

Abstract
Background:
Oxidative stress and neuronal apoptosis play important roles in the pathogenesis of early brain injury (EBI) after subarachnoid hemorrhage (SAH). The activation of TGR5, a novel membrane-bound bile acid receptor, possesses anti-oxidative stress and anti-apoptotic effects in hepatobiliary disease and kidney disease. The present study aimed to explore the neuroprotective effect of TGR5 activation against EBI after SAH and the potential underlying mechanisms.
Methods:
The endovascular perforation model of SAH was performed on 199 Sprague Dawley rats to investigate the beneficial effects of TGR5 activation after SAH. INT-777, a specific synthetic TGR5 agonist, was administered intranasally at 1 h after SAH induction. TGR5 CRISPR and ALDH2 CRISPR were administered intracerebroventricularly at 48 h before SAH to illuminate potential mechanisms. The SAH grade, short-term and long-term neurobehavioral tests, TUNEL staining, Fluoro-Jade C staining, Nissl staining, immunofluorescence staining, and western blots were performed at 24 h after SAH.
Results:
The expressions of endogenous TGR5 and ALDH2 gradually increased and peaked at 24 h after SAH. TGR5 was expressed primarily in neurons, as well as in astrocytes and microglia. The activation of TGR5 with INT-777 significantly improved the short-term and long-term neurological deficits, accompanied by reduced the oxidative stress and neuronal apoptosis at 24 h after SAH. Moreover, INT-777 treatment significantly increased the expressions of TGR5, cAMP, phosphorylated PKCε ALDH2, HO-1, and Bcl-2, while downregulated the expressions of 4-HNE, Bax, and Cleaved Caspase-3. TGR5 CRISPR and ALDH2 CRISPR abolished the neuroprotective effects of TGR5 activation after SAH.
Conclusions:
In summary, the activation of TGR5 with INT-777 attenuated oxidative stress and neuronal apoptosis via the cAMP/PKCε/ALDH2 signaling pathway after SAH in rats. Furthermore, TGR5 may serve as a novel therapeutic target to ameliorate EBI after SAH.
Keywords: Subarachnoid hemorrhage, Early brain injury, TGR5, INT-777, ALDH2, Oxidative stress, Neuronal apoptosis
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) accounts for 5-10% of all stroke types [1]. It is a life threatening cerebrovascular disease, with a fatality rate ranged from 27% to 44% worldwide [2]. Early brain injury (EBI) is the leading cause of mortality and delayed neurological deficits after SAH [3, 4]. However, the specific molecular mechanism of EBI after SAH remains unknown. Oxidative stress and neuronal apoptosis have been shown to be key devastating processes in EBI induced by SAH [5–8]. Therefore, alleviating oxidative stress injury and neuronal apoptosis are particularly important to improve the prognosis of SAH.
Mitochondrial dysfunction is a common cause of oxidative stress injury and neuronal apoptosis in neurological diseases, including neurodegenerative disease, intracerebral hemorrhage (ICH), ischemic stroke, epilepsy, and SAH [7–13]. Mitochondrial dysfunction is considered to be a crucial therapeutic target for EBI following SAH [8, 14, 15].
Trans-membrane G protein-coupled receptor-5 (TGR5), also known as GPBAR1 or GPR131, has been identified as a membrane-bound bile acid receptor expressed in the gall bladder, kidney, brown adipose tissue, liver, intestine, and brain [16, 17]. Recent studies suggested that the activation of TGR5 promotes mitochondrial biogenesis and suppresses oxidative stress in diabetes and obesity related kidney disease [18]. TGR5 activation also protects cholangiocytes from CD95 death receptor-mediated apoptosis, and attenuates ischemia/reperfusion induced hepatocellular apoptosis [19, 20]. In the central nervous system (CNS), TGR5 activation exerts a beneficial effect on fat metabolism in mouse brains [21], alleviates microglial activation in hepatic encephalopathy and acute brain injury [22, 23], and mitigates the deterioration of function in amyotrophic lateral sclerosis patients [24]. Interestingly, a recent study has indicated that the activation of TGR5 provides a beneficial effect against cognitive impairment, neuronal apoptosis, and synaptic dysfunction in an animal model of Alzheimer’s disease [25]. As a novel potent and specific semisynthetic TGR5 agonist, 6R-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777), has been shown to induce mitochondrial biogenesis, decrease oxidative stress, and increase fatty acid oxidation in kidney disease [18, 26]. In animal models of brain injury, INT-777 administration modulated neuronal apoptosis and neuroinflammation via TGR5 signaling [25, 27]. However, the effects of TGR5 activation in EBI after SAH have never been reported. Furthermore, the activation of mitochondrial aldehyde dehydrogenase two (ALDH2), significantly preserved the mitochondrial function via PKCε phosphorylation, which has been shown to provide significant protection against oxidative stress and apoptosis in the setting of cardiac and cerebral ischemia/reperfusion injury [28–31]. PKCε phosphorylation raised in response to increased intracellular cAMP activity [32]. Given that the TGR5-cAMP-PKA axis underlays the bile acid-mediated anti-inflammatory effect [17], it is likely that ALDH2 may be the important downstream protein contributing to the TGR5 mediated anti-oxidative stress and anti-apoptotic effects. To date, the regulation and molecular mechanism of ALDH2 in SAH has never been investigated.
In this present study, we explored the role of TGR5 in SAH. We showed for the first time that the TGR5 activation with INT-777 attenuated EBI after SAH in rats through the suppression of the oxidative stress injury and neuronal apoptosis in part via cAMP/PKCε/ALDH2 signaling pathway (shown in Fig. s1).
Materials and methods
Animals
Adult male Sprague-Dawley rats weighing 280–320g were used in this project. Rats were housed in a 12 h light and dark cycle, in an environment with controlled humidity and temperature, with free access to food and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Loma Linda University, which comply with the National Institutes of Health Guidelines for the Care and Use of laboratory Animals in Neuroscience Research and ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Experimental design
A total of four separated experiments were performed as shown in Fig. s2.
Experiment 1
We characterized the time course of expressions and cellular localization of endogenous TGR5 receptors and ALDH2 in the ipsilateral (left) hemisphere after SAH. A total of 36 rats were randomly assigned to six groups (n=6 per group): Sham, SAH-3 h, SAH-6 h, SAH-12 h, SAH-24 h, SAH-72 h. Western blot analysis was performed to assess the protein levels of TGR5 and ALDH2. To determine cellular localization, an additional 4 rats (n=2 per group) in the Sham and SAH-24 h groups were used for double immunofluorescence staining of TGR5 and ALDH2 with neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP) or calcium-binding adaptor molecule 1 (Iba-1).
Experiment 2
To evaluate the treatment effects of TGR5 activation with INT-777 on short-term neurological outcome after SAH, 36 rats were randomly assigned to five groups (n=6 per group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777 (10 μg/kg), SAH+INT-777 (30 μg/kg), and SAH+INT-777 (90 μg/kg) for neurological tests. The SAH grade, and neurobehavioral performance of modified Garcia test and beam balance test were assessed at 24 h after SAH in all groups. An additional 12 rats (n=4 per group) in the Sham, SAH+vehicle, and SAH+INT-777 (best dose) groups were used for terminal deoxynucleotidyl transferase dUTP (TUNEL) staining, Fluoro-Jade C staining, immunofluorescence staining, and western blot to evaluate oxidative stress markers and neuronal apoptosis in the ipsilateral (left) hemisphere at 24 h after SAH. The best dose of INT-777 was selected based on the short-term neurological outcomes, which was also used for the following long-term outcome and mechanism experiments.
Experiment 3
To evaluate the treatment effects of TGR5 activation with INT-777 on long-term neurological outcomes after SAH, 30 rats were randomly assigned to three groups (n=10 per group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777. The Rotarod test was performed on day 7, day 14, and day 21 after SAH. Morris water maze was performed on day 23–27 after SAH. Nissl staining was performed to assess the neuronal degeneration on day 28 after SAH.
Experiment 4
To explore the neuroprotective mechanism of TGR5 activation, 36 rats were randomly assigned to six groups (n=6 per group): Sham, SAH+vehicle (10% dimethyl sulfide), SAH+INT-777, SAH+INT-777+TGR5 CRISPR, SAH+INT-777+scrambled CRISPR, SAH+INT-777+ALDH2 CRISPR. In addition, to assess the knockout efficiency of CRISPR, an additional 24 rats (n=4 per group) were subjected to six groups: naive+scrambled CRISPR, naive+TGR5 CRISPR, naive+ALDH2 CRISPR, SAH+scrambled CRISPR, SAH+TGR5 CRISPR, and SAH+ALDH2 CRISPR. The ipsilateral hemisphere of each brain was collected for western blot analysis after neurological performance and SAH grade evaluation at 24 h after SAH.
SAH model
The endovascular perforation model of SAH was performed as previously described [33]. Isoflurane (4% induction, 2.5% maintenance) anesthetized rats were intubated and then connected to the ventilator in the supine position. A sharp 4–0 monofilament nylon suture was inserted into the left internal carotid artery from the external carotid artery and perforated the bifurcation of the anterior and middle cerebral arteries. Following the perforation, isoflurane was reduced to 1.5%, the nylon suture was cut through the wall of the blood vessel, and withdrawn immediately. Rats in the Sham group underwent the same procedures, except for blood vessel wall puncture. All animals were monitored intraoperatively; the heart rate, respiration, skin color, and pedal reflex were assessed intraoperatively every 5 minutes to confirm the anesthetic status and prevent distress. After the completion of surgery and weaning from ventilation, the animals were transferred into a heated chamber, maintained at a temperature of 37.5 °C, for recovery. Respiration, heart rate and skin color were monitored postoperatively every 15 minutes until normal behavior resumed.
SAH grading
The severity of SAH was evaluated by a blinded investigator using the SAH grading scale after euthanasia as previously reported [34]. The basal cistern was divided into six segments, each with a score from 0 to 3 according to the subarachnoid blood clot. The total score was calculated by adding all area scores (maximum SAH grade=18). Rats with a score of 8 or less were excluded from this study.
Drug administration
Intranasal drug administration
Intranasal drug administration was performed at 1 h after SAH as previously described [35]. Animals were placed in a supine position with 2% isoflurane anesthesia. A total volume of 30 μL vehicle (10% dimethyl sulfide) or INT-777 (MedChem Express, NJ, USA) at three different doses (10 μg/kg, 30 μg/kg, and 90 μg/kg) were administered into the left and right nares, alternating 10 μL in one naris every 5 minutes for a period of 15 minutes.
Intracerebroventricular drug administration
As described previously [5], rats were anesthetized with isoflurane (4% induction, 2.5% maintenance) and placed in a stereotaxic frame. The needle of a 10 μL Hamilton syringe (Microliter 701, Hamilton Company, USA) was inserted through a burr hole perforated through the skull into the left lateral ventricle at the following coordinates relative to bregma: 0.9 mm posterior, 1.5 mm lateral, and 3.2 mm beneath the horizontal plane of the skull. For the mechanism experiment, we used an engineered form of CRISPR-associated (Cas9) protein system. In this system, TGR5 or ALDH2 CRISPR was used to knockdown TGR5 and ALDH2 gene expression in the rat brain. A total of 2 μg per animal of knockout CRISPR was given intracerebroventricularly (i.c.v) at a rate of 1 μL/min using a pump. TGR5 CRISPR (Santa Cruz Biotechnology, Dallas, TX, USA), ALDH2 CRISPR (Santa Cruz Biotechnology, Dallas, TX, MA, USA), or scrambled CRISPR (Santa Cruz Biotechnology, Dallas, TX, USA) was injected into the ipsilateral (left) ventricle at 48 h before SAH. To prevent possible leakage, the needle was kept in situ for 5 minutes after completing the injection and then withdrawn slowly for 5 minutes. The burr hole was sealed with bone wax immediately after removal of the needle, the incision was sutured, and the rats were awakened from anesthesia.
Mass spectrometry
To detect the presence of INT-777 in brain tissue after the intranasal administration, we used the liquid chromatography mass spectrometry (LC-MS/MS) system (Agilent Technologies, CA, USA) with atmospheric pressure chemical ionization source. Brain tissues were prepared as previous described [36]. Briefly, 150 mg brain tissue was fully homogenized for 2 minutes, and 1.5 mL acetonitrile (Sigma-Aldrich, USA) was added. It was centrifuged at 14000 g for 30 minutes at 4 °C. The supernatant was transferred into a tube and dried under a negative pressure (below 2.0 kPa) for 7 h. The residue was reconstituted with 150 μL 50% acetonitrile in water, then centrifuged again at 14000 g centrifuge for 20 minutes, at 4 °C. The 20 μL of the supernatant was placed into the LC-MS/MS system. MS spectra were collected in m/z 100-700 negative reflection mode. MassHunter Software Version B.08.00 (Agilent Technologies, CA, USA) was used to analyze the LC-MS/MS data.
Short-term neurological performance
The short-term neurobehavioral outcomes were assessed blindly at 24h after SAH by an investigator. The 18 points modified Garcia score and 4 points beam balance test were used as previously described [15]. Higher scores indicated better neurological performance.
Long-term neurological performance
Rotarod test
The rotarod test was performed to evaluate sensorimotor coordination and balance on days 7, 14, and 21 after SAH as previously described [37]. The rotating speed began at 5 revolutions per minute (RPM) or 10 RPM, followed by gradual acceleration of 2 RPM every 5 seconds. The duration that rats were able to stay on the accelerating rotating cylinder was recorded by a photo beam circuit.
Morris water maze
The Morris water maze test was performed on day 23–27 after SAH to evaluate spatial learning capacity and memory ability as previously shown [38]. Briefly, the animal was then taken to the platform, and kept there for 5 seconds on the first day of cueing test. During the following days of spatial learning test, animals were placed in a semi-random set of starting positions and were tasked with finding the submerged platform. The time limit for each animal to find the platform was 60 seconds. The probe test was performed on day 27 after SAH, in which animals were allowed to search for platform areas after removing the actual platform. A video recording system traced the activities of the animals and the swim patterns, from which swimming distance, escape latency, and swimming path were quantified by Computer Tracking System (San Diego Instruments Inc., CA, USA).
Histological analysis
Animals were deeply anesthetized, then underwent a trans-cardiac perfusion of 100 mL of chilled phosphate buffered saline (PBS, 0.01 M, pH 7.4), followed by 100 mL of 4% paraformaldehyde (PFA). Whole brains were rapidly harvested and fixed in 4% PFA at 4 °C for 24 h, then dehydrated with 30% sucrose for 72 h. After being embedded into OCT (Scigen Scientific Gardena, CA, USA) and frozen at − 80 °C, the 8-15 μm coronal brain sections were cut on a cryostat (LM3050S, Leica Microsystems, Bannockburn, Germany) and mounted on normal Poly-L-Lysine coated slides for double immunofluorescence [39], TUNEL staining [40], Fluoro-Jade C (FJC) staining [15], 8-OHdG staining [41], Mitosox staining [42], and Nissl staining [7]. The slides were visualized and photographed with a fluorescence microscope (DMi8, Leica Microsystems, Germany).
Immunofluorescence staining
Slices were washed with 0.01 M of PBS three times for 5-10 min then incubated in 0. 3% Triton X-100 in 0.01 M of PBS for 10 min at room temperature. After being blocked with 5% donkey serum in 0.01 M of PBS for 2 h at room temperature, the sections were incubated at 4 °C overnight with primary antibody including: anti-TGR5 (1:200, ab72608, Abcam, Cambridge, MA, USA), anti-ALDH2 (1:200, ab108306, Abcam, Cambridge, MA, USA), anti-NeuN (1:200, ab104224, Abcam, Cambridge, MA, USA), anti-GFAP (1:200, ab53554, Abcam, Cambridge, MA, USA), and anti-Iba-1 (1:200, ab5076, Abcam, Cambridge, MA, USA). Then, the slices were washed with 0.01 M of PBS and incubated with fluorescence-conjugated secondary antibodies (1:500, Jackson ImmunoResearch, PA, USA) for 1h at room temperature.
TUNEL staining
Double staining of neuron marker NeuN and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using situ Apoptosis Detection Kit (Roche, USA) according to the manufacturer’s instructions at 24 h after SAH to evaluate the quantification of neuronal apoptosis. The numbers of TUNEL-positive neurons were counted in the ipsilateral cortex. Six random sections per slice over a microscopic field of 400x magnification were averaged. Data were presented as the ratio of TUNEL-positive neurons (%).
Fluoro-Jade C staining
FJC staining was performed using a modified FJC Ready-to-Dilute Staining Kit (Biosensis, USA) to detect degenerating neurons according to the manufacturer’s instructions at 24 h after SAH. The number of FJC-positive neurons were counted and averaged in six random brain sections per slice over a microscopic field of 400x magnification using ImageJ software (ImageJ 1.5, NIH, USA) to evaluate the extent of the neuronal damage. The data were presented as the average number of FJC-positive neurons as per mm2 in selected the fields.
Nissl staining
Nissl staining was performed to evaluate hippocampus injury as previously described [7]. The prepared slices (15 μm) were viewed for degenerating neurons in different hippocampal regions. The mean number of surviving neurons at 200x magnification within the hippocampus CA1 was counted and the neuronal density loss was estimated.
8-OHdG and Mitosox immunohistochemistry
To assess the brain oxidative stress DNA damage and mitochondrial superoxide level, the freshly prepared frozen 8-10 μm brain slices were produced on normal Poly-L-Lysine coated slides. The slides were immersed in antigen retrieval solution (pH 6.0) and heated in a microwave for 15 min to unmask antigens. The slices were then dipped in 3% H2O2 for 10 min to block endogenous peroxidase. The slices were incubated with 8-hydroxy-2’-deoxyguanosine (8-OHdG) antibody (1:200, ab62623, Abcam, Cambridge, MA, USA), and Mitosox antibody (1:1000, M36008, Thermo Fisher, CA, USA) at room temperature. Six randomly selected tubules were examined under microscopic field of 400x and 200x magnification. The fluorescence intensity was quantified by ImageJ software (ImageJ 1.5, NIH, USA).
Western blot analysis
Western blot was performed as previously described [43]. Rats were anesthetized with isoflurane at 24 h after SAH and trans-cardially perfused with 100 mL of chilled PBS (0.01 M, pH7.4) followed by decapitation. The ipsilateral brain hemispheres were instantly collected and snap frozen in liquid nitrogen, then stored in −80 °C freezer for storage until use. Brain samples were homogenized in RIPA lysis buffer (sc-24948, Santa Cruz Biotechnology, TX, USA) and further centrifuged at 14,000 g at 4 °C for 30 min. Equal amounts of protein (30 μg) were loaded onto 7.5%-12.5% SDS-PAGE gel, then electrophoresed and transferred to nitrocellulose membranes (0.2 μm), which were blocked with 5% non-fat blocking grade milk (Bio-Rad, Hercules, CA, USA) and incubated with the following primary antibodies overnight at 4 °C: anti-TGR5 (1:1000, ab72608, Abcam, MA, USA), anti-cAMP (1:1000, ab76238, Abcam, MA, USA), anti-p-PKCε (1:1000, ab63387, Abcam, MA, USA), anti-PKCε (1:1000, Cell Signaling Technology Inc., MA, USA), anti-ALDH2 (1:1000, ab108306, Abcam, MA, USA), anti-Bcl-2 (1:1000, ab59348, Abcam, MA, USA), anti-Bax (1:4000, ab182734, Abcam, MA, USA), anti-4-HNE (1:1000, ab46545, Abcam, MA, USA), anti-HO-1 (1:1000, ab68477, Abcam, MA, USA), anti-Cleaved Caspase-3 (1:1000, D175, Cell Signaling Technology, MA, USA), and anti-β-actin (1:5000, sc-47778, Santa Cruz Biotechnology, TX, USA). On the following day, the membranes were incubated with the appropriate secondary antibody (1:5000, Santa Cruz Biotechnology, Dallas, TX, USA) and (1:5000, Millipore Sigma, Temecula, CA, USA) at room temperature for 2 h. Immunoblots were then visualized with ECL Plus chemiluminescence reagent kit (Amersham Bioscience, PA, USA) and quantified with optical methods using the ImageJ (ImageJ 1.5, NIH, USA). The results were normalized using β-actin as an internal control.
Statistical analysis
All data were expressed as the mean and standard deviation (mean ± SD). Statistical analysis was performed using GraphPad Prism (Graph Pad Software, San Diego, CA, USA). One-way ANOVA followed by Tukey’s post hoc test was used for comparison among multiple groups. Two-way ANOVA was used to analyze the long-term neurological functions. P < 0.05 was considered statistically significant.
Results
Mortality and SAH grade
Of the total 199 rats used, 146 rats underwent SAH induction of which 20 (13.70%) rats died within 24 h after SAH. 7 rats were excluded from this project due to mild SAH. There was no mortality in the Sham group (Fig. 1A). Subarachnoid blood clots were present around the circle of Willis and ventral brain stem with a significant difference in SAH grade between Sham and SAH groups (Fig. 1B). There were no significant differences in SAH grading scores between all SAH groups (Fig. 1C).
Figure 1. Mortality and subarachnoid hemorrhage (SAH) grade.
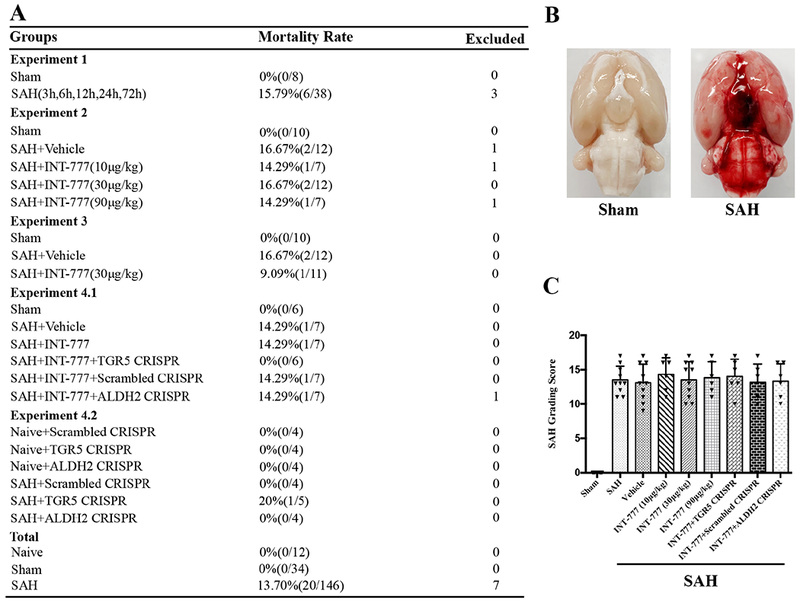
(A) Animal usage and mortality of all experiment groups. (B) Representative pictures showed that subarachnoid blood clots were mainly presented around the circle of Willis in the rat brain at 24 h after SAH. (C) SAH grading scores of all SAH groups. Vehicle, 10% dimethyl sulfide.
Expression of endogenous TGR5 and ALDH2 in ipsilateral hemisphere after SAH
The time course of TGR5 receptor and ALDH2 protein levels were measured by western blot. The results showed that the expression of both TGR5 and ALDH2 started increasing as early as 3 h after SAH, peaked at 24 h after SAH, compared with Sham group (P < 0.01, Fig. 2, 3A–B).
Figure 2. Times course of TGR5 expressions as well as cellular localization of TGR5 receptor after SAH.
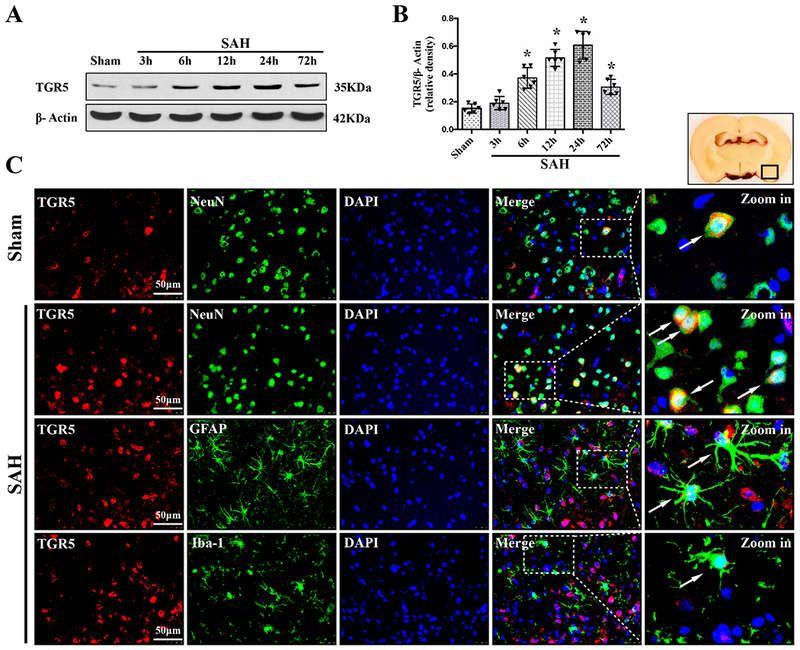
Representative western blot bands of time course (A) and densitometric quantification of TGR5 (B) in the ipsilateral hemisphere after SAH. Data was represented as mean ± SD. n=6 per group. *P<0.05 vs. Sham group; One-way ANOVA, Tukey’s post hoc test. (C) Representative microphotographs of co-immunofluorescence staining of TGR5 (red) with neurons (NeuN, green), astrocytes (GFAP, green), and microglia (Iba-1, green) in the ipsilateral basal cortex at 24 h after SAH. Nuclei were stained with DAPI (blue). A small black square within coronal section of brain indicated the location of where the immunofluorescence staining images were taken. n=2 per group.
Figure 3. Times course of ALDH2 expressions as well as cellular localization of ALDH2 after SAH.
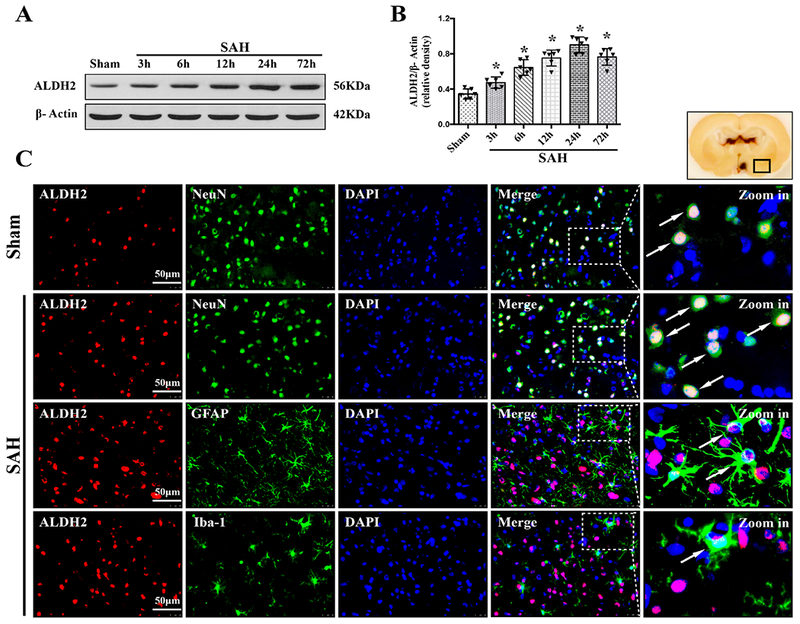
Representative western blot bands of time course (A) and densitometric quantification of ALDH2 (B) in the ipsilateral hemisphere after SAH. Data was represented as mean ± SD. n=6 per group. *P<0.05 vs. Sham group; One-way ANOVA, Tukey’s post hoc test. (C) Representative microphotographs of co-immunofluorescence staining of ALDH2 (red) with neurons (NeuN, green), astrocytes (GFAP, green), and microglia (Iba-1, green) in the ipsilateral basal cortex at 24 h after SAH. Nuclei were stained with DAPI (blue). A small black square within coronal section of brain indicated the location of where the immunofluorescence staining images were taken. n=2 per group.
Double immunofluorescence staining of TGR5 receptors and ALDH2 with NeuN (a marker for neurons), GFAP (a marker for astrocytes), and Iba-1 (a marker for microglia) was performed in the Sham group and 24 h-SAH group after the injury. The results showed that TGR5 receptors and ALDH2 were primarily expressed in neurons and some in astrocytes, and microglia in the rats, respectively; the number of TGR5-positive neurons and ALDH2-positive neurons were increased in the SAH (24 h) group compared with the Sham group (Fig. 2, 3C).
INT-777 detection in the brain after SAH by intranasal administration
LC-MS/MS was used to detect INT-777 in the brain tissues after intranasal administration. MS/MS spectra of INT-777 in the rat’s brain showed a characteristic peak at m/z 449 (Fig. s3C), which was similar to the INT-777 standard ion pattern (Fig. s3A–B), indicating the intraparenchymal entry of INT-777 delivered via intranasal route.
Activation of TGR5 improved short-term neurological deficits at 24 h after SAH
Both the modified Garcia scale and beam balance test showed that significant neurological impairments were observed in the SAH+vehicle group at 24 h after SAH when compared with the Sham group (Fig. 4A–B). INT-777 treatment at a dose of 30μg/kg and 90μg/kg significantly improved the neurological performance of the modified Garcia test and beam balance test at 24 h after SAH compared with the vehicle treated SAH rats (Fig. 4A–B). Based on this result, the dose of 30 μg/kg was determined as the best dose of INT-777 in the subsequent experiments.
Figure 4. Effects of TGR5 activation with INT-777 on short-term neurobehavioral outcome and neuronal damage at 24 h after SAH.
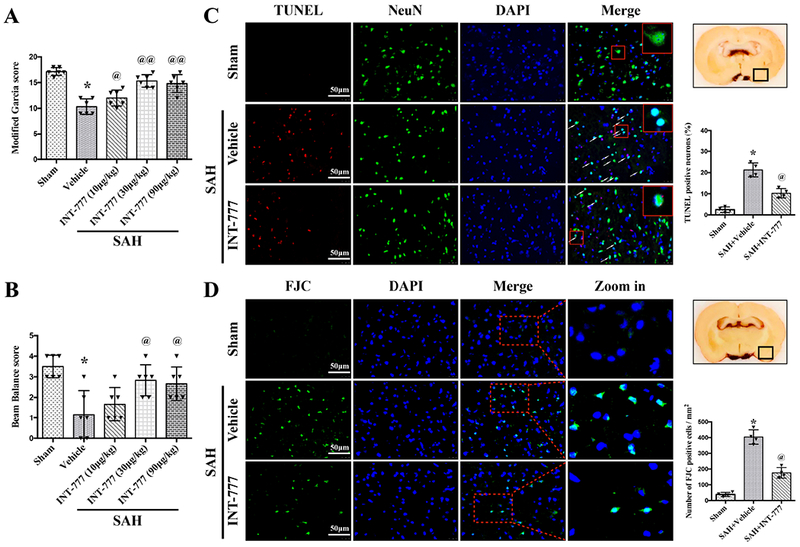
INT-777 improved the modified Garcia score (A) and beam balance score (B) at 24 h after SAH. n=6 per group. (C) Representative micrographs and quantitative analysis of TUNEL-positive neurons within the ipsilateral basal cortex at 24 h after SAH. A small black square in the coronal section of brain indicated the area used for TUNEL-positive neurons. n=4 per group. (D) Representative micrographs and quantitative analysis of Fluoro-Jade C (FJC)-positive cells within the ipsilateral basal cortex at 24 hours after SAH. A small black square in the coronal section of brain indicated the area used for counting FJC-positive cells. n=4 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05, @@P<0.01 vs. SAH+Vehicle group; One-way ANOVA, Tukey’s post hoc test.
Activation of TGR5 reduced neuronal apoptosis and neuronal degeneration at 24 h after SAH
TUNEL staining and FJC staining were performed to evaluate neuronal apoptosis and neuronal degeneration in the ipsilateral basal cortex at 24 h after SAH. The TUNEL-positive and FJC-positive neurons in the SAH+vehicle group were significantly increased compared with the Sham group at 24 h after SAH (Fig. 4C–D). The activation of TGR5 with INT-777 treatment significantly reduced the neuronal damage (Fig. 4C–D). Consistently, western blot test data showed that anti-apoptotic protein Bcl-2 was significantly decreased, while pro-apoptotic protein Bax was significantly increased after SAH. INT-777 treatment resulted in a higher protein level of Bcl-2 and lower protein level of Bax than vehicle treated SAH rats (Fig. 5B, G–H).
Figure 5. Effects of TGR5 activation with INT-777 on oxidative stress level and neuronal apoptosis at 24 h after SAH.
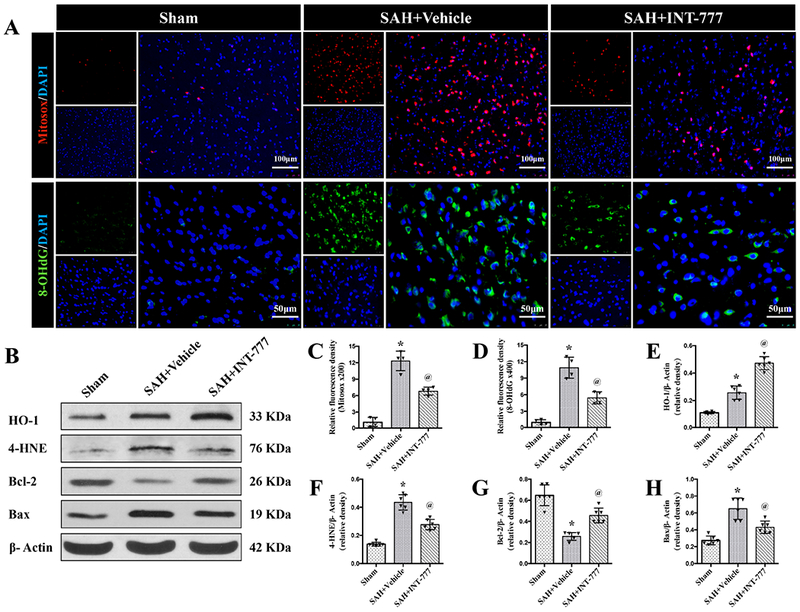
(A) Representative micrographs of Mitosox (red) and 8-OHdG (green) immunofluorescence staining in the ipsilateral basal cortex at 24 h after SAH. Nuclei were stained with DAPI (blue). (C-D) Quantitative analysis of Mitosox and 8-OHdG fluorescence intensity. A small black squares in the coronal section of brain indicated the area used for counting 8-OHdG and Mitosox-positive cells, n=4 per group. (B) Representative western blot bands and densitometric quantification of HO-1, 4-HNE, Blc-2, and Bax (E-H) in the ipsilateral hemisphere at 24 h after SAH, n=6 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group; One-way ANOVA, Tukey’s post hoc test.
Activation of TGR5 reduced ipsilateral hemisphere oxidative stress injury at 24 h after SAH
The immunofluorescence staining of 8-OHdG and Mitosox as well as the western blot assay of HO-1 and 4-HNE protein levels within the ipsilateral hemisphere were performed to evaluate the oxidative stress injury at 24 h after SAH. The fluorescence intensities of 8-OHdG (a marker of oxidative stress to DNA) and Mitosox (a marker of mitochondrial superoxide) were evident in the SAH+vehicle group compared with the Sham group at 24 h after SAH (Fig. 5A, C–D). The activation of TGR5 with INT-777 significantly reduced the fluorescence intensities of 8-OHdG and Mitosox compared with the SAH+vehicle group (Fig. 5A, C–D). Western blot data showed that 4-hydroxynonenal (4-HNE, a marker of oxidative stress) and heme oxygenase-1 (HO-1, a marker of anti-oxidative stress response) were remarkably increased at 24 h after SAH compared with the Sham group (Fig. 5B, E–F). INT-777 treatment further increased the level of HO-1 significantly while reduced the level of 4-HNE (Fig. 5B, E–F).
Activation of TGR5 improved long-term neurological deficits at 28 d after SAH
Rotarod test showed that SAH remarkably induced persistent neurologic dysfunctions in the SAH+vehicle group compared to the Sham group on both 5 RPM and 10 RPM acceleration velocity tests (Fig. 6A–B). INT-777 treatment significantly improved falling latency 1 week and 2 weeks after SAH on both 5 RPM and 10 RPM acceleration velocity tests compared with the SAH+vehicle group. However, there was no significant difference of latency in rotarod test between SAH+INT-777 and SAH+vehicle groups at 3 weeks after SAH (Fig. 6A–B).
Figure 6. Effects of TGR5 activation with INT-777 on long-term (28 d) neurobehavioral outcome after SAH.
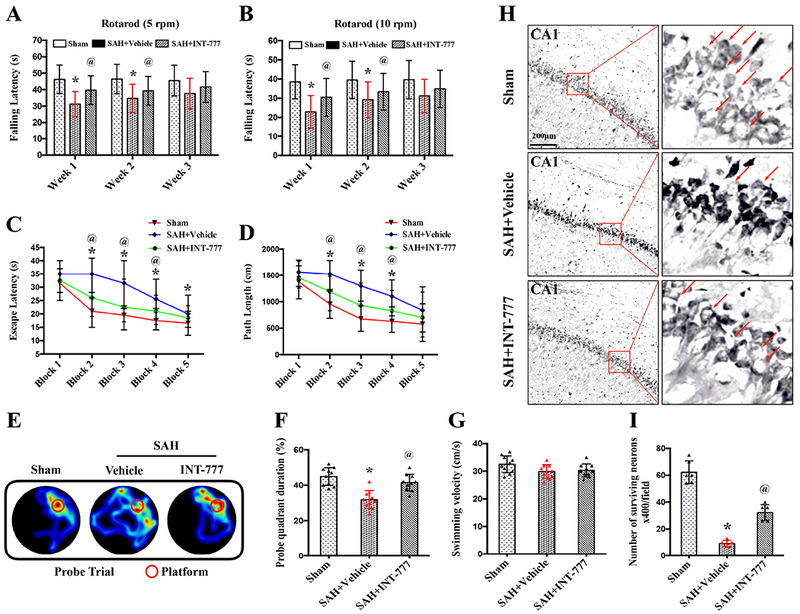
(A-B) Rotarod test on the first, second, third week after SAH. (C-D) Escape latency and swimming distance of Morris water maze test on days 23 to 27 after SAH. (E) Representative thermal imaging pictures of the probe trial. The red circles indicated the probe platform. (F) Quantification of probe quadrant duration in the probe trial on day 27 after SAH. (G) Swimming velocities of different groups in the probe trial. (H-I) Representative micrographs and neuronal quantifications of Nissl staining in hippocampal CA1 region. Arrows indicated the normal neurons. n=10 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group; Two-way repeated measures ANOVA, Tukey’s post hoc test (A-D), and Oneway ANOVA, Tukey’s post hoc test (F-G).
Additionally, the Morris water maze test revealed that spatial memory and learning ability were impaired in the SAH+vehicle group compared with the Sham group, resulting in prolonged escape latency and longer swimming distance to platform as well as less time spent in the probe quadrant on 23-27 d after SAH (Fig. 6C–F). Compared with the SAH+vehicle group, the INT-777 treatment group significantly improved the memory and spatial learning deficits, in terms of the shorter escape latency and swimming distance to platform as well as more time spent in probe quadrant (Fig. 6C–F). There were no significant differences in the swimming speed among all groups (Fig. 6G).
Activation of TGR5 reduced neuronal degeneration at 28 d after SAH
Nissl staining was performed in brain slices at the level of hippocampus at 28 d after SAH. Within the CA1 region of ipsilateral hippocampus, there was significantly more neuron loss and shrinkage morphology of neurons in the SAH+vehicle group compared with the Sham group. INT-777 treatment significantly reversed such neuronal damage in CA1 of the ipsilateral hippocampus when compared to the SAH+vehicle group (Fig. 6H–I).
TGR5 CRISPR and ALDH2 CRISPR reversed the protective effects of INT-777 on neurobehavioral outcomes after SAH
The i.c.v injection of TGR5 CRISPR or ALDH2 CRISPR at 48 h before SAH significantly decreased the endogenous expressions of TGR5 or ALDH2 in naive and SAH rats, demonstrating the knockout efficacy of TGR5 and ALDH2 CRISPRs in the present study (Fig. 7C–E). The pretreatment of TGR5 CRISPR or ALDH2 CRISPR significantly reversed the neurobehavioral benefits of INT-777 on the modified Garcia score and the beam balance score (Fig. 7A–B).
Figure 7. Effects of CRISPR knockout of TGR5 and ALDH2 on neurobehavioral outcome at 24 h after SAH.
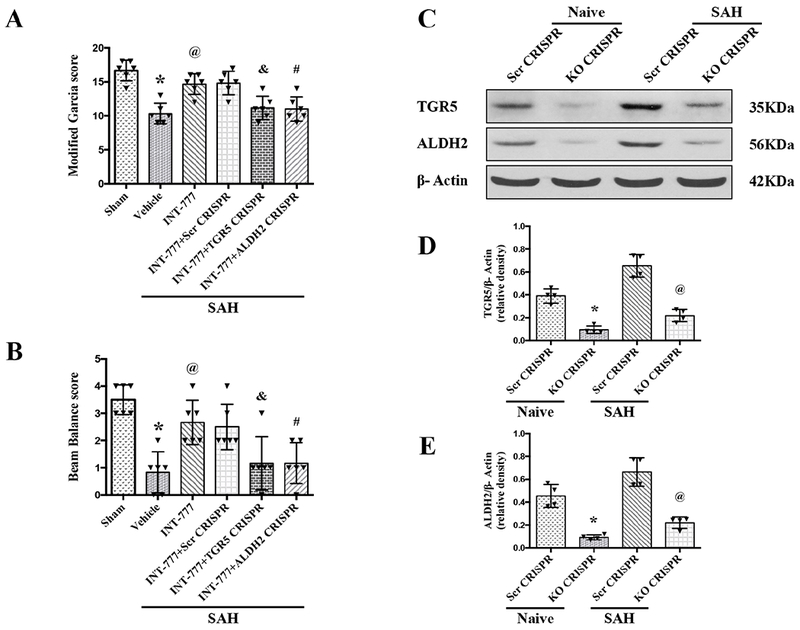
(A-B) TGR5 CRISPR and ALDH2 CRISPR pretreatment reversed the neurobehavioral benefits of INT-777 at 24 h after SAH, n=6 per group. (C-E) Representative western blot bands and densitometric quantification of TGR5 and ALDH2 demonstrated the efficacy of CRISPR knockout in naive and SAH rats, n=4 per group. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group; @P<0.05 vs. SAH+Vehicle group; &P<0.05 vs. SAH+scr CRISPR group; #P<0.05 vs. SAH+scr CRISPR group; One-way ANOVA, Tukey’s post hoc test; scr CRISPR, scrambled CRISPR; KO CRISPR, Knockout CRISPR.
Activation of TGR5 with INT-777 attenuated oxidative stress and neuronal apoptosis via cAMP/PKCε/ALDH2 signaling pathway at 24 h after SAH
Western blots were performed to evaluate the activation of TGR5 and its downstream signaling molecules at 24 h after SAH. The results showed that the expressions of TGR5, cAMP, p-PKCε ALDH2, Bax, Cleaved Caspase-3, HO-1, and 4-HNE were significantly increased, while, the expressions of Bcl-2 remarkably decreased at 24 h after SAH compared with Sham group (Fig. 8 and 9). Moreover, INT-777 treatment further increased the expression levels of TGR5, cAMP, p-PKCε ALDH2 and HO-1, but the expression of Bax, Cleaved Caspase-3 and 4-HNE were decreased in the SAH+INT-777 group when compared with the SAH+vehicle group (Fig. 8A–J). TGR5 knockdown by TGR5 CRISPR pretreatment significantly decreased the downstream protein levels including cAMP, p-PKCε, ALDH2, HO-1 and Bcl-2 in the SAH+INT-777+TGR5 CRISPR group compared with those in the SAH+INT-777+scrambled CRISPR group. Consistently, significant overexpression of Bax, Cleaved Caspase-3 and 4-HNE were observed in the SAH+INT-777+TGR5 CRISPR group when compared with the SAH+INT-777+scrambled CRISPR group (Fig. 8A–J).
Figure 8. TGR5 CRISPR abolished the anti-oxidative stress and anti-apoptosis effects of INT-777 at 24 h after SAH.

(A) Representative western blot bands. (B-J) Densitometric quantification of TGR5, cAMP, p-PKCε/PKCε, ALDH2, HO-1, 4-HNE, Bcl-2, Bax, and Cleaved Caspase-3 (CC3) in the ipsilateral hemisphere at 24 h after SAH. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group, @P<0.05 vs. SAH+Vehicle group, &P<0.05 vs. SAH+scr CRISPR group; One-way ANOVA, Tukey’s post hoc test, n=6 per group; scr CRISPR, scrambled CRISPR.
Figure 9. ALDH2 CRISPR abolished the anti-oxidative stress and anti-apoptosis effects of INT-777 at 24 h after SAH.
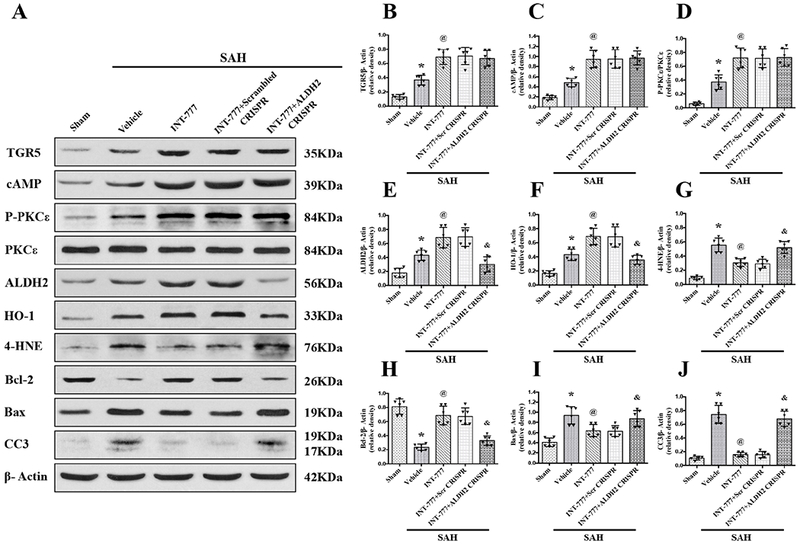
(A) Representative western blot bands. (B-J) Densitometric quantification of TGR5, cAMP, p-PKCε/PKCε, ALDH2, HO-1, 4-HNE, Bcl-2, Bax, and Cleaved Caspase-3 (CC3) in the ipsilateral hemisphere at 24 h after SAH. Vehicle: 10% dimethyl sulfide. Data was represented as mean ± SD. *P<0.05 vs. Sham group, @P<0.05 vs. SAH+Vehicle group, &P<0.05 vs. SAH+scr CRISPR group; One-way ANOVA, Tukey’s post hoc test, n=6 per group; scr CRISPR, scrambled CRISPR.
ALDH2 knockdown by ALDH2 CRISPR pretreatment significantly suppressed the expression of ALDH2, Bcl-2 and HO-1, but increased protein levels of Bax, Cleaved Caspase-3 and 4-HNE at 24 h after SAH in the SAH+ INT-777+ALDH2 CRISPR group compared with the SAH+INT-777+scrambled CRISPR group. ALDH2 CRISPR did not change the expression of cAMP nor the phosphorylation of PKCε (Fig. 9A–J).
Discussion:
Substantial studies revealed that oxidative stress injury and neuronal apoptosis are closely associated with poor outcomes due to EBI after SAH [5, 8, 15, 40]. Hence, inhibition of oxidative stress and neuronal apoptosis would be effective therapeutic strategies to mitigate EBI after SAH. In the present study, we, for the first time, demonstrated that the endogenous protein levels of TGR5 and ALDH2 were increased after SAH in rats, and peaked at 24 h after SAH. The TGR5 receptors were primarily expressed in neurons at 24 h after SAH. In addition, the activation of TGR5 with INT-777 significantly improved the short-term and long-term neurological deficits, accompanied by the decreases in oxidative stress injury and neuronal apoptosis after SAH. Mechanistically, TGR5 activation upregulated the protein levels of cAMP, p-PKCε, ALDH2, HO-1, Bcl-2 and down-regulated the protein levels of 4-HNE, Bax and Cleaved Caspase-3 within ipsilateral hemisphere at 24 h after SAH. The knockdown of TGR5 or ALDH2 by specific TGR5 CRISPR and ALDH2 CRISPR abolished the beneficial effects of TGR5 activation on neurological deficits, oxidative stress and neuronal apoptosis. Taken together, our findings suggested that the activation of TGR5 with INT-777 alleviated oxidative stress and neuronal apoptosis of EBI, leading to the improvement of neurobehavioral impairments after SAH in rats. This neuroprotection at least in part, was attributed to the cAMP/PKCε/ALDH2 signaling pathway.
Oxidative stress injury followed by SAH exacerbated neuronal apoptosis and degradation within the cerebral cortex and hippocampus, leading to the neurological dysfunctions in EBI after SAH [15, 44–46]. TGR5 has been shown to mediate anti-oxidative stress and anti-apoptotic effects [18, 19, 27]. In our study, there was a notable elevation of endogenous TGR5 levels in the ipsilateral brain hemisphere after SAH which peaked at 24 h after the injury. Furthermore, double immunofluorescence staining indicated that the TGR5 was primarily localized in neurons and some in astrocytes and microglia cells. These findings are consistent with previous studies in which 1) TGR5 receptors were identified in neurons, astrocytes, and microglia cells in the rodent brain [16]; and 2) there was an upregulation of brain TGR5 following hepatic encephalopathy in mice [22]. To be noted, the downregulation of brain TGR5 was also reported in other types of experimental brain injury models including LPS induced neuroinflammmation and Aβ1-42 induced Alzheimer’s disease [25, 27]. Such discrepancy may be due to different injury mechanisms across these animal models.
Additionally, ALDH2 is one of the important mitochondrial matrix proteins that can regulate oxidative stress and apoptosis associated with mitochondrial dysfunction [47–49]. It has also been reported to provide significant protection against oxidative stress and apoptosis by facilitating the clearance of subsequent toxic aldehydes, such as 4-HNE [50, 51]. Activating ALDH2 with Alda-1 administration also alleviated hepatic fibrosis involving activation of Nrf2/HO-1 antioxidant pathway [52]. For neurodegenerative diseases, it was suggested that enhancing ALDH2 enzyme activity could attenuate neuronal apoptosis by decreasing ROS accumulation, reversing mitochondrial membrane depolarization, and inhibiting mitochondrial apoptotic pathways [48, 53, 54]. In addition, previous studies found that the overexpression level of ALDH2 would be neuroprotective in Parkinson’s disease, Alzheimer’s disease, post-stoke epilepsy, depression, and ischemic cerebral injury [47, 48, 54–56]. In the present study, there were significantly increases in endogenous ALDH2 along with TGR5, and it also peaked at 24 h after SAH. The upregulation of TGR5 and ALDH2 may suggest an activation of an endogenous neuroprotective mechanism after SAH. However, the extent of these increases were not sufficient to override the injury.
INT-777, as a novel potent and selective TGR5 agonist with remarkable activity in vivo, has been shown to result in side-chain modification of TGR5 receptor (methylation or sulphation), leading to the increasing activity of TGR5 receptor [26, 57], which caused an elevation of intracellular cAMP levels and the subsequent activation signaling pathways [58–60]. Previous studies showed that the activation of TGR5 with INT-777 protected against renal oxidative stress and lipid accumulation in a mouse model of kidney disease associated with obesity and diabetes [18, 61]. Moreover, INT-777 also attenuated hepatocellular apoptosis and cholangiocyte apoptosis following ischemia-reperfusion injury [19, 20]. Interestingly, TGR5 protein up-regulations by INT-777 have been previously reported in other animal model of diseases. In a mouse model of Alzhemier’s disease, INT-777 up-regulated TGR5 protein levels via INT-777 in a dose-dependent manner, which ameliorated cognitive impairment [25, 27]. In a mice model of liver ischemia/reperfusion injury, INT-777 up-regulated TGR5 protein levels and attenuated liver ischemia/reperfusion injury [62]. Consistently, we found that there was an increased cAMP levels associated with INT-777-induced TGR5 protein up-regulation, suggesting the TGR5 activation. Although the underlying molecular mechanisms of which the activation of TGR5 provided its protective effects are not definite, several studies have focused on its modulation of oxidative stress and apoptosis. Wang et al demonstrated that TGR5 activation stimulated the AMPK-SIRT1-PGC1 axis, resulting in increased mitochondrial biogenesis, while preventing renal oxidative stress and lipid accumulation [18]. TGR5 signaling also induced mitochondrial fission through the ERK/DRP1 pathway, thus improving mitochondrial respiration mediated metabolic disorders [63]. Furthermore, the activation of TGR5 could induce anti-apoptotic effects through serine phosphorylation of the CD95 receptor in murine cholangiocytes [19]. Zhuang et al. also reported that ischemic preconditioning could trigger TGR5 activation, which reduced inflammatory responses and inhibited hepatocellular apoptosis in ischemia/reperfusion injury of the liver [20]. The neuroprotective roles of TGR5 activation and its agonist in CNS has become popular in recent years. TGR5 activation could alleviate neuroinflammation in hepatic encephalopathy and acute brain injury [22]. TGR5 signaling also ameliorated memory deficits, this effect being attributed to the blockade of neuronal apoptosis, neuroinflammation, and synaptic toxicity within the hippocampus and frontal cortex in mouse model of Alzhemier’s disease [27]. Together, these studies revealed a strong link between TGR5 activation and anti-oxidative stress, anti-apoptosis, and anti-inflammation characteristics.
In the present study, intranasal administration of INT-777 1 h after SAH further upregulated TGR5 protein levels and improved short-term and long-term neurological functions. In INT-777 treated SAH rats, there were significantly attenuated oxidative stress levels and neuronal apoptosis, evidenced in reduced positive staining of Mitosox, 8-OHdG, TUNEL, and FJC at 24 h after SAH. The western blots assays consistently showed that INT-777 treatment significantly down-regulated the protein levels of 4-HNE, Bax, and Cleaved Caspase-3, while up-regulated HO-1, and Bcl-2 at 24 h after SAH. Therefore, we speculated that the activation of TGR5 with INT-777 alleviated neurological impairments after SAH, possibly by suppressing oxidative stress injury and neuronal apoptosis.
We further explored the signaling pathway underlying the anti-oxidative stress and anti-neuronal apoptosis of TGR5 activation after SAH. The TGR5-activated cAMP-PKA signaling pathway was identified as a unique mechanism in regulating renal AQP2 mediated water homeostasis, glucose-stimulated insulin secretion, inhibiting NLRP3 inflammasome activation, inhibiting bacterial infection due to cholestasis and inhibiting neuroinflammation in acute brain injury [17, 23, 64–66]. Intriguingly, an elevation of intracellular cAMP levels can also result in the phosphorylation of PKCε [32, 67]. Coincidentally, PKCε was proposed as an upstream regulatory molecule of ALDH2 providing protective effects in ischemic heart injury and ischemic stroke via phosphorylation of PKCε [28–31, 68]. In the present study, we found that the TGR5 activation with INT-777 significantly increased the protein levels of cAMP and p-PKCε as well as ALDH2 in the ipsilateral brain hemisphere at 24 h after SAH. The knockdown of TGR5 reduced cAMP levels and its downstream molecules of p-PKCε, ALDH2, HO-1, and Bcl-2, but increased the expressions of 4-HNE, Bax, and Cleaved Caspase-3. Moreover, although the knockdown of ALDH2 had no effects on the expression of cAMP and PKCε phosphorylation, it significantly decreased the expressions of HO-1, and Bcl-2, while increasing the expressions of 4-HNE, Bax, and Cleaved Caspase-3. Thus, our data suggested that TGR5 activation upregulated the ALDH2 expression at least in part via cAMP/p-PKCε signaling, leading to the effects of anti-oxidative stress and anti-neuronal apoptosis after SAH.
There are several limitations in this study. First, previous studies and our own data showed that the TGR5 receptor was also expressed in astrocytes and microglial cells [16, 22]. The anti-inflammatory effects and blood-brain barrier protection of TGR5 activation after SAH need further investigation. Second, previous studies have demonstrated that TGR5 activation could 1) regulate mitochondrial biogenesis by stimulating the AMPK-SIRT1-PGC1 axis [18]; 2) induce mitochondrial fission through the ERK/DRP1 pathway [63]; 3) alleviate inflammation and apoptosis via cAMP/PKA axis and downstream regulatory molecules [17, 19, 23, 64]. Thus, we could not exclude the contribution of other signaling pathways to the anti-oxidative stress and anti-apoptotic effects of TGR5 activation in SAH model. Third, we only evaluated the neuroprotective effect of TGR5 activation in male rats. Future studies are warranted to validate its treatment efficacy against SAH in female rats.
In conclusion, the activation of TGR5 with INT-777 improved neurological deficits, by attenuating oxidative stress and neuronal apoptosis after SAH in rats. The anti-oxidative and anti-apoptotic effects were, at least in part, through the cAMP/PKCε/ALDH2 signaling pathway. Thus, TGR5 may serve as a novel therapeutic target to ameliorate EBI after SAH.
Figure S1. Activation of TGR5 with INT-777 attenuated oxidative stress and neuronal apoptosis via cAMP/PKCε/ALDH2 pathway after subarachnoid hemorrhage in rats.
Figure S2. Experimental design. SAH, subarachnoid hemorrhage; WB , western blot; IHC, immunohistochemistry; i.n, intranasal; i.c.v, intracerebroventricular.
Figure S3. Detection of INT-777 in brain after intranasal administration. (A) Mass spectra of INT-777 detected with full scan mass spectrometry (MS). (B) MS/MS spectra of precursor ion at m/z 449 from INT-777 standard. (C) MS/MS spectra of precursor ion at m/z 449 from the brain of dosed rats.
Supplementary Material
The expressions of endogenous TGR5 receptors and ALDH2 gradually increased after SAH. The TGR5 receptors were primarily expressed in neurons after SAH in rats.
The activation of TGR5 with INT-777 significantly improved the short-term and long-term neurological deficits, accompanied by reduced the oxidative stress and neuronal apoptosis after SAH in rats.
Activation of TGR5 with INT-777 attenuated oxidative stress and neuronal apoptosis, at least in part, via cAMP/PKC /ALDH2 signaling pathway at 24 h after SAH.
Acknowledgements
This study was supported by grants from the National Institutes of Health (NS081740 and NS082184) to John H. Zhang, and a grant from Jiangsu provincial medical talents project fund (QNRC2016263) to Dr. G. Zuo.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
There is no conflict of interest.
Data availability statement
The data support the findings of this study and are available from the corresponding author upon reasonable request.
Reference
- 1.Lawton MT and Vates GE, Subarachnoid Hemorrhage. N Engl J Med, 2017. 377(3): p. 257–266. [DOI] [PubMed] [Google Scholar]
- 2.Nieuwkamp DJ, et al. , Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol, 2009. 8(7): p. 635–42. [DOI] [PubMed] [Google Scholar]
- 3.Conzen C, et al. , The Acute Phase of Experimental Subarachnoid Hemorrhage: Intracranial Pressure Dynamics and Their Effect on Cerebral Blood Flow and Autoregulation. Transl Stroke Res, 2018. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H and Nakano F, To Improve Translational Research in Subarachnoid Hemorrhage. Transl Stroke Res, 2018. 9(1): p. 1–3. [DOI] [PubMed] [Google Scholar]
- 5.Shi L, et al. , PCMT1 Ameliorates Neuronal Apoptosis by Inhibiting the Activation of MST1 after Subarachnoid Hemorrhage in Rats. Transl Stroke Res, 2017. [DOI] [PubMed] [Google Scholar]
- 6.Jimenez-Xarrie E, et al. , Uric Acid Treatment After Stroke Prevents Long-Term Middle Cerebral Artery Remodelling and Attenuates Brain Damage in Spontaneously Hypertensive Rats. Transl Stroke Res, 2018. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, et al. , Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury. Transl Stroke Res, 2018. 9(1): p. 74–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fumoto T, et al. , The Role of Oxidative Stress in Microvascular Disturbances after Experimental Subarachnoid Hemorrhage. Transl Stroke Res, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Petro M, et al. , Tissue plasminogen activator followed by antioxidant-loaded nanoparticle delivery promotes activation/mobilization of progenitor cells in infarcted rat brain. Biomaterials, 2016. 81: p. 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burbulla LF, et al. , Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science, 2017. 357(6357): p. 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pauletti A, et al. , Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain, 2017. 140(7): p. 1885–1899. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Narayanan SV, Dave KR, and Perez-Pinzon MA, Ischemic Preconditioning Protects Astrocytes against Oxygen Glucose Deprivation Via the Nuclear Erythroid 2-Related Factor 2 Pathway. Transl Stroke Res, 2018. 9(2): p. 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, et al. , Sustained release of bioactive hydrogen by Pd hydride nanoparticles overcomes Alzheimer’s disease. Biomaterials, 2019. 197: p. 393–404. [DOI] [PubMed] [Google Scholar]
- 14.Cobley JN, Fiorello ML, and Bailey DM, 13 reasons why the brain is susceptible to oxidative stress. Redox Biol, 2018. 15: p. 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mo J, et al. , AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats. Redox Biol, 2019. 20: p. 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keitel V, et al. , The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia, 2010. 58(15): p. 1794–805. [DOI] [PubMed] [Google Scholar]
- 17.Guo C, et al. , Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity, 2016. 45(4): p. 802–816. [DOI] [PubMed] [Google Scholar]
- 18.Wang XX, et al. , G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J Am Soc Nephrol, 2016. 27(5): p. 1362–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reich M, et al. , TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut, 2016. 65(3): p. 487–501. [DOI] [PubMed] [Google Scholar]
- 20.Zhuang L, et al. , Ischemic Preconditioning protects hepatocytes from ischemia-reperfusion injury via TGR5-mediated anti-apoptosis. Biochem Biophys Res Commun, 2016. 473(4): p. 966–972. [DOI] [PubMed] [Google Scholar]
- 21.Eggink HM, et al. , Chronic infusion of taurolithocholate into the brain increases fat oxidation in mice. J Endocrinol, 2018. 236(2): p. 85–97. [DOI] [PubMed] [Google Scholar]
- 22.McMillin M, et al. , TGR5 signaling reduces neuroinflammation during hepatic encephalopathy. J Neurochem, 2015. 135(3): p. 565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yanguas-Casas N, et al. , TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 With Anti-Inflammatory Effects in Microglial Cells. J Cell Physiol, 2017. 232(8): p. 2231–2245. [DOI] [PubMed] [Google Scholar]
- 24.Elia AE, et al. , Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol, 2016. 23(1): p. 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, et al. , Inhibitory effect of INT-777 on lipopolysaccharide-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Prog Neuropsychopharmacol Biol Psychiatry, 2019. 88: p. 360–374. [DOI] [PubMed] [Google Scholar]
- 26.Pellicciari R, et al. , Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem, 2009. 52(24): p. 7958–61. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, et al. , Neuroprotective effects of INT-777 against Abeta1-42-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Brain Behav Immun, 2018. 73: p. 533–545. [DOI] [PubMed] [Google Scholar]
- 28.Guo JM, et al. , ALDH2 protects against stroke by clearing 4-HNE. Cell Res, 2013. 23(7): p. 915–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aldi S, et al. , Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C epsilon-dependent aldehyde dehydrogenase type-2 activation. J Pharmacol Exp Ther, 2014. 349(3): p. 508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marino A, et al. , S1P receptor 1-Mediated Anti-Renin-Angiotensin System Cardioprotection: Pivotal Role of Mast Cell Aldehyde Dehydrogenase Type 2. J Pharmacol Exp Ther, 2017. 362(2): p. 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, et al. , Mitochondrial PKC-epsilon deficiency promotes I/R-mediated myocardial injury via GSK3beta-dependent mitochondrial permeability transition pore opening. J Cell Mol Med, 2017. 21(9): p. 2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, et al. , Exchange factor directly activated by cAMP-PKCepsilon signalling mediates chronic morphine-induced expression of purine P2X3 receptor in rat dorsal root ganglia. Br J Pharmacol, 2018. 175(10): p. 1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng J, et al. , LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol, 2019. 21: p. 101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugawara T, et al. , A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods, 2008. 167(2): p. 327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Enkhjargal B, et al. , Intranasal administration of vitamin D attenuates blood-brain barrier disruption through endogenous upregulation of osteopontin and activation of CD44/P-gp glycosylation signaling after subarachnoid hemorrhage in rats. J Cereb Blood Flow Metab, 2017. 37(7): p. 2555–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang Y, et al. , Sensitive liquid chromatography/mass spectrometry methods for quantification of pomalidomide in mouse plasma and brain tissue. J Pharm Biomed Anal, 2014. 88: p. 262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan F, et al. , ErbB4 protects against neuronal apoptosis via activation of YAP/PIK3CB signaling pathway in a rat model of subarachnoid hemorrhage. Exp Neurol, 2017. 297: p. 92–100. [DOI] [PubMed] [Google Scholar]
- 38.Dubue JD, et al. , Intrahippocampal Anisomycin Impairs Spatial Performance on the Morris Water Maze. J Neurosci, 2015. 35(31): p. 11118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu F, et al. , Recombinant milk fat globule-EGF factor-8 reduces oxidative stress via integrin beta3/nuclear factor erythroid 2-related factor 2/heme oxygenase pathway in subarachnoid hemorrhage rats. Stroke, 2014. 45(12): p. 3691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pang J, et al. , Apolipoprotein E Exerts a Whole-Brain Protective Property by Promoting M1? Microglia Quiescence After Experimental Subarachnoid Hemorrhage in Mice. Transl Stroke Res, 2018. 9(6): p. 654–668. [DOI] [PubMed] [Google Scholar]
- 41.Dai Y, et al. , Calcitriol inhibits ROS-NLRP3-IL-1beta signaling axis via activation of Nrf2-antioxidant signaling in hyperosmotic stress stimulated human corneal epithelial cells. Redox Biol, 2019. 21: p. 101093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma T, et al. , Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci, 2011. 31(15): p. 5589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahmood T and Yang PC, Western blot: technique, theory, and trouble shooting. N Am J Med Sci, 2012. 4(9): p. 429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ayer RE and Zhang JH, Oxidative stress in SAH. Acta Neurochir Suppl, 2008. 104: p. 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhan Y, et al. , Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats. Crit Care Med, 2012. 40(4): p. 1291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan LF, et al. , Mdivi-1 ameliorates early brain injury after subarachnoid hemorrhage via the suppression of inflammation-related blood-brain barrier disruption and endoplasmic reticulum stress-based apoptosis. Free Radic Biol Med, 2017. 112: p. 336–349. [DOI] [PubMed] [Google Scholar]
- 47.Wang W, et al. , Heavy ethanol consumption aggravates the ischemic cerebral injury by inhibiting ALDH2. Int J Stroke, 2015. 10(8): p. 1261–9. [DOI] [PubMed] [Google Scholar]
- 48.Stachowicz A, et al. , Proteomic Analysis of Mitochondria-Enriched Fraction Isolated from the Frontal Cortex and Hippocampus of Apolipoprotein E Knockout Mice Treated with Alda-1, an Activator of Mitochondrial Aldehyde Dehydrogenase (ALDH2). Int J Mol Sci, 2017. 18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao L, et al. , Mitochondria-targeted ubiquinone (MitoQ) enhances acetaldehyde clearance by reversing alcohol-induced posttranslational modification of aldehyde dehydrogenase 2: A molecular mechanism of protection against alcoholic liver disease. Redox Biol, 2018. 14: p. 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duan Y, et al. , Mitochondrial aldehyde dehydrogenase 2 protects gastric mucosa cells against DNA damage caused by oxidative stress. Free Radic Biol Med, 2016. 93: p. 165–76. [DOI] [PubMed] [Google Scholar]
- 51.Gu X, et al. , Effect of ALDH2 on High Glucose-Induced Cardiac Fibroblast Oxidative Stress, Apoptosis, and Fibrosis. Oxid Med Cell Longev, 2017. 2017: p. 9257967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma X, et al. , Aldehyde dehydrogenase 2 activation ameliorates CCl4 -induced chronic liver fibrosis in mice by up-regulating Nrf2/HO-1 antioxidant pathway. J Cell Mol Med, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song K, et al. , Rutin attenuates ethanol-induced neurotoxicity in hippocampal neuronal cells by increasing aldehyde dehydrogenase 2. Food Chem Toxicol, 2014. 72: p. 228–33. [DOI] [PubMed] [Google Scholar]
- 54.Chiu CC, et al. , Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp Neurol, 2015. 263: p. 244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang H, et al. , The ALDH2 rs671 polymorphism affects post-stroke epilepsy susceptibility and plasma 4-HNE levels. PLoS One, 2014. 9(10): p. e109634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stachowicz A, et al. , The impact of mitochondrial aldehyde dehydrogenase (ALDH2) activation by Alda-1 on the behavioral and biochemical disturbances in animal model of depression. Brain Behav Immun, 2016. 51: p. 144–53. [DOI] [PubMed] [Google Scholar]
- 57.Schaap FG, Trauner M, and Jansen PL, Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol, 2014. 11(1): p. 55–67. [DOI] [PubMed] [Google Scholar]
- 58.Watanabe M, et al. , Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature, 2006. 439(7075): p. 484–9. [DOI] [PubMed] [Google Scholar]
- 59.Thomas C, et al. , TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab, 2009. 10(3): p. 167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pols TW, et al. , TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab, 2011. 14(6): p. 747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang XX, et al. , FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J Am Soc Nephrol, 2018. 29(1): p. 118–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang H, et al. , Plasma membrane-bound G protein-coupled bile acid receptor attenuates liver ischemia/reperfusion injury via the inhibition of toll-like receptor 4 signaling in mice. Liver Transpl, 2017. 23(1): p. 63–74. [DOI] [PubMed] [Google Scholar]
- 63.Velazquez-Villegas LA, et al. , TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat Commun, 2018. 9(1): p. 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haselow K, et al. , Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J Leukoc Biol, 2013. 94(6): p. 1253–64. [DOI] [PubMed] [Google Scholar]
- 65.Vettorazzi JF, et al. , The bile acid TUDCA increases glucose-induced insulin secretion via the cAMP/PKA pathway in pancreatic beta cells. Metabolism, 2016. 65(3): p. 54–63. [DOI] [PubMed] [Google Scholar]
- 66.Li S, et al. , Bile Acid G Protein-Coupled Membrane Receptor TGR5 Modulates Aquaporin 2-Mediated Water Homeostasis. J Am Soc Nephrol, 2018. 29(11): p. 2658–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu Y, Li G, and Huang LYM, Inflammation induces Epac-protein kinase C alpha and epsilon signaling in TRPV1-mediated hyperalgesia. Pain, 2018. 159(11): p. 2383–2393. [DOI] [PubMed] [Google Scholar]
- 68.Chen CH, et al. , Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science, 2008. 321(5895): p. 1493–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.