

Abstract
Aims
A population pharmacokinetic (PK) analysis was conducted to quantify the impact of patient‐specific and concurrent medication factors on pevonedistat PK.
Methods
Data were pooled from 6 clinical studies consisting of 335 patients with solid tumours or haematological malignancies administered pevonedistat alone or in combination with azacitidine, docetaxel, carboplatin + paclitaxel, or gemcitabine. Model development and covariate analysis followed standard methods. Parameters and bootstrap 95% confidence intervals were estimated using nonlinear mixed‐effects modelling. The final model was evaluated using visual predictive checks and other goodness‐of‐fit criteria.
Results
A linear 2‐compartment model best described pevonedistat PK. The final model included the effect of body surface area (BSA) on clearance (CL and Q) and volume of distribution of pevonedistat, effect of concomitantly administered carboplatin + paclitaxel on CL, and effect of albumin on Q. Race, sex, age, tumour type (haematological vs solid), mild or moderate renal impairment (creatinine clearance ≥30 mL/min), or mild hepatic impairment, had no impact on pevonedistat PK.
Conclusions
The clinical PK profile of pevonedistat is comparable in patients with solid tumours or haematological malignancies. All PK parameters exhibited ≥20% change over the observed BSA range (1.38–3 m2) with CL ranging from 75.5 to 208% of the reference value, with simulations supporting BSA‐based dosing to minimize interindividual variability in drug exposures. Concurrent administration of carboplatin + paclitaxel decreased pevonedistat CL by approximately 44%, while coadministration with azacitidine, gemcitabine or docetaxel did not alter pevonedistat CL. No other factors were identified as influencing pevonedistat PK.
Keywords: anticancer drugs, pharmacokinetics, population analysis
What is already known about this subject
Pevonedistat (TAK‐924; formerly MLN4924) is an investigational, first‐in‐class, small‐molecule NEDD8‐activating enzyme inhibitor in development for treatment of solid tumours or haematological malignancies.
Several clinical studies have characterized the pharmacokinetics of pevonedistat administered alone or in combination with standard of care therapies in patients with solid tumours or haematological malignancies.
What this study adds
We provide an integrated model‐based analysis of pevonedistat pharmacokinetics across multiple studies and cancer patient populations.
The population model quantitatively describes the impact of patient characteristics and comedications on pevonedistat pharmacokinetics (single agent or coadministered with standard‐of‐care chemotherapy). It also supports body surface area‐based dosing in the target patient populations.
1. INTRODUCTION
Protein degradation by the ubiquitin proteasome system (UPS) is necessary for the regulation of a broad array of cellular processes, including: cell‐cycle division; DNA repair, growth and differentiation; quality control; and regulation of membrane receptors and ion channels. Defects in the system result in the pathogenesis of many cancers and numerous other diseases, including Alzheimer's disease, Parkinson's disease, Huntington disease, prion‐like lethal disorders, cystic fibrosis, Angelman's syndrome and Liddle syndrome.1 Drugs targeting the UPS have the potential to improve the outcome of many of these diseases, especially malignant conditions.
The neddylation pathway that conjugates neural precursor cell‐expressed, developmentally downregulated 8 (NEDD8) to the cullin‐RING ligases (CRLs), the largest subfamily of the https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1023, is controlled by NEDD8‐activating enzyme (NAE).2 CRLs control the proteasomal degradation of proteins with roles in cell cycle progression, DNA replication and signal transduction.3 CRL ligase function is dependent on NEDD8 conjugation, and therefore NAE is critical in regulating CRL activity.2 Pevonedistat (TAK‐924; formerly MLN4924) is an investigational, first‐in‐class, small‐molecule NAE inhibitor.4 Pevonedistat covalently binds with NEDD8 to form a pevonedistat–NEDD8 adduct that prevents NEDD8 conjugation to CRLs and thus results in CRL substrate accumulation and apoptotic cell death.4, 5
Single‐agent pevonedistat has been studied in Phase I clinical trials in patients with advanced nonhaematological malignancies (Study C15001, NCT00677170); lymphoma and multiple myeloma (Study C15002, NCT00722488); acute myelogenous lymphoma (AML), myelodysplastic syndromes (MDS), or acute lymphoblastic leukaemia (Study C15003, NCT0091106); and melanoma (Study C15005, NCT01011530). Combination therapy with azacitidine was studied in AML patients (Study C15009, NCT01814826), and patients with solid tumours in combination with either docetaxel, carboplatin + paclitaxel, or gemcitabine (Study C15010, NCT01862328).
The pharmacokinetics (PK) of pevonedistat, following single and multiple intravenous (IV) administrations, has been evaluated based on data from 4 of the Phase I studies reported here.6, 7, 8, 9 Plasma concentrations of pevonedistat declined in a bi‐exponential manner at the end of a 1‐hour IV infusion, with little or no notable drug accumulation following once‐daily dosing for 5 consecutive days or intermittent dosing. This observation is consistent with a mean terminal elimination half‐life of approximately 5–8 hours estimated across doses and schedules. Pevonedistat PK is linear over the dose range studied based on a daily area under the plasma concentration vs time curve (AUC) from time 0 to 24 hours that increased proportionately with dose from 25 to 278 mg/m2.
The clinical development programme for pevonedistat has evaluated its safety and efficacy across a range of cancer indications, including solid tumours and haematological malignancies. Clinical trials have evaluated pevonedistat both as a single agent and in combination with standard‐of‐care agents. In solid tumours, combinations with 3 distinct chemotherapy regimens were evaluated (docetaxel, carboplatin + paclitaxel, gemcitabine) aimed at possible future development in solid tumour indications where these chemotherapy backbones may represent standards of care.10 In AML and MDS, pevonedistat has been evaluated in combination with the hypomethylating agent azacitidine,11, 12 and is currently in the pivotal phase of clinical investigation in higher risk MDS and low blast AML in the PANTHER Phase III trial (NCT 03268954).
The objectives of this population PK (PPK) analysis were to describe IV pevonedistat PK using nonlinear mixed effects modelling in patients with solid or haematological malignancies and identify potential predictors of interpatient variability in pevonedistat exposures when given alone or in combination with standard‐of‐care chemotherapy.
2. METHODS
2.1. Clinical studies and patients
Adult patients from 4 single‐agent Phase I and 2 combination Phase Ib dose‐escalation clinical studies were included in the analysis (Table 1). In the 4 Phase I studies in patients with solid tumours or haematological malignancies, single‐agent pevonedistat was administered via a 1‐hour IV infusion at dose levels of 25–278 mg/m2, across 6 different dosing schedules, in 21‐day cycles.6, 7, 8, 13 In the Phase Ib study C15009 in treatment‐naïve elderly (≥60 years) AML patients considered unlikely to benefit from conventional chemotherapy, 20 or 30 mg/m2 pevonedistat was administered via a 1‐hour IV infusion on days 1, 3 and 5 in combination with 75 mg/m2 azacitidine (IV/subcutaneous) on days 1–5, 8 and 9 in 28‐day cycles.14 In the Phase Ib study C15010, 15, 20 or 25 mg/m2 pevonedistat was administered via a 1‐hour IV infusion on days 1, 3 and 5 in combination with docetaxel (Arm 1), carboplatin + paclitaxel (Arm 2), or gemcitabine (Arm 3) on day 1 in 21‐day cycles in patients with solid tumours felt to be appropriate for treatment with 1 of the 3 chemotherapy regimens in this study. The clinical protocols were approved by the Institutional Review Board or Independent Ethics Committee for each site, and informed consent was obtained from all patients prior to enrolment.
Table 1.
Summary of pevonedistat clinical studies included in the population pharmacokinetics analysis
| Study | NCT | Cancer type | n | Pevonedistat regimen | Pevonedistat doses | Pevonedistat treatment schedule | Pharmacokinetic sampling | Total number samples per study |
|---|---|---|---|---|---|---|---|---|
| C15001 | NCT00677170 | Solid tumour | 60 | Single agent | 25–196 mg/m2 | Once per day for 5 days | Day 1—predose, end of infusion, 30 min, 1, 2, 4, 8 h postinfusion; day 2—predose, end of infusion; day 5—predose, end of infusion, 30 min, 1, 2, 4, 6, 10, 24, 72, 120 h postinfusion | 849 |
| C15002 | NCT00722488 | Haematological | 56 | Single agent | 25–261 mg/m2 | Cycle 1—days 1, 2, 8, 9 | Day 1—predose, end of infusion, 30 min, 1, 2, 4, 8 h postinfusion; day 2—predose, end of infusion; day 8—predose, end of infusion; day 9—predose, end of infusion, 30 min, 1, 2, 4, 6, 9, 24, 72, 144 h postinfusion | 850 |
| C15003 | NCT00911066 | Haematological | 72 | Single agent | 25–83 mg/m2 | Cycle 1—days 1, 3, 5 | Day 1—predose, end of infusion, 30 min, 1, 2, 4, 8, 24 h postinfusion; day 3—predose; day 5—predose, end of infusion, 3 to 6 h postinfusion | 571 |
| C15005 | NCT01011530 | Solid tumour | 36 | Single agent | 50–278 mg/m2 | Days 1, 4, 8, 11 | Day 1—predose, end of infusion, 30 min, 1, 4, 7 h postinfusion; day 2–24 h; day 3–48 h; day 4—predose, end of infusion, 3 to 6 h; day 8—predose, end of infusion, 4, 7 h; day 11—predose, end of infusion, 4, hours; day 15 (at clinic visit). | 337 |
| C15009 | NCT01814826 | Haematological | 64 | Combination with azacitidine | 20, 30, 40, 50 mg/m2 | Days 1, 3, 5 | Day 1—predose, end of infusion, 1, 2, 4, 6 h postinfusion; day 2–24 h postinfusion; day 3—predose; day 5—predose, end of infusion, 1, 2, 4, 6 h postinfusion; day 6–24 h postinfusion; day 7–48 h postinfusion | 831 |
| C15010 | NCT01862328 | Solid tumour | 58 | Combination with docetaxel (arm 1), carboplatin + paclitaxel (arm 2), gemcitabine (arm 3) | 15, 25, 37, 50 mg/m2 | Days 1, 3, 5 | Day 1—predose, end of infusion, 1.5, 3 h postdose; day 2–20 h postdose; day 3—predose | 330 |
Data are presented for the 335 evaluable subjects with observed pharmacokinetic data.
2.2. Bioanalytical methods
Blood samples were collected before and at predetermined specific time points after the start of pevonedistat infusion during the first treatment cycle of each study. Samples were analysed for pevonedistat plasma concentrations using validated liquid chromatography–tandem mass spectrometry methods as described previously.7 The dynamic range was 0.0500–25.0 ng/mL for the low range method (precision [%CV]: 3.1–5.1%; accuracy [%bias]: −1.8% to −0.5%) and 1.00–500 ng/mL for the medium range method (precision: 2.5–3.1%; accuracy: −1.5 to 0.0%).
2.3. Population PK model development and validation
Population modelling was performed using NONMEM Version 7.3 (Icon Development Solutions, Dublin, Ireland)15 with Intel Visual Fortran Intel 64 Compiler XE, Version 12.0.0.104 Build 20101006 (Santa Clara, CA, USA). The R data analysis language (Version 2.3.0 or higher) was used for most graphical output and data manipulation,16 making use of the ggplot2 graphing package (Version 0.9.1 or higher) and the doBy data aggregation package (Version 4.5–3 or higher) or plyr data aggregation package (Version 1.7.1 or higher). The remaining graphical output and data manipulation were performed using Microsoft Excel 2003 or later. The PK of pevonedistat was characterized using nonlinear mixed effects (population) compartmental models.
Structural PK model selection was guided by visual inspection of semi‐logarithmic plots of concentration–time profiles and the results of a previous preliminary population PK analysis. The base PPK model consisted of the following components: a structural model that described plasma concentrations of pevonedistat as a function of time, an interindividual variability (IIV) model that described random variability among individuals in the study population, and a residual error model that characterized the random variability in observed data within an individual. IIV was included on all parameters for the PPK model and the Ω matrix was evaluated to develop the most complete matrix possible. Residual variability was modelled using the log transform both sides approach with an additive error model. The first‐order conditional estimation method was used.
For the covariate model development, covariates were plotted against the individual estimated IIV (eta plots) for all covariates. Plots that showed a trend or were expected to influence pevonedistat PK (e.g. renal function, liver function or body size) were tested as single covariate models. For the evaluation of creatinine clearance (CrCL) as a covariate, values >150 mL/min were capped at 150 mL/min based on physiological plausibility considerations. The χ2 test (P < .05) for the log‐likelihood difference in the objective function value (OFV) between nested models with degrees of freedom equal to the difference in the number of parameters between models was used to declare superiority of 1 model over another. This corresponds to a reduction in OFV of 3.84 for comparison of models that differ by 1 parameter. The covariance step was implemented with each NONMEM run, and standard errors for parameter estimates as well as correlation between parameters were evaluated. Models that resulted in structural PK model parameter estimates with high associated standard error (>35% of the parameter estimate), models with a high degree of correlation between parameters (>90%), and models that included a covariate(s) whose effect on the estimated parameter value was negligible (e.g. a <20% change in the parameter over the range of covariate values in the dataset), were not considered further. Covariates that met the criteria for selection were incorporated into a full model. Lastly, the final PPK model was chosen using backwards elimination by retaining only the statistically significant (P < .01) covariate effects. A continuous covariate was considered clinically relevant if its inclusion resulted in >20% change in point estimates over the range from low (5th percentile) to high (95th percentile) values of the covariate. For a categorical covariate, clinical relevance was defined as 20% change in point estimates compared to the typical parameter values of the reference population.
Visual predictive checks (VPCs) were performed on the final PPK model.17 The parameter estimates were assumed to have a multivariate normal distribution with the mean vector set to the population parameter estimates and the covariance matrix set to the covariance matrix of the estimates from the final model. The final model was used to simulate 1000 replicates using the design, dose regimen and covariates of the original dataset. Relevant summary measures were generated for both the observed and simulated data. The observed summary measure was then compared to selected percentiles (lower 5th, median 50th and upper 95th) of the 1000 simulated summary measures.
A nonparametric unstratified bootstrap18 was conducted on the PPK model using the full dataset to determine the confidence intervals of the parameters for the final models. Parameters were evaluated to ensure the 95% confidence intervals did not include zero.
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,19 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.20
3. RESULTS
A total of 6 clinical studies consisting of 346 adult patients (59% male; 87% Caucasian) contributed 3768 observations; 11 of 346 patients did not have PK data (Table 1). Patients were aged 23–90 years (mean age: 62.1 years). Body weight ranged from 43.5–180 kg (mean weight: 80.3 kg). Forty‐six percent of the patients had normal renal function, 36 and 18% had mild (estimated CrCL 60–89 mL/min) to moderate (CrCL 30–59 mL/min) renal impairment, respectively, and 1 patient had severe renal impairment (Table 2).
Table 2.
Characteristics of the study populations
| Study | Age (y) | Body weight (kg) | BMI (kg/m2) | BSA (m2) | ALB (g/L) | CrCLa (mL/min) | ALT (IU/L) | AST (IU/L) | BILI (μmol/L) | HCT | Hb (g/L) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C15001 | 59.5 (34–84) | 81.1 (45.7–148) | 28.8 (20.2–45.8) | 1.98 (1.38–2.72) | 35 (21–50) | 99.1 (45.9–251) | 25.5 (10–87) | 26 (12–60) | 8.55 (1.71–18.8) | 0.346 (0.214–0.485) | 116 (9.9–163) |
| C15002 | 60 (26–90) | 80.1 (51.4–141) | 27.3 (18.9–41.5) | 1.93 (1.48–2.69) | 39 (26–50) | 94.3 (42.3–175) | 18 (6–134) | 25 (13–110) | 6.84 (1.71–17.1) | 0.342 (0.239–0.435) | 112 (83–153) |
| C15003 | 65 (23–84) | 74.1 (48.6–180) | 26 (15.7–55.5) | 1.86 (1.43–3) | 37 (25–48) | 79 (42.1–301) | 27 (8–98) | 25 (7–76) | 8.55 (3.42–18.8) | 0.266 (0.196–0.383) | 91 (70–127) |
| C15005 | 62.1 (32.6–79.4) | 83.5 (52–129) | 28.2 (18.1–41.6) | 2.01 (1.49–2.54) | 39.6 (29–44) | 104 (58.3–216) | 15.5 (6–49) | 19.5 (10–55) | 6.84 (3.42–17.1) | 0.36 (0.255–0.5) | 120 (86–172) |
| C15009 b | 75 (61–89) | 71.9 (43.5–128) | 25.6 (17.6–40.3) | 1.84 (1.38–2.54) | NR | 65.8 (26.3–186) | 25 (6–114) | 21.5 (11–95) | 11.1 (1.71–37.6) | NR | NR |
| C15010 | 61 (26–84) | 77.1 (44.7–150) | 25.6 (16.9–43.8) | 1.92 (1.42–2.78) | NR | 97.9 (49.8–250) | 23 (7–67) | 25 (10–81) | 6.84 (1.71–18.8) | NR | NR |
ALB, albumin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BILI, total bilirubin; BMI, body mass index; BSA, body surface area; CrCL, creatinine clearance; Hb, haemoglobin; HCT, haematocrit; NR, not reported.
Data are presented as median (range).
Cockcroft–Gault formula was used to estimate creatinine clearance: for men (140 – age) (weight in kg)/72 × serum creatinine; for women, this result is multiplied by the factor of 0.85.
Only 1 subject was severely renally impaired at baseline, and renal function worsened to severe status in 4 other subjects.
The first step in development of the current PPK model was selection of a base model without consideration of covariate effects, which was developed in a previous preliminary PPK analysis.21 Additional evaluations of stochastic models were conducted. Previous pooled analyses had shown that the PK profile of pevonedistat can be characterized with a 2‐compartment disposition model with linear elimination, also supported by visual inspection of log‐linear concentration–time plots. The base model included terms describing IIV on clearance (CL), central volume of distribution (Vc), distributional clearance (Q) and peripheral volume of distribution (Vp). The PPK model also included a covariance block that included covariance between CL, Q and Vp. This model was brought into the current model analysis as the best base. Examination of IIV vs IIV plots suggested strong correlation between Vc, Vp and Q, but not CL. This was supported by evaluations that included covariance between these parameters, while CL was not found to have strong correlation with the other PK parameters. The residual error was set to be an additive function (exponential when back transformed). There was a substantial drop in the OFV when a full covariance block was allowed between all 4 PK parameters. However, the condition number rose dramatically from 12.51 for the model with a diagonal OMEGA matrix to 99.93 with the inclusion of all 4 parameters in the covariance block, suggesting over‐parameterization. The OMEGA matrix was then simplified to remove CL from the full block, which converged well with acceptable parameter precision and a condition number of 16.38, which was acceptable. This model was selected as the base model for the combined dataset.
Then, the effects of demographic or physiological covariates were evaluated on the base PPK model. A full covariate model was developed by testing covariate‐parameter relationships graphically and as single‐covariate models. The effects of body size were important predictors of pevonedistat distribution volumes (Vc and Vp) and clearance (CL and Q) parameters. Albumin (ALB) was a significant covariate on Q only. The primary laboratory indicators of hepatic function (alanine aminotransferase [ALT], aspartate aminotransferase [AST], bilirubin [BILI]) tested over the range represented in the dataset did not explain variability in CL in the univariate testing. Renal function as determined by CrCL was also not found to affect pevonedistat CL. In addition, sex, race and age did not affect the PK parameters. Because the initial final model had high standard errors on the body surface area (BSA) effect factors, this model was simplified to allow the effect of BSA on CL and Q, and the effect of BSA on Vc and Vp to be the same. This reduced the model and improved standard errors but also resulted in only a 1‐point objective function change, suggesting minimal impact on model performance. The magnitude and consistency of the identified covariate effects were also examined using the bootstrap results. Vectors of bootstrapped parameters were used to compute the mean effects of identified covariates on CL, Vc, Q and Vp with 95% confidence interval and plotted along with the observed individual parameters. All the parameters exhibited at least a 20% change in parameter values over the observed BSA range in the present analysis dataset. Over the range of BSA (1.38–3 m2), CL ranges from 75.5 to 208% of the reference value.
The parameter estimates of the final model are shown in Table 3 along with the results of a nonparametric bootstrap. The parameter estimates were made relative to the hypothetical reference patient with a BSA of 1.73 m2 and ALB of 40 g/L, and not receiving concurrent carboplatin and paclitaxel. For the typical patient, this suggests an α phase (distribution) half‐life of 1.27 hours and a β‐phase (elimination) half‐life of 7.85 hours. The parameters are generally estimated with good precision (standard errors <20%). Shrinkage was low for CL (8.4%), and generally reasonable for Vc, Q and Vp (15, 20.5 and 10.9%, respectively). The condition number, calculated as the square root of the ratio of the highest to lowest eigenvalue, was 20.1, which is acceptable and suggests low collinearity of the parameters in the model. Goodness‐of‐fit plots stratified by dose (≤160 mg and > 160 mg, where 160 mg was the approximate median dose) are provided in Figure S1. VPCs were performed using the final PPK model and model parameters to simulate concentrations using the same sample times as were used in the clinical studies. The simulated concentration data were binned by time after dose to provide smoother curves. Median and upper and lower prediction intervals of simulated concentrations were determined within each of the various time intervals for each of the 1000 simulated datasets, and overall medians and confidence intervals were then computed and used to compare the predicted distributions to the observed dataset. The simulated and observed data were dose normalized to account for the wide range of doses in the dataset. Figure 1 is the VPC for the full analysis dataset. The observed data are generally well‐contained within the prediction intervals, and the data not contained within the prediction intervals appear to be equally distributed above and below these intervals. The observed medians and upper and lower percentiles agree well with the simulated data. The VPC plot shown in Figure 2 demonstrates that the PPK model was robust when the simulated data are compared with observed data from combination with azacitidine or carboplatin + paclitaxel. Based on evaluation of goodness‐of‐fit plots, shrinkage, condition number, parameter performance and VPC inspection, it was concluded that the model could simulate the observed data with acceptable accuracy and could therefore be used for the simulation of pevonedistat PK alone or when administered in combination with carboplatin + paclitaxel.
Table 3.
Final model parameters
| Parameter (units) | Typical value | SE (%CV) | Lower 2.5 percentile CI | Bootstrap median | Upper 97.5 percentile CI |
|---|---|---|---|---|---|
| CL (L/h) | 31.5 | 6.4 | 27.3 | 31.4 | 35 |
| BSA on CL a | 1.33 | 12.3 | 0.991 | 1.33 | 1.66 |
| Carboplatin + paclitaxel on CL | −0.441 | 8.8 | −0.51 | −0.437 | −0.352 |
| Q (L/h) | 21.9 | 6.6 | 19.7 | 22 | 24.6 |
| BSA on Q a | 1.33 | 12.3 | 0.991 | 1.33 | 1.66 |
| Albumin on Q | 1.45 | 34.7 | 0.603 | 1.45 | 2.3 |
| Vc (L) | 117 | 8.4 | 96.7 | 115 | 136 |
| BSA on Vc b | 1.39 | 12.5 | 1.05 | 1.39 | 1.72 |
| Vp (L) | 122 | 3.9 | 112 | 121 | 131 |
| BSA on Vp b | 1.39 | 12.5 | 1.05 | 1.39 | 1.72 |
| IIV_CL (%CV) | 36.6 | 20.8 | 21.7 | 35.9 | 55.9 |
| IIV_Vc(%CV) | 36.6 | 20.4 | 26.6 | 36.6 | 52 |
| IIV_Q (%CV) | 60.5 | 8.3 | 51.3 | 60.2 | 68.6 |
| IIV_Vp (%CV) | 36.9 | 6.3 | 32.2 | 36.7 | 41.5 |
| Corr (Vc, Q) | 0.691 | NE | 0.386 | 0.663 | 0.955 |
| Corr (Vc,Vp) | 0.941 | NE | 0.767 | 0.943 | 1.001 |
| Corr (Q,Vp) | 0.515 | NE | 0.36 | 0.524 | 0.676 |
| Residual error (%CV) | 33.2 | 6.4 | 29.2 | 32.9 | 36.8 |
BSA, body surface area; CI, confidence interval (2.5th and 97.5th percentiles); CL, clearance; Corr, correlation; CV, coefficient of variation; IIV, interindividual variability; Q, clearance of distribution; SE, standard error; Vc, central volume of distribution; Vp, peripheral volume of distribution;
Figure 1.
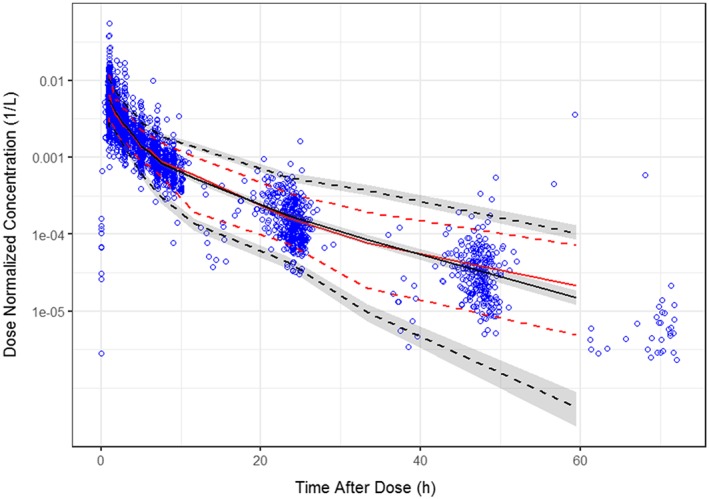
Visual predictive checks results for final model—observed and simulated pevonedistat dose‐normalized concentrations vs time after dose—all data. Blue open circles are the observed data; solid red line is the median of the observed data; red dashed lines are the 5th and 95th percentiles of the observed data; black solid line is the median of the simulated data; black dashed lines are the 5th and 95th percentiles of the simulated data; grey shaded areas are the 95% confidence intervals associated with the simulated lower and upper percentiles. The confidence intervals (CIs) are not computed beyond 60 hours as the number of observations is insufficient to determine confidence intervals
Figure 2.
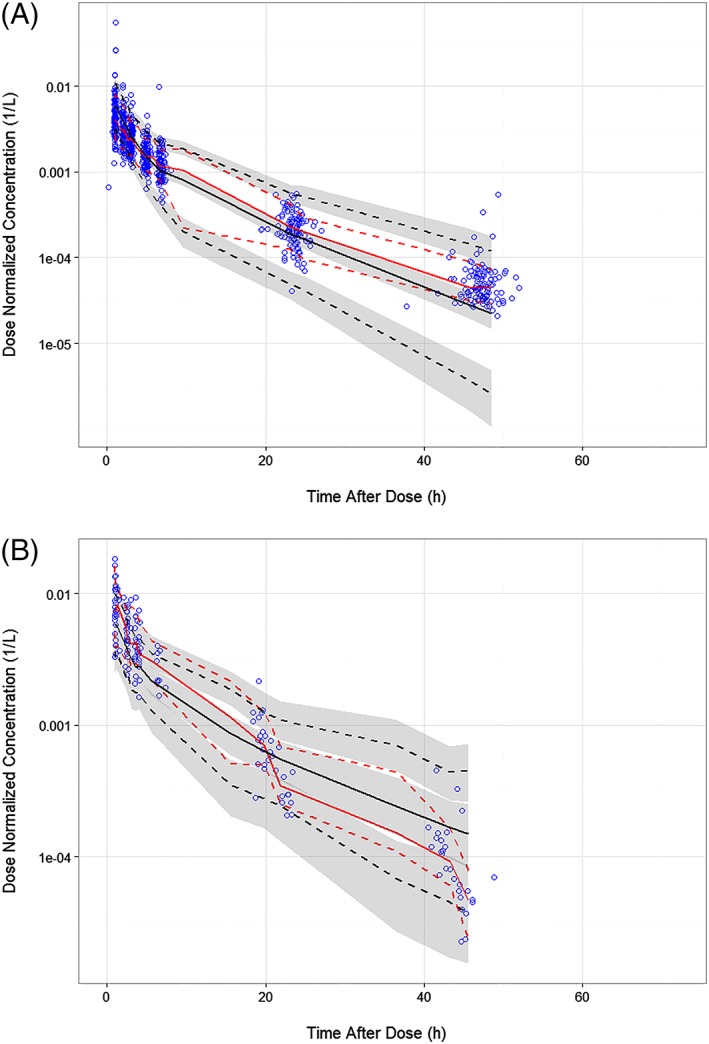
Visual predictive checks of dose‐normalized concentration–time data of pevonedistat coadministered with azacitidine (A) or carboplatin plus paclitaxel (B). Blue open circles are the observed data; solid red line is the median of the observed data; red dashed lines are the 5th and 95th percentiles of the observed data; black solid line is the median of the simulated data; black dashed lines are the 5th and 95th percentiles of the simulated data; grey shaded areas are the 95% confidence intervals associated with the simulated lower and upper percentiles
Panel A in Figure 3 shows the comparison of CL by patient sex. There was no evidence of any effect of patient sex on pevonedistat CL. Panel B shows the comparison of CL by age category, which also showed no trend. Panel C shows the impact of categorized CrCL on pevonedistat CL. CrCL appeared to have an effect on pevonedistat CL in the univariate covariate assessment, but was removed from the backward testing of the full model (P < .01). Panel D shows the comparison of CL between patients with normal and elevated AST. There were no apparent effects of elevated AST on pevonedistat CL. Panel E shows the impact of BSA on CL, and there was a positive correlation between increasing BSA and increasing CL. Panel F shows the impact of race on pevonedistat CL, which was not found to be a predictor. Panel G shows the comparison of pevonedistat CL between patients who were treated for solid tumours (studies 15001, 15005, 15010) against that of patients treated for haematological malignancies (studies 15002, 15003, 15009). Patients in Study 15010 who received pevonedistat with carboplatin + paclitaxel were excluded from this panel, since this combination was identified as having an independent effect upon pevonedistat CL. There was no significant difference in pevonedistat CL in patients treated for haematological vs solid tumours (haematological vs nonhaematological). Panel H shows the effect of concurrent chemotherapeutic agents on pevonedistat CL. Concomitant administration of carboplatin + paclitaxel was identified to impact CL and was retained in the backwards covariate elimination. The estimated covariate effect of −0.441 on CL with concurrent administration of carboplatin + paclitaxel shows that this combination therapy reduces pevonedistat CL by 44.1%, resulting in an average increase in the pevonedistat exposure (AUC) of 79% (1.79‐fold). An overlay plot of observed concentrations vs time after dose for patients dosed with pevonedistat and those who received pevonedistat and carboplatin + paclitaxel is provided in Figure 4. In this figure, the dose‐normalized pevonedistat concentrations with concomitant carboplatin + paclitaxel are high relative to the other observations. Concurrent gemcitabine was identified as a possible covariate in the initial univariate testing, but was subsequently eliminated in the stepwise removal of covariates. Azacitidine and docetaxel did not enter the PK model as covariates and it was concluded that these coadministered agents did not affect pevonedistat CL.
Figure 3.
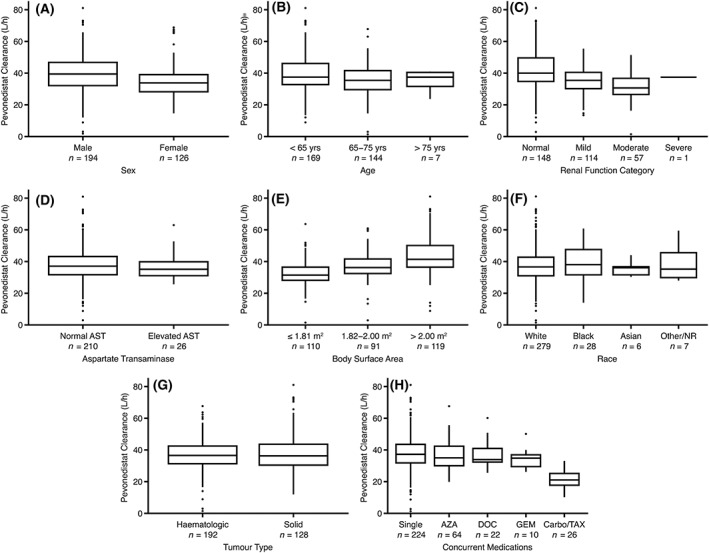
Pevonedistat clearance by patient covariates. Horizontal lines comprising the box are the 25th, 50th (median) and 75th percentiles. The whisker ends denote 1.5 times the difference between the 25th and 75th percentiles and the symbols beyond the whiskers are the outliers
Figure 4.
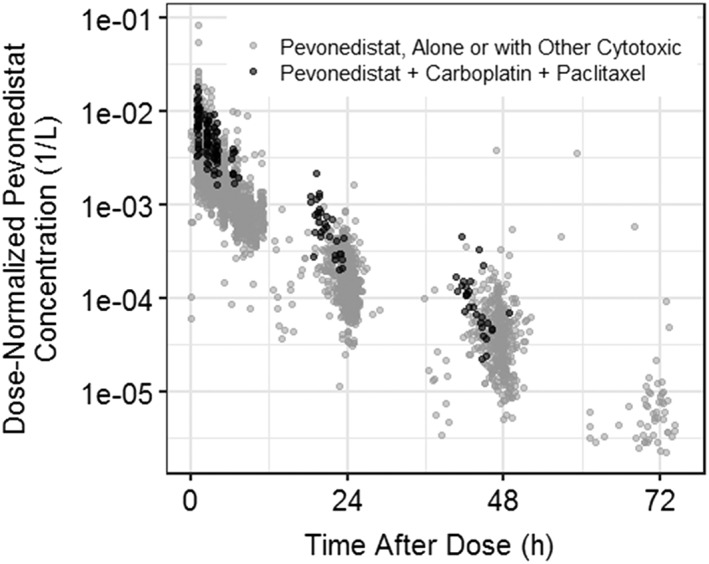
Pevonedistat concentration vs time with and without concomitant administration of carboplatin + paclitaxel
Pevonedistat is currently in the phase 2/3 clinical development for the treatment of haematological malignancies and the recommended clinical dose is 20 mg/m2 administered on days 1, 3 and 5 in combination with azacitidine in a 28‐day cycle. Given the identified impact of BSA on pevonedistat CL, simulations comparing the expected ranges of AUC following a dose of 20 mg/m2 vs a fixed dose of 34.6 mg (the dose that would be administered to a patient with a BSA of 1.73 m2) were performed and the results are presented in Figure 5. This figure shows that a dose of 20 mg/m2 provides uniform exposure across a wide range of body sizes, while a fixed dose results in low exposures for patients with high BSA, and high exposures for low BSA patients. Simulations of expected concentrations vs time following a 20 mg/m2 dose of pevonedistat on days 1, 3 and 5 are shown in Figure 6 (semi‐log scale in upper panel and linear scale in the lower panel), which shows little accumulation, consistent with the estimated terminal half‐life of approximately 8 hours.
Figure 5.
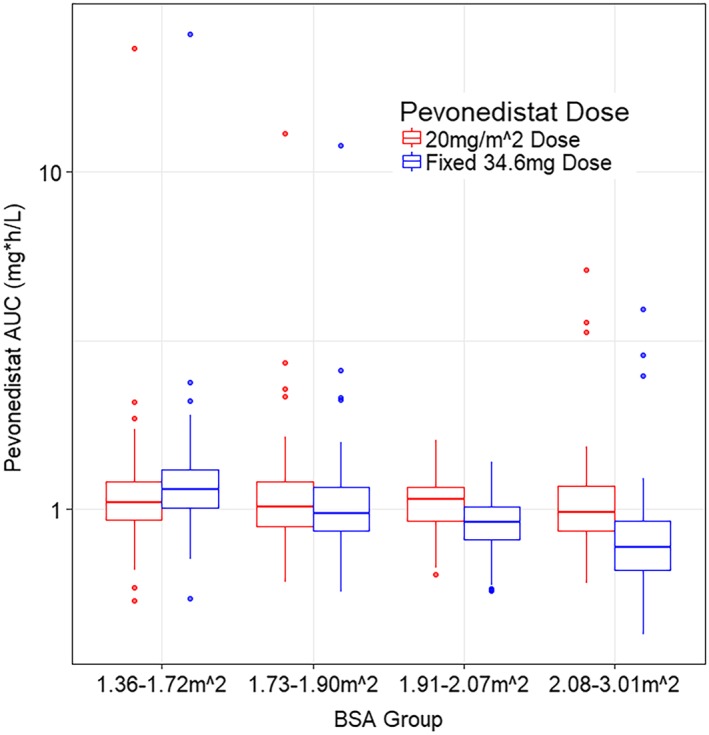
Simulated pevonedistat exposures following fixed (mg) vs BSA based dosing (mg/m2). AUC, area under the concentration–time curve; BSA, body surface area
Figure 6.
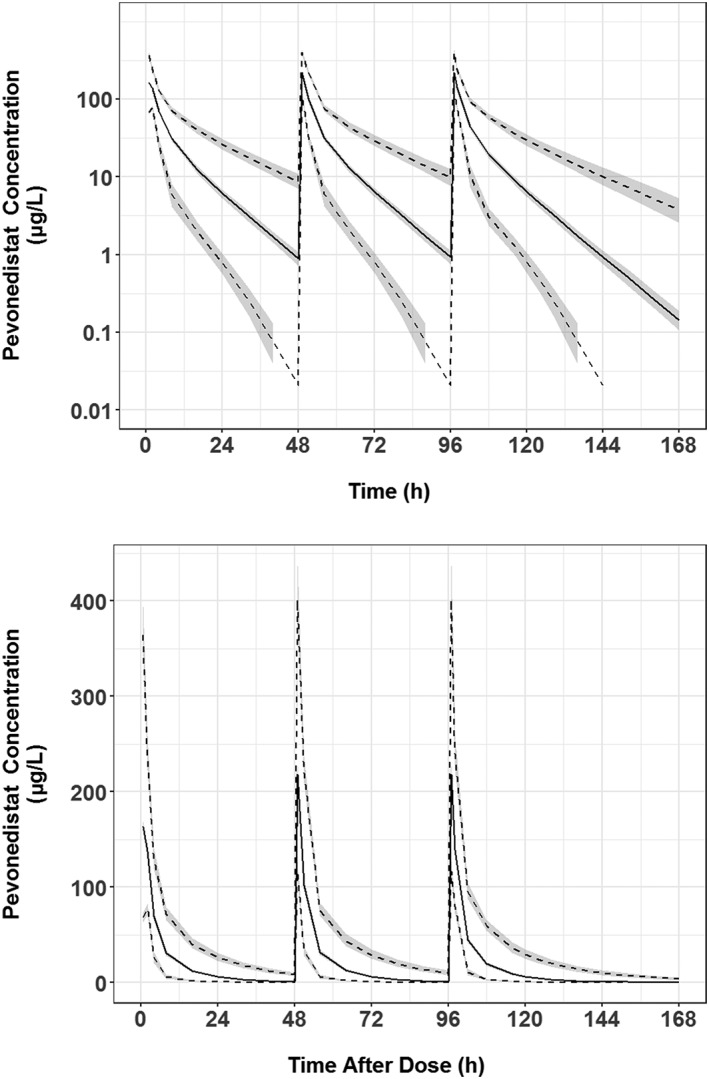
Simulated pevonedistat concentration–time profiles at the recommended clinical dose of 20 mg/m2 administered on days 1, 3 and 5. Note: Black solid line is the median simulated concentration; black dashed lines are the lower 5th and upper 95th percentiles of the simulated data; grey shaded areas are the 95% confidence intervals for each percentile
4. DISCUSSION
Novel treatments are needed for cancer patients considered unfit for conventional chemotherapy. Pevonedistat is an investigational, first‐in‐class, small‐molecule NAE inhibitor that is being studied for treatment of patients with solid or haematological malignancies. Characterization of the extent and sources of PK variability is a key objective in anticancer drug development, as it is important to inform posology across contexts of clinical use and patient‐specific factors.22 To this end, this analysis was conducted to identify the sources of variability in pevonedistat PK using a population modelling approach that provides a quantitative understanding of the impact of the covariate factors on drug CL, thereby supporting dosing decisions in clinical development. Data from 346 patients with solid tumours or haematological malignancies enrolled in 6 pevonedistat clinical studies contributed to this analysis. Pevonedistat PK were adequately described by a 2‐compartment PPK model with first‐order elimination. Effects of selected demographic and physiological covariate factors were evaluated.
4.1. Justification of BSA‐based dosing
The effects of BSA were important predictors of pevonedistat PK, affecting all parameters (CL, Vc, Q and Vp). Over the range of BSA in the present dataset (1.38–3 m2), CL ranges from 75.5% to 208% of the reference value. The effect of using a BSA‐adjusted dose of 20 mg m2 in patients of differing BSA ranges was evaluated through simulation of both BSA based dosing and flat dosing (Figure 5). Demographic factors from all the patients included in this PPK analysis, except those from Study C15010 who received concurrent carboplatin + paclitaxel, were used in this simulation. In each box and whisker plot, the expected AUC for BSA‐adjusted dose of 20 mg/m2 (blue box and points) is compared to the expected AUC if a flat pevonedistat dose of 36.6 mg was administered (red box and red crosses for outlier points). Dosing at 20 mg/m2 shows that the expected ranges of AUC values show a much narrower range of variability, from 107% of reference at the low end of the BSA range to 83.4% of reference at the high end of the BSA range. This suggests that the dosing of the drug should be adjusted to BSA to minimize differences in drug exposure (AUC) across a patient population of differing body size.
4.2. Pevonedistat in combination with other chemotherapy
Standard of care therapies (azacitidine, docetaxel, carboplatin + paclitaxel, or gemcitabine) were combined with pevonedistat in studies C15009 and C15010. The PPK model evaluated the effect of these agents on the PK of pevonedistat. Only the combination of carboplatin and paclitaxel was found to have a significant effect on pevonedistat PK by decreasing pevonedistat CL by 44% on average. Pevonedistat CL was unaffected by dosing in combination with azacitidine, docetaxel and gemcitabine.
The concomitant administration of pevonedistat with carboplatin and paclitaxel resulted in an average 44% reduction in pevonedistat CL in a typical individual. A dose reduction of approximately 45% would be expected to adjust the range of systemic exposures of pevonedistat when combined with carboplatin + paclitaxel to that observed when pevonedistat is administered as a single agent. Based on the dose‐proportional PK of pevonedistat, it can be inferred that pevonedistat systemic exposures at the maximum tolerated dose (MTD) of 20 mg/m2,10 in combination with carboplatin + paclitaxel on the days of coadministration, would be similar to exposures achieved as a single agent at 36 mg/m2, which is within the pharmacodynamically active range based on previous clinical experience where pharmacodynamic effects downstream of NAE inhibition were observed at ≥25 mg/m2 doses.7 The mechanism underlying this observed reduction of pevonedistat clearance by the combination of carboplatin and paclitaxel is currently unknown. From a safety perspective, it is to be noted that the MTD/RP2D of this combination was determined based on confirmation of suitability of 20 mg/m2 pevonedistat for multicycle administration in this combination setting. Accordingly, the observed effect of carboplatin plus paclitaxel on pevonedistat PK is not expected to be clinically relevant when administered at the RP2D determined in this combination setting. Neither carboplatin nor paclitaxel are established perpetrators of drug–drug interactions. The mechanisms of pevonedistat CL are currently under investigation. Although in vitro studies pointed to a potentially important role of CYP3A‐mediated metabolism, the results of clinical drug–drug interaction studies with the strong and moderate CYP3A inhibitors itraconazole and fluconazole, respectively, did not reveal clinically meaningful increases in pevonedistat systemic exposure.10 A radiolabelled mass balance study of pevonedistat has recently been completed (NCT 03057366) and is anticipated to further evaluate the CL mechanisms of pevonedistat, which will be important in further investigating potential mechanisms of the observed effect of carboplatin + paclitaxel on pevonedistat CL. Importantly, as the MTD and recommended Phase II dose of pevonedistat of 20 mg/m2 for investigational use in combination with carboplatin and paclitaxel has been qualified through clinical safety experience in the context of dosing over multiple treatment cycles,10 the clinical evaluation of this combination is nevertheless supported by the totality of safety and clinical pharmacokinetic experience.
4.3. Effects of renal and hepatic impairment
There were few patients with any abnormal liver function tests at baseline. Mildly abnormal liver function tests at baseline did not affect the clearance of pevonedistat. Decreased ALB concentrations are predicted to decrease Q but had no impact on systemic clearance. Over the range of ALB (20–50 g/L), Q varied from 8.01 to 30.27 L/h. Thus, ALB impacted the shape of the concentration time profile but did not impact AUC. None of the other laboratory values of liver function (ALT, AST, BILI) were influential covariates on pevonedistat PK over the range of values represented in the dataset. However, the dataset did not include representation of patients with BILI >1.5 times the upper limit of normal per protocol‐specified exclusion criteria, and as such this analysis does not allow conclusions regarding exposures of pevonedistat in patients with moderate or severe hepatic impairment.
The dose‐normalized concentration–time profiles for all renal function categories were comparable, demonstrating similar PK behaviour in patients with normal, mildly, or moderately impaired renal function. Renal function (CrCL range 26.3–301 mL/min) was also not identified as a significant covariate in the final model. During the univariate covariate assessment, CrCL met the prespecified forward selection criteria to be incorporated in the full model (P < .05), consistent with the observed trend for lower pevonedistat CL with lower CrCL from graphical inspection of the data. However, CrCL was removed from the full model during backward elimination (P < .01), confirming that CrCL could not explain more of the IIV of pevonedistat CL than was already described by BSA. The lack of effect of mild to moderate renal impairment on pevonedistat clearance is also consistent with expectations for a drug that is primarily cleared by hepatic metabolism. The renal elimination pathway played a minor role in the clearance of pevonedistat (2.5% of total plasma clearance) based on the recently completed clinical radiolabelled mass balance study (Takeda Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited data on file). However, it is to be noted that the effect of severe renal impairment on pevonedistat PK is currently unknown and warrants evaluation as severe renal impairment can potentially impact nonrenal clearance of metabolized drugs.23 Taken together, it can be concluded from this model‐based analysis that mild or moderate renal impairment did not impact pevonedistat clearance, such that no dose modifications should be required for pevonedistat in these specific patient populations based on PK considerations. It should, however, be noted that there were only 5 patients with severe renal impairment in the analysis dataset (one patient was classified as severe at baseline, 4 other patients were classified as severe later), and as such the present analysis does not permit a definitive assessment of the effect of severe renal impairment on pevonedistat PK. The observed lack of effect of mild or moderate renal impairment on pevonedistat PK is consistent with the lack of appreciable renal clearance of pevonedistat, with a < 5% contribution to total systemic clearance based on the clinical mass balance study ( Millennium Pharmaceuticals, Inc. data on file).
4.4. Effects of other covariates
Body weight was identified as an important predictor for pevonedistat clearance early in the covariate model development, but was supplanted by the normalized BSA from a practical standpoint, given that body weight and BSA are highly correlated and that BSA‐based dosing was used in the clinical development program as traditionally utilized for cytotoxic intravenously administered small molecule anticancer agents like pevonedistat. Furthermore, the selection of BSA over body weight as the covariate was also supported by the good control of PK variability across the patient population demonstrated via simulation with BSA‐based dosing (Figure 5). Age (range 23–90 years) was also not identified as being predictive of pevonedistat PK. Sex and race had no apparent effects on pevonedistat PK. Disease type (haematological vs nonhaematological tumour) also had no apparent effects on pevonedistat PK.
5. CONCLUSIONS
The PK pevonedistat in adult cancer patients was adequately described by a 2‐compartment model with first order elimination. BSA was an important predictor of all pevonedistat PK parameters (CL, Vc, Q and Vp). The impact of BSA on CL justifies the use of BSA‐based dosing. ALB was found to impact Q, but did not affect the systemic clearance or AUC of pevonedistat. Concurrent administration of carboplatin + paclitaxel decreased pevonedistat CL by approximately 44%. Coadministration with azacitidine, gemcitabine or docetaxel did not appear to affect the clearance of pevonedistat. Race, sex, age, tumour type (haematological vs solid), mild or moderate renal impairment (CrCL ≥30 mL/min), or mildly impaired liver function, to the extent represented in this dataset (i.e. total bilirubin ≤ upper limit of normal [ULN] and alanine aminotransferase [ALT]/aspartate aminotransferase [AST] ≤2.5 × ULN), had no impact on pevonedistat PK.
COMPETING INTERESTS
H.M.F., X.Z., D.V.F., F.S. and K.V. are employees of Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. D.R.M is a consultant for Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
CONTRIBUTORS
H.M.F., X.Z., D.F., F.S. and K.V. were responsible for evaluation conception and design. All authors were responsible for collection and assembly of data, and data analysis and interpretation. All authors contributed to the writing and critical revision of the manuscript, reviewed and approved the final version, and agreed to be accountable for all aspects of the work.
6.
Supporting information
FIGURE S1
Goodness‐of‐fit of final population pharmacokinetics model all data. Note: Dashed green line is a loess smooth, solid grey line is line of identity or unity, observations from doses <160 mg are presented as open red triangles, observations from doses >160 mg are presented as open blue circles
ACKNOWLEDGEMENTS
The authors thank all patients and their families, and the principal investigators and staff at study centres. The authors also acknowledge Projections Research Inc. for medical writing assistance. All clinical data were generated in studies funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Faessel HM, Mould DR, Zhou X, Faller DV, Sedarati F, Venkatakrishnan K. Population pharmacokinetics of pevonedistat alone or in combination with standard of care in patients with solid tumours or haematological malignancies. Br J Clin Pharmacol. 2019;85:2568–2579. 10.1111/bcp.14078
http://Clinicaltrials.gov identifiers: NCT00677170, NCT00722488, NCT0091106, NCT01011530, NCT01814826, NCT01862328.
Data Availability Statement:Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com/for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
DATA AVAILABILITY STATEMENT
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com/for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
REFERENCES
- 1. Reinstein E, Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann Intern Med. 2006;145(9):676‐684. [DOI] [PubMed] [Google Scholar]
- 2. Soucy TA, Smith PG, Rolfe M. Targeting NEDD8‐activated cullin‐RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15(12):3912‐3916. [DOI] [PubMed] [Google Scholar]
- 3. Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1(7):708‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8‐activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732‐736. [DOI] [PubMed] [Google Scholar]
- 5. Brownell JE, Sintchak MD, Gavin JM, et al. Substrate‐assisted inhibition of ubiquitin‐like protein‐activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8‐AMP mimetic in situ. Mol Cell. 2010;37(1):102‐111. [DOI] [PubMed] [Google Scholar]
- 6. Swords RT, Erba HP, Deangelo DJ, et al. Pevonedistat (MLN4924), a first‐in‐class NEDD8‐activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534‐543. [DOI] [PubMed] [Google Scholar]
- 7. Sarantopoulos J, Shapiro GI, Cohen RB, et al. Phase I study of the investigational NEDD8‐activating enzyme inhibitor pevonedistat (TAK‐924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res. 2016;22(4):847‐857. [DOI] [PubMed] [Google Scholar]
- 8. Shah JJ, Jakubowiak AJ, O'connor OA, et al. Phase I study of the novel investigational NEDD8‐activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22(1):34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhatia S, Pavlick AC, Boasberg P, et al. A phase I study of the investigational NEDD8‐activating enzyme inhibitor pevonedistat (TAK‐924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34(4):439‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lockhart AC, Bauer TM, Aggarwal C, et al. Phase Ib study of pevonedistat, a NEDD8‐activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs. 2019;37(1):87‐97. 10.1007/s10637-018-0610-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swords RT, Coutre S, Maris MB, et al. Pevonedistat, a first‐in‐class NEDD8‐activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood. 2018;131(13):1415‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu S‐J, Cheong J‐W, Onishi Y, Iida H, Kobayashi Y, Kim H‐J, et al. Phase 1/1b study of pevonedistat (PEV) as a single agent or combined with azacitidine (AZA) in east asian patients (pts) with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) EHA Learning Center 2018; Abstract PS997.
- 13. Bhatia S, Hamid O, Pavlick AC, et al. MLN4924, an investigational NEDD8‐activating enzyme (NAE) inhibitor, in patients (pts) with metastatic melanoma: results of a phase I study. J Clin Oncol. 2011;29(Suppl):Abstract 8529. [Google Scholar]
- 14. Swords RT, Savona MR, Maris MB, et al. Pevonedistat (MLN4924), an investigational, first‐in‐class NAE inhibitor, in combination with azacitidine in elderly patients with acute myeloid leukemia (AML) considered unfit for conventional chemotherapy: updated results from the phase 1 C15009 trial. Blood. 2014;124(21):Abstract 2313. [Google Scholar]
- 15. Boeckmann AJ, Bea LSL, Sheiner LB. In: NONMEM Users Guide ‐ Parts I to VIII , ed. NONMEM Installation Guide, NONMEM Project Group. San Francisco: University of California; March 1998. [Google Scholar]
- 16. Ortmann O, Weiss JM, Diedrich K. Gonadotrophin‐releasing hormone (GnRH) and GnRH agonists: mechanisms of action. Reprod Biomed Online. 2002;5(Suppl 1):1‐7. [DOI] [PubMed] [Google Scholar]
- 17. Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35(2):185‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yafune A, Ishiguro M. Bootstrap approach for constructing confidence intervals for population pharmacokinetic parameters. I: a use of bootstrap standard error. Stat Med. 1999;18(5):581‐599. [DOI] [PubMed] [Google Scholar]
- 19. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: enzymes. Br J Pharmacol. 2017;174:S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faessel HM, Mould DR, Dezube BJ, Santillana S, Venkatakrishnan K. Population pharmacokinetics (PK) of the investigational NEDD8‐activating enzyme (NAE) inhibitor pevonedistat (TAK‐924/MLN4924) administered alone or in combination with azacitidine in patients (pts) with solid or hematologic malignancies. Mol Cancer Ther. 2015;14(12 Suppl 2):Abstract B148. [Google Scholar]
- 22. Venkatakrishnan K, Friberg LE, Ouellet D, et al. Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin Pharmacol Ther. 2015;97(1):37‐54. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85(3):305‐311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1
Goodness‐of‐fit of final population pharmacokinetics model all data. Note: Dashed green line is a loess smooth, solid grey line is line of identity or unity, observations from doses <160 mg are presented as open red triangles, observations from doses >160 mg are presented as open blue circles
Data Availability Statement
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com/for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.