

Abstract
The opposing activities of phosphatases and kinases determine the phosphorylation status of proteins, yet kinases have received disproportionate attention in studies of cellular processes, with the roles of phosphatases remaining less understood. This paper describes the use of phosphotyrosine-containing peptide arrays together with MALDI mass spectrometry to directly profile phosphatase substrate selectivities. Twenty-two phosphotyrosine phosphatases were characterized with the arrays to give a profile of their specificities. An analysis of the data revealed that certain residues in the substrates had a conserved effect on activity for all enzymes tested, including the general rule that inclusion of a basic lysine or arginine residue on either side of the phosphotyrosine decreased activity. This insight also provides a new perspective on the role of a R1152Q mutant in the insulin receptor, which is known to exhibit a lower phosphorylation level, and which this work suggests may be due to an increased activity towards phosphatase enzymes. The use of SAMDI-MS to provide a rapid and quantitative assay of phosphatase enzymes will be important to gaining a more complete understanding of the biochemistry and biology of this important enzyme class.
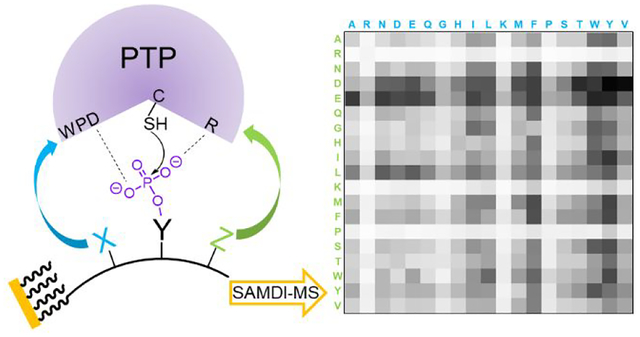
Graphical Abstract
INTRODUCTION
While the phosphorylated states of proteins are determined by the balance of opposing kinase and phosphatase activities, the overwhelming majority of work has addressed the roles of kinases and their substrates in regulating phosphorylation, and has generally assumed that phosphatases serve a non-regulatory housekeeper role.1 However, this assumption lacks justification and appears inconsistent with the roughly equal numbers of tyrosine kinases (PTK) and phosphotyrosine phosphatases (PTP) in the human proteome (90 PTKs and 107 PTPs).2–3 Further, recent work has illustrated a regulatory role for PTPs and sophisticated modes of regulation.4–7 Their dysregulated activities have also been directly linked to disease and cancer; SHP2 (PTPN11), for example, has been identified as the first oncogenic phosphatase.8–10 Advancing our understanding of the roles that PTPs play in signaling would benefit from determining the substrate specificities of different members of the family. Here we use peptide arrays and SAMDI-MS (self-assembled monolayers for matrix-assisted laser desorption/ionization mass spectrometry) to profile twenty-two phosphatases and we report distinct classes of substrate specificities for members of the PTP family.
Assays of phosphatase activity are quite challenging, and largely not well-suited to the direct determination of phosphatase specificity. One approach uses bottom-up proteomics or ELISA (enzyme-linked immunosorbent assay) to observe dephosphorylation of a sample that has first been enriched in phosphoproteins.11–12 Approaches for directly assaying enzymatic phosphatase activities frequently use generic and non-specific substrates—commonly, para-nitrophenylphosphate (pNPP) or 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP)—which release products that can be measured with absorbance or fluorescence spectroscopy. Alternatively, the malachite green assay measures the phosphate by-product, but is difficult to apply to cell lysates that have significant background levels of phosphate ions.13–15 Hence, these assays involve tedious sample preparation, are not high throughput, and do not easily permit the use of a large number of substrates; these limitations have hindered studies of PTP specificity, particularly as compared to numerous studies of kinase specificity and activity.16–19 MS-based phosphatase activity assays have gradually become popular in the field for their easier workflow and the high-throughput nature. MALDI-MS has been used in several recent examples to screen for PTP inhibitors.20–22
Combinatorial libraries and peptide arrays are powerful tools for studying the substrate specificities of a variety of enzymes.23–24 To determine the specificity of PTPs, for example, Pei and coworkers used a combinatorial bead-based peptide library that was prepared by split-pool synthesis. They developed a colorimetric labeling method to identify non-phosphorylated tyrosine residues that result from PTP activity and could then sequence those beads to identify active substrates for the PTPs.25–26 Their work showed acidic residues are generally favored over basic residues in the substrates for 14 PTPs, and basic residues decrease activity.27 However the combinatorial methods have the limitation that they can isolate and sequence a small fraction of the peptides in the pool, and therefore while they provide general trends for activity, they do not give a nuanced understanding of specificity. Cesareni and coworkers prepared an array of phosphotyrosine-containing peptides. Because they were unable to directly measure the extent of dephosphorylation of each peptide, they instead used a mutant PTP that could bind the substrate but was catalytically impaired, and used this binding activity as a proxy for enzyme activity. They measured ‘activity’ profiles for sixteen PTPs using an assay where the PTP was conjugated to GST (glutathione S-transferase), which was then labeled with an anti-GST-Cy5 conjugate. However, there remains the possibility that the mutant enzyme has an altered substrate-specificity and there is the limitation that catalytically impaired mutants are not available for all phosphatases.28–31 Other work has used phosphopeptide arrays to profiles PTPs, with an anti-phosphotyrosine antibody to identify active substrates.32
Our development of the SAMDI-MS method provides a label-free and high throughput assay for measuring a broad range of enzyme activities.33–37 This method uses self-assembled monolayers that present the peptide substrate against a background of tri(ethylene glycol) groups, which are effective at preventing the non-specific adsorption of proteins and play an important role in biological assays.38 Treatment of the monolayer with a solution containing the enzyme may lead to a post-translational modification of the substrate, which is accompanied by a corresponding change in mass. This reaction product can then be quantitated with matrix-assisted laser desorption-ionization mass spectrometry, which reveals peaks corresponding to both the substrate and product (and any intermediates or additional products). The SAMDI-MS method is compatible with the common 384 and 1,536 spot formats and has been used to profile enzymes with peptide arrays.23, 31, 39–43 We also recently demonstrated SAMDI could be used to profile the activity of a PTP on a phosphopeptide array.44 Becker and coworkers’ recent advance in studying protein-protein interactions using protein arrays and MALDI-MS also demonstrates the power of combining these technologies.45–46
Here, we describe the use of a peptide array based on a sequence previously used in earlier studies of SHP2 activity: Ac-TRDXpYZTC-NH2, and where the X and Z positions are variable.47 We profiled 22 phosphatases and generally found that the specificities of those investigated previously were consistent with earlier reports, but our studies also revealed that many of the PTPs have unique and highly selective activities. Our data provides general rules of how charge, steric bulk and hydrophobic character affect enzyme activity for the various PTPs. Finally, our data confirm that all of the phosphatases lack activity towards substrates that have an arginine or lysine residue to either side of the phosphotyrosine, and we discuss the possible relevance of this dependence in molecular mechanisms of diabetes.
RESULTS AND DISCUSSION
Preparation of Phosphopeptide Array.
We prepared an array having 361 peptides with the sequence Ac-TRDXpYZTC-NH2, where the X and Z positions comprise each of the 19 canonical amino acids except for cysteine. In this way, we can determine which residues, when present adjacent to the phosphotyrosine, promote and inhibit activity. We used an array plate having 384 gold spots arranged in the geometry of the common microwell plate, where each was modified with a self-assembled monolayer presenting maleimide groups at a density of 10% against a background of tri(ethylene glycol) groups as described previously.44 The peptides were synthesized using standard protocols with FMOC-protected amino acids and then stored in a 384 multi-well plate as described previously.44 The peptides were transferred to the monolayer array plate using a robotic liquid handler, where each peptide underwent immobilization to the monolayer in its spot via conjugate addition of cysteine thiol to the maleimide group (Figure 1).
Figure 1.

Profiling phosphatase activities using peptide arrays and SAMDI mass spectrometry. (Left) Peptides are immobilized on a self-assembled monolayer surface presenting 10% maleimide against a background of tri(ethylene glycol) gruops. (Middle) The array is treated with a phosphatase and the extent of dephosphorylation of each peptide is analyzed with a MALDI-TOF mass spectrometer. (Right) A SAMDI spectrum of the initial monolayer has a peak at m/z = 1972 corresponding to the phosphotyrosine peptide−alkyldisulfide conjugate and a spectrum of the monolayer after treatment with a phosphate reveals a new peak at 80 Da lower mass, which corresponds to the dephosphorylated product.
Profiling Activities of DEP1 (PTPRJ).
We first describe an experiment to profile the specificity of the transcriptional regulatory phosphatase DEP1 on the peptide array. We prepared a solution of the phosphatase (1.2 nM in 100 mM Tris, pH 7.5, 50 mM NaCl and 100 μM TCEP) and used a robotic liquid dispenser to rapidly apply 2 μL of this solution to each spot on the array plate. The array was placed in a humidified chamber at 37°C for one hour and then rinsed first with water and then ethanol, and finally treated with THAP (2,4,6-trihydroxyacetophenone) matrix. The plate was analyzed using an AbSciex 5800 MALDI-TOF mass spectrometer to acquire mass spectra for each spot, which revealed separate peaks corresponding to the substrate and product of the reaction. The conversion of phosphopeptide to its product was characterized by integration of the corresponding peaks and is given by Activity = AUCproduct / (AUCsubstrate + AUCproduct) × 100 % where AUC refers to the area under the curve (Figure 1). The ionization efficiencies of the substrate and product are not identical and therefore these nominal conversions are not calibrated, but the quantities do provide a relative measure of activity and therefore are useful in the following studies.
The activities for each peptide sequence are represented in a 19 × 19 heatmap where each row defines the amino acid in the Z position (+1), and each column defines the amino acid in the X (−1) position. The percent dephosphorylation is represented in greyscale with white corresponding to 0% activity and black to 100% activity. The heatmap of DEP1 (Figure 2, upper left) shows that peptides containing a glycine in the Z position have higher activity, and similarly those having the aromatic residues phenylalanine and tyrosine, and to a lesser extent the hydrophobic residues isoleucine and leucine, in the X position are more active.
Figure 2.

Activity heatmaps for each of 22 phosphatases profiled on the peptide array having 361 sequences of form Ac-TRDXpYZTC-NH2. The columns represent 19 different amino acids at X position and the rows represent the amino acids at the Z position. The activities are represented in grey scale where white corresponds to 0 % activity and black to 100 %.
Profiling Activities of 22 Phosphatases.
We repeated the experiment described above for 21 additional phosphatases—ten non-receptor protein tyrosine phosphatases (NRPTPs, classical PTPs), eight receptor protein tyrosine phosphatases (RPTPs, classical PTPs), three regenerating liver phosphatases (PRLs, VH1-like PTPs) and one alkaline phosphatase (ALP). The heatmaps for each of these phosphatases are shown in Figure 2. Initial inspection of the heatmaps reveals several significant observations. First, the phosphatases show a range in specificity, with some having a clear preference for a smaller number of peptides within the array. Second, there are multiple classes of specificity that are shared by a subset of the enzymes. Third, there are positions in the substrate where certain residues affect the activity of the substrate in a similar way for all of the enzymes, and other specificities that are shared by a subset of the enzymes.
Quantitative Analysis of Heatmaps.
We first analyzed the heatmaps to quantitate the influence that a particular residue has on activity of the substrate for each phosphatase. For each amino acid in the X position, we determined the average activity of all peptides in the row corresponding to that residue in the heatmap. We also determined the average activity for all peptides in the array (which we refer to as the global average). The ratio of the difference (Δ) of the average activity for the particular residue (AAx) and the global average (GA) was then determine according to the equation Δ = (AAx − GA)/GA × 100 %. We similarly repeated this analysis for residues in the Z position (AAz). The numerical values of these ratios for all 38 rows and columns (19 amino acids at either the X or Z position) for each of the 22 phosphatases are shown in Figure 3. This analysis gives insight into the sequence determinants of activity. The residues that contribute to more than 10% activation or inhibition at X (−1) and Z (+1) positions are summarized in Table 1. The Table reveals that certain residues consistently activate or inhibit activity of the substrate across the phosphatases we profiled. Finally, we also generated histograms to identify the residues that activate and inhibit phosphatase activities at X and Z positions (Figure 4). The x-axis represents amino acids and the y-axis is the total number of PTPs found from Table 1 that activates/inhibits dephosphorylation activities at either X or Z positions. We found that the determinants of sequence selectivity depend significantly on the chemical properties of the amino acid including charge, steric bulk, hydrophobic character as well as the PTP active site structure, and we discuss several of these observations below.
Figure 3.

Heatmap illustrating the average role than an amino acid residue in either the X (AAX) or Z (AAZ) position has on the activity of the substrate for each phosphatase enzyme. For each amino acid and position, the ratio of the difference of the average activity of peptides having that residue and the average of all peptides over the latter were determined and represented both in a red-to-green color scale and with the percent. Green indicates that the residue on average increases the phosphatase activity and red indicates a reduction.
Table 1.
Summary of phosphatase selectivity on pY-library Ac-TRDXpYZTC-NH2
| Phosphatase | activate X | inhibit X | activate Z | inhibit Z | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 % | 20 % | >30 % | 10 % | 20 % | >30 % | 10 % | 20 % | >30 % | 10 % | 20 % | >30 % | |
| PTP1B (PTPN1) | I,L,Y,V | F | R,G,K | P | E,L,S,T,W,V | G,M,Y | R,H,K | P | ||||
| TC-PTP (PTPN2) | M,S,T | K,W | R | D,I,M,T | S | H,Y | P | R | ||||
| MEG1 (PTPN4) | D,E,M | W | H,K | R | S,T | G,M | W | Q,H,I,K,P | R,Y | |||
| SHP1 (PTPN6) | V | I,L,F,W,Y | D,Q | A,N,G,H | R,K,P | Y | D,E,W | Q,H | R,K | |||
| PTPN7 | E,M | D,F | W,Y | A,N,G,H,P,T | Q | R,K | G | E,F,Y | D,W | Q,H,S,T | V | R,K,P |
| MEG2 (PTPN9) | I,S,T,V | N,Q | M | K,P | D,E,G | R | F,W | A,N,I,Y | Q,G,M | R,L,T | S | D,E,P,V |
| SHP2 (PTPN11) | E,W | V | I,L,F,Y | Q | G | R,K,P | N,G,L,F,W | E | D,Y | P | R,K,V | |
| PTPN12 | V | I,L,F,W,Y | A,D | Q,H,S | R,G,K,P | L,F,Y | D,E | A,Q,T,V | H | R,K,P | ||
| PTPN13 | E,V | I,L,W | F,Y | A,N,Q | G | R,K,P | E,G,F | W,Y | A,N,QH | R,K | ||
| PTPN14 | V | I,L,F,W,Y | A,D,E | Q,H | R,N,G,K,P | F | G,W,Y | R,H,I,M,P,S,T | N,Q | K | ||
| PTPRA | L,V | I,F,W,Y | A,Q,H,S | N,D | R,G,K,P | G | F,W,Y | A,N,P | R,K | |||
| PTPbeta (PTPRB) | I,L,M | F,W,Y | A,R,N,D,E,Q,P | G | K | F | W,Y | G | N,D,Q,T | R,K | ||
| CD45 (PTPRC) | E,L,M,T,V | I | F,Y | P | D | R,G,K,P | H,I,Y | S,V | D,M,F | N | R,K,P | |
| PTPRE | M | L | F,W,Y | N,D,E,Q,G,T | R,K | L,F | A,G,W | Y | R,N,D,E | P | K | |
| RPTPG(PTPRG) | I,L,F,T,V | Y | D,K,P,W | R,G | E,G,I,L | R,K,W,Y,V | ||||||
| DEP1 (PTPRJ) | W,V | I,L | F,Y | D,E,Q | R,G,H | K,P | F | P | G,Y | R,E,H,L,K | N,D,Q,I | |
| PTPmu (PTPRM) | R,L | I,F.W.Y | N,H,K,T | A,D,E,Q,S | G,P | W,Y | R,G | A,N,D,E,Q,I,S,V | ||||
| PTPsigma (PTPRS) | M | I,L,F,W,Y,V | T | R,N | A,D,E,Q,G,K,P,S | L | G,F,W,Y | A,N,D,E,Q,I | K,S,T,V | |||
| PRL1 | E,M | D | W | H,V | K | R | E,Y | A,D,M | W | H,I,P,S,T | L,K | R |
| PRL2 | D,L,F | W,Y | N,Q,G,H,S,T | R,K | F | N,E | D,W | S | P,T,V | R,K | ||
| PRL3 | E,L,F | Y | W | Q,T | R,K | D,E,M | Y | W | P | K | R | |
| hALP | G,T | F,P,Y | E | R | H,K | F,V | A | G,Y | R,I,L,T | H | K,P | |
Figure 4.

Histograms showing the role of residues found in phosphatase specificities. The x-axis represents amino acids and the y-axis is the total number of PTPs that activates/inhibits dephosphorylation activities at either X or Z positions. The colors correspond to different degrees of activation/inhibition in activities assayed by SAMDI-MS. (A) Aromatic and hydrophobic residues are activating at the X position. (B) Basic residues, G and P are inhibitory at X position. (C) Aromatic residues are favored at the Z position while G is also favored in contrary to X position. (D) Basic residues and P, again, are inhibitory at the Z position.
Basic Residues Generally Reduce Substrate Activity.
Among the most consistent trends in our data is that basic residues (R, K, H) at either X or Z position very frequently decrease the activity of the substrate for all but one of the phosphatases studied here, consistent with a previous report.27 Among 22 phosphatases, 21 exhibit a preference against basic residues adjacent to pY on their substrate. For example, the oncogenic phosphatase SHP2 shows an average decrease in activity of 64% and 53% when arginine is present at the X and Z positions, respectively. This trend may be explained by electrostatic interactions in the pY binding pocket. Most PTP catalytic sites have a signature motif HCXXGXXRS(T) or HC(X)5R where a cysteine residue acts as the nucleophile and the conserved arginine residue folds back toward the phosphate-binding pocket to assist in substrate binding and catalysis.48 The presence of a second positively-charged residue nearby may reduce the electrostatic attraction between the phosphatase and its substrate and would therefore lower the binding energy of the substrate. Our observation that arginine (R) and lysine (K) have a more significant effect in decreasing activity than does histidine (H) is consistent with their basicities, because the lower pKa of protonated imidazole means that it can be deprotonated with less energetic penalty. Below, we return to the significance of the basic residue in regulating phosphorylation states of proteins and possible relevance to disease.
We did find, however, one phosphatase, PTPmu, that favored an arginine residue in both the X and Z positions. This enzyme, though, does not tolerate a lysine residue in this position, suggesting that a specific hydrogen bonding interaction is involved and not just electrostatics. We studied the active site sequence using UniProtKB protein sequence alignment tool49 and found that PTPmu has two PTP domains (Supporting Information). The first one with catalytic motif HCSAGVGRT is conserved across all assayed RPTPs. The second domain has a catalytic motif HCLNGGGRS that is different from others. This second catalytic domain might contribute to the arginine preference with extra hydrogen bonding interactions.
Acidic Residues Play a Complex Role.
Unlike the influence of basic residues described above, the presence of acidic residues (D and E) have varied effects on the activities of the peptide substrates for the phosphatases. Inclusion of an acidic residue can either increase, decrease or not affect the activity of the peptide, depending on the particular phosphatase. We found SHP1, SHP2, PTPN7, PTPN12, PTPN13 and PRLs showed increased activities for peptides having an acidic residue next to the pY substrate; PTPN14, MEG2, PTPRA, PTPRB, PTPRE, DEP1, PTPsigma, PTPmu showed lower activities on substrates having acidic residues in these positions; PTP1B, TC-PTP, MEG1, RPTPg and hALP were less affected by the presence of acidic residues in these positions. CD45 is an interesting case because an aspartic acid (D) at the X position reduces PTP activity but when present at the Z position, results in an increased activity. Our results reveal that acidic residues play a more complex role than commonly described as generally being favored.27, 50–51 The quantitative SAMDI-MS assay allows us to see the whole picture of PTP activity profiles and understand how each amino acid contributes to the selectivities.
Glycine Has Different Effects at X and Z Positions.
Glycine (G) is a unique residue because of its wider conformational space in protein structure52. We found that glycine plays very different roles in affecting substrate activity when present at X (−1) and Z (+1) positions. At the Z position, glycine is often an activator (observed for 15 out of 22 phosphatases, Figure 4C) and we find it never inhibits activity relative to the average activity for all peptides (Figure 4D). This trend suggests that the binding pocket at the +1 position of the substrate does not participate in strong recognition of the sidechain. At the X position, in contrast, glycine frequently serves to decrease activity (observed for 16 out of 22 phosphatases, Figure 4C). Clearly, side chain interactions of the substrate with the active site are important in this position.
Proline Is a Strong Inhibitor for PTPs.
Proline (P), because of the secondary amide, has a turn conformation that often disrupts binding of a substrate to the active sites of enzymes.53 We found that in no case is the presence of a proline residue at either the X or the Z position favorable for PTP activity. All 21 PTPs disfavor proline residues at both X or Z positions on their substrates (Figure 4B and 4D). ALP has a slight preference for a proline at the X position though we note it is not a classic cysteine-based protein tyrosine phosphatase and the active site structure is very different.54
Aromatic and Hydrophobic Residues Are General Activators.
We found that aromatic residues (Y, F, W) are among the most consistent activators for PTP activity (Figure 4A and 4C). This observation is consistent with previous work that analyzed substrate selectivity of SHP1, SHP2, PTP1B and PTPRA26. We found the trend is universal to all 21 PTPs assayed. A crystal structure provides some structural insight into the preference for aromatic residues.55 In SHP2, two aromatic groups (Y279, H426) occupy either side of the PTP active site, providing π-π interactions to pY and its neighboring residues on the substrate. The analogous residues in PTP1B are Y46 and F182. From sequence alignment of all the PTPs assayed (Supporting Information), we found that the role for these two residues in substrate recognition are highly conserved. All PTPs have the Y residue (except PTPN14, which has an I) in the first position and either an H, Y or F in the second position (except RPTPg which has an M). These residues also provide hydrophobic interactions to the hydrophobic residues (A, I, L, V) while they are less activating compared to aromatic residues perhaps because the interaction is weaker and less specific.
Polar Neutral Residues Are Less Involved in PTP Specificities.
Unlike residues having side chains with charge, aromatic or steric non-polar groups, polar neutral residues (N, Q, S, T) have fewer features for molecular recognition and play a minor role in enhancing or decreasing the activities of the peptide substrates. Amides (N, Q) are more frequently found to be slightly disfavored, but even this trend is inconsistent across the phosphatases.
Comparisons to Prior Studies.
Pei and coworkers’ pioneering studies of PTP specificity used combinatorial peptide libraries prepared by split-pool synthesis and had more than 106 peptides.27 Beads that had active peptide sequences for the PTPs were identified with a colorimetric labeling method and then sequenced by partial Edman degradation-mass spectrometry (PED-MS). This approach has the benefit that it rapidly identifies the most active sequences from a very large number of peptides, but the limited throughput of PED-MS only allows a very small fraction of the beads to be sequenced. In their experiments as low as 0.0008% of peptides were sequenced and therefore the overall specificity and the nuanced preferences for the PTPs are not revealed. The approach also relies on a qualitative isolation of beads based on intensely, medium and lightly red beads and does not directly provide a more quantitative ranking of activities. In these respects, our work with peptide arrays and mass spectrometry provides a complementary insight into the PTP specificity. We note that the expense associated with peptide synthesis currently limits our array sizes to hundreds (not thousands or millions) of peptides, but it does have the benefit of providing relative activities for each peptide substrate in the array.
Pei and cowokers first discovered that PTPs generally have a preference of substrates having an abundance of acidic residues and disfavor substrates with basic residues. Our work confirms this finding that basic residues are indeed the most disfavored for PTPs in their substrates, and reveals that acidic residues promote activity, through they play more complex roles when compared to all other amino acids. SHP1, SHP2, PTPN7, PTPN12, PTPN13 and PRLs prefer acidic residues whereas PTPN14, MEG2, PTPRA, PTPRB, PTPRE, DEP1, PTPsigma, PTPmu disfavor them; and they are not particularly preferred when compared to all other amino acids for PTP1B, TC-PTP, MEG1, RPTPg and hALP. For those PTPs that prefer acidic residues, we found that the preference is more obvious at Z (+1) position. For example, a glutamic acid (E) at X (−1) leads to a small −2% activity decrease for PTPN12, however at Z (+1) it increases activity by +113% for this phosphatase, and was consistent with results reported by Pei and coworkers. Similarly, we also find that aromatic residues in the substrate promote activity for the PTPs. While the combinatorial screening approach taken by Pei and coworkers successfully captured the most obvious characteristics of PTP substrate selectivites, our use of peptide arrays gave a more complete understanding of specificity. We are able to better assess those residues that generally contribute to lower PTP activities and understand their roles. We also found that glycine has an opposite effect when present in the X and Z positions; prolines are disfavored in general; non-aromatic hydrophobic residues are also activators and polar residues are less involved in PTP specificities.
Categorizing PTPs According to Their Substrate Selectivities.
We next asked whether the different specificities of the PTPs are related to the phylogenetic tree and sequence similarity of the enzymes. In Figure 5, we show how the presence or absence of five amino acids (R, D, E, G, P) can be used as filters to partition the PTPs into distinct classes. Starting at the left of the tree we divide the enzymes into groups according to each of eight features (listed at the top of the figure), where the upper group has the feature and the lower group does not. We started by selecting the enzymes that favor a P residue in the X position and followed by those that favor an R in either X or Z positions; these rules isolated a single phosphatase (hALP). We then selected PTPs that favor a R residue in both the X and Z positions (again, isolating one enzyme PTPmu). Because acidic residues play a complex role in PTP selectivity—where they can either increase, decrease or not affect the activity on the peptide—we then separated PTPs that either favor D and E in both the X and Y positions (D,EX,Y) or disfavor these residues. Finally, noting that glycine often promotes activity when present in the Z position but decreases activity in X position, we used this filter to further separate the PTPs. Finally, we separated the PTPs according to role of proline in decreasing activity.
Figure 5.

Specificity tree of the 22 phosphatases profiled with peptide arrays and SAMDI-MS. We use specificity features based on five amino acid residues (P, R, D, E, G) at X and Z position to differentiate the phosphatases. A well-separated tree indicates that phosphatases are unique in their substrate specificity.
We used the UniProtKB tool to compare and align the PTP sequences (Supporting Information) and we generally found that PTPs that are evolutionarily related have similar specificities. For example, SHP1 and SHP2 share >55% sequence identity (60.8 similarity in PTP domain), a common backbone fold, and a common regulatory mechanism.56 We found that their substrate specificities were indistinguishable by the criteria we used in preparing the selectivity tree (Figure 5). Similarly, PTPN13 shares a common substrate specificity with SHP1/2 in our selectivity tree and also has a high sequence similarity to SHP1 and SHP2 in the protein sequence. Three PRLs are very similar in both sequence and substrate selectivity and yet different from classic PTPs. The three phosphatases PTPRA, DEP1 and PTPbeta all belong to the same receptor-like PTP family and exhibited a similar specificity. The enzyme hALP is serine-based phosphatase that uses Zn2+ and Mg2+ activation, and its catalytic mechanism is very different from the cysteine-based PTPs. The sequence is completely unique, and this is the only enzyme we find that prefers a P residue in the X position. We also note that these observations agree with an evolutionary tree analysis previously reported by Cesareni and coworkers.57
A Potential Mutation/Modification Crosstalk Involving PTP1B.
The phosphatase specificities determined in this work can contribute to our understanding of the mechanisms by which mutations contribute to pathological phenotypes. For example, Beguinot and coworkers reported a mutant of the insulin receptor (INSR, R1152Q) that was found in patients with non-insulin-dependent diabetes (Figure 6A).58 This mutant is interesting because it is adjacent to a tyrosine that is known to be autophosphorylated (Y1151) by the insulin-bound receptor and that participates in the tyrosine triplet that activates the receptor tyrosine kinase (RTK) activity of INSR and regulates the downstream cellular signal transduction.59 This study also reported that the mutant had a higher level of basal kinase activity on various endogenous and exogenous substrates, even though phosphorylation of Y1151 is significantly lower than that in the wild-type receptor. The phosphorylation of the mutant by the wild-type receptor was also impaired, leading to the conclusion that the mutation led to poor phosphorylation of Y1151 because it is a less active substrate for its own kinase domain, and in turn had a lower sensitivity to insulin, which in turn contributes to diabetes. It is interesting that this study did not consider the possibility that the mutation made the site more active for its PTP; because the level of phosphorylation depends on the dynamic balance of two opposing activities, it can be decreased either by a lower kinase activity or a higher phosphatase activity. In this example, PTP1B is specifically responsible for dephosphorylating this site.60
Figure 6.

Dephosphoraylation of peptides corresponding to WT and mutant INSR by the PTP1B phosphatase. (A) The mutation R1152Q occurs in the triple phosphorylation site of INSR and is adjacent to the Y1151 phosphorylation site. (B) SAMDI spectra following treatment of each peptide with PTP1B shows significantly more activity on the peptide corresponding to the mutant.
Our profiling experiments revealed that replacing an arginine with a glutamine on a peptide substrate leads to an increase in PTP1B activity. This insight would suggest that mutant INSR shows less phosphorylation at Y1151 both because it is a poorer substrate for the kinase and because the corresponding phosphorylated peptide is a better substrate for PTP1B. We gained support for this possibility by comparing the relative activities of PTP1B on 2 peptides—Ac-TRDYpYRTC-NH2 (YpYR) and Ac-TRDYpYQTC-NH2 (YpYQ)—where we found the latter was significantly more active as a substrate for PTP1B. A crystal structure of PTP1B and the triple-phosphorylated INSR peptide reveals how the arginine residue is repelled by this highly basic triple-pY-binding pocket.61
CONCLUSION
This work addresses the long-standing challenges in characterizing phosphatase activities. Our use of peptide arrays and SAMDI-MS is the first approach to rapidly determine the specificity profiles of phosphatases using hundreds of peptide substrates. By applying this method to 22 phosphatases, we observed trends in sequence-dependent activity that will be useful in developing hypotheses for phosphorylation-dependent signaling activities in cells; many of these trends agreed with previous work based on combinatorial screening, but gave a more complete assessment of specificity. The most general trend—that the presence of a basic residue next to the phosphotyrosine site decreases PTP activity on the substrate—will likely have broad relevance (such as our description of a mutant in non-insulin dependent diabetes), and we expect these data will be valuable in many more contexts. Equally important, we believe this approach will lead to more complete understandings of the biochemistry and biology of phosphatase enzymes.
EXPERIMENTAL SECTION
General.
Laboratory chemicals and reagents were purchased from MilliporeSigma and used without additional purification unless specified. Peptide synthesis reagents, including Fmoc amino acids and Rink-amide resin, were purchased from Anaspec. Phosphatases were purchased from MilliporeSigma. Self-assembled monolayers with matrix-assisted laser desorption-ionization (SAMDI) mass spectrometry were performed on a 5800 MALDI TOF/TOF mass spectrometer (AbSciex) using either manual or automated protocols. A detailed protocol of monolayer plate preparation, peptide synthesis and phosphatase assay can be found in a previously published method paper.44
Peptide Synthesis.
Solid-phase peptide synthesis was performed on Rink-amide resin (10 mg) housed in 96-well filter plates. N-terminal fluorenylmethyloxycarbonyl (Fmoc) protecting groups were deprotected with 20% piperidine in dimethylformamide (DMF) at room temperature for 20 min. The resin was filtered and rinsed 5 times with DMF on a multiscreen vacuum manifold. Amino acids were coupled to the resin with PyBop and N-methylmorpholine (NMM) in DMF at a 4:4:8 molar excess for 30 minutes, twice. The deprotection and coupling were repeated for each residue. Following deprotection of the final residue, the N-terminus was acetylated with 10% acetic anhydride in DMF for 60 minutes. Peptides were cleaved and deprotected in 95% trifluoroacetic acid (TFA), 2.5% triethylsilane (TES), and 2.5% water for 16 h. The cleavage solution was evaporated with N2 gas flow. The peptides were resuspended in 0.1% TFA in water, lyophilized and resuspended again in 0.1%TFA in water to a final concentration of 200 μM and stored at −80 °C.
Preparing Peptide Arrays on SAMDI Plates.
Steel array plates evaporated with 384 gold spots (with a diameter of 3.0 mm) were soaked at 4 °C in a solution of 1 mM total disulfide with 0.8 mM tri(ethylene glycol)-terminated C11-alkane disulfide and 0.2 mM C11-alkane disulfide with one terminal tri(ethylene glycol) and one terminal maleimide in ethanol for 2 days to allow assembly of the monolayer (10% maleimide coverage). Peptides were diluted to a final concentration of 20 μM with 100 mM Tris buffer (pH 7.5) in a 384-well plate with 5 μL TCEP beads (Thermo Scientific) in each well. Using a TECAN robotic liquid handler, 2 μL of peptide solution from each well was transferred on to the corresponding gold spot on the monolayer-presenting plate and incubated at 37 °C in a humidified chamber for 1 h to allow peptide immobilization.
Phosphatase Assays by SAMDI Mass Spectrometry.
Phosphatases were diluted in PTP buffer (100 mM Tris, pH 7.5, 50 mM NaCl, 100 μM TCEP) and 2 μL was applied to each gold spot on the peptide array plate with a Multidrop Combi (Thermo Scientific). The reactions were incubated at 37 °C in a humidified chamber. (See Supporting Information for concentration and incubation time for each phosphatase.) After the reaction was complete, the plate was rinsed with water and ethanol, treated with 1 μL matrix (10 mg/mL THAP, 5 mg/mL ammonium citrate dibasic in 50% acetonitrile, 50% water and 0.1 % phosphoric acid) to each spot and dried in air for 20 min. The spots were analyzed by MALDI-TOF MS to obtain a mass spectrum for each reaction. Enzymatic activities were quantified by measuring the areas under the curve (AUCs) for the dephosphorylate product peak and the substrate peak and determining the activity (%) = AUCproduct / (AUCsubstrate + AUCproduct) × 100 %. Activity heatmaps were generated by Microsoft Excel.
Supplementary Material
ACKNOWLEDGEMENTS
This work has been supported by the National Cancer Institute of the National Institute of Health under Award Number U54CA199091, the Department of Defense, Defense Threat Reduction Agency under Award Number HDTRA1-15-1-0052, the Air Force Research Laboratory under Award Number FA8650-15-2-5518, and the NU-NTU Institute for Nano Medicine located at the International Institute for Nanotechnology, Northwestern University, USA, and the Nanyang Technological University, Singapore, under Award Number Agmt10/20/14.
Footnotes
Supporting Information. Reaction conditions, phosphatase sequence alignment.
M.M. is the founder and chairman of SAMDI Tech Inc., which uses SAMDI-MS to assist clients in the pharmaceutical industry in early phase drug discovery. The authors declare that they have no other competing interests.
REFERENCE
- 1.Tonks NK; Neel BG, Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 2001, 13 (2), 182–195. [DOI] [PubMed] [Google Scholar]
- 2.Alonso A; Sasin J; Bottini N; Friedberg I; Friedberg I; Osterman A; Godzik A; Hunter T; Dixon J; Mustelin T, Protein tyrosine phosphatases in the human genome. Cell 2004, 117 (6), 699–711. [DOI] [PubMed] [Google Scholar]
- 3.Beck JR; Lawrence A; Tung AS; Harris EN; Stains CI, Interrogating Endogenous Protein Phosphatase Activity with Rationally Designed Chemosensors. ACS Chem. Biol 2016, 11 (1), 284–90. [DOI] [PubMed] [Google Scholar]
- 4.Hale AJ; Ter Steege E; den Hertog J, Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev. Biol 2017, 428 (2), 283–292. [DOI] [PubMed] [Google Scholar]
- 5.Hendriks WJ; Elson A; Harroch S; Pulido R; Stoker A; den Hertog J, Protein tyrosine phosphatases in health and disease. FEBS J. 2013, 280 (2), 708–30. [DOI] [PubMed] [Google Scholar]
- 6.Tonks NK, Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7 (11), 833–46. [DOI] [PubMed] [Google Scholar]
- 7.Tonks NK, Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280 (2), 346–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan G; Kalaitzidis D; Neel BG, The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008, 27 (2), 179–92. [DOI] [PubMed] [Google Scholar]
- 9.Loh ML; Vattikuti S; Schubbert S; Reynolds MG; Carlson E; Lieuw KH; Cheng JW; Lee CM; Stokoe D; Bonifas JM; Curtiss NP; Gotlib J; Meshinchi S; Le Beau MM; Emanuel PD; Shannon KM, Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103 (6), 2325–31. [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia M; Niemeyer CM; Fragale A; Song X; Buechner J; Jung A; Hahlen K; Hasle H; Licht JD; Gelb BD, Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet 2003, 34 (2), 148–50. [DOI] [PubMed] [Google Scholar]
- 11.Tsai CF; Wang YT; Yen HY; Tsou CC; Ku WC; Lin PY; Chen HY; Nesvizhskii AI; Ishihama Y; Chen YJ, Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat Commun 2015, 6, 6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue L; Wang WH; Iliuk A; Hu L; Galan JA; Yu S; Hans M; Geahlen RL; Tao WA, Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (15), 5615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geladopoulos TP; Sotiroudis TG; Evangelopoulos AE, A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem 1991, 192 (1), 112–116. [DOI] [PubMed] [Google Scholar]
- 14.Takai A; Mieskes G, Inhibitory effect of okadaic acid on the p-nitrophenyl phosphate phosphatase activity of protein phosphatases. Biochem. J 1991, 275 (Pt 1), 233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welte S; Baringhaus KH; Schmider W; Muller G; Petry S; Tennagels N, 6,8-Difluoro-4-methylumbiliferyl phosphate: a fluorogenic substrate for protein tyrosine phosphatases. Anal. Biochem 2005, 338 (1), 32–8. [DOI] [PubMed] [Google Scholar]
- 16.Zhu H; Klemic JF; Chang S; Bertone P; Casamayor A; Klemic KG; Smith D; Gerstein M; Reed MA; Snyder M, Analysis of yeast protein kinases using protein chips. Nat. Genet 2000, 26, 283. [DOI] [PubMed] [Google Scholar]
- 17.Fasolo J; Sboner A; Sun MG; Yu H; Chen R; Sharon D; Kim PM; Gerstein M; Snyder M, Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011, 25 (7), 767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang ZY; Thieme-Sefler AM; Maclean D; McNamara DJ; Dobrusin EM; Sawyer TK; Dixon JE, Substrate specificity of the protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U. S. A 1993, 90 (10), 4446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang ZY; Dixon JE, Protein tyrosine phosphatases: mechanism of catalysis and substrate specificity. Adv. Enzymol. Relat. Areas Mol. Biol 1994, 68, 1–36. [DOI] [PubMed] [Google Scholar]
- 20.Alchab F; Sibille E; Ettouati L; Bana E; Bouaziz Z; Mularoni A; Monniot E; Bagrel D; Jose J; Le Borgne M; Chaimbault P, Screening of indeno[1,2-b]indoloquinones by MALDI-MS: a new set of potential CDC25 phosphatase inhibitors brought to light. J. Enzyme Inhib. Med. Chem 2016, 31 (sup3), 25–32. [DOI] [PubMed] [Google Scholar]
- 21.Winter M; Bretschneider T; Kleiner C; Ries R; Hehn JP; Redemann N; Luippold AH; Bischoff D; Buttner FH, Establishing MALDI-TOF as Versatile Drug Discovery Readout to Dissect the PTP1B Enzymatic Reaction. SLAS Discov 2018, 23 (6), 561–573. [DOI] [PubMed] [Google Scholar]
- 22.Winter M; Ries R; Kleiner C; Bischoff D; Luippold AH; Bretschneider T; Buttner FH, Automated MALDI Target Preparation Concept: Providing Ultra-High-Throughput Mass Spectrometry-Based Screening for Drug Discovery. SLAS Technol 2018, 2472630318791981. [DOI] [PubMed] [Google Scholar]
- 23.Szymczak LC; Kuo HY; Mrksich M, Peptide Arrays: Development and Application. Anal. Chem 2018, 90 (1), 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng X; Wei J; Wang Y; Zhang H; Wang Z, The role of peptide microarrays in biomedical research. Analytical Methods 2018, 10 (38), 4614–4624. [Google Scholar]
- 25.Garaud M; Pei D, Substrate profiling of protein tyrosine phosphatase PTP1B by screening a combinatorial peptide library. J. Am. Chem. Soc 2007, 129 (17), 5366–7. [DOI] [PubMed] [Google Scholar]
- 26.Ren L; Chen X; Luechapanichkul R; Selner NG; Meyer TM; Wavreille AS; Chan R; Iorio C; Zhou X; Neel BG; Pei D, Substrate specificity of protein tyrosine phosphatases 1B, RPTPalpha, SHP-1, and SHP-2. Biochemistry 2011, 50 (12), 2339–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selner NG; Luechapanichkul R; Chen X; Neel BG; Zhang ZY; Knapp S; Bell CE; Pei D, Diverse levels of sequence selectivity and catalytic efficiency of protein-tyrosine phosphatases. Biochemistry 2014, 53 (2), 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palma A; Tinti M; Paoluzi S; Santonico E; Brandt BW; Hooft van Huijsduijnen R; Masch A; Heringa J; Schutkowski M; Castagnoli L; Cesareni G, Both Intrinsic Substrate Preference and Network Context Contribute to Substrate Selection of Classical Tyrosine Phosphatases. J. Biol. Chem 2017, 292 (12), 4942–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun H; Tan LP; Gao L; Yao SQ, High-throughput screening of catalytically inactive mutants of protein tyrosine phosphatases (PTPs) in a phosphopeptide microarray. Chem. Commun. (Camb.) 2009, (6), 677–9. [DOI] [PubMed] [Google Scholar]
- 30.Weeks AM; Wells JA, Engineering peptide ligase specificity by proteomic identification of ligation sites. Nat. Chem. Biol 2018, 14 (1), 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wood SE; Sinsinbar G; Gudlur S; Nallani M; Huang CF; Liedberg B; Mrksich M, A Bottom-Up Proteomic Approach to Identify Substrate Specificity of Outer-Membrane Protease OmpT. Angew. Chem. Int. Ed. Engl 2017, 56 (52), 16531–16535. [DOI] [PubMed] [Google Scholar]
- 32.Kohn M; Gutierrez-Rodriguez M; Jonkheijm P; Wetzel S; Wacker R; Schroeder H; Prinz H; Niemeyer CM; Breinbauer R; Szedlacsek SE; Waldmann H, A microarray strategy for mapping the substrate specificity of protein tyrosine phosphatase. Angew. Chem. Int. Ed. Engl 2007, 46 (40), 7700–3. [DOI] [PubMed] [Google Scholar]
- 33.Berns EJ; Cabezas MD; Mrksich M, Cellular Assays with a Molecular Endpoint Measured by SAMDI Mass Spectrometry. Small 2016, 12 (28), 3811–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan W; Ban L; Cai L; Li L; Chen W; Liu X; Mrksich M; Wang PG, Combining carbochips and mass spectrometry to study the donor specificity for the Neisseria meningitidis beta1,3-N-acetylglucosaminyltransferase LgtA. Bioorg. Med. Chem. Lett 2011, 21 (17), 5025–8. [DOI] [PubMed] [Google Scholar]
- 35.Kornacki JR; Stuparu AD; Mrksich M, Acetyltransferase p300/CBP associated Factor (PCAF) regulates crosstalk-dependent acetylation of histone H3 by distal site recognition. ACS Chem. Biol 2015, 10 (1), 157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min DH; Su J; Mrksich M, Profiling kinase activities by using a peptide chip and mass spectrometry. Angew. Chem. Int. Ed. Engl 2004, 43 (44), 5973–7. [DOI] [PubMed] [Google Scholar]
- 37.Mrksich M, Mass spectrometry of self-assembled monolayers: a new tool for molecular surface science. ACS Nano 2008, 2 (1), 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prime KL; Whitesides GM, Self-assembled organic monolayers: model systems for studying adsorption of proteins at surfaces. Science 1991, 252 (5009), 1164–7. [DOI] [PubMed] [Google Scholar]
- 39.Gurard-Levin ZA; Kilian KA; Kim J; Bahr K; Mrksich M, Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem. Biol 2010, 5 (9), 863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurard-Levin ZA; Kim J; Mrksich M, Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. ChemBioChem 2009, 10 (13), 2159–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kightlinger W; Lin L; Rosztoczy M; Li W; DeLisa MP; Mrksich M; Jewett MC, Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat. Chem. Biol 2018, 14 (6), 627–635. [DOI] [PubMed] [Google Scholar]
- 42.Kuo HY; DeLuca TA; Miller WM; Mrksich M, Profiling deacetylase activities in cell lysates with peptide arrays and SAMDI mass spectrometry. Anal. Chem 2013, 85 (22), 10635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue AY; Szymczak LC; Mrksich M; Bagheri N, Machine Learning on Signal-to-Noise Ratios Improves Peptide Array Design in SAMDI Mass Spectrometry. Anal. Chem 2017, 89 (17), 9039–9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szymczak LC; Huang CF; Berns EJ; Mrksich M, Combining SAMDI Mass Spectrometry and Peptide Arrays to Profile Phosphatase Activities. Methods Enzymol. 2018, 607, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker CF; Wacker R; Bouschen W; Seidel R; Kolaric B; Lang P; Schroeder H; Muller O; Niemeyer CM; Spengler B; Goody RS; Engelhard M, Direct readout of protein-protein interactions by mass spectrometry from protein-DNA microarrays. Angew. Chem. Int. Ed. Engl 2005, 44 (46), 7635–9. [DOI] [PubMed] [Google Scholar]
- 46.Gogolin L; Schroeder H; Itzen A; Goody RS; Niemeyer CM; Becker CF, Protein-DNA arrays as tools for detection of protein-protein interactions by mass spectrometry. ChemBioChem 2013, 14 (1), 92–9. [DOI] [PubMed] [Google Scholar]
- 47.Yu B; Liu W; Yu WM; Loh ML; Alter S; Guvench O; Mackerell AD Jr.; Tang LD; Qu CK, Targeting protein tyrosine phosphatase SHP2 for the treatment of PTPN11-associated malignancies. Mol. Cancer Ther. 2013, 12 (9), 1738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Denu JM; Dixon JE, Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol 1998, 2 (5), 633–41. [DOI] [PubMed] [Google Scholar]
- 49.Pundir S; Martin MJ; O’Donovan C; UniProt C, UniProt Tools. Curr Protoc Bioinformatics 2016, 53, 1 29 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao BM; Keasey SL; Tropea JE; Lountos GT; Dyas BK; Cherry S; Raran-Kurussi S; Waugh DS; Ulrich RG, Phosphotyrosine Substrate Sequence Motifs for Dual Specificity Phosphatases. PLoS One 2015, 10 (8), e0134984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turner AH; Lebhar MS; Proctor A; Wang Q; Lawrence DS; Allbritton NL, Rational Design of a Dephosphorylation-Resistant Reporter Enables Single-Cell Measurement of Tyrosine Kinase Activity. ACS Chem. Biol 2016, 11 (2), 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan BX; Sun YQ, Glycine residues provide flexibility for enzyme active sites. J. Biol. Chem 1997, 272 (6), 3190–4. [DOI] [PubMed] [Google Scholar]
- 53.Lu KP; Liou YC; Zhou XZ, Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002, 12 (4), 164–72. [DOI] [PubMed] [Google Scholar]
- 54.Andrews LD; Zalatan JG; Herschlag D, Probing the origins of catalytic discrimination between phosphate and sulfate monoester hydrolysis: comparative analysis of alkaline phosphatase and protein tyrosine phosphatases. Biochemistry 2014, 53 (43), 6811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hof P; Pluskey S; Dhe-Paganon S; Eck MJ; Shoelson SE, Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92 (4), 441–50. [DOI] [PubMed] [Google Scholar]
- 56.Neel BG; Chan G; Dhanji S, Chapter 98 - SH2 Domain-Containing Protein-Tyrosine Phosphatases In Handbook of Cell Signaling (Second Edition), Bradshaw RA; Dennis EA, Eds. Academic Press: San Diego, 2010; pp 771–809. [Google Scholar]
- 57.Sacco F; Perfetto L; Castagnoli L; Cesareni G, The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586 (17), 2732–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Formisano P; Sohn KJ; Miele C; Di Finizio B; Petruzziello A; Riccardi G; Beguinot L; Beguinot F, Mutation in a conserved motif next to the insulin receptor key autophosphorylation sites de-regulates kinase activity and impairs insulin action. J. Biol. Chem 1993, 268 (7), 5241–8. [PubMed] [Google Scholar]
- 59.Hubbard SR, The insulin receptor: both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol 2013, 5 (3), a008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White MF, Insulin signaling in health and disease. Science 2003, 302 (5651), 1710–1. [DOI] [PubMed] [Google Scholar]
- 61.Salmeen A; Andersen JN; Myers MP; Tonks NK; Barford D, Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol. Cell 2000, 6 (6), 1401–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.