

Abstract
Aims
Type 1 diabetes can be complicated with neuropathy that involves immune‐mediated and inflammatory pathways. Glucagon‐like peptide‐1 receptor agonists such as liraglutide, have shown anti‐inflammatory properties, and thus we hypothesized that long‐term treatment with liraglutide induced diminished inflammation and thus improved neuronal function.
Methods
The study was a randomized, double‐blinded, placebo‐controlled trial of adults with type 1 diabetes and confirmed symmetrical polyneuropathy. They were randomly assigned (1:1) to receive either liraglutide or placebo. Titration was 6 weeks to 1.2–1.8 mg/d, continuing for 26 weeks. The primary endpoint was change in latency of early brain evoked potentials. Secondary endpoints were changes in proinflammatory cytokines, cortical evoked potential, autonomic function and peripheral neurophysiological testing.
Results
Thirty‐nine patients completed the study, of whom 19 received liraglutide. In comparison to placebo, liraglutide reduced interleukin‐6 (−22.6%; 95% confidence interval [CI]: −38.1, −3.2; P = .025) with concomitant numerical reductions in other proinflammatory cytokines. However neuronal function was unaltered at the central, autonomic or peripheral level. Treatment was associated with −3.38 kg (95% CI: −5.29, −1.48; P < .001] weight loss and a decrease in urine albumin/creatinine ratio (−40.2%; 95% CI: −60.6, −9.5; P = .02).
Conclusion
Hitherto, diabetic neuropathy has no cure. Speculations can be raised whether mechanism targeted treatment, e.g. lowering the systemic level of proinflammatory cytokines may lead to prevention or treatment of the neuroinflammatory component in early stages of diabetic neuropathy. If ever successful, this would serve as an example of how fundamental mechanistic principles are translated into clinical practice similar to those applied in the cardiovascular and nephrological clinic.
Keywords: anti‐inflammation, diabetic neuropathy, glucagon‐like peptide‐1 agonist, interleukin‐6, liraglutide
What is already known about this subject
There is no known cure to diabetic neuropathy, which may affect the central, autonomic or peripheral nervous systems and consequently reduces life expectancy and quality of life.
In preclinical trials, glucagon‐like peptide‐1 agonist treatment has shown anti‐inflammatory and neuroprotective effects, and thus possesses a justified treatment option in diabetic neuropathy.
Heightened levels of interleukin‐6 have been associated with reduced heart rate variability in type 1 diabetes, potentially indicating a specific importance of this cytokine in autonomic neuropathy.
What this study adds
Liraglutide elicited a reduction in interleukin‐6 in patients with long time type 1 diabetes and confirmed symmetrical polyneuropathy.
There was a numerical reduction in other cytokines indicating anti‐inflammatory actions of liraglutide, however no improvement was shown in neural function, plausibly because the polyneuropathy had reached a point beyond reversibility.
Clinically, liraglutide induced weight loss and improved microalbuminuria, confirming compliance to the study drug.
1. INTRODUCTION
Diabetes is a chronic metabolic disorder associated with chronic low‐grade inflammation, oxidative stress and changes in endoneurial integrity, which can result in several complications such as polyneuropathy. In type 1 diabetes, the pancreatic islet β cells are the target of an autoimmune response, which destroys the insulin producing capacity. Different cytokines have been associated with islet mononuclear infiltrate and cell toxicity.1 Systemic inflammation is typically quantified by assessing serum levels of proinflammatory cytokines such as tumour necrosis factor‐α (TNF‐α) and interleukin (IL)‐1 and IL‐6. Such concomitant inflammation has been associated with marked neuronal loss leading to widespread disturbances in neuronal function in neurodegenerative diseases e.g. in Alzheimer's disease.2 In addition, in vitro and in vivo studies provide convincing evidence that glucagon‐like peptide‐1 (GLP‐1) agonists possess anti‐inflammatory and antioxidative effects, putatively mediated alterations in monocyte function, diminished macrophage infiltrations and subsequent inhibition of proinflammatory pathways.3 Furthermore, preclinical studies of the effect of GLP‐1 agonists on the central nervous system have demonstrated beneficial effects on memory, synaptic brain plasticity and glucose metabolism.4 In addition, experimental animal studies have indicated that GLP‐ 1 agonists (exenatide) may also exert direct neuroprotective and neurotrophic effects independent of its glycaemic effects.5, 6, 7 For example, in diabetic and nondiabetic mice, GLP‐1 receptors are present on sensory neurons, axons, Schwann cells and dorsal root ganglia.
Diabetic polyneuropathies are common and present in approximately 50% of adults with long‐term diabetes, the pathogenesis of which is multifactorial including vascular, metabolic, immune mediated and inflammatory pathways.8 The clinical manifestations of polyneuropathy are pleomorphic and are associated with substantial socioeconomic burdens, and reduced quality of life. Polyneuropathy affects classically the peripheral axons in a length dependent manner leading to classical stocking‐and‐glove representation and is termed diabetic symmetric polyneuropathy (DSPN). These neuropathic changes coexists often with diabetic autonomic neuropathy and alterations of the structural and functional brain processing, which may be evident as a reduction in regional grey matter volume, microstructural damage within nerve tracts, and alterations in processing of somatosensory evoked potentials (SEPs).9, 10, 11 Despite a negative proof‐of‐concept study evaluating the effect of exenatide, a GLP‐1 agonist, on measures of DSPN and cardiac autonomic neuropathy (CAN) in patients with type 2 diabetes, it is, however, plausible that GLP‐1 agonists via an anti‐inflammatory mechanism could target the neuroinflammatory component of polyneuropathy, potentially leading to improved neuronal function of the central and peripheral nervous system in diabetes.12, 13
Therefore, we hypothesized that treatment with a GLP‐1 agonist, improves neuronal function through diminished inflammation in type 1 diabetes, independent of glucose metabolism. The primary objective was to explore the neuronal function in response to anti‐inflammatory actions of liraglutide treatment and thus the secondary aim was to investigate the effect on inflammatory parameters.
2. METHODS
2.1. Study population
We performed a prospective, randomized, double‐blinded, parallel‐group, placebo‐controlled trial at Aalborg University Hospital from June 2014 to January 2017. Patients were recruited at the Department of Endocrinology, and potential eligible patients were prescreened on the basis of a recorded vibration perception threshold above 18 V. All patients underwent nerve conduction tests to diagnose DSPN according to the Toronto criteria not more than 4 weeks prior to study entering.14 Prior to entering the study, prescribed medication e.g. type of antihypertensive treatment was registered and within the safety window it was intended to limit alterations in such medication throughout the study. Additional inclusion criteria were age over 18 years, a verified diagnosis of type 1 diabetes for a minimum of 2 years: (glycated haemoglobin [HbA1c] ≥6.5% [>48 mmol/mol]), stable hyperglycaemic medication insuring that patients as minimum had received the given treatment (long acting and fast acting insulin or insulin pump with dosing adjustments according to regimens) for at least 3 months prior to study entrance, body mass index > 22 kg/m2 and written consent. Exclusion criteria included type 2 diabetes, other neurological disorders than DSPN, estimated glomerular filtration rate < 60 mL/min/1.73 m2; calcitonin >25 ng/L, HbA1c level < 6.5%, use of GLP‐1 agonists or DPP‐4 inhibitors. Treatments were masked and appeared identical and were randomly assigned 1:1 liraglutide or placebo in blocks of 8, generated from a randomization list provided by the drug supplier. The intervention was titrated over a 6‐week period to a dose between 1.2–1.8 mg/d, depending on tolerability. The treatment was continued for a further 26 weeks. Dropouts/withdrawals were mirror randomized. Ethical approval was granted by Region Nordjylland, Denmark (N‐20130077) and all participants gave written informed consent prior to entry. The study was registered in public databases (EUDRA CT, ref 2013–004375‐12; and http://clinicaltrials.gov, ref NCT02138045) and performed in accordance with International Council for Harmonization Good Clinical Practice and the Declaration of Helsinki. All participants underwent multiple tests over a period of 3 study days at baseline. All experimental procedures were performed between 8 AM and noon. Follow‐up was performed after 26 weeks of intervention.
2.1.1. Central neuronal assessment—electrical stimulation and evoked potentials
Electrical stimulation was applied using surface electrodes (15 × 15 mm, Neuroline 700, Ambu A/S, Ballerup, Denmark) at the right median nerve (medial to the palmaris longus tendon and directly proximal to the right wrist) in order to elicit somatosensory evoked potential (SEP). A computer‐controlled electrical stimulator (Noxitest IES 230, Aalborg, Denmark) using bespoke software (LabVIEW, Custom made at Aalborg University, Denmark) delivered the electrical stimulus. The threshold at which thumb twitching was evident was found by slowly increasing the stimulus intensity in increments of 1 mA. Stimulus intensity was set at 1 mA above the twitch threshold and were delivered at 1 mA above the threshold in 2 trials of 1000 square pulses (0.2 ms, 2.3 Hz) at baseline and after 26 weeks of treatment at final dose.
The peripheral and spinal neuronal activity was recorded through surface Ag/AgCl electrodes (Neuroline 710; Ambu A/S, Ballerup, Denmark). The recording electrodes were located on the ipsilateral Erb's point and referenced to the contralateral Erb's point. Additionally, recording electrodes were located over the C7 cervical spinous process (Cv7) referenced to an electrode placed above the jugular notch but below the glottis. The skin was lightly scratched with sandpaper at the location of each electrode to remove dead skin cells, in order to ensure the best possible recordings and the electrode impedance was kept below 2 kΩ. The data were recorded continuously at 20 kHz (SynAmp, Neuroscan, El Paso, TX, USA) and stored offline for analysis. Electroencephalographic signals were recorded with the Neuroscan System (Version 4.5; Compumedics, Charlotte, NC, USA) from 62 surface cylindrical Ag/AgCl scalp electrodes, by use of the extended 10–20 system montage (Quik‐Cap International; Compumedics). All participants were seated in supine position with their legs supported and eyes open during the entire recording. Conductive electrode gel was applied in each electrode to reduce the impedance below 10 kΩ, which was monitored using proprietary software (Neuroscan, Version 4.3.1; Compumedics). The electroencephalograph was recorded as continuous files with open online filters with a sampling rate of 1 kHz and stored for further analysis of electrically elicited evoked potentials. Assessment of SEPs is a reliable objective method for following upstream activity from the periphery to the spinal cord, to the brainstem and finally to the cortex. The reference electrode was situated between AFz and Fz, and therefore the polarity is inverted (e.g. our N14 corresponds to is P14 in other work15). Early cortical activation was analysed at the negative potential (N20) and the positive potential (P22), at the centro‐parietal CP5 electrode contralateral to the stimulation, and the peak‐to‐peak amplitude was used. In a similar way the latencies and amplitude of the subcortical response (N14‐P18) was analysed at the occipital midline electrode (Oz), and late cortical response (N60‐P80) was assessed at the central midline electrode C1.16
2.1.2. Systemic biochemistry, inflammatory profile and macrophage function
Routine laboratory tests to monitor e.g. HbA1c, cholesterol and triglycerides were analysed at the Department of Clinical Biochemistry, Aalborg University Hospital. Fasting venous blood was collected and subsequently centrifuged at 800 g for 10 minutes at room temperature. Plasma was immediately frozen at −80°C. Levels of proinflammatory cytokines (IL1β, TNF‐α, IL‐6, IL‐8, IL‐10) were analysed by using a multiplex cytokine assay (Meso‐Scale Discovery, Rockville, MD, USA) and specific macrophage markers (sCD163 and sCD206) were analysed by enzyme‐linked immunosorbent assay at Department of Clinical Biochemistry at Aarhus University Hospital.
2.1.3. Autonomic neuronal assessment—cardiometric derived autonomic measures
Twenty‐four‐hour Holter monitoring electrocardiographic recordings were undertaken (Lifecard CF; Del Mar Reynolds, Spacelabs Healthcare Inc., Snoqualmie, WA, USA), according to internationally recommended standard.17 The initial recording period comprised a 10‐minute epoch where participants were instructed to relax followed by 2 × 15‐minute periods of guided respiratory rate of 15 breaths/min in the supine position followed by the same respiration rate in standing position. Blood pressure was simultaneously measured noninvasively in these 2 positions (Omron M4, Hoofddorp, Netherlands). Orthostatic hypotension was defined as >20 mmHg reduction in systolic blood pressure on standing. Twenty‐four‐hour heart rate variability (HRV) was assessed (Impresario Software version 3; Spacelabs Healthcare Inc., Snoqualmie, WA, USA), deriving HRV indices from within 5‐minute cycles of R‐R‐intervals. The following time domain HRV indices were obtained: standard deviation of the averages of NN intervals in all 5‐minute segments of a 24‐hour recording, reflecting autonomic imbalance (both sympathetic and parasympathetic tone); and the root mean square of difference of successive normal R‐R intervals reflecting parasympathetic tone.18 In addition, fast Fourier transformation provided the following frequency domain HRV indices: derived total power, very low frequency, low frequency, high frequency and the low/high frequency ratio (reflecting sympatico–vagal balance). All HRV indices were adjusted for baseline heart rate.19 Furthermore, validated measures of cardiac vagal tone and cardiac sensitivity to the baroreflex were derived.20 Both cardiac vagal tone and cardiac sensitivity to the baroreceptor are real time measures of efferent and afferent brainstem influence on the heart (Neuroscope, MediFit Instruments, Enfield, Essex, UK). The complementarity of these measures in diabetes are described in detail elsewhere.18
2.1.4. Peripheral neuronal assessment—peripheral nerve function
Nerve conduction studies was evaluated by use of standardized neurophysiological testing by trained neurophysiologists, according to American Association of Electrodiagnostic Medicine.21 Assessments of nerve conduction velocities, amplitudes and F‐waves were performed on the motor nerves (peroneal, tibial and ulnar nerves) and sensory nerves (sural, radial and median). To avoid influence of skin‐temperature on the conduction velocity, appropriate warming measures were used to ensure that the testing room did not allow skin temperatures below 32°C. Spring‐ring electrodes were used to record digital sensory nerve action potential. A plastic bar electrode was used for all other nerves. The results were processed according to reference values accepted by our EMG laboratories. To evaluate the severity of large fibre neuropathy, a composite score consisting of numerical ratings from 5 components of the sural, peroneal and tibial nerve assessment was used (range 0–15), where a total of 3 points indicate DSPN.22
Small fibre neuropathy was measured by a computerized Thermo Tester (TSA II NeuroSensory analyser; Medoc Ltd, Ramat Yishai, Israel). The temperature increased from a baseline of 32°C to a maximum of 52°C with increments of 1°C/s. The thermode was positioned on the skin on the right volar forearm, 10 cm proximal from the wrist. At pain tolerance threshold, the participants were told to press a button and the average of 3 successive stimulations were used for further analysis.
Loss of protective sensation was assessed by applying a standardized 26‐g monofilament perpendicular to the plantar side of the first toe and a reference area on the forearm. The size of the monofilament (gram) was noted at the pain detection threshold. If no pain was evoked at the maximum size, 300 g was notified for further calculation.
2.2. Study endpoints
The primary endpoint was the change in the early precortical N20 latency of the primary SEP elicited in response to electrical stimuli delivered to the median nerve before and after 26 weeks of placebo‐controlled intervention at the titrated dose.23 The secondary outcomes were changes in systemic proinflammatory cytokines (IL‐1β, TNF‐α, IL‐6, IL‐8 and IL‐10) and specific macrophage markers (sCD163 and sCD206), subcortical evoked potentials (N14) and late cortical potentials (N60), measures of autonomic function, and standardized neurophysiological testing. Tertiary outcomes were alterations in weight, HbA1c, heart rate, blood pressure and insulin utilization.
2.3. Statistics
The sample size was determined according to previously reported electrophysiological latencies from our laboratory. However, these results were not specific to liraglutide‐induced changes. This study was powered to detect a minimal difference of 1.0 standard deviation (SD) between liraglutide and placebo in the N20 latency and amplitude. With 80% power and a 2‐sided significance level of 0.05 the sample size needed to detect this change is 16 in each group or 32 patients in total, assuming a 20% SD in recorded latencies. Factoring in an attrition rate of 25% we set a sample size of 20 patients per group.
Patient characteristics are presented as means with SD or as medians with interquartile range (IQR) according to whether data followed the normal distribution or not. Group differences at baseline were assessed by Student t test and χ2 test for categorical variables.
Effects of liraglutide versus placebo were modelled by linear regression adjusting for baseline variables of study outcomes. To fulfil the requirement of a normal distribution of the model residuals, outcomes were log‐transformed when applicable. Consequently, estimates for these models are given in percentages. Where outcomes where not log transformed estimates are reported as absolute values. HRV indices were adjusted for resting heart rate at the time of testing. Statistical significance was inferred at a 2‐tailed P‐value <.05. Analyses were performed prior to unblinding, using SAS version 9.4 (SAS Institute, Cary, NC, USA).
3. RESULTS
In total, 602 patients with type 1 diabetes were identified in the electronic patient records; however, 488 did not meet the specific inclusion criteria (Figure 1).
Figure 1.
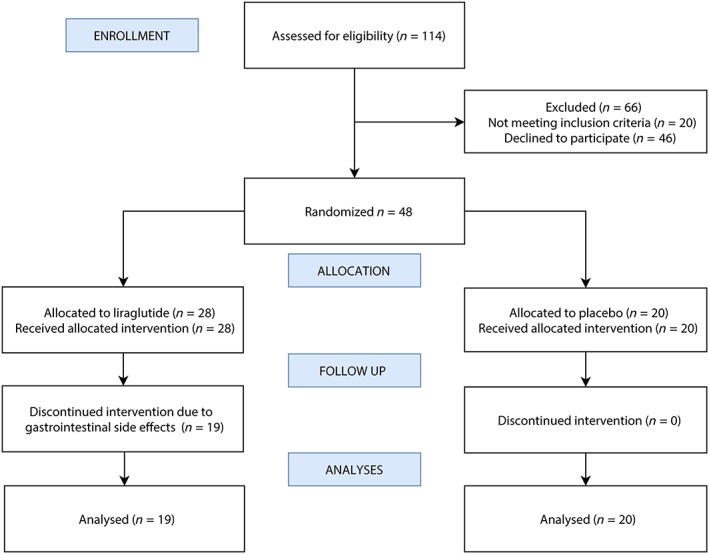
The consort flow chart shows the progress through this prospective, randomized, double‐blind, parallel‐group, placebo‐controlled trial
The study personnel assessed the 114 remaining patients for additional eligibility. Of these, 66 patients were excluded (n = 20, did not have abnormal nerve conduction velocities and n = 46 declined to participate). Ultimately, 48 patients were randomized, 9 of whom withdrew from the study due to adverse effects (7/9: severe nausea; 5/9: vomiting; 3/9: reflux; 3/9: decreased appetite). In total 19 patients randomized to liraglutide and 20 to placebo completed the trial. Overall, 80% of patients were male, had a mean (SD) age of 50.4 years (8.6), a diabetes duration of 32.4 years (9.3), a HbA1c of 66 mmol/mol (IQR 58–73) ~8.2% (IQR 7.5–8.8) and 35 patients (90%) had orthostatic hypotension. The baseline characteristics of the 2 study groups are shown in Table 1. No between group differences were identified in the clinical evaluation of severity of DSPN, conduction velocities or amplitudes. Moreover, no differences between groups were shown in baseline characteristics including sex distribution, age, HbA1c, body mass index, diabetes duration, blood pressure, kidney function, cholesterol, smoking habits or use of medication except intermediate‐acting insulin and insulin pump therapy.
Table 1.
Baseline characteristics
| Liraglutide (n = 19) | Placebo (n = 20) | P‐values (group difference) | |
|---|---|---|---|
| Demographics | |||
| Sex (male) | 17/90 | 14/70 | .121 |
| Age (y) | 51 (10) | 50 (8) | .577 |
| Weight (kg) | 93 (17) | 92 (17) | .945 |
| Body mass index (kg/m2) | 29 (4) | 29 (5) | .779 |
| Diabetes duration (y), range | 31 (24–43) | 32 (26–40) | .963 |
| Regular smoking | 4/25 | 4/22 | .849 |
| Distal symmetrical polyneuropathy | |||
| Sural nerve conduction velocity (m/s) | 42 (41–44) | 43 (39–47) | .914 |
| Sural nerve amplitude (mV) | 2.2 (1.7–3) | 3.0 (1.8–4.0) | .821 |
| Tibial nerve conduction velocity (m/s) | 42 (35–45) | 39 (35–43) | .243 |
| Tibial nerve amplitude (mV) | 3.3 (1.6–4.7) | 1.2 (0.8–1.7) | .255 |
| Median nerve conduction velocity (m/s) | 51 (47–53) | 49 (46–51) | .365 |
| Median nerve amplitude (mV) | 9.0 (7.8–10.2) | 8.3 (7.3:9.3) | .342 |
| Severity neuropathy composite score | 9.7 (7.1–12.2) | 8.7 (6.5–10.8) | .523 |
| Michigan neuropathy screening instrument | 2.8 (1.9–3.7) | 2.9 (1.7:4.0) | .933 |
| Biothesiometry (V) | 38.3 (13.8) | 34.2 (13.7) | .350 |
| Thermal pain tolerance threshold (°C) | 48.3 (2.2) | 46.6 (4.3) | .132 |
| Monofilament size to evoke pain (g) | 271 (20) | 294 (6) | .267 |
| Cardiac derived parameters | |||
| Systolic blood pressure (mmHg) | 153 (16) | 150 (16) | .475 |
| Diastolic blood pressure (mmHg) | 84 (13) | 84 (10) | .851 |
| Orthostatic hypotension | 18/95 | 17/85 | .310 |
| Clinical biochemistry | |||
| eGFR (CKD‐epi) | 85 (65–90) | 85 (72–90) | .867 |
| HbA1c (mmol/mol) | 69 (59–80) | 63 (57–71) | .122 |
| HbA1c (%) | 8.5 (7.5–9.5) | 7.9 (7.3–8.6) | .122 |
| Urinary albumin excretion rate (mg/24‐hour) | 78 (23–554) | 33 (27–235) | .413 |
| Total cholesterol (mmol/L) | 4.4 (0.8) | 4.6 (0.6) | .493 |
| HDL cholesterol (mmol/L) | 1.5 (0.5) | 1.6 (0.4) | .541 |
| LDL cholesterol (mmol/L) | 2.4 (0.6) | 2.5 (0.5) | .248 |
| Triglycerides (mmol/L) | 0.8 (0.7–1.2) | 0.9 (0.7–1.1) | .431 |
| Medication | |||
| Fast‐acting insulin | 17/90 | 17/85 | .675 |
| Intermediate‐acting insulin | 3/16 | 0/0 | n/a |
| Long‐acting insulin | 9/47 | 9/45 | .882 |
| Insulin pump therapy | 1/5 | 6/30 | .034 |
| Statins | 7/37 | 10/50 | .403 |
| Diuretics | 3/16 | 3/15 | .946 |
| Beta blocker | 4/21 | 2/10 | .333 |
| RAAS blockade | 10/53 | 8/40 | .425 |
Data are means (standard deviation), median (interquartile range) or n/%. eGFR = estimated glomerular filtration rate (mL/min/1.73 m2); RAAS = renin angiotensin aldosterone system; HbA1c = glycated haemoglobin.
3.1. Primary study endpoint
3.1.1. Central neuronal assessment—evoked potentials
Twenty‐six weeks of intervention with liraglutide did not demonstrate changes in early precortical N‐20 latency from electrically evoked brain potentials with a difference of −1.5% (95% confidence interval [CI]: −10.0, 8.0), see Figure 2 and Supplementary Table S1.
Figure 2.
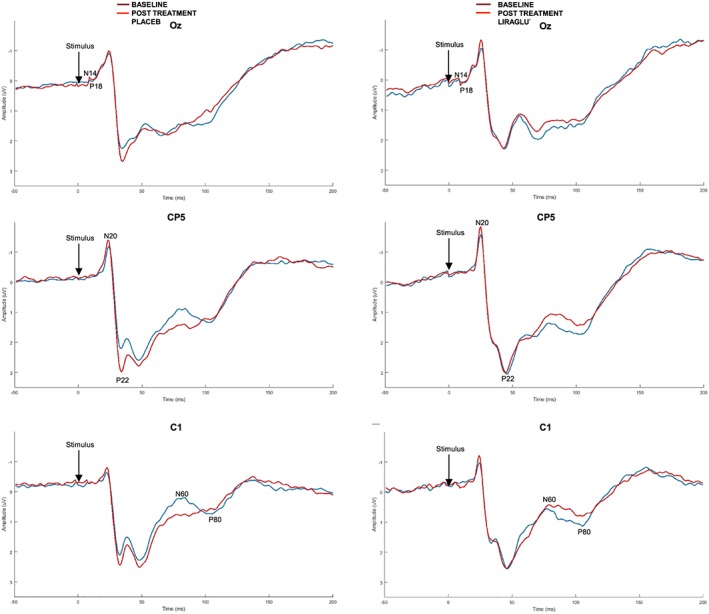
Grand mean averages of the cortical evoked potentials from 3 different electrodes, representing the subcortical activation (Oz), the early cortical activation (CP5) and the late cortical activation (C1). It can be seen that there is no difference between liraglutide and placebo in latencies and amplitudes
3.2. Secondary endpoints
3.2.1. Inflammatory profile
Liraglutide significantly reduced IL‐6 (−22.6% (95%CI −38.1, −3.2) compared to placebo with a numerical reduction in other proinflammatory cytokines and markers of macrophage function (Figure 3 and Table 2A).
Figure 3.
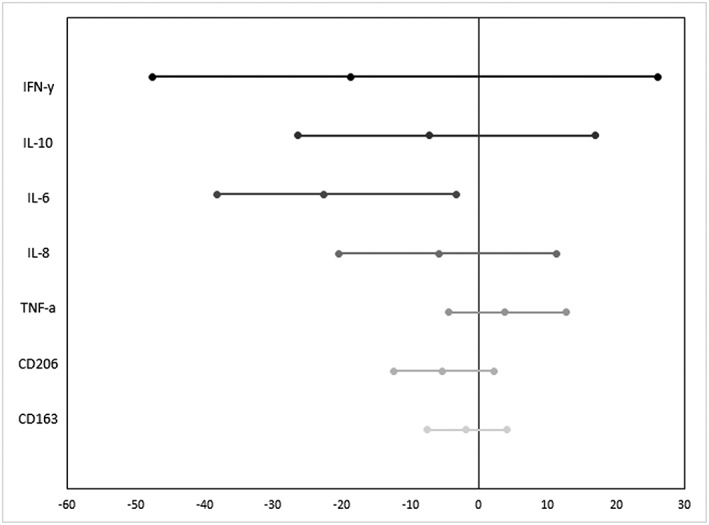
Liraglutide reduced systemic proinflammatory markers, here shown as relative differences in comparison to placebo. The reduction reached significance for interleukin (IL)‐6; however, a similar numerical trend was present for other proinflammatory cytokines, suggesting a potential systemic anti‐inflammatory effect of the glucagon‐like peptide‐1‐agonist. The error bars illustrate the 95% confidence intervals. IFN = interferon; TNF = tumour necrosis factor
Table 2A.
Inflammatory profile
| Randomization | Week 26 group differences | P‐value | ||||
|---|---|---|---|---|---|---|
| Liraglutide | Placebo | Liraglutide | Placebo | Liraglutide vs placebo | ||
| IFN‐y | 11.6 (7.6, 21.55) | 7.81 (5.42, 16.45) | 7.67 (5.96, 13.8) | 9.47 (5.21, 12.6) | −18.7% (−47.6, 26.1) | .354 |
| IL‐10 | 0.49 (0.31, 0.64) | 0.39 (0.35, 0.59) | 0.38 (0.32, 0.63) | 0.41 (0.3, 0.63) | −7.2% (−26.4, 17.0) | .528 |
| IL‐6 | 1.18 (0.73, 2.06) | 0.94 (0.79, 1.45) | 0.84 (0.64, 1.12) | 1.00 (0.79, 1.44) | −22.6% (−38.1, −3.2) | .025 |
| IL‐8 | 13.6 (11.85, 16.8) | 13.43 (11.43, 17.65) | 14.35 (10.25, 16.1) | 14.35 (12, 20.35) | −5.8% (−20.3, 11.3) | .483 |
| TNF‐a | 3.27 (3, 4.01) | 2.91 (2.67, 3.58) | 3.21 (2.95, 3.87) | 3.01 (2.64, 3.42) | 3.8% (−4.4, 12.8) | .369 |
| CD206 | 0.3 (0.23, 0.33) | 0.21 (0.18, 0.26) | 0.27 (0.25, 0.35) | 0.21 (0.19, 0.25) | −5.3% (−12.3, 2.3) | .167 |
| CD163 | 2.03 (1.63, 2.81) | 1.88 (1.5, 2.51) | 1.88 (1.6, 2.56) | 1.87 (1.58, 2.39) | −1.8% (−7.5, 4.1) | .536 |
The proinflammatory cytokines and macrophages markers in response 26 weeks of liraglutide treatment are secondary endpoints. Data are expressed as mean (standard deviation) or median and (interquartile range). Estimates of test for treatment effect are in % or absolute values (95%CL) [P values for group difference]. IFN = interferon; IL = interleukin; TNF = tumour necrosis factor; UACR = urine albumin/creatinine ratio; eGFR = estimated glomerular filtration rate. Concentration for IFN‐γ, IL‐10, IL‐6, IL‐8 and TNF‐α are pg/mL. Concentration of CD206 and CD163 are mg/L.
3.3. Central neuronal assessment—evoked potentials
There were no differences in either latency or amplitude in the upstream activation of the brain across a number of levels including sub‐, early‐ and late‐cortical activation, see Figure 2 and supplementary material.
3.4. Autonomic neuronal assessment—cardiometric derived autonomic measures
Liraglutide treatment did not elicit any alterations in cardiometrically derived autonomic measures compared to placebo (Supplementary material).
3.5. Peripheral neuronal assessment—peripheral nerve function
Liraglutide treatment was not associated with changes in peripheral nerve function, (Table 2B).
Table 2B.
Inflammatory profile adjusted for confounding
| Controlled for weight | Controlled for HbA1C | Controlled for UACR | Controlled for eGFR | |||||
|---|---|---|---|---|---|---|---|---|
| Group difference** | P‐value** | Group difference* | P‐value* | Group difference** | P‐value** | Group difference | P‐value* | |
| IFN‐y | 7.0% (−33.3, 71.6) | .780 | −21.3% (−49.2, 22.0) | .284 | −22.8% (−51.1, 21.9) | .267 | −20.1% (−48.6, 24.1) | .317 |
| IL‐10 | −2.4% (−25.0, 27.) | .857 | −9.9% (−28.6, 13.5) | .376 | −8.2% (−28.3, 17.4) | .495 | −8.4% (−27.7, 15.9) | .465 |
| IL‐6 | −18.0% (−36.4, 5.7) | .125 | −25.7% (−39.7, –8.4) | .005 | −24.1% (−40, −4.1) | .021 | −23.8% (−39.2, −4.5) | .018 |
| IL‐8 | −2.5% (−19.4, 18.1) | .799 | −7.9% (−22.2, 9.1) | .343 | −4.6% (−19.9, 13.6) | .595 | −8.1% (−22, 8.2) | .308 |
| TNF‐a | 9.1% (−0.4, 19.4) | .060 | 3.4% (−4.5, 12.0) | .408 | 3.9% (−4.3, 12.8) | .366 | 3.2% (−4.6, 11.7) | .431 |
| CD206 | −2.0% (−10.1, 6.8) | .639 | −5.7% (−12.7, 1.8) | .135 | −0.9% (−6.6, 5.1) | .755 | −2.2% (−7.8, 3.8) | .465 |
| CD163 | 1.3% (−5.0, 7.9) | .699 | −1.9% (−7.6, 4.1) | .525 | −3.6% (−10.7, 4) | .340 | −5.6% (−12.5, 1.8) | .137 |
Models have been adjusted for baseline values of the given outcome, and adjusted for: (i) change in weight; (ii) change in HbA1C; (iii) change in UACR; and (iv) change in eGFR during trial. IFN = interferon; IL = interleukin; TNF = tumour necrosis factor; UACR = urine albumin/creatinine ratio.
*P < 0.05; **P < 0.01.
Liraglutide treatment resulted in a weight reduction of 3.38 kg (95%CI −5.29, −1.48, P < .001) when compared to placebo regardless of any effect on HbA1c or total insulin use. Furthermore liraglutide reduced urine albumin‐to‐creatinine ratio by 40.3% (95%CI: −60.6, −9.5, P = .015; for more details see Table 3).
Table 3.
Clinical outcome
| Randomization | Week 26 group differences | P‐value | ||||
|---|---|---|---|---|---|---|
| Liraglutide | Placebo | Liraglutide | Placebo | Liraglutide vs placebo | ||
| Weight (kg) | 92.5 (16.5) | 92.1 (17.2) | 88.5 (17.1) | 91.6 (16.1) | −3.38 (−5.29, −1.48) | <.001 |
| HbA1c (mmol/mol) | 69.0 (59.0, 80.0) | 63.0 (56.5, 71.0) | 72.0 (66.0, 74.) | 66.0 (60.0, 73.5) | 1.18% (−4.31, 6.98) | .681 |
| Total insulin use (IU/d) | 55.0 (44.0, 82.0) | 44.0 (390.0, 68.0) | 50.0 (39.0, 73.0) | 43.0 (38.5, 63.0) | −1.57% (−18.14, 18.34) | .866 |
| UACR | 17.99 (7.55, 53.88) | 8.5 (4.42, 13.87) | 12.41 (8.91, 25.24 | 8.53 (6.5, 14.8) | −40.25% (−60.57, −9.48) | .015 |
| eGFR | 85 (65, 90) | 85 (71.5, 90) | 90 (63, 90) | 90 (71, 90) | −1.49% (−6.78, 4.11) | .595 |
Tertiary endpoints, showing the data are median (interquartile range). Estimates of test for treatment effect are in % or absolute values (95% confidence interval); P‐values for group difference. Models have been adjusted for baseline values of the given outcome. UACR = urine albumin/creatinine ratio; eGFR = estimated glomerular filtration rate; HbA1c = glycated haemoglobin.
4. DISCUSSION
Twenty‐six weeks of liraglutide treatment reduced the systemic level of the proinflammatory cytokine IL‐6, and a similar numerical trend for other proinflammatory cytokines was found, suggesting a potential systemic anti‐inflammatory effect. The liraglutide induced in average 3 kg weight loss and reduced urine albumin‐to‐creatinine ratio by 40%. Changes in IL‐6 were interestingly independent of glycaemic control as well as renal function, however associated to the weight loss. No treatment group‐differences with regards to improvement or decline in any electrophysiological measures to assess the neuronal function was shown.
To the best of our knowledge, this is the first time that a randomized placebo‐controlled study has shown significant decrease in IL‐6 in response to liraglutide treatment in humans. Although IL‐6 primarily is regarded as a proinflammatory cytokine, its actions are not limited to the immune system and its regenerative properties are increasingly recognized including neuronal differentiation and regeneration, metabolic processes, and liver regeneration.24 The current findings indicate a GLP‐1‐mediated reduction in macrophage infiltration and inhibition of inflammatory pathways, although it is not clear why there was a preferential effect on IL‐6 rather than other proinflammatory cytokines. It is known that macrophages residing in adipose tissue are the major sources for elevated plasma IL‐6 in obesity, and therefore the observed reduction in IL‐6 seems directly influenced by the weight loss itself.25 Nevertheless, heightened levels of IL‐6 have been associated with reduced heart rate variability in type 1 diabetes, potentially indicating a specific importance of this cytokine in autonomic neuropathy.26 Furthermore, elevated levels of IL‐6 have been identified as an independent predictor of type 2 diabetes and associated cardiovascular events.27, 28 In addition, it has recently been suggested that the inflammatory cascade from CRP to IL‐6 provides a novel therapeutic opportunity of atheroprotection by targeting the central IL‐6 signalling system and thus ultimately inhibits the IL‐1β producing inflammasome.29 Concomitantly with the reduction in IL‐6 were numerical trends present in other cytokines. This is in line with the presence of GLP‐1 receptors demonstrated on monocytes/macrophages and supports the reported 12% reduction of TNF‐α in liraglutide treated patients with type 2 diabetes and microalbuminuria.30 The shown reduction of microalbuminuria in the liraglutide treated group is in accordance with previous data from the larger outcome studies.31, 32 In addition, recent findings advocate that the IL‐6 pathway seems overactive in 40% of type 1 diabetes patients and that this previously has been implicated in the initiation and progression of microalbuminuria.33 In that context, our data show a concomitantly reduction in IL‐6 and microalbuminuria in type 1 diabetes and confirmed DSPN and thus may contribute to a mechanistic understanding of the liraglutide induced nephroprotection.
The pathogenesis of diabetic neuropathies is complex and poorly understood, involving vascular, metabolic, immune‐mediated and inflammatory pathways, which taken together leads to ischaemia, oxidative stress, nonenzymatic glycation of neural structures and heightened inflammatory response.8 The microglia produces proinflammatory cytokines such as IL‐6 and free radicals, which have been shown to be neurotoxic in e.g. Alzheimer's disease.34 Furthermore, increased levels of TNF‐α has been correlated to heart rate variability and CAN,35 and are speculated to play a pathogenic role in the development and maintenance of diabetic neuropathy.36 It is therefore intriguing to target the anti‐inflammatory component of neurodegeneration,37 which is emphasized by the promising results in Parkinson's disease, where activation of the GLP‐1 axis led to positive effects on practically defined off‐medication motor scores.38 The neuronal damage in diabetes is arguably being the final cumulative downstream effect and thus subtle changes in neuronal function may not be detectable with current techniques.
To investigate the anti‐inflammatory effect of liraglutide on peripheral and central neuronal function, we used the latency of the primary SEPs to median nerve stimulation—the N20 peak—as the primary outcome, because it the most consistent peak reaching the pre‐cortical level, encompassing neuronal transduction, transmission, neuronal communication and central synaptic plasticity. Furthermore, this peak also shows low latency variability between subjects. In addition, the absence of the N20 component is considered the most reliable indicator of unfavourable prognosis in stroke and post‐cardiopulmonary arrest. To investigate where and when the anti‐inflammatory actions influence the neuronal communication within the neuroaxis, detailed neurophysiological information is needed. However, the data in this study do not support a differentiated interpretation. Given that the severity score ranged from 3–15, we postulate that some of the patients had late‐stage neuropathy characterized by de‐myelinization and axonal loss beyond the point of possible reversibility, which may have hampered the assessment. We cannot explain the over‐representation of men, as the literature report ambiguous results on the influence of sex on development of DSPN39, 40; however, the onset of DSPN is earlier in men plausibly due to lifestyle and depletion of androgens, which exerts specific neuroprotective effects.41, 42 Therefore, the skewed sex distribution rather represents selection bias, possibly because men have favoured participating in a resource‐demanding study with many applied technologies.
Similarly, liraglutide failed to have any beneficial effect on the secondary outcomes of cardiometrically derived parameters of autonomic function and standardized neurophysiological peripheral nerve testing. Our HRV results repeated after 26 weeks are in contrast to the findings of a previous studies, which demonstrated a reduction in HRV in type 2 diabetic patients treated for 12 weeks with liraglutide.43 It was proposed that the reduction in HRV was secondary to GLP‐1 receptor activation by liraglutide in the sinoatrial node, resulting in a more chronometric heart rhythm.43 However, reduced HRV has been shown to be associated with altered central processing within the operculum–insular network, underlining the systemic influence of diabetic neuropathy.9 This discordance in findings may reflect differences in diabetic disease phenotype and study design. In addition, patient enrolled in the present study may suffer from manifest and irreversible CAN as 35/39 participants were diagnosed with orthostatic hypotension, suggesting co‐existence of severe CAN. This finding is in accordance with a similar trial performed in adults with type 2 diabetes and mild neuropathy.13 Finally, the tertiary outcomes showed that liraglutide in comparison to placebo induced weight loss, which confirms the drug compliance. Weight loss is considered as anti‐inflammatory in itself, and it has previously been shown that substantially weight reduction in morbidly obese patients induces a significant decrease in IL‐6, whereas TNF‐α remains unaltered.44 In this study, the average weight loss was only approximately 3 kg; however, this reduction seemed associated with the reduction in IL‐6. The novel aspect of this study was the utilization of a broad range of techniques to evaluate the effect of liraglutide in a multi‐level neurophysiological model. Taken together, liraglutide treatment consistently failed to demonstrate any reversibility or attenuation of progression of polyneuropathy in this cohort but then again, patients were recruited based on severe DSPN, which may not be at a reversible state.
There are several limitations to our study. First, the extern validity or generalizability to other phenotypes of diabetes is unclear and with a relatively short intervention of 26 weeks the study design does not allow for differentiating between the drug effect in either the short or long term. Consequently, there is a risk of obscuring a real positive long‐term neuroprotective effect of liraglutide. If that is the case, the conclusion of the paper has major implications, both generally (publication bias in preclinical trials) and specifically on the future studies of diabetic neuropathy. Second, these patients had in average–long duration (>31 years) of diabetes, which was present prior to guidelines advocating for intensive glycaemic control and therefore longstanding and more severe neuropathy may reasonably be expected in this subpopulation. Even though experimental evidence indicates that GLP‐1 agonists may have direct neurotrophic effects, we acknowledge that the design of including adults with established neuropathy, expectedly represents disease progression where neuronal changes are at irreversible state. Third, choice of measuring the net neuronal function by the use of SEPs and anticipating a clinical difference of a standard deviation may have opposed our hypothesis, and have obscured a true effect (Type I error). Fourth, this study did not investigate neuroregenerative or neuroprotective response to liraglutide, as for instance investigating thin fibre regeneration, density or morphology in skin biopsies or corneal confocal microscopy. These measures could, however, in contrast to the current study, yield the potential to identify the warranted anti‐inflammatory effect in early stage neuropathy. Fifth, the plasma levels of cytokines are continuously influenced by different cascades of signalling transduction and thus the plasma levels are susceptible to changes in glycaemic control, metabolism and medication. However severe events including major infections and hospitalizations are ruled out due to systematic reports of adverse events. Finally, despite of an overall goal of optimizing treatment, all participants in this study had relatively high levels of HbA1c in both placebo‐ and liraglutide‐treated patients. Such hyperglycaemic levels are, in themselves, proinflammatory and thus could counteract a liraglutide‐induced anti‐inflammatory effect.
5. CONCLUSION
Liraglutide reduced IL‐6 but did not improve neuronal function, possibly because the polyneuropathy had reached a point beyond reversibility. Hitherto, diabetic neuropathy has no cure. Thus, speculations can be raised whether mechanism targeted treatment, e.g. lowering the systemic level of proinflammatory cytokines may lead to prevention or treatment of the neuroinflammatory component in early stages of diabetic neuropathy. If ever successful, this would serve as an example of how fundamental mechanistic principles are translated into clinical practice similar to those applied in the cardiovascular and nephrological clinic for the benefit of future patients.
COMPETING INTERESTS
The study sponsor was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication.
CONTRIBUTORS
Study design and original idea: C.B., A.M.D., H.H.L. and B.B. C.B., P.E.J., A.J. and J.K. collected data. C.B., C.S.H., S.R., A.J., H.J.M., A.M.D. and B.B. analysed and interpreted the data. C.B., C.S.H. and B.B. wrote first draft, but all authors contributed to the final manuscript. C.B., A.M.D., P.E.J. and B.B critically reviewed the manuscript for intellectual content. C.B. is the guarantor of the work, and as such had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
6.
Supporting information
TABLE S1
Multilevel neurophysiological outcomes
ACKNOWLEDGEMENTS
The Novo Nordisk Scandinavia AS and Empowering Industry and Research (EIR) Northern Jutland supported this investigator‐initiated and ‐driven trial. C.B. received funding from the Talent Programme, Aalborg University. No funding source had any role in study design, data collection, data analysis, data interpretation or the preparation of this article. The authors had full access to all data in the study and final responsibility for the decision to submit for publication.
Brock C, Hansen CS, Karmisholt J, et al. Liraglutide treatment reduced interleukin‐6 in adults with type 1 diabetes but did not improve established autonomic or polyneuropathy. Br J Clin Pharmacol. 2019;85:2512–2523. 10.1111/bcp.14063
TODINELI study: (EUDRA CT: 2013–004375‐12, Ethics Ref: N‐20130077). Clinical trial registration number: http://clinicaltrials.gov NCT02138045.
Data Availability Statement:The datasets generated and/or analysed during the current study are available from the corresponding author on request.
DATA AVAILABILITY STATEMENT
The datasets generated and/or analysed during the current study are available from the corresponding author on request.
REFERENCES
- 1. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta‐cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5(4):219‐226. 10.1038/nrendo.2009.21 [DOI] [PubMed] [Google Scholar]
- 2. Hong H, Kim BS, Im H‐I. Pathophysiological role of Neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int Neurourol J. 2016;20(Suppl 1):S2‐S7. 10.5213/inj.1632604.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang SY, Kim DB, Ryu GR, et al. Exendin‐4 inhibits iNOS expression at the protein level in LPS‐stimulated Raw264.7 macrophage by the activation of cAMP/PKA pathway. J Cell Biochem. 2013;114(4):844‐853. 10.1002/jcb.24425 [DOI] [PubMed] [Google Scholar]
- 4. Gejl M, Gjedde A, Egefjord L, et al. In Alzheimer's disease, 6‐month treatment with GLP‐1 analog prevents decline of brain glucose metabolism: randomized, placebo‐controlled, double‐blind clinical trial. Front Aging Neurosci. 2016;8:108 10.3389/fnagi.2016.00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kan M, Guo G, Singh B, Singh V, Zochodne DW. Glucagon‐like peptide 1, insulin, sensory neurons, and diabetic neuropathy. J Neuropathol Exp Neurol. 2012;71(6):494‐510. 10.1097/NEN.0b013e3182580673 [DOI] [PubMed] [Google Scholar]
- 6. Himeno T, Kamiya H, Naruse K, et al. Beneficial effects of exendin‐4 on experimental polyneuropathy in diabetic mice. Diabetes. 2011;60(9):2397‐2406. 10.2337/db10-1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamamoto H, Lee CE, Marcus JN, et al. Glucagon‐like peptide‐1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. J Clin Invest. 2002;110(1):43‐52. 10.1172/JCI15595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol. 2012;11(6):521‐534. 10.1016/S1474-4422(12)70065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brock C, Søfteland E, Gunterberg V, et al. Diabetic autonomic neuropathy affects symptom generation and brain‐gut axis. Diabetes Care. 2013;36(11):3698‐3705. 10.2337/dc13-0347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drewes AM, Søfteland E, Dimcevski G, et al. Brain changes in diabetes mellitus patients with gastrointestinal symptoms. World J Diabetes. 2016;7(2):14‐26. 10.4239/wjd.v7.i2.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frøkjær JB, Andersen LW, Brock C, et al. Altered brain microstructure assessed by diffusion tensor imaging in patients with diabetes and gastrointestinal symptoms. Diabetes Care. 2013;36(3):662‐668. 10.2337/dc12-1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP‐1: theoretical and practical applications. Curr Med Res Opin. 2011;27(3):547‐558. 10.1185/03007995.2010.549466 [DOI] [PubMed] [Google Scholar]
- 13. Jaiswal M, Martin CL, Brown MB, et al. Effects of exenatide on measures of diabetic neuropathy in subjects with type 2 diabetes: results from an 18‐month proof‐of‐concept open‐label randomized study. J Diabetes Complications. 2015;29(8):1287‐1294. 10.1016/J.JDIACOMP.2015.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tesfaye S, Boulton AJ, Dyck PJ, et al. Toronto diabetic neuropathy expert group. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33(10):2285‐2293. 10.2337/dc10-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cruccu G, Aminoff MJ, Curio G, et al. Recommendations for the clinical use of somatosensory‐evoked potentials. Clin Neurophysiol. 2008;119(8):1705‐1719. 10.1016/j.clinph.2008.03.016 [DOI] [PubMed] [Google Scholar]
- 16. Lelic D, Valeriani M, Fischer IWD, Dahan A, Drewes AM. Venlafaxine and oxycodone have different effects on spinal and supraspinal activity in man: a somatosensory evoked potential study. Br J Clin Pharmacol. 2017;83(4):764‐776. 10.1111/bcp.13177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Camm AJ, Malik M, Bigger JT, et al. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Task force of the European Society of Cardiology and the north American Society of Pacing and Electrophysiology. Circulation. 1996;93(5):1043‐1065. [PubMed] [Google Scholar]
- 18. Brock C, Jessen N, Brock B, et al. Cardiac vagal tone, a non‐invasive measure of parasympathetic tone, is a clinically relevant tool in type 1 diabetes mellitus. Diabet Med. 2017;34(10):1428‐1434. 10.1111/dme.13421 [DOI] [PubMed] [Google Scholar]
- 19. Novak V, Saul JP, Eckberg DL. Task force report on heart rate variability. Circulation. 1997;96(3):1056‐1057. [PubMed] [Google Scholar]
- 20. Farmer AD, Coen SJ, Kano M, et al. Normal values and reproducibility of the real‐time index of vagal tone in healthy humans: a multi‐center study. Ann Gastroenterol. 2014;27(4):362‐368. [PMC free article] [PubMed] [Google Scholar]
- 21. Nilsson M, Piasco A, Nissen TD, et al. Reproducibility of psychophysics and electroencephalography during offset analgesia. Eur J Pain (United Kingdom). 2014;18(6):824‐834. 10.1002/j.1532-2149.2013.00424.x [DOI] [PubMed] [Google Scholar]
- 22. Dyck PJ, Davies JL, Litchy WJ, O'Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester diabetic neuropathy study cohort. Neurology. 1997;49(1):229‐239. [DOI] [PubMed] [Google Scholar]
- 23. Kane N, Oware A. Somatosensory evoked potentials aid prediction after hypoxic–ischaemic brain injury. Pract Neurol. 2015;15(5):352‐360. 10.1136/practneurol-2015-001122 [DOI] [PubMed] [Google Scholar]
- 24. Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S. The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta ‐ Mol Cell Res. 2011;1813(5):878‐888. 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- 25. Ziegler D, Dannehl K, Mühlen H, Spüler M, Gries FA. Prevalence of cardiovascular autonomic dysfunction assessed by spectral analysis, vector analysis, and standard tests of heart rate variation and blood pressure responses at various stages of diabetic neuropathy. Diabet Med. 1992;9(9):806‐814. [DOI] [PubMed] [Google Scholar]
- 26. González‐Clemente J‐M, Vilardell C, Broch M, et al. Lower heart rate variability is associated with higher plasma concentrations of IL‐6 in type 1 diabetes. Eur J Endocrinol. 2007;157(1):31‐38. 10.1530/EJE-07-0090 [DOI] [PubMed] [Google Scholar]
- 27. Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population‐based European prospective investigation into cancer and nutrition (EPIC)‐Potsdam study. Diabetes. 2003;52(3):812‐817. 10.2337/diabetes.52.3.812 [DOI] [PubMed] [Google Scholar]
- 28. Lowe G, Woodward M, Hillis G, et al. Circulating inflammatory markers and the risk of vascular complications and mortality in people with type 2 diabetes and cardiovascular disease or risk factors: the ADVANCE study. Diabetes. 2014;63(3):1115‐1123. 10.2337/db12-1625 [DOI] [PubMed] [Google Scholar]
- 29. Ridker PM. From C‐reactive protein to Interleukin‐6 to Interleukin‐1: moving upstream to identify novel targets for Atheroprotection. Circ Res. 2016;118(1):145‐156. 10.1161/CIRCRESAHA.115.306656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Scholten BJ, Persson F, Rosenlund S, et al. Effects of liraglutide on cardiovascular risk biomarkers in patients with type 2 diabetes and albuminuria: a sub‐analysis of a randomized, placebo‐controlled, double‐blind, crossover trial. Diabetes Obes Metab. 2017;19(6):901‐905. 10.1111/dom.12884 [DOI] [PubMed] [Google Scholar]
- 31. Mann JFE, Ørsted DD, Brown‐Frandsen K, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med. 2017;377(9):839‐848. 10.1056/NEJMoa1616011 [DOI] [PubMed] [Google Scholar]
- 32. von Scholten BJ, Persson F, Rosenlund S, et al. The effect of liraglutide on renal function: a randomized clinical trial. Diabetes Obes Metab. 2017;19(2):239‐247. 10.1111/dom.12808 [DOI] [PubMed] [Google Scholar]
- 33. Purohit S, Sharma A, Zhi W, et al. Proteins of TNF‐α and IL6 pathways are elevated in serum of Type‐1 diabetes patients with microalbuminuria. Front Immunol. 2018;9:154 10.3389/fimmu.2018.00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ayasolla K, Khan M, Singh AK, Singh I. Inflammatory mediator and beta‐amyloid (25‐35)‐induced ceramide generation and iNOS expression are inhibited by vitamin E. Free Radic Biol Med. 2004;37(3):325‐338. 10.1016/j.freeradbiomed.2004.04.007 [DOI] [PubMed] [Google Scholar]
- 35. Jung C‐H, Kim B‐Y, Kim C‐H, Kang S‐K, Jung S‐H, Mok J‐O. Association of serum adipocytokine levels with cardiac autonomic neuropathy in type 2 diabetic patients. Cardiovasc Diabetol. 2012;11(1):24 10.1186/1475-2840-11-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. González‐Clemente JM, Mauricio D, Richart C, et al. Diabetic neuropathy is associated with activation of the TNF‐alpha system in subjects with type 1 diabetes mellitus. Clin Endocrinol (Oxf). 2005;63(5):525‐529. 10.1111/j.1365-2265.2005.02376.x [DOI] [PubMed] [Google Scholar]
- 37. Aharoni R, Arnon R. Linkage between immunomodulation, neuroprotection and neurogenesis. Drug News Perspect. 2009;22(6):301‐312. 10.1358/dnp.2009.22.6.1395253 [DOI] [PubMed] [Google Scholar]
- 38. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2017;390(10103):1664‐1675. 10.1016/S0140-6736(17)31585-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bansal D, Gudala K, Muthyala H, Esam HP, Nayakallu R, Bhansali A. Prevalence and risk factors of development of peripheral diabetic neuropathy in type 2 diabetes mellitus in a tertiary care setting. J Diabetes Investig. 2014;5(6):714‐721. 10.1111/jdi.12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kautzky‐Willer A, Harreiter J, Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. 2016;37(3):278‐316. 10.1210/er.2015-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kamenov ZA, Parapunova RA, Georgieva RT. Earlier development of diabetic neuropathy in men than in women with type 2 diabetes mellitus. Gend Med. 2010;7(6):600‐615. 10.1016/j.genm.2010.11.001 [DOI] [PubMed] [Google Scholar]
- 42. Aaberg ML, Burch DM, Hud ZR, Zacharias MP. Gender differences in the onset of diabetic neuropathy. J Diabetes Complications. 2008;22(2):83‐87. 10.1016/j.jdiacomp.2007.06.009 [DOI] [PubMed] [Google Scholar]
- 43. Kumarathurai P, Anholm C, Larsen BS, et al. Effects of Liraglutide on heart rate and heart rate variability: a randomized, double‐blind, placebo‐controlled crossover study. Diabetes Care. 2017;40(1):117‐124. 10.2337/dc16-1580 [DOI] [PubMed] [Google Scholar]
- 44. Kopp HP, Kopp CW, Festa A, et al. Impact of weight loss on inflammatory proteins and their association with the insulin resistance syndrome in morbidly obese patients. Arterioscler Thromb Vasc Biol. 2003;23(6):1042‐1047. 10.1161/01.ATV.0000073313.16135.21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1
Multilevel neurophysiological outcomes
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on request.