2014. ‘Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure’, Hypertension, 63: 1033–40. [DOI] [PubMed] [Google Scholar]
- Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, Avino S, De Marco P, Bussolati B, Maggiolini M, and De Francesco EM 2016. ‘GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells’, Oncotarget, 7: 94–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossello X, Ariti C, Pocock SJ, Ferreira JP, Girerd N, McMurray JJV, Van Veldhuisen DJ, Pitt B, and Zannad F 2019. ‘Impact of mineralocorticoid receptor antagonists on the risk of sudden cardiac death in patients with heart failure and left-ventricular systolic dysfunction: an individual patient-level meta-analysis of three randomized-controlled trials’, Clin Res Cardiol, 108: 477–86. [DOI] [PubMed] [Google Scholar]
- Rossignol P, Claggett BL, Liu J, Vardeny O, Pitt B, Zannad F, and Solomon S 2018. ‘Spironolactone and Resistant Hypertension in Heart Failure With Preserved Ejection Fraction’, Am J Hypertens, 31: 407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira-Llopis S, Banuls C, de Maranon AM, Diaz-Morales N, Jover A, Garzon S, Rocha M, Victor VM, and Hernandez-Mijares A 2017. ‘Low testosterone levels are related to oxidative stress, mitochondrial dysfunction and altered subclinical atherosclerotic markers in type 2 diabetic male patients’, Free Radic. Biol. Med, 108: 155–62. [DOI] [PubMed] [Google Scholar]
- Ruparelia N, Chai JT, Fisher EA, and Choudhury RP 2017. ‘Inflammatory processes in cardiovascular disease: a route to targeted therapies’, Nat Rev Cardiol, 14: 133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht R, Reul JM, van Steensel B, Spengler D, Soder M, Berning B, Holsboer F, and Damm K 1993. ‘Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands’, Eur J Pharmacol, 247: 145–54. [DOI] [PubMed] [Google Scholar]
- Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM, and Alcaide P 2016. ‘Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure’, J Am Heart Assoc, 5: e003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador AM, Moss ME, Aronovitz M, Mueller KB, Blanton RM, Jaffe IZ, and Alcaide P 2017. ‘Endothelial mineralocorticoid receptor contributes to systolic dysfunction induced by pressure overload without modulating cardiac hypertrophy or inflammation’, Physiol Rep, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer N, Lohmann C, Winnik S, van Tits LJ, Miranda MX, Vergopoulos A, Ruschitzka F, Nussberger J, Berger S, Luscher TF, Verrey F, and Matter CM 2013. ‘Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity’, Eur. Heart J, 34: 3515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes KK, Jaffe IZ, Iyer L, Dabreo A, Aronovitz M, Newfell B, Hansen U, Rosano G, and Mendelsohn ME 2008. ‘Rapid recruitment of temporally distinct vascular gene sets by estrogen’, Mol Endocrinol, 22: 2544–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz UD, and Abraham GE 1975. ‘Corticosterone and aldosterone levels during the menstrual cycle’, Obstet Gynecol, 45: 339–42. [PubMed] [Google Scholar]
- Seeger H, Wallwiener D, and Mueck AO 2009. ‘Effects of drospirenone on cardiovascular markers in human aortic endothelial cells’, Climacteric, 12: 80–7. [DOI] [PubMed] [Google Scholar]
- Sequeira SJ, Loughlin T, Cunningham S, Culliton MT, Hannon S, Heffernan A, and McKenna TJ 1986. ‘Evaluation of an aldosterone radioimmunoassay: the renin-angiotensin-aldosterone axis as a function of sex and age’, Ann Clin Biochem, 23 ( Pt 1): 65–75. [DOI] [PubMed] [Google Scholar]
- Sharma G, and Prossnitz ER 2017. ‘G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis’, Adv Exp Med Biol, 1043: 427–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen ZX, Chen XQ, Sun XN, Sun JY, Zhang WC, Zheng XJ, Zhang YY, Shi HJ, Zhang JW, Li C, Wang J, Liu X, and Duan SZ 2017. ‘Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Atherosclerosis by Affecting Foam Cell Formation and Efferocytosis’, J. Biol. Chem, 292: 925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H, Sunamura S, and Satoh K 2016. ‘RhoA/Rho-Kinase in the Cardiovascular System’, Circ Res, 118: 352–66. [DOI] [PubMed] [Google Scholar]
- Shiota M, Fujimoto N, Higashijima K, Imada K, Kashiwagi E, Takeuchi A, Inokuchi J, Tatsugami K, Kajioka S, Uchiumi T, and Eto M 2018. ‘Mineralocorticoid receptor signaling affects therapeutic effect of enzalutamide’, Prostate. [DOI] [PubMed] [Google Scholar]
- Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, Seely EW, Adler GK, Jaffe IZ, Karas RH, Williams GH, and Romero JR 2018. ‘Biological Sex Modulates the Adrenal and Blood Pressure Responses to Angiotensin II’, Hypertension, 71: 1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A, Fornari L, Naftolin F, and Genazzani AR 2006. ‘Estrogen receptor alpha interacts with Galpha13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway’, Mol Endocrinol, 20: 1756–71. [DOI] [PubMed] [Google Scholar]
- Sowers JR 1998. ‘Diabetes mellitus and cardiovascular disease in women’, Arch Intern Med, 158: 617–21. [DOI] [PubMed] [Google Scholar]
- Stewart PM, Penn R, Holder R, Parton A, Ratcliffe JG, and London DR 1993. ‘The hypothalamo-pituitary-adrenal axis across the normal menstrual cycle and in polycystic ovary syndrome’, Clin Endocrinol (Oxf), 38: 387–91. [DOI] [PubMed] [Google Scholar]
- Studen KB, Sebestjen M, Pfeifer M, and Prezelj J 2011. ‘Influence of spironolactone treatment on endothelial function in non-obese women with polycystic ovary syndrome’, Eur J Endocrinol, 164: 389–95. [DOI] [PubMed] [Google Scholar]
- Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, Zhang WC, Liu Y, Zhu M, Kong D, Zhou L, Lu L, Shen ZX, Yi Y, Du L, Qin M, Liu X, Hua Z, Sun S, Yin H, Zhou B, Yu Y, Zhang Z, and Duan SZ 2017. ‘T-Cell Mineralocorticoid Receptor Controls Blood Pressure by Regulating Interferon-Gamma’, Circ Res, 120: 1584–97. [DOI] [PubMed] [Google Scholar]
- Sundd P, Pospieszalska MK, Cheung LS, Konstantopoulos K, and Ley K 2011. ‘Biomechanics of leukocyte rolling’, Biorheology, 48: 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Iwai M, Mogi M, Oshita A, Yoshii T, Higaki J, and Horiuchi M 2006. ‘Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation’, Arterioscler. Thromb. Vasc. Biol, 26: 917–21. [DOI] [PubMed] [Google Scholar]
- Syngle A, Vohra K, Kaur L, and Sharma S 2009. ‘Effect of spironolactone on endothelial dysfunction in rheumatoid arthritis’, Scand J Rheumatol, 38: 15–22. [DOI] [PubMed] [Google Scholar]
- Szmuilowicz ED, Adler GK, Williams JS, Green DE, Yao TM, Hopkins PN, and Seely EW 2006. ‘Relationship between aldosterone and progesterone in the human menstrual cycle’, J Clin Endocrinol Metab, 91: 3981–7. [DOI] [PubMed] [Google Scholar]
- Szymanski P, Klisiewicz A, Lubiszewska B, Lipczynska M, Kowalski M, Janas J, and Hoffman P 2011. ‘Gender differences in angiotensin II and aldosterone secretion in patients with pressure overloaded systemic right ventricles are similar to those observed in systemic arterial hypertension’, Int J Cardiol, 147: 366–70. [DOI] [PubMed] [Google Scholar]
- Tada Y, Yagi K, Kitazato KT, Tamura T, Kinouchi T, Shimada K, Matsushita N, Nakajima N, Satomi J, Kageji T, and Nagahiro S 2010. ‘Reduction of endothelial tight junction proteins is related to cerebral aneurysm formation in rats’, J Hypertens, 28: 1883–91. [DOI] [PubMed] [Google Scholar]
- Takeda AN, Pinon GM, Bens M, Fagart J, Rafestin-Oblin ME, and Vandewalle A 2007. ‘The synthetic androgen methyltrienolone (r1881) acts as a potent antagonist of the mineralocorticoid receptor’, Mol Pharmacol, 71: 473–82. [DOI] [PubMed] [Google Scholar]
- Tang F 1985. ‘Effect of sex and age on serum aldosterone and thyroid hormones in the laboratory rat’, Horm Metab Res, 17: 507–9. [DOI] [PubMed] [Google Scholar]
- Tomaschitz A, Pilz S, Ritz E, Grammer T, Amrein K, Merger S, Meinitzer A, Winkelmann BR, Boehm BO, and Marz W 2011. ‘Relationship between plasma aldosterone concentration and soluble cellular adhesion molecules in patients referred to coronary angiography’, Exp Clin Endocrinol Diabetes, 119: 649–55. [DOI] [PubMed] [Google Scholar]
- Tsujimoto T, and Kajio H 2017. ‘Abdominal Obesity Is Associated With an Increased Risk of All-Cause Mortality in Patients With HFpEF’, J Am Coll Cardiol, 70: 2739–49. [DOI] [PubMed] [Google Scholar]
- Tyree CM, Zou A, and Allegretto EA 2002. ‘17beta-Estradiol inhibits cytokine induction of the human E-selectin promoter’, J Steroid Biochem Mol Biol, 80: 291–7. [DOI] [PubMed] [Google Scholar]
- Upadhya B, Hundley WG, Brubaker PH, Morgan TM, Stewart KP, and Kitzman DW 2017a. ‘Effect of Spironolactone on Exercise Tolerance and Arterial Function in Older Adults with Heart Failure with Preserved Ejection Fraction’, J Am Geriatr Soc, 65: 2374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya B, Pisani B, and Kitzman DW 2017b. ‘Evolution of a Geriatric Syndrome: Pathophysiology and Treatment of Heart Failure with Preserved Ejection Fraction’, J Am Geriatr Soc, 65: 2431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, Lumeng CN, and Mortensen RM 2010. ‘Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice’, J. Clin. Invest, 120: 3350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM, Zhao Y, Xu A, and Leung SW 2016. ‘Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator’, Circ Res, 119: 375–96. [DOI] [PubMed] [Google Scholar]
- Vasan RS, Evans JC, Benjamin EJ, Levy D, Larson MG, Sundstrom J, Murabito JM, Sam F, Colucci WS, and Wilson PW 2004. ‘Relations of serum aldosterone to cardiac structure: gender-related differences in the Framingham Heart Study’, Hypertension, 43: 957–62. [DOI] [PubMed] [Google Scholar]
- Vecchiola A, Lagos CF, Fuentes CA, Allende F, Campino C, Valdivia C, Tapia-Castillo A, Ogishima T, Mukai K, Owen G, Solari S, Carvajal CA, and Fardella CE 2013. ‘Different effects of progesterone and estradiol on chimeric and wild type aldosterone synthase in vitro’, Reprod Biol Endocrinol, 11: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizzardi E, Pina PD, Caretta G, Bonadei I, Sciatti E, Lombardi C, D’Aloia A, Curnis A, and Metra M 2015. ‘The effect of aldosterone-antagonist therapy on aortic elastic properties in patients with nonischemic dilated cardiomyopathy’, J Cardiovasc Med (Hagerstown), 16: 597–602. [DOI] [PubMed] [Google Scholar]
- Vukusich A, Kunstmann S, Varela C, Gainza D, Bravo S, Sepulveda D, Cavada G, Michea L, and Marusic ET 2010. ‘A randomized, double-blind, placebo-controlled trial of spironolactone on carotid intima-media thickness in nondiabetic hemodialysis patients’, Clin J Am Soc Nephrol, 5: 1380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu YH, Yang XP, Rhaleb NE, Xu J, Peterson E, Rudolph AE, and Carretero OA 2004. ‘Role of a selective aldosterone blocker in mice with chronic heart failure’, J Card Fail, 10: 67–73. [DOI] [PubMed] [Google Scholar]
- Wehling M 2018. ‘Rapid actions of aldosterone revisited: Receptors in the limelight’, J Steroid Biochem Mol Biol, 176: 94–98. [DOI] [PubMed] [Google Scholar]
- Williams GH, Burgess E, Kolloch RE, Ruilope LM, Niegowska J, Kipnes MS, Roniker B, Patrick JL, and Krause SL 2004. ‘Efficacy of eplerenone versus enalapril as monotherapy in systemic hypertension’, Am J Cardiol, 93: 990–6. [DOI] [PubMed] [Google Scholar]
- Wilson PW, D’Agostino RB, Sullivan L, Parise H, and Kannel WB 2002. ‘Overweight and obesity as determinants of cardiovascular risk: the Framingham experience’, Arch Intern Med, 162: 1867–72. [DOI] [PubMed] [Google Scholar]
- Xu J, Murphy SL, Kochanek KD, Bastian B, and Arias E 2018. ‘Deaths: Final Data for 2016’, Natl Vital Stat Rep, 67: 1–76. [PubMed] [Google Scholar]
- Yanes LL, and Romero DG 2009. ‘Dihydrotestosterone stimulates aldosterone secretion by H295R human adrenocortical cells’, Mol Cell Endocrinol, 303: 50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Fuller PJ, Morgan J, Shibata H, McDonnell DP, Clyne CD, and Young MJ 2014. ‘Use of phage display to identify novel mineralocorticoid receptor-interacting proteins’, Mol Endocrinol, 28: 1571–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, and Young MJ 2016. ‘Mineralocorticoid receptor antagonists-pharmacodynamics and pharmacokinetic differences’, Curr Opin Pharmacol, 27: 78–85. [DOI] [PubMed] [Google Scholar]
- Yugar-Toledo JC, Modolo R, de Faria AP, and Moreno H 2017. ‘Managing resistant hypertension: focus on mineralocorticoid-receptor antagonists’, Vasc Health Risk Manag, 13: 403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B, and Group, Emphasis-Hf Study. 2011. ‘Eplerenone in patients with systolic heart failure and mild symptoms’, N Engl J Med, 364: 11–21.21073363 [Google Scholar]