

Abstract
Depletion of glutathione (GSH) is considered a critical pathogenic event promoting alcohol-induced lipotoxicity. We recently show that systemic GSH deficiency in mice harboring a global disruption of the glutamate-cysteine ligase modifier subunit (Gclm) gene confers protection against alcohol-induced steatosis. While several molecular pathways have been linked to the observed hepatic protection, including nuclear factor erythroid 2-related factor 2 and AMP-activated protein kinase pathways, the precise mechanisms are yet to be defined. In this study, to gain insights into the molecular mechanisms underpinning the protective effects of loss of GCLM, global profiling of hepatic polar metabolites combined with liver microarray analysis was carried out. These inter-omics analyses revealed both low GSH- and alcohol-driven changes in multiple cellular pathways involving the metabolism of amino acids, fatty acid, glucose and nucleic acids. Notably, several metabolic changes were uniquely present in alcohol-treated Gclm-knockout mouse livers, including acetyl-CoA enrichment and diversion of acetyl-CoA flux from lipogenesis to alterative metabolic pathways, elevation in glutamate concentration, and induction of the glucuronate pathway and nucleotide biosynthesis. These metabolic features reflect low GSH-elicited cellular response to chronic alcohol exposure, which is beneficial for the maintenance of hepatic redox and metabolic homeostasis. The current study indicates that fine-tuning of hepatic GSH pool may evoke metabolic reprogramming to cope with alcohol-induced cellular stress.
Keywords: Glutathione, glutamate cysteine ligase, acetyl-CoA, alcoholic steatosis, metabolomics, transcriptomics
Graphical Abstract
1. Introduction
Excessive consumption of alcohol is a major cause of chronic liver disease. Globally, alcoholic liver disease (ALD) 1 accounts for 0.9% of total mortality and 0.6% of disability-adjusted life years and remains a public health problem worldwide [1]. ALD initially manifests as simple fatty liver (steatosis), which may progress to steatohepatitis, liver fibrosis and cirrhosis, and eventually hepatocellular carcinoma. To date, the management of ALD remains challenging due to the lack of detailed understanding of determinants of its pathogenesis and progression.
Chronic alcohol consumption modulates numerous cellular pathways in the liver and other organs, among which alcohol-induced redox perturbation appears to play a critical role in the pathogenesis of ALD [2]. Over-production of reactive molecules, including electrophiles (e.g. acetaldehyde and lipid peroxidation byproducts), reactive oxygen species (ROS) and reactive nitrogen species (RNS), may arise from ethanol metabolism, CYP2E1 induction, mitochondrial dysfunction, and proinflammatory processes [2]. In addition, ethanol exposure was shown to reduce the antioxidant capacity of the liver through depleting antioxidants and inactivating antioxidant enzymes [2]. The pathophysiological consequences of oxidative insults include, among others, impaired lipid metabolism leading to steatosis, hepatocyte injury and activation of liver fibrosis, all of which are features of ALD [3].
It is well established that glutathione (GSH), the most abundant cellular non-protein thiol, plays a pivotal role in maintaining redox homeostasis and contributes significantly to protection against oxidative insults [4]. The rate-limiting step in the GSH biosynthesis pathway is catalyzed by the glutamate-cysteine ligase (GCL), a heterodimer comprising a catalytic (GCLC) and a modifier (GCLM) subunit [5]. Disruption of the mouse Gclc gene in hepatocytes results in >95% depletion of hepatic GSH and induces liver pathologies characteristic of various clinical stages of fatty liver disease [6, 7]. Global disruption of mouse Gclm gene generates a mouse model (Gclm-knockout) that exhibits normal liver functioning in spite of the hepatic GSH concentration being only ~15% of normal [8]. Intriguingly, following 6 week of ethanol intake with the Lieber-DeCarli (LD) diet, Gclm-knockout mice were protected from alcohol-induced steatosis [9]. Such protection appeared to be associated with redox activation of nuclear factor erythroid 2-related factor 2 (NRF2) and AMP-activated protein kinase (AMPK), which are key regulators of cellular stress response, cellular metabolism and biogenesis [9]. Although these results revealed the hepatoprotective effect of low GSH, the mechanistic details remain largely unknown.
It was documented in ALD patients and experimental animals that alcohol consumption impacts multiple biochemical pathways [10]. In addition to aberrant lipid metabolism, alcohol-related dysregulation of other cellular metabolisms contributes to ALD pathogenesis [10]. In the current study, we aimed to gain insights into the mechanism(s) by which GSH deficiency (due to loss of GCLM) regulates hepatic metabolic homeostasis in response to chronic ethanol consumption. This was accomplished through global profiling of hepatic polar metabolome. Liver microarray analysis was conducted in parallel to explore molecular mechanisms underlying protection from steatosis through integrated pathway analysis.
2. Materials and methods
2.1. Reagents
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
2.2. Animals and chronic ethanol feeding
All animal experiments were performed at the University of Colorado Anschutz Medical Campus, as reported previously [9]. All animal procedures were approved by and conducted in compliance with Institutional Animal Care and Use Committee (IACUC) of the same institution. Briefly, 10~12 week male C57BL/6J wild-type (WT) and Gclm-null (KO) mice were fed a modified LD diet (Bio-Serv, Frenchtown, NJ) for 6 weeks. Ethanol-fed (EtOH) mice received the LD diet containing 2-5% (v/v) ethanol (increased weekly by 1% until 5% reached). Pair-fed (CON) mice received LD diet containing equivalent calories derived from carbohydrates replacing ethanol. At the end of feeding period, mice were euthanized and their livers were flash frozen in liquid nitrogen and stored at −80°C for latter metabolomics, gene expression and biochemical analyses.
2.3. Tissue extraction for metabolomics analyses
Frozen liver tissues (~30 mg) were homogenized using a Precellys 24 homogenizer (Bertin technologies, France) and extracted using modified Bligh-Dyer method [11] to separate the polar and non-polar metabolites. α-Aminopimelic acid (10 μM) was added to each sample before extraction to normalize differences in metabolite extraction efficiency. The aqueous layer containing polar metabolites was vacuum-dried and reconstituted in acetonitrile/water/methanol (65:30:5) mixture containing 5 μM difluoromethylornithine.
2.4. Global metabolite profiling
Hydrophilic interaction liquid chromatography (HILIC) was coupled with electrospray ionization mass spectrometry (ESI-MS) to analyze changes in metabolic signature. Deproteinated tissue extracts were analyzed in Xevo G2 ESI-QTOFMS coupled with ultraperformance liquid chromatography (UPLC) using a HILIC BEH amide column (Waters Corp, Milford, MA) as described previously [12]. Chromatogram quality and retention time reproducibility across the run were manually inspected. Deconvolution, binning and integration was performed using MarkerLynx software (Waters Corp, Milford, MA). The intensity of each ion was normalized to total ion counts or liver weights to generate a data matrix consisting of m/z value, retention time, and the normalized peak area. Data quality inspection, unsupervised and supervised analysis of metabolomic signatures were performed using SIMCA-P12+ software (Umetrics, Kinnelon, NJ). Unsupervised segregation of samples on global metabolomic space was analyzed by principal components analysis (PCA). The supervised orthogonal projection to latent structures (OPLS) model was used to identify ions contributing to discrimination (indicated by their distance from origin along Y-axes) of metabolic traits as described [12]. A list of ions showing significant (P < 0.05) difference in abundance among groups was generated from the loading S-plot and used for further identification and quantitation. Data mining for metabolite identification was performed using MassTRIX (http://metabolomics.helmholtz-muenchen.de/masstrix/) as previously reported [12].
2.5. Targeted metabolite quantitation
Selected metabolites in the liver extract were quantitated by multiple reaction monitoring using ESI triple-quad platform coupled with 2.1 × 50 mm Acquity UPLC HILIC BEH amide column (1.7 μM) as previously described [12]. An internal standard α-aminopimelic acid (5 μM) was used to normalize area under the peak (response). The concentration of each metabolite was determined from a calibration curve derived from serially diluted solutions of an authentic standard and normalized to liver weight. Results are expressed as mole per gram liver weight. The ratio of abundances of metabolites biochemically connected was calculated using the IS-normalized response of respective metabolites. Data are presented as floating bars (min to max, line at mean) showing individual data points from each experimental group (N = 4/group).
2.6. Liver microarray analysis
Total RNA was isolated from frozen liver tissues using TRIzol Reagent™ (ThermoFisher Scientific Inc., MA) according to the manufacturer's protocol. Complementary DNA was dye-coupled and hybridized to Agilent 44K mouse 60-mer oligonucleotide microarrays (Agilent Technologies, Santa Clara, CA). Microarray data were processed and analyzed using Genespring GX 11.5.1 software (Agilent Technologies, Santa Clara, CA).
2.7. Quantitative real-time PCR (Q-PCR)
Total RNA was isolated from frozen liver tissues using TRIzol Reagent™ according to the manufacturer’s protocol. cDNA was synthesized using iScript cDNA synthesis kit (BioRad, Hercules, CA) according to the manufacturer’s instructions using 1 μg total RNA in a 20 μl reaction volume. Q-PCR reaction mixtures contained 0.5 μl cDNA, 1x SYBR Green Supermix (BioRad, Hercules, CA), and 0.15 μM gene-specific primer sets in a total volume of 10 μl Sequences of Q-PCR primers can be found in Table S1. Reactions were run using the CFX96 Touch Detection System (BioRad, Hercules, CA). Expression of β−2-microglobulin (B2m) was used for normalization of CT data according to the ΔΔCT method [13]. Relative mRNA levels of individual genes were reported as fold of expression in pair-fed control WT mice (WT-CON). Data are presented as mean ± S.D. (N = 4/ group).
2.8. Measurements of nucleotide cofactors
Total liver concentrations of NAD, NADH, NADP, and NADPH were measured using biochemical assay kits (Abcam, Cambridge, MA) according to the manufacturer’s protocol. Results are reported in mole per milligram total proteins. Total protein levels of liver extracts were quantified using the Bradford assay (BioRad, Hercules, CA) according to the manufacturer’s protocol. Data are presented as floating bars (min to max, line at mean) showing individual data points from each experimental group (N = 4/group).
2.9. Statistic analysis
Qlucore Omics Explorer (QOE) program version 3.4 (Qlucore AB, Sweden) was used for differential analysis of microarray gene expression among four experimental groups by one-way ANOVA (N = 4/group); q < 0.05 were used as the significance cut-off criteria and the q-values were generated based on the Benjamini–Hochberg False Discovery Rate (FDR) method [14]. Group differences in metabolite quantitation, gene expression by Q-PCR, and biochemical assays were analyzed using Graphpad Prism software (San Diego, CA) by two-way ANOVA, where the two factors were loss of GCLM (Gclm-KO) and EtOH feeding. The post-hoc Holm-Sidak test was performed for multiple comparisons test. P < 0.05 was considered significant. Two-way ANOVA P values can be found in Table S2. Hierarchical clustering of significant metabolites or genes was performed by QOE.
2.10. Bioinformatics Analysis
Functional enrichment analysis of differentially expressed genes by hierarchical clusters was performed using the Ingenuity Pathway Analysis (IPA) Knowledge Base (version 39408507, Ingenuity Systems, QIAGEN). Top canonical pathways and cellular functions involving differentially expressed transcripts (molecule number > 5) were calculated based on the Fisher’s right-tailed exact test. Inter-omics analyses combining metabolomic and transcriptomic signatures were performed and visualized using the Cytoscape Software version 3.5 [15] for biological network enrichment analysis.
3. Results
3.1. Liver metabolic profiling reveals a distinct metabolic signature associated with loss of GCLM and ethanol consumption
During the 6-week feeding period, the daily intake of CON or EtOH diet by WT and Gclm-KO mice were no different (data not shown). The overall liver polar metabolic signature was analyzed by principal components analysis. The scores-scatter plot for liver weight-normalized metabolic signature recorded in positive (Fig. 1A, left panel) and negative (Fig. 1A, right panel) ESI modes showed that mice segregate along first principal component according to their genotypes, irrespective of EtOH administration. The respective loadings plots showed that the highest contribution to this segregation was from ions with m/z = 308.092+ in the positive ion mode (Fig. 1B, dashed circle in left panel) and ions with m/z = 306.077- in the negative ion mode (Fig. 1B, dashed circle in right panel). A similar pattern of segregation was also observed when total ion count normalization was used (data not shown). These ions were more abundant in the WT liver metabolome and putatively represent protonated and deprotonated GSH, respectively. These results demonstrate that the global metabolic profiling correctly captured the biochemical phenotype caused by lack of GCLM expression. However, the high abundance of aforementioned ions might mask small but significant changes due to EtOH administration. Thus, orthogonal projections to latent structures discriminant analysis (OPLS-DA) in the positive (Fig. S1) and negative (Fig. S2) modes were used for supervised analysis of changes in the metabolic signature specifically associated with genotype or EtOH exposure. Ions showing a significant difference in abundance were selected from the S-plots (Figs. S1&S2) and the corresponding metabolites were identified. A total of 77 metabolites including these as well as others belonging to related pathways were quantitated using authentic standards and 29 metabolites showed differential abundance among the four experimental groups (Fig. 1C). Pairwise inter-correlation analysis of these molecules revealed three major clusters of highly correlated metabolites (Fig. 1D), suggesting each cluster may be represented by metabolites from biochemically linked pathways.
Fig. 1. Global and targeted metabolic profiling of the hepatic polar metabolome.

Metabolomic analyses were conducted in the livers of wild-type (WT) and Gclm-KO (KO) mice pair-fed control (CON) or ethanol-containing (EtOH) diet for six weeks. (A-B) Untargeted metabolic profiling by HILIC/ESI-MS. Scores-scatter plots (A) and loadings plots (B) of principal component analysis of liver weight-normalized metabolic data recorded in positive (left panels) and negative (right panels) modes. (C-D) Targeted metabolite quantitation by UPLC-MS/MS. 29 metabolites showed differential abundance among the four experimental groups. (C) Hierarchy clustering heat map of 29 metabolites. The color key represents values of relative abundance. (D) Pair-wise inter-correlation heat map of 29 metabolites. The color key represents Pearson correlation r values (P < 0.05). WT-CON, wild-type mice fed the control diet; KO-CON, Glcm-KO mice fed the control diet; WT-EtOH, wild-type mice fed the ethanol-containing diet; KO-EtOH, Glcm-KO mice fed the ethanol-containing diet.
3.2. Inter-omics analysis identified derangement of selective biochemical pathways associated with loss of GCLM and ethanol consumption
Liver microarray analysis detected 480 significant gene mRNAs that were differentially expressed due to loss of GCLM or EtOH consumption. Based on the trend of change, these genes fell into three clusters (Fig. 2A): cluster I contained 108 genes that were primarily upregulated by loss of GCLM, cluster II contained 306 genes that were induced synergistically by loss of GCLM and EtOH consumption, and cluster III contained 66 genes that were suppressed by either loss of GCLM or EtOH consumption. The significant genes include genes encoding metabolic enzymes (31%), followed by transporters (9%), kinase/phosphatase (7%), peptidase (5.1%), and transcription factors (4.9%) (Fig. 2B). IPA functional enrichment analysis revealed that cluster I and II genes are largely enriched in overlapping top canonical pathways, including NRF2-mediated oxidative stress, xenobiotic metabolism signaling, nicotine/serotonin degradation and protein ubiquitination (Fig. 2B). Among top five cellular functions, drug and lipid metabolism are both enriched by cluster I and II genes. In addition, while cluster I genes are enriched in metabolism of carbohydrates, nucleic acids and amino acids, cluster II genes are enriched in energy production, small molecule biochemistry and protein synthesis. The cluster III representing suppressed genes appears to be involved primarily in steroid metabolism, lipid biosynthesis and fatty acid metabolism. Network enrichment analysis was subsequently conducted by combining panels of significant metabolites and significant genes encoding metabolic enzymes. This inter-omics analysis yielded seven major metabolic networks that are enriched by input metabolites and genes (Fig. 2C), including (i) fatty acid and central carbon metabolism, (ii) amino acid metabolism, (iii) nucleic acid metabolism, (iv) amino sugar metabolism, (v) pentose phosphate pathway, (vi) glycerophospholipid metabolism, and (vii) xenobiotic metabolism. This result is suggestive of functional associations between observed changes in the liver metabolome and transcriptome. Detailed changes in respective pathways are presented in following sections.
Fig. 2. Overview of liver microarray analysis and inter-omics network enrichment analysis.

(A-B) Microarray gene expression assay was performed in the livers of wild-type (WT) and Gclm-KO (KO) mice pair-fed control (CON) or ethanol-containing (EtOH) diet for six weeks. (A) Hierarchy clustering heat map of 480 genes that were differentially expressed among four experimental groups. The color key represents values of relative abundance. (B) Three clusters of genes were noted based on their expression patterns (details are presented in 3.2). For each gene cluster, the proportion of genes encoding proteins of a specific function (as a percentage of the total number of genes in the cluster) is presented as a pie chart. Top five canonical pathways and cellular functions identified by IPA enrichment analysis are shown as bar charts; their - Log(P-value) are presented on the x-axis of the bar charts. (C) Inter-omics network enrichment analysis was performed using the Cytoscape Software by combining significant metabolites and metabolic enzyme-encoding genes (inputs). Seven major metabolic networks were identified, representing metabolic pathways of: (1) fatty acid and central carbon, (2) amino acids, (3) nucleotides, (4) amino sugars, (5) pentose phosphate pathway, (6) glycerol phospholipids, and (7) xenobiotics.
3.3. Alterations in metabolites and gene expression involved in GSH metabolism and related pathways
In the liver, GSH biosynthesis is biochemically linked to the trans-methylation and trans-sulfuration pathways and thereby is linked to the metabolism of S-adenosylmethionine (SAM) and sulfur-containing amino acids, respectively [16]. Additionally, GSH serves as a cellular reservoir of its constituent amino acids via the γ-glutamyl cycle [17]. A simplified scheme of selected metabolic reactions and intermediates that are involved in these pathways is presented in Fig. 3A. There was a main effect of Gclm-KO on hepatic concentrations of GSH, GSSG, glutamate and threonine, whereas EtOH feeding altered the levels of SAM and glutamate; no interaction between the two main effects was noted for any of these metabolites (Table S2). In particular, by UPLC quantitation, liver GSH and GSSG concentrations in Gclm-KO mice were ~1% and 20%, respectively, of WT levels, irrespective of EtOH administration (Fig. 3B); the resulting GSH/GSSG ratios were much lower in Gclm-KO vs. WT mice (Fig. 3B), indicating a more oxidized GSH pool in the Gclm-KO liver. Among quantified amino acids or derivatives (Table 1 and Fig. 3C), glutamate and threonine in Gclm-KO liver were twice that of WT liver; EtOH feeding further increased glutamate levels in Gclm-KO livers by 60% (Fig. 3C). In the trans-methylation pathway [18]. EtOH feeding induced a reduction in hepatic SAM concentration and SAM/S-adenosylhomocysteine (SAH) ratio only in Gclm-KO mice (Fig. 3D), suggesting that low GSH or low GSH/GSSG ratio may trigger alcohol-associated dysregulation of cellular methylation [19].
Fig. 3. Alterations in metabolites and gene expression involved in GSH metabolism and related pathways.

(A) Scheme of selected biochemical reactions and metabolites involved in GSH metabolism (via γ-glutamyl cycle) and trans-methylation and trans-sulfuration pathways. Solid lines with arrow indicate single biochemical reactions. Solid lines terminated by solid diamond indicates cellular source. Hepatic concentrations of (B) GSH and GSSG, (C) glutamate and threonine, and (D) SAM and SAH. Results are expressed in mole per gram (g) liver weight. Data are presented as floating bars (min to max, line at mean; N = 4/group) showing individual data points from wild-type (WT, closed circles) and Gclm-KO (KO, open circles) mice. *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. (E) Relative mRNA abundance measured by Q-PCR. Results are expressed as mean ± S.D. (N = 4/group). *Compared to WT-CON, ^compared to WT-EtOH, or #compared to KO-CON: P < 0.05 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. WT-CON, wild-type mice fed control diet; KO-CON, Glcm KO mice fed control diet; WT-EtOH, wild-type mice fed ethanol-containing diet; KO-EtOH, Glcm KO mice fed ethanol-containing diet. Cys, cysteine; γ-GC, γ-glutamylcysteine; Gln, glutamine; Glu, glutamate; Gly, glycine; Hcy, homocysteine; Met, methionine; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; GCL, glutamate-cysteine ligase; GCLC, catalytic subunit of GCL; GCLM, modifier subunit of GCL; GLS, glutaminase; GLUD, glutamate dehydrogenase; GSH, reduced glutathione; GSSG, oxidized glutathione; GPX, glutathione peroxidase; GSR, glutathione disulfide reductase; GST, glutathione S-transferase.
Table 1.
Hepatic metabolite concentrations
| Experimental group | WT-CON | KO-CON | WT-EtOH | KO-EtOH |
|---|---|---|---|---|
| Amino acids and derivatives (nmol/g liver)1 | ||||
| Arginine | 87 ± 22.7 | 76 ± 16.8 | 98 ± 10.2 | 73 ± 13.9 |
| Aspartate | 471 ± 108.4 | 564 ± 162.9 | 509 ± 62.6 | 686 ± 260.9 |
| Betaine | 195 ± 36 | 102 ± 35a | 111 ± 57 | 139 ± 67 |
| Carnitine | 265 ± 24.2 | 240 ± 43.1 | 230 ± 44.0 | 277 ± 73.1 |
| Choline | 3748 ± 1896 | 3831 ± 1240 | 8944 ± 2726b | 8013 ± 2617b |
| Citruline | 11 ± 1.9 | 15 ± 2.9 | 28 ± 21.5 | 31 ± 35.6 |
| Creatine | 689 ± 101.8 | 603 ± 72.6 | 665 ± 158.6 | 816 ± 399.6 |
| Creatinine | 17 ± 1.0 | 17 ± 2.0 | 19 ± 3.5 | 19 ± 7.2 |
| Glutamine | 4082 ± 472.0 | 4335 ± 1016.6 | 4295 ± 252.6 | 4848 ± 1412.1 |
| Histidine | 381 ± 25.0 | 419 ± 75.0 | 410 ± 46.2 | 457 ± 54.4 |
| Lysine | 611 ± 71.1 | 614 ± 103.6 | 672 ± 240.5 | 644 ± 174.7 |
| Methionine | 51 ± 10.2 | 51 ± 6.1 | 69 ± 19.5 | 69 ± 9.5 |
| N-acetylglutamine | 805 ± 167.8 | 873 ± 180.4 | 733 ± 268.2 | 937 ± 512.2 |
| Nε-acetyllysine | 2 ± 0.4 | 3 ± 0.4 | 3 ± 1.0 | 4 ± 1.9 |
| N-acetylcysteine | 1.0 ± 0.3 | 0.7 ± 0.1 | 1.6 ± 0.7 | 0.7 ± 0.1 |
| Ornithine | 279 ± 28.8 | 375 ± 79.7 | 359 ± 124.9 | 449 ± 131.7 |
| Proline | 160 ± 22.0 | 202 ± 38.5 | 158 ± 61.4 | 239 ± 59.1 |
| Taurine | 11284 ± 980 | 14808 ± 1260a | 10609 ± 1546 | 9967 ± 4418b |
| Glycolysis and TCA cycle (nmol/g liver)1 | ||||
| ADP | 369 ± 33 | 486 ± 109 | 514 ± 56 | 606 ± 144 |
| ATP | 205 ± 13 | 226 ± 54 | 215 ± 45 | 210 ± 50 |
| Isocitrate | 306 ± 67.7 | 273 ± 33.8 | 311 ± 60.5 | 281 ± 111.9 |
| Lactate | 57 ± 11 | 69 ± 7 | 36 ± 4b | 41 ± 7b |
| Malate | 4948 ± 1062.8 | 4725 ± 1348.7 | 4354 ± 793.3 | 5197 ± 1211.8 |
| Succinic acid | 408 ± 83.5 | 617 ± 242.6 | 350 ± 90.8 | 629 ± 333.4 |
| Nucleic acid metabolism (nmol/g liver)1 | ||||
| Adenosine | 197 ± 29 | 224 ± 68 | 243 ± 46 | 344 ± 86b |
| AMP | 34 ± 12.7 | 34 ± 12.9 | 42 ± 18.1 | 47 ± 20.5 |
| CMP | 72 ± 10.2 | 98 ± 27.7 | 97 ± 22.1 | 90 ± 31.2 |
| Cytidine | 2 ± 0.5 | 3 ± 0.7 | 2 ± 0.3 | 3 ± 0.6 |
| deoxycytidine | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 |
| deoxyuridine | 7117 ± 4393.5 | 8117 ± 3693.1 | 6490 ± 4275.5 | 5793 ± 2604.3 |
| GMP | 898 ± 34 | 900 ± 216 | 1166 ± 125 | 1464 ± 513b |
| Hypoxanthine | 689 ± 101.8 | 603 ± 72.6 | 665 ± 158.6 | 816 ± 399.6 |
| IMP | 1125 ± 74.8 | 1049 ± 395.1 | 1055 ± 229.8 | 1245 ± 538.5 |
| TMP | 7 ± 1.1 | 6 ± 0.6 | 7 ± 0.7 | 7 ± 1.4 |
| UMP | 11085 ± 128 | 15801 ± 4001 | 13013 ± 2758 | 17652 ± 5690 |
| Uracil | 3 ± 0.9 | 2 ± 0.5 | 2 ± 1.1 | 3 ± 2.8 |
| UTP | 54 ± 7.5 | 52 ± 5.7 | 55 ± 4.4 | 53 ± 8.6 |
| Xanthine | 203 ± 18.6 | 150 ± 38.3 | 157 ± 31.4 | 157 ± 77.8 |
| Xanthosine | 181 ± 44 | 182 ± 66 | 88 ± 21b | 104 ± 44b |
| Nucleotide cofactors (nmol/mg proteins)2 | ||||
| NADPH | 1.37 ± 0.42 | 1.10 ± 0.29 | 1.39 ± 0.39 | 1.24 ± 0.27 |
| NADP | 0.33 ± 0.07 | 0.30 ± 0.07 | 0.38 ± 0.11 | 0.40 ± 0.1 |
| NADPH/NADP ratio | 4.2 ± 0.7 | 3.8 ± 1.3 | 3.8 ± 1.2 | 3.2 ± 0.9 |
Selected metabolites in the liver extract were quantitated by UPLC-MS/MS. The concentration of each metabolite was determined from respective calibration curve and normalized by liver weight. Results are reported in nmole per gram liver weight.
Concentrations of NADH and NADPH were measured using a biochemical assay kit (Abcam, Cambridge, MA); results are reported in nmole per milligram total proteins.
Data are presented as mean ± S.D. (N = 4/group). Group differences were analyzed using Graphpad Prism software by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test.
P < 0.05, when compared with diet-matched WT mice;
P < 0.05, when compared with pair-fed mice of the same genotype.
Microarray analysis and mRNA quantification revealed inductions of genes encoding selective enzymes involved in GSH metabolism [Gclc, glutathione peroxidase 4 (Gpx4), glutathione S-transferases Mu1 and Mu 4 (Gstml and Gstm4) and glutathione disulfide reductase (Gsr)] and glutamate metabolism [glutaminase 1 (Gls1) and glutamate dehydrogenase 1 (Glud1)] in Gclm-KO mice irrespective of EtOH administration (Fig. 3E). GPX and GST enzymes consume GSH for detoxifying electrophilic compounds, whereas GSR reduces GSSG to GSH. Therefore, concomitant induction of these genes would be expected to facilitate a fast turnover of GSH in the Gclm-KO liver. Upregulation of Gls1 and Glud1 genes is in line with higher glutamate concentrations in the Gclm-KO liver, as both enzymes can generate glutamate.
3.4. Alterations in metabolites and gene expression involved in acetyl-CoA metabolic flux
Acetyl-CoA is a key metabolic node connecting cellular catabolic and anabolic reactions [20]. A simplified scheme of selected metabolic reactions and intermediates that are involved in acetyl-CoA metabolic flux is presented in Fig. 4A. Numerous metabolites involved in these pathways were altered due to a main effect of Gclm-KO and/or EtOH feeding (Table S2). While hepatic acetyl-CoA levels declined following EtOH administration, the levels in Gclm-KO mice were twice that of diet-matched WT mice (Fig. 4B). A similar trend was also observed for acetylcarnitine and butyrylcarnitine, two short-chain fatty acylcarnitines generated during mitochondrial fatty acid β-oxidation (Fig. 4C). Among five quantified N-acetylated amino acids, EtOH feeding elevated the level of Nα-acetyllysine in WT and Gclm-KO mice by approximately 2-fold (Fig. 4D) without increasing the lysine per se (Table 1); no difference in Nε-acetyllysine levels was observed (Table 1). In contrast, there was a significant interaction between the effects of Gclm-KO and EtOH feeding on hepatic concentrations of N-acetylglutamate, which was elevated exclusively in EtOH-fed Gclm-KO mice by 4-fold (Fig. 4D); this appeared to be due to the increased influx of glutamate, as evidenced by unchanged N-acetylglutamate/glutamate ratios (Fig. 4D). While no changes in intermediates of glycolysis or tricarboxylic acid (TCA) cycle were noted (Table 1), thiamine, in its diphosphate form serving as an essential cofactor for several key enzyme complexes in these pathways (Fig. 4A), was attenuated by EtOH feeding in WT (~40%) and more dramatically in Gclm-KO mice (~60%) (Fig. 4E). Interestingly, 2-hydroxyglutarate, a rare metabolite derived from α-ketoglutarate by the action of isocitrate dehydrogenase (IDH) in normal cells [21], was found doubled in Gclm-KO mice in both diet groups (Fig. 4E). No difference in the steady state concentrations of ADP or ATP was observed among four experimental groups (Table 1).
Fig. 4. Alterations in metabolites and gene expression involved in acetyl-CoA metabolic flux.

(A) Scheme of selected biochemical reactions and metabolites involved in acetyl-CoA metabolic flux. Acetyl-CoA can be generated from (1) fatty acid oxidation, (2) pyruvate oxidation, (3) amino acids catabolism, and (4) ethanol-derived acetate. Acetyl-CoA can be consumed for energy production via (5) tricarboxylic acid (TCA) cycle, (6) fatty acid biosynthesis, (7) cholesterol and steroid biosynthesis, and (8) acetylation process. Solid or dashed lines with arrow indicate single or multiple biochemical reactions, respectively. Hepatic concentrations of (B) acetyl-CoA, (C) short-chain acylcarnitines, (D) N-acetyl amino acids, and (E) thiamine and 2-hydroxyglutarate. Results are expressed in mole per gram (g) liver weight. Data are presented as floating bars (min to max, line at mean; N = 4/group) showing individual data points from wild-type (WT, closed circles) and Gclm-KO (KO, open circles) mice. *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. (F) Relative mRNA abundance measured by Q-PCR. Results are expressed as mean ± S.D. (N = 4/group). *Compared to WT-CON, ^compared to WT-EtOH, or #compared to KO-CON: P < 0.05 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. WT-CON, wild-type mice fed control diet; KO-CON, Glcm KO mice fed control diet; WT-EtOH, wild-type mice fed ethanol-containing diet; KO-EtOH, Glcm KO mice fed ethanol-containing diet; Glu, glutamate; TG, triglycerides; Thr, threonine; ACC, acetyl-CoA carboxylase; ACOX, acyl-CoA oxidase; ACSL, long-chain acyl-CoA synthetase; CYP17A1, cytochrome P450 17a1; DLD, dihydrolipoamide dehydrogenase; EBP, emopamil binding protein; IDH, isocitrate dehydrogenase; LPL, lipoprotein lipase; OGDC, oxoglutarate dehydrogenase complex; PCCA, propionyl-CoA carboxylase; PDC, pyruvate dehydrogenase complex; PDHA1, pyruvate dehydrogenase α1 subunit; SLC22A5, solute carrier family 22 member 5; SLC25A17, solute carrier family 25 member 17.
In a previous study, we reported expression changes in genes encoding lipid metabolizing enzymes, which are featured by suppression of lipogenic genes and induction of fatty acid oxidation genes in Gclm-KO mouse liver [9]. Such a pattern of gene expression change would be predicated to result in a net increase in acetyl-CoA in the Gclm-KO liver as was observed in the present study (Fig. 4B). Liver microarray and mRNA quantification identified changes in additional genes involved in acetyl-CoA metabolic flux or associated pathways (Fig. 4F). First, the lipogenic gene acetyl-CoA carboxylase beta (Accb) was suppressed by 50% in Gclm-KO mice and two genes involved in steroid biosynthesis [emopamil binding protein (Ebp) and cytochrome 17a1 (Cyp 17 a1)] were drastically suppressed in Gclm-KO mice and by EtOH feeding. Genes involved in lipolysis [lipoprotein lipase Lpl] and fatty acid oxidation [acyl-CoA oxidase 2 (Acox2)] were upregulated in Gclm-KO mice. Second, genes encoding transporters that facilitate uptake of carnitine (Slc22a5) and the transport of cofactors for fatty acid oxidation (Slc25a17) were upregulated in Gclm-KO livers. Third, genes encoding the pyruvate dehydrogenase α1 subunit (Pdha1) and dihydrolipoamide dehydrogenase (Dld), two components of the pyruvate dehydrogenase complex (PDC) that produces acetyl-CoA from pyruvate, were upregulated in Gclm-KO mice irrespective of EtOH intake. Lastly, the gene encoding the mitochondrial isoform of IDH (Idh2) was induced by EtOH feeding exclusively in Gclm-KO liver.
3.5. Alterations in metabolites and gene expression involved in amino sugar metabolism, pentose phosphate pathway and glucuronic acid pathway
Changes in metabolites involved in several pathways of glucose metabolism were noted as a main effect of Gclm-KO and/or EtOH feeding (Table S2). A simplified scheme of selected metabolic reactions and intermediates is presented in Fig. 5A. In amino sugar metabolism, also known as hexosamine biosynthesis pathway (HBP), N-acetylglucosamine-6-phosphate (GlcNAc-6P) was elevated by EtOH feeding in Gclm-KO mice (Fig. 5B). In contrast, UDP-acetylglucosamine (UDP-GlcNAc), a precursor of cellular glycosylation, was ~50% lower in EtOH-fed Gclm-KO mice vs. WT counterparts (Fig. 5B). Gluconate-6P, an intermediate metabolite of the oxidative pentose phosphate pathway (PPP), was ~2-fold abundant in Gclm-KO vs. WT mice irrespective of EtOH intake (Fig. 5C). While there was an overall ethanol-induced reduction in hepatic concentrations of UDP-glucose (UDP-Glc), the precursor for glycogen biosynthesis, such reduction was only significant in Gclm-KO mice (Fig. 5D). In the glucuronic acid pathway, [22][23]hepatic levels of UDP-glucuronic acid (UDP-GlcUA) and glucuronic acid (GlcUA) were profoundly elevated by EtOH feeding exclusively in Gclm-KO mice (Fig. 5D). In this panel of metabolites, there was a significant interaction between the effects of Gclm-KO and EtOH feeding on hepatic levels of UDP-GlcNAc and UDP-GlcUA (Table S2).
Fig. 5. Alterations in metabolites and gene expression involved in amino sugar metabolism, pentose phosphate pathway and glucuronic acid pathway.

(A) Scheme of selected biochemical reactions and metabolites involved in these pathways. Solid or dashed lines with arrow indicate single or multiple biochemical reactions, respectively. Hepatic concentrations of metabolites of (B) hexosamine biosynthesis, (C) oxidative pentose phosphate pathway, and (D) glucuronic acid pathway. Results are expressed in mole per gram (g) liver weight. Data are presented as floating bars (min to max, line at mean; N = 4/group) showing individual data points from wild-type (WT, closed circles) and Gclm-KO (KO, open circles) mice. *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. (E) Relative mRNA abundance measured by Q-PCR. Results are expressed as mean ± S.D. (N = 4/group). *Compared to WT-CON, åcompared to WT-EtOH, or #compared to KO-CON: P < 0.05 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. WT-CON, wild-type mice fed control diet; KO-CON, Glcm KO mice fed control diet; WT-EtOH, wild-type mice fed ethanol-containing diet; KO-EtOH, Glcm KO mice fed ethanol-containing diet; 6PGL, phosphogluconolactonase; Fru-6P, fructose-6-phosphate; Glc, glucose; Glc-1P, glucose-1-phosphate; Glc-6P, glucose-6-phosphate; GlcN-6P, glucosamine-6-phosphate; GlcNAc, N-acetylglucosamine; GlcNAc-1P, N-acetylglucosamine-1-phosphate; GlcNAc-6P, N-acetylglucosamine-6-phosphate; GlcUA, glucuronic acid; Gln, glutamine; Glu, glutamate; PRPP, 5-phosphoribosyl-1-pyrophosphate; UDP-Glc, UDP-glucose; UDP-GlcNAc, UDP-acetylglucosamine; UDP-GlcUA, UDP-glucuronic acid; GCK, glucokinase; GNPDA, glucosamine-6-phosphate deaminase; NAGK, N-acetylglucosamine kinase; PGM, phosphoglucomutase 3; UGDH, UDP-glucose 6-dehydrogenase; UGP, UDP-glucose pyrophosphorylase 2; UGTs, UDP-glucuronosyltransferases.
The hepatic expression of several genes involved in these metabolic pathways was altered by the loss of GCLM and/or by EtOH consumption (Fig. 5E). Glucokinase (Gck) and phosphoglucomutase 3 (Pgm3) were drastically suppressed in Gclm-KO vs. WT mice when fed the CON diet; EtOH feeding induced these genes in Gclm-KO mice, but attenuated Gck expression in WT mice. On the other hand, genes encoding HBP enzymes [glucosamine-6-phosphate deaminase 2 (Gnpda2) and N-acetylglucosamine kinase (Nagk)], GlcUA pathway enzymes [UDP-glucose pyrophosphorylase 2 (Ugp2) and UDP-glucose 6-dehydrogenase (Ugdh)] and UDP-glucuronosyltransferases (Ugt2b5 and Ugt2b35) were all upregulated in Gclm-KO mice fed either CON or EtOH diet and in general correlate well with changes in metabolic profile.
3.6. Alterations in metabolites and gene expression involved in nucleic acids metabolism and nucleotide cofactors
A simplified scheme of selected biochemical pathways in purine and pyrimidine metabolism is presented in Fig. 6A. Two metabolites involved in these pathways were found altered (Table S2). Uric acid, the end-product of purine degradation, was reduced by EtOH feeding in general; albeit it was dramatically depleted in EtOH-fed Gclm-KO mice when compared with CON-fed Gclm-KO mice (Fig. 6B). Ratios of uric acid to its precursors xanthine and hypoxanthine were both decreased by EtOH feeding exclusively in Gclm-KO mice (Fig. 6B), suggesting suppressed purine degradation in Gclm-KO livers. With regard to pyrimidines (Fig. 6C), the ratio of UMP versus glutamine, the precursor amino acid for de novo pyrimidine biosynthesis, was much higher in Gclm-KO mice indicating augmented salvage biosynthesis. There was a significant interaction between the effects of Gclm-KO and EtOH feeding on the UDP concentration (Table S2), which was approximately doubled in EtOH-fed Gclm-KO mice relative to other experimental groups. The nucleotide cofactors NAD/NADH and NADP/NADPH are redox couples intimately involved in cellular metabolism. Hepatic NADH concentrations in CON-fed Gclm-KO mice were twice that seen in WT counterparts; EtOH feeding increased NADH levels only in WT mice (Fig. 6D). No changes in NAD concentrations were noted. This resulted in doubling of NADH/NAD ratio in Gclm-KO vs. WT mice when fed the CON diet and an increase in NADH/NAD ratio in WT mice following EtOH consumption (Fig. 6D). No differences in NADP and NADPH were observed (Table 1).
Fig. 6. Alterations in metabolites and gene expression involved in nucleic acids metabolism and nucleotide cofactors.

(A) Scheme of selected biochemical reactions and metabolites involved in purine and pyrimidine metabolism. Solid or dashed lines with arrow indicate single or multiple biochemical reactions, respectively. Hepatic concentrations of metabolites of (B) purine metabolism and (C) pyrimidine metabolism; results are expressed in mole per gram (g) liver weight. (D) Hepatic concentrations of NAD and NADH; results are expressed in mole per milligram (mg) total proteins. All data are presented as floating bars (min to max, line at mean; N = 4/group) showing individual data points from wild-type (WT, closed circles) and Gclm-KO (KO, open circles) mice. *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. (E) Relative mRNA abundance measured by Q-PCR. Results are expressed as mean ± S.D. (N = 4/group). *Compared to WT-CON, ^compared to WT-EtOH, or #compared to KO-CON: P < 0.05 by two-way ANOVA with post-hoc Holm-Sidak's multiple comparisons test. WT-CON, wild-type mice fed control diet; KO-CON, Glcm KO mice fed control diet; WT-EtOH, wild-type mice fed ethanol-containing diet; KO-EtOH, Glcm KO mice fed ethanol-containing diet; AK4, Adenylate kinase 4; CTPS, CTP synthase; HPRT, hypoxanthine phosphoribosyltransferase. PRPP, 5-phosphoribosyl-1-pyrophosphate
Liver microarray and mRNA quantitation identified only three metabolic genes that were altered at the transcriptional level (Fig. 6E). Hypoxanthine phosphoribosyltransferase (HPRT) plays an important role in the purine salvage pathway by converting hypoxanthine to IMP and guanine to GMP [24]. CTP synthase (CTPS) plays a central role in de novo synthesis of cytosine nucleotides by converting UTP to CTP [25]. Adenylate kinase 4 (AK4) is involved in maintaining cellular nucleotides homeostasis by catalyzing the reversible transfer of phosphate group among adenine and guanine nucleotides [26]. These nucleotide-metabolizing genes were induced by 2-4-fold in Gclm-KO mice fed either CON or EtOH diet.
4. Discussion
There is a large body of evidence indicating that low hepatic GSH content is pathogenically involved in liver diseases of various etiologies [27-30]. This is expected given the crucial function of GSH in maintaining cellular redox homeostasis and in detoxifying electrophiles. New knowledge derived from redox biology research, however, provides convincing evidence that chronic oxidative and nitrosative stress under physiological conditions trigger cellular mechanisms that promote survival and damage repair [31-33]. In line with this notion, utilizing a genetic mouse model (Gclm-KO mice), we demonstrated that chronic oxidative stress due to GSH deficiency confers protection against fatty liver injuries induced by several environmental and dietary insults, including alcohol intake [34]. In the context of alcohol-related liver damage, Gclm-KO mice manifest enhanced ethanol metabolism and show gene expression changes that suppress lipogenesis and promote fatty acid oxidation [9]. In the current study, global metabolic profiling of liver identified a wide range of metabolic changes that reflect a mixture of low GSH-and ethanol-driven effects on the metabolism of amino acids, fatty acids, glucose and nucleic acids. In comparison with a limited number of reports on the liver polar metabolome in experimental models of alcoholic liver damage, the current study recaptured some principal metabolic perturbations (or similar trends) associated with ethanol intake in WT mice, for instance attenuation of fatty acid oxidation [35, 36], impairment of glucose metabolism [35-37], depletion of thiamine [38], and elevation of N-acetyl amino acids [35, 39]. More importantly, this study identified metabolic signatures that are unique in the liver from ethanol-fed Gclm-KO mice.
One striking difference between WT and Gclm-KO mice is the high abundance of glutamate in Gclm-KO livers, which is further elevated following ethanol consumption. Apart from possible accumulation of glutamate due to impaired GSH biosynthesis, upregulation of genes encoding glutamate-producing enzymes (viz., Gls, Gludl and Gnpda) likely also contributes to this increase. Enhanced glutamate production may confer several biochemical advantages in the Gclm-KO liver: (i) it compensates for the high Km of GCLC towards glutamate [5], thereby serving to augment GSH biosynthesis; (ii) it can fuel ATP production through TCA cycle via GLUD-mediated α-ketoglutarate production; and (iii) it serves as an alternative metabolic sink for ethanol-derived carbons via the formation of N-acetylglutamate that is found in high levels exclusively in ethanol-exposed Gclm-KO liver [40]. It is interesting to note that Gclm-KO liver also has higher levels of threonine, which are unaffected by ethanol feeding. Threonine is an essential amino acid being both glucogenic and ketogenic. The biochemical mechanism underlying increased threonine in the Gclm-KO liver is unknown and warrants future studies. One can speculate that it may result from reduced catabolism or merely from increases in intestinal and hepatic uptake of threonine.
Acetyl-CoA enrichment represents another metabolic feature in the liver of Gclm-KO mice. In the liver, acetyl-CoA is generated primarily in mitochondria from fatty acid β-oxidation, glycolysis and catabolism of branched-chain amino acids. Under alcohol consumption, acetyl-CoA can be derived from acetate, the end metabolite of ethanol metabolism. Mitochondrial acetyl-CoA can feed the TCA cycle to generate ATP or can be shuttled to the cytosol, where it serves as the precursor for biosynthesis of fatty acids, cholesterol, steroids, or specific amino acids. Acetyl-CoA is also the substrate for cellular acetylation process. Our metabolomic and transcriptomic analyses support augmented production of acetyl-CoA from pyruvate oxidation and fatty acid oxidation, and reduced acetyl-CoA consumption for lipid and steroid biosynthesis in Gclm-KO mice relative to WT mice when fed control diet [9]. Despite expected acetyl-CoA flux from ethanol-derived acetate, ethanol intake for 6 weeks resulted in reduced acetyl-CoA levels in Gclm-KO and WT mice. This ethanol-induced effect, based on a previous work [9] and this study, involves both common and distinct biochemical pathways in WT and Gclm-KO mice. Firstly, while fatty acid oxidation in the Gclm-KO liver remained at a higher level, ethanol-intake appears to inhibit this process in both WT and Gclm-KO livers as evidenced by a decrease in acylcarnitine intermediates of fatty acid beta-oxidation. Secondly, lipogenesis consumes acetyl-CoA in the WT liver, whereas this pathway remains suppressed in the Gclm-KO liver [9]. Thirdly, in ethanol-exposed Gclm-KO livers, there is a unique elevation of N-acetylglutamate and GlcNAc-6P, which aids in the redistribution of acetyl-CoA pool. Taken together, it may be concluded that chronic ethanol consumption leads to differential changes in hepatic acetyl-CoA metabolism between WT and Gclm-KO mice, which reflects a diversion from lipogenesis to alternative biochemical pathways in Gclm-KO mice.
Shift in the intracellular NADH/NAD+ ratio is a key biochemical event responsible for fatty liver induced by excessive alcohol consumption [41, 42]. Elevation in the NADH/NAD+ ratio resulting from alcohol dehydrogenase (ADH)- and aldehyde dehydrogenase (ALDH)-mediated ethanol metabolism is believed to cause inhibition of gluconeogenesis, fatty acid oxidation and the TCA cycle [41, 42]. As expected, ethanol feeding increased liver concentrations of NADH and NADH/NAD+ ratio in WT mice. In contrast, the liver NADH and NADH/NAD+ ratio were intrinsically high in Gclm-KO mice fed a control diet, likely due to increased production of NADH from upregulated pyruvate dehydrogenase complex and fatty acid oxidation pathway. Furthermore, ethanol intake caused no change in NADH concentration and no shift in NADH/NAD+ ratio in Gclm-KO liver. This result suggests that NADH derived from oxidative ethanol metabolism may only have a modest impact on total hepatic NADH pool in the Gclm-KO mice. A previous study demonstrated that the Gclm-KO liver has a higher capacity for ethanol and acetaldehyde metabolism without showing notable changes in the activity of ethanol and acetaldehyde metabolizing enzymes [9]. Seemingly in line with this observation, profound hepatic induction of the glucuronidation pathway was observed in the Gclm-KO mice following chronic ethanol consumption, as evidenced by abundant precursor UDP-glucuronate and induction of UGT enzymes. Glucuronidation is considered a minor non-oxidative pathway for ethanol metabolism [43] and its product, ethyl glucuronide, has been proposed as a stable urine marker for alcohol exposure [44, 45]. One may speculate that the unique and profound induction of this pathway contributes significantly to ethanol elimination in the Gclm-KO liver and thereby spare NADH loading from oxidative metabolism of ethanol. Future metabolomics analyses in serum and urine would help to test this hypothesis.
A clear message derived from these metabolomic and transcriptomic studies is that only a small proportion of metabolic changes can be linked to gene expression changes at the transcriptional level. This panel of differentially expressed genes can be classified broadly into three groups: (i) targets of NRF2, including antioxidant genes (e.g. Gpx and Gsr), xenobiotic metabolizing genes (e.g. Gst and Ugt) and molecular transporter genes (e.g. Slc22a5 and Slc25a17); (ii) targets of the AMPK signaling pathway [46], including lipid metabolizing genes regulated by sterol regulatory element-binding protein 1 (SREBP1) (e.g. Fasn and Acc) [47] or by peroxisome proliferator-activated receptor alpha (PPARα) (e.g. Acsl and Acox) [48]; and (iii) genes encoding mitochondrial metabolism enzymes that are regulated by cellular nutrient and energy status (e.g. Pdha and Idh2) [49]. Interestingly, most of these genes respond primarily to the loss of GCLM, implying a dominant role of low GSH-elicited transcriptional mechanism in mediating long-term adaptations in cellular metabolism. On the other hand, these findings imply that post-transcriptional mechanisms may play a larger role in mediating metabolic adaptations upon ethanol exposure. It is an intriguing finding that UDP-GlcNAc, the donor substrate for O-GlcNAcylation of proteins, is depleted exclusively in ethanol-fed Gclm-KO mice. Since UDP-GlcNAc is synthesized via HBP, which integrates several metabolic pathways of glucose, fatty acids, pyrimidine and glutamine, O-GlcNAc modification of proteins represents an important regulatory mechanism for nutrient and stress sensing and subsequent regulation of liver metabolism [50-52]. Aberrant O-GlcNAcylation has been implicated in hepatic insulin resistance and fibrosis; it is proposed that a “safe zone” of global O-GlcNAcylation must be maintained for normal liver functioning [53, 54]. To date, little is known about the role of O-GlcNAcylation that takes place in the cytosol, the nucleus and the mitochondria, in the pathogenesis of ALD. Our finding raised the plausibility that low GSH-associated decrease in UDP-GlcNAc production, and by extension modulation of protein O-GlcNAcylation, may contribute to the protection against alcohol-induced steatosis. Future post-translational modification proteomics studies are warranted to elucidate these molecular processes in a greater detail.
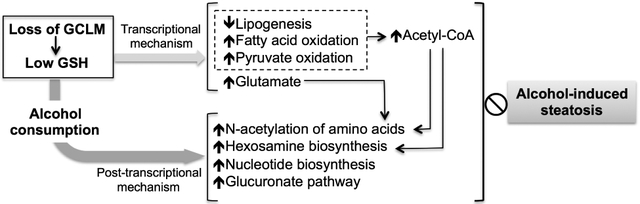
5. Conclusion
The present study has identified new hepatic metabolic signatures intrinsically linked to the loss of GCLM (and by extension low hepatic GSH) and modulated by alcohol consumption, the combination of which are, at least in part, causally associated with hepatic protection against alcohol-induced steatosis in Gclm-KO mice (Fig. 7). These metabolic changes cover multiple biochemical pathways and represent a metabolic network that appears to be reprogramed by both transcriptional and post-transcriptional mechanisms (Fig. 7). Compared with the WT liver, the Gclm-KO liver is enriched with acetyl-CoA as a result of increased production from pyruvate oxidation and fatty acid oxidation, and reduced utilization for lipogenesis. Following chronic ethanol consumption, these intrinsic metabolic adaptations remain functional and are supplemented by increased acetyl-CoA flux to hexosamine biosynthesis and N-acetylation of amino acids. Thus, the net effect of observed changes in acetyl-CoA metabolism in the Gclm-KO liver appears to be the diversion of acetyl-CoA flux (including acetyl-CoA derived from ethanol metabolism) from lipogenesis to alternative biochemical pathways. In addition, several beneficial metabolic changes are uniquely present in the ethanol-exposed Gclm-KO liver (Fig. 7), including elevated production of glutamate (that boosts GSH biosynthesis and serves as an alternative sink for ethanol-derived acetyl group), induction of the glucuronate pathway (that contributes to ethanol metabolism), and induction of nucleotide biosynthesis (that feeds into hexosamine biosynthesis and glucuronate pathways). Thus, we demonstrate that our inter-omics study is robust in capturing molecular machineries responsible for GSH deficiency-elicited protection against alcohol-induced steatosis. Equally importantly, it reveals potentially new targets to manipulate the process for preventive and therapeutic interventions of ALD.
Fig. 7. Scheme of GSH deficiency-elicited reprogramming of hepatic metabolic machinery in response to alcohol consumption.

Compared with the WT liver, the GSH-deficient (due to loss of GCLM) liver exhibits intrinsic metabolic changes (green shaded area) involving the metabolism of amino acids, lipid, fatty acids, and glucose. They represent metabolic adaptations largely mediated by transcriptional mechanisms. The GSH-deficient (Gclm-KO) liver is acetyl-CoA enriched, as a result of increased production from pyruvate oxidation and fatty acid oxidation, and reduced flux to de novo lipogenesis. Following chronic alcohol consumption, these metabolic adaptations remain functional and are supplemented by unique metabolic changes occurring in the ethanol-exposed Gclm-KO liver (orange shaded area), including increased acetyl-CoA flux to N-acetylation of amino acids and hexosamine biosynthesis, elevated production of glutamate (that boosts GSH biosynthesis and serves as an alternative sink for ethanol-derived acetyl group), induction of the glucuronate pathway (that contributes to ethanol metabolism), and induction of nucleotide biosynthesis (that feeds into hexosamine biosynthesis and glucuronic acid pathways). Post-transcriptional mechanisms likely play a larger role in mediating metabolic adaptations upon ethanol exposure. Such coordinate reprograming of hepatic metabolic machinery serves to protect Gclm-KO mice against alcohol-induced steatosis. Hepatic levels of metabolites (circles) or activities of metabolic pathways (boxes) in Gclm-KO mice that are higher (red outlined) or lower (blue outlined) than WT mice are shown. Grey bands with arrows indicate metabolic flux of metabolites. Lines terminated by solid diamonds indicate cellular source for downstream metabolic pathways.
Supplementary Material
Highlights.
GSH-deficient (Gclm-KO) liver is acetyl-CoA and glutamate enriched.
Low GSH promotes pyruvate and fatty acid oxidation and inhibits lipogenesis.
Ethanol-exposed WT and KO liver show differential changes in acetyl-CoA metabolism.
Transcriptional and post-transcriptional mechanisms underlie metabolic adaptions.
Acknowledgement
We thank Dr. Peng Gang (Yale School of Public Health) for valuable input into our statistical analysis.
Funding sources:
This work was supported in part by the National Institutes of Health grants K01AA025093 (YC), R24AA022057 (VV), U01AA021724 (VV), the NIH Intramural Research Program (FJG) and the Department of Atomic Energy, Government of India (SKM).
Footnotes
Abbreviations: ACC, acetyl-CoA carboxylase; ACOX, acyl-CoA oxidase; ACSL, long-chain acyl-CoA synthetase; ADH, alcohol dehydrogenase; AK4, adenylate kinase 4; ALD, alcoholic liver disease; ALDH, aldehyde dehydrogenase; AMPK, AMP-activated protein kinase; ANOVA, analysis of variance; CTPS, CTP synthase; CYP17A1, cytochrome P450 17a1; Cys, cysteine; DLD, dihydrolipoamide dehydrogenase; EBP, emopamil binding protein; ESI-MS, electrospray ionization mass spectrometry; FDR, False Discovery Rate; Fru-6P, fructose-6-phosphate; γ-GC, γ-glutamylcysteine; GCK, Glucokinase; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase modifier subunit; Glc, glucose; Glc-1P, glucose-1-phosphate; Glc-6P, glucose-6-phosphate; GlcN-6P, glucosamine-6-phosphate; GlcNAc, N-acetylglucosamine; GlcNAc-1P, N-acetylglucosamine-1-phosphate; GlcNAc-6P, N-acetylglucosamine-6-phosphate; GlcUA, glucuronic acid; Gln, glutamine; GLS1, glutaminase 1; Glu, glutamate; GLUD1, glutamate dehydrogenase 1; Gly, glycine; GNPDA, glucosamine-6-phosphate deaminase; GPX, glutathione peroxidase; GSH, reduced glutathione; GSR, glutathione disulfide reductase; GSSG, oxidized glutathione; GST, glutathione S-transferase; GSTM, glutathione S-transferases Mu; HBP, hexosamine biosynthesis pathway; Hcy, homocysteine; HILIC, hydrophilic interaction liquid chromatography; HPRT, hypoxanthine phosphoribosyltransferase; IACUC, Institutional Animal Care and Use Committee; IDH, isocitrate dehydrogenase; IPA, Ingenuity Pathway Analysis; LPL, lipoprotein lipase; Met, methionine; NAGK, N-acetylglucosamine kinase; NRF2, nuclear factor erythroid 2-related factor 2; OGDC, oxoglutarate dehydrogenase complex; OPLS-DA, orthogonal projection to latent structures discriminant analysis; PCA, principal components analysis; PCCA, propionyl-CoA carboxylase; PDC, pyruvate dehydrogenase complex; PDHA1, pyruvate dehydrogenase γ1 subunit; 6PGL, 6-phosphogluconolactonase; PGM3, phosphoglucomutase 3; PPARα, peroxisome proliferator-activated receptor alpha; PRPP, 5-phosphoribosyl-1-pyrophosphate; Q-PCR, quantitative real-time PCR; QOE, Qlucore Omics Explorer; RNS, reactive nitrogen species; ROS, reactive oxygen species; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SLC22A5, solute carrier family 22 member 5; SLC25A17, solute carrier family 25 member 17; SREBP1, sterol regulatory element-binding protein 1; TCA, tricarboxylic acid; TG, triglycerides; Thr, threonine; UDP-Glc, UDP-glucose; UDP-GlcNAc, UDP-acetylglucosamine; UDP-GlcUA, UDP-glucuronic acid; UGDH, UDP-glucose 6-dehydrogenase; UGP2, UDP-glucose pyrophosphorylase 2; UGTs, UDP-glucuronosyltransferases; UPLC, ultra-performance liquid chromatography.
Conflict of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol. 2015;62:S38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Albano E Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Molecular aspects of medicine. 2008;29:9–16. [DOI] [PubMed] [Google Scholar]
- [3].Neuman MG, French SW, French BA, Seitz HK, Cohen LE, Mueller S, et al. Alcoholic and non-alcoholic steatohepatitis. Experimental and molecular pathology. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–26. [DOI] [PubMed] [Google Scholar]
- [5].Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005;280:33766–74. [DOI] [PubMed] [Google Scholar]
- [6].Chen Y, Yang Y, Miller ML, Shen D, Shertzer HG, Stringer KF, et al. Hepatocyte-specific Gclc deletion leads to rapid onset of steatosis with mitochondrial injury and liver failure. Hepatology. 2007;45:1118–28. [DOI] [PubMed] [Google Scholar]
- [7].Chen Y, Johansson E, Yang Y, Miller ML, Shen D, Orlicky DJ, et al. Oral N-acetylcysteine rescues lethality of hepatocyte-specific Gclc-knockout mice, providing a model for hepatic cirrhosis. Journal of Hepatology. 2010;53:1085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse. Novel model system for a severely compromised oxidative stress response. Journal of Biological Chemistry. 2002;277:49446–52. [DOI] [PubMed] [Google Scholar]
- [9].Chen Y, Singh S, Matsumoto A, Manna SK, Abdelmegeed MA, Golla S, et al. Chronic Glutathione Depletion Confers Protection against Alcohol-induced Steatosis: Implication for Redox Activation of AMP-activated Protein Kinase Pathway. Sci Rep. 2016;6:29743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Harrigan GG, Maguire G, Boros L. Metabolomics in alcohol research and drug development. Alcohol Res Health. 2008;31:26–35. [PubMed] [Google Scholar]
- [11].Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. [DOI] [PubMed] [Google Scholar]
- [12].Manna SK, Tanaka N, Krausz KW, Haznadar M, Xue X, Matsubara T, et al. Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology. 2014;146:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- [14].Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- [15].Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jung YS. Metabolism of Sulfur-Containing Amino Acids in the Liver: A Link between Hepatic Injury and Recovery. Biol Pharm Bull. 2015;38:971–4. [DOI] [PubMed] [Google Scholar]
- [17].Meister A, Griffith OW, Novogrodsky A, Tate SS. New aspects of glutathione metabolism and translocation in mammals. Ciba Found Symp. 1979:135–61. [DOI] [PubMed] [Google Scholar]
- [18].Kharbanda KK. Role of transmethylation reactions in alcoholic liver disease. World J Gastroenterol. 2007;13:4947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mandrekar P Epigenetic regulation in alcoholic liver disease. World J Gastroenterol. 2011;17:2456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–21. [DOI] [PubMed] [Google Scholar]
- [21].Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37:1949–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol. 2013;45:1121–32. [DOI] [PubMed] [Google Scholar]
- [23].Adeva-Andany MM, Perez-Felpete N, Fernandez-Fernandez C, Donapetry-Garcia C, Pazos-Garcia C. Liver glucose metabolism in humans. Biosci Rep. 2016;36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yamaoka T, Itakura M. [Metabolism of purine nucleotides and the production of uric acid]. Nihon Rinsho. 1996;54:3188–94. [PubMed] [Google Scholar]
- [25].Williams JC, Kizaki H, Weber G, Morris HP. Increased CTP synthetase activity in cancer cells. Nature. 1978;271:71–3. [DOI] [PubMed] [Google Scholar]
- [26].Noma T, Fujisawa K, Yamashiro Y, Shinohara M, Nakazawa A, Gondo T, et al. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem J. 2001;358:225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Botta D, White CC, Vliet-Gregg P, Mohar I, Shi S, McGrath MB, et al. Modulating GSH synthesis using glutamate cysteine ligase transgenic and gene-targeted mice. Drug Metab Rev. 2008;40:465–77. [DOI] [PubMed] [Google Scholar]
- [28].Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free radical biology & medicine. 2010;49:1406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Paracha UZ, Fatima K, Alqahtani M, Chaudhary A, Abuzenadah A, Damanhouri G, et al. Oxidative stress and hepatitis C virus. Virol J. 2013;10:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu W, Baker SS, Baker RD, Zhu L. Antioxidant Mechanisms in Nonalcoholic Fatty Liver Disease. Curr Drug Targets. 2015;16:1301–14. [DOI] [PubMed] [Google Scholar]
- [31].Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang K, Zhang T, Dong Q, Nice EC, Huang C, Wei Y. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013;4:e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen Y, Dong H, Thompson DC, Shertzer HG, Nebert DW, Vasiliou V. Glutathione defense mechanism in liver injury: insights from animal models. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2013;60:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bradford BU, O'Connell TM, Han J, Kosyk O, Shymonyak S, Ross PK, et al. Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease. Toxicol Appl Pharmacol. 2008;232:236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jang ZH, Chung HC, Ahn YG, Kwon YK, Kim JS, Ryu JH, et al. Metabolic profiling of an alcoholic fatty liver in zebrafish (Danio rerio). Mol Biosyst. 2012;8:2001–9. [DOI] [PubMed] [Google Scholar]
- [37].Nicholas PC, Kim D, Crews FT, Macdonald JM. 1H NMR-based metabolomic analysis of liver, serum, and brain following ethanol administration in rats. Chem Res Toxicol. 2008;21:408–20. [DOI] [PubMed] [Google Scholar]
- [38].Laforenza U, Patrini C, Gastaldi G, Rindi G. Effects of acute and chronic ethanol administration on thiamine metabolizing enzymes in some brain areas and in other organs of the rat. Alcohol Alcohol. 1990;25:591–603. [DOI] [PubMed] [Google Scholar]
- [39].Shi X, Yao D, Chen C. Identification of N-acetyltaurine as a novel metabolite of ethanol through metabolomics-guided biochemical analysis. J Biol Chem. 2012;287:6336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].de Cima S, Polo LM, Diez-Fernandez C, Martinez AI, Cervera J, Fita I, et al. Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis. Sci Rep. 2015;5:16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res. 2009;33:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rasineni K, Casey CA. Molecular mechanism of alcoholic fatty liver. Indian J Pharmacol. 2012;44:299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kaeferstein H Forensic relevance of glucuronidation in phase-II-metabolism of alcohols and drugs. Leg Med (Tokyo). 2009;11 Suppl 1:S22–6. [DOI] [PubMed] [Google Scholar]
- [45].Gika HG, Ji C, Theodoridis GA, Michopoulos F, Kaplowitz N, Wilson ID. Investigation of chronic alcohol consumption in rodents via ultra-high-performance liquid chromatography-mass spectrometry based metabolite profiling. J Chromatogr A. 2012;1259:128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, et al. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. The Journal of physiology. 2006;574:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes & development. 2000;14:2819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sugden MC, Caton PW, Holness MJ. PPAR control: it's SIRTainly as easy as PGC. The Journal of endocrinology. 2010;204:93–104. [DOI] [PubMed] [Google Scholar]
- [49].Hollinshead KE, Tennant DA. Mitochondrial metabolic remodeling in response to genetic and environmental perturbations. Wiley Interdiscip Rev Syst Biol Med. 2016;8:272–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chatham JC, Marchase RB. Protein O-GlcNAcylation: A critical regulator of the cellular response to stress. Curr Signal Transduct Ther. 2010;5:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao L, Feng Z, Yang X, Liu J. The regulatory roles of O-GlcNAcylation in mitochondrial homeostasis and metabolic syndrome. Free Radic Res. 2016;50:1080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci. 2003;60:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang K, Yin R, Yang X. O-GlcNAc: A Bittersweet Switch in Liver. Front Endocrinol (Lausanne). 2014;5:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.