

Abstract
Background
Patients with metastatic colorectal cancer can benefit from anti-EGFR therapy, such as cetuximab and panitumumab. However, colorectal cancers harboring constitutive activating mutations in KRAS, NRAS and BRAF genes are not responsive to anti-EGFR therapy. To select patients for appropriate treatment, genetic testing of these three genes is routinely performed.
Methods
We applied bridged nucleic acid-clamp real-time PCR (BNA-clamp PCR) to detect somatic hotspot mutations in KRAS, NRAS and BRAF. PCR products from BNA-clamp PCR were subsequently analyzed Sanger sequencing. We then compared results with those from the PCR–reverse sequence-specific oligonucleotide probe (PCR-rSSO) method, which has been used as in vitro diagnostic test in Japan. To validate the mutation status, we also performed next generation sequencing using all samples.
Results
In 50 formalin-fixed paraffin-embedded tissues, KRAS mutations were detected at frequencies of 50% (25/50) and 52% (26/50) by PCR-rSSO and BNA-clamp PCR with Sanger sequencing, respectively, and NRAS mutations were detected at 12% (6/50) and 12% (6/50) by PCR-rSSO and BNA-clamp PCR with Sanger sequencing, respectively. The concordance rate for detection of KRAS and NRAS mutations between the two was 94% (47/50). However, there were three discordant results. We validated these three discordant and 47 concordant results by next generation sequencing. All mutations identified by BNA-clamp PCR with Sanger sequencing were also identified by next generation sequencing. BNA-clamp PCR detected BRAF mutations in 6% (3/50) of tumor samples.
Conclusions
Our results indicate that BNA-clamp PCR with Sanger sequencing detects somatic mutations in KRAS, NRAS and BRAF with high accuracy.
Keywords: KRAS, NRAS, BRAF, Colorectal cancer, BNA
Background
The incidence of colorectal cancer has been increasing. Metastatic colorectal cancers have a high mortality rate with a five-year survival rate of less than 10%. Genetic alterations in RAS–MAPK and PI3K–AKT pathway are common in colorectal cancers. The most recurrently mutated genes in these pathways are KRAS (Kirsten rat sarcoma viral oncogene homolog, OMIM: 190070), NRAS (neuroblastoma RAS viral oncogene homolog, OMIM: 164790) and BRAF (v-raf murine sarcoma viral oncogene homolog B1, OMIM: 164757). Monoclonal antibodies against the epidermal growth factor receptor (EGFR), including cetuximab and panitumumab, have been used to treat patients with metastatic colorectal cancer. These antibodies bind to the extracellular domain of EGFR and inhibit its downstream signaling, which mainly affects cell proliferation and survival via RAS-RAF-MEK-ERK and PI3K-AKT pathways.
Anti-EGFR therapy is beneficial in approximately 15% of patients with wild-type KRAS metastatic colorectal tumors, whereas patients with KRAS-mutated tumors show little response [1–5]. Furthermore, anti-EGFR therapy is more beneficial to patients with wild-type NRAS and BRAF [6–8]. It is well-known that somatic hotspot mutations are located in codons 12 and 13 (exon 2), codons 59 and 61 (exon 3), and codons 117 and 146 (exon 4) of KRAS and NRAS genes, and in codon 600 (exon 15) in BRAF. In colorectal cancers, KRAS mutations are observed in 42% of cases, while mutations in NRAS (10%) and BRAF (10%) are less frequent [9, 10]. KRAS, NRAS and BRAF mutations occur in colorectal cancers in a mutually exclusive manner [10].
Genetic analysis of somatic hotspot mutations in KRAS, NRAS and BRAF is now standard practice for selecting patients for anti-EGFR therapies. To simultaneously detect different types of mutations by real-time PCR, we tested the bridged nucleic acid (BNA)-clamp technique [11]. A BNA is an artificial nucleic acid that strongly binds to a complementary DNA structure [12]. BNA-clamp PCR enables mutations to be detected because the melting temperature of a perfectly matched BNA-DNA duplex is much higher than that of DNA-DNA duplex [13–15]. Furthermore, mutated alleles can be selectively amplified because the BNA clamp oligonucleotide inhibits amplification of the wild-type allele.
In this study, we examined the clinical utility of the BNA-clamp PCR technique to detect KRAS, NRAS and BRAF mutations. To this end, we determined the mutation status of 50 patients with colorectal cancer from formalin-fixed paraffin embedded (FFPE) tissues and compared these results with those from the PCR–reverse sequence-specific oligonucleotide probe (PCR-rSSO) method, which is approved for in vitro diagnostic test for analyzing KRAS, NRAS and BRAF in patients with colorectal cancer in Japan [16]. To validate the mutations status in three genes, we conducted panel sequencing by next generation sequencing (NGS) [17–27].
Methods
Samples and study design
We collected tumor tissues from 50 patients with colorectal cancer between November 2010 and February 2016 and prepared FFPE samples from these tissues. The samples were analyzed by SRL Inc. (Tokyo, Japan) using an in-vitro diagnostic PCR-rSSO kit. We analyzed the same tumor samples by the BNA-clamp method.
To estimate the concordant and discordant results were obtained by the two methods, validation was performed by next generation sequencing (NGS) analysis using an Ion PGM system (Thermo Fisher Scientific, MA, USA) [17, 18, 24, 28]. The Institutional Review Board of clinical research and genome research committee at Yamanashi Central Hospital approved this retrospective study and written informed consent was obtained from patients. Patients had the opportunity to refuse to participate in the study.
PCR-rSSO
Five 10-μm thick sections from each of the 50 FFPE tissues were analyzed by PCR-rSSO using a MEBGEN™ RASKET kit (MBL, Nagoya, Japan) [29, 30]. This analysis was performed by SRL Inc. (Tokyo, Japan). The PCR-rSSO kit detected 48 types of mutation in KRAS and NRAS, but did not target BRAF. In brief, multiplex PCR amplified codons 12, 13, 59, 61, 117 and 146 in KRAS and NRAS using eight sets of biotinylated primer pairs. PCR products were hybridized with complementary mutated probes immobilized on fluorescent-beads. After washing, the hybridized beads were mixed with phycoerythrin-labeled streptavidin (SA-PE) solution. Fluorescence was detected on a Luminex100/200 instrument (Luminex) and the types of mutation Identified.
BNA-clamp PCR
For each of the 50 samples, DNA was extracted from two 10-μm thick FFPE sections and from five 10-μm thick tumor biopsy sections using an Agencourt FormaPure DNA kit (Beckman Coulter, CA, USA) according to the manufacturer’s protocol. DNA concentration was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). If tumor purity was less than 10%, we performed laser capture microdissection to enrich for tumor cells. To this end, tumor tissues were stained with hematoxylin and eosin, and then microdissected using an ArcturusXT laser capture microdissection system (Thermo Fisher Scientific).
To detect mutations in KRAS, NRAS and BRAF, we used a BNA Real-time PCR Mutation Detection Kit Extended RAS (Riken Genesis, Tokyo, Japan). This kit contains nine types of BNA-probe, primers and PCR enzymes for detecting mutations by quantitative real-time PCR. Nine types of primer/probes were designed in house to target KRAS at codons 12/13, 59/61, 117 and 146, NRAS at codons 12/13, 59/61, 117 and 146, and BRAF at codon 600. The BNA is an artificial nucleic acid that hybridizes to a perfectly matched template with high affinity. A BNA clamp selectively inhibits PCR of the wild-type template, but does not influence a mutated template [12]. According to the manufacturer’s protocol, the reaction mixture comprised 12.5 μL 2x Master Mix, 2.5 μL 10x Oligo mix, 0.4 μL 25 μM ROX™ Reference Dye, 0.25 μL Uracil-N-glycosylase (UNG), and 20–100 ng FFPE DNA in a 25 μL total volume. Real-time PCR was conducted on a ViiA7 Real Time System (Thermo Fisher Scientific) with the following cycling conditions: 50 °C for 3 min, 95 °C for 2 min, and 40 cycles of 95 °C for 30 s and 60 °C for 45 s. The data were analyzed using ViiA7 software v2.2.2 (Thermo Fisher Scientific). The threshold line was set at 0.04. The threshold cycle (Ct) value was assigned to each PCR reaction and amplification curve was visually assessed. When amplification plot did not reach to threshold line, we examined whether the sample harbored mutations by Sanger sequencing using BNA-clamp PCR products (Additional file 4: Table S1).
Sanger sequencing
To further determine nucleotide changes and to characterize the deduced amino acid changes, we performed Sanger sequencing on samples, in which PCR amplification plot was observed but did not reached to threshold line by BNA-clamp PCR. BNA-clamp PCR products were purified using ExoSAP-IT Express PCR Cleanup Reagent (Thermo Fisher Scientific) [31, 32]. Purified products were used as templates and Sanger sequencing was performed using the BNA Real-time PCR Extended RAS Mutation Sequencing Primer (Riken Genesis) and the BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Sequencing reactions consisted of 1.0 μL template PCR product, 0.5 μL 3.2 μM forward primer or 0.5 μL 3.2 μM reverse primer, 2 μL Big Dye Buffer, 1 μL BigDye v3.1 and 5.5 μL nuclease free water. PCR was conducted on a Veriti Thermal Cycler (Thermo Fisher Scientific) with the following cycling conditions (BigDye_Kit _Fast): 96 °C for 1 min, 25 cycles of 96 °C for 10 s, 50 °C for 5 s and 60 °C for 75 s, and hold at 4 °C. PCR products were purified with a BigDye XTerminator Purification Kit (Thermo Fisher Scientific) and subsequently sequenced on a 3500 Genetic Analyzer (Thermo Fisher Scientific). The data were analyzed by Sequencing Analysis Software v5.4 (Thermo Fisher Scientific) [18, 25, 33].
Validation by NGS
Sequencing libraries were prepared using the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific) as previously described [17, 19, 23, 28, 34]. Briefly, multiplex PCR was performed using the Ion AmpliSeq™ Cancer Hotspot Panel v2 (Thermo Fisher Scientific), which targets the hotspot regions of 50 oncogenes and tumour suppressor genes [17–28, 32–37]. PCR products were partially digested with FuPa reagent and subsequently ligated to adaptors and barcodes using the Ion Xpress Barcode Adapters Kit (Thermo Fisher Scientific). The ligated library was purified with Agencourt AMPure XP reagent (Beckman Coulter, Brea, CA), and the library concentration was determined using an Ion Library Quantitation Kit (Thermo Fisher Scientific). Each library was diluted and the same amount of each was pooled. Emulsion PCR and chip loading was performed on an Ion Chef with the Ion PGM Hi-Q View Chef Kit. Sequencing was performed using an Ion PGM Hi-Q View Sequencing Kit on the Ion PGM (Thermo Fisher Scientific). Variant calling and annotation were performed using an Ion Reporter Server System (Thermo Fisher Scientific). We identified nonsynonymous mutations with the AmpliSeq CHPv2 single sample workflow (version 5.10) and used the following filtering parameters: (i) a minimum count of ≥10 for mutant allele reads, (ii) coverage depth ≥ 20 at the somatic variant site, (iii) variant allele faction ≥5%, and p-value cut-off of 0.05, and (iv) variants present in the dbSNP database (version 138) were filtered out (UCSC Common SNPs = Not In) [24, 27]. Binary SAM (BAM) files were visualized by Ion Reporter™ Genomic Viewer.
Sensitivity determination
To examine sensitivity testing experiments, TaqMan™ Control Genomic DNA (human) (Thermo Fisher Scientific) was spiked with different amounts of Horizon Tru-Q 7 (1.3% Tier) Reference Standard (Horizon Discovery, Cambridge, UK) harboring engineered mutations. The mixtures represented 1–33% and 0.4–12% variant allele fraction range in KRAS at codon 12/13 and BRAF at codon 600, respectively. The total number of DNA molecules was kept in constant.
Results
KRAS and NRAS mutations detected by PCR-rSSO
We analyzed FFPE tissues from 50 patients with colorectal cancer. DNA was extracted from sections and subjected to PCR-rSSO (Fig. 1). Of 50 samples, at least one mutation in either KRAS or NRAS was detected in 31 samples (Table 1). Twenty five mutations were found in KRAS (17 at codon 12, 5 at codon 13, 1 at codon 61, and 2 at codon 146). Six mutations were detected in NRAS (2 at codon 12, 1 at codon 13, and 3 at codon 61).
Fig. 1.

Experimental design. We tested FFPE samples from 50 patients with colorectal cancer. Tissue sections were subjected to PCR-rSSO and BNA-clamp PCR. BNA-clamp PCR involved real-time PCR and amplified the mutated allele. The amplification plot was verified using mutation-positive samples and an internal control was amplified to confirm assay integrity. PCR products from BNA-clamp PCR were subsequently analyzed by Sanger sequencing to confirm the nucleotide changes
Table 1.
Comparison of KRAS and NRAS mutation status determined by PCR-rSSO, BNA-clamp PCR and NGS
| PCR-rSSO | BNA-clamp PCR | Deep sequencing by NGS | ||||
|---|---|---|---|---|---|---|
| Sample No. | Nucleotide changes | Deduced amino acid changes | Nucleotide changes | Deduced amino acid changes | Nucleotide changes | Deduced amino acid changes |
| #1 | Wild-type | Wild-type | c.180_181delTCinsAA | KRAS Q61K | c.180_181delTCinsAA | KRAS Q61K |
| #2 | c.34G > T | NRAS G12C | Wild-type | Wild-type | Wild-type | Wild-type |
| #3 | c.436G > A | KRAS A146T | c.436G > A | KRAS A146T | c.436G > A | KRAS A146T |
| Wild-type | Wild-type | c.35G > A | NRAS G12D | c.35G > A | NRAS G12D | |
| #4 | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A |
| #5 | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A |
| #6 | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A | c.35G > C | KRAS G12A |
| #7 | c.34G > T | KRAS G12C | c.34G > T | KRAS G12C | c.34G > T | KRAS G12C |
| #8 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #9 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #10 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #11 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #12 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #13 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #14 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #15 | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D | c.35G > A | KRAS G12D |
| #16 | c.34G > A | KRAS G12S | c.34G > A | KRAS G12S | c.34G > A | KRAS G12S |
| #17 | c.34G > A | KRAS G12S | c.34G > A | KRAS G12S | c.34G > A | KRAS G12S |
| #18 | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V |
| #19 | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V |
| #20 | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V | c.35G > T | KRAS G12 V |
| #21 | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D |
| #22 | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D |
| #23 | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D |
| #24 | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D |
| #25 | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D | c.38G > A | KRAS G13D |
| #26 | c.182A > T | KRAS Q61L | c.182A > T | KRAS Q61L | c.182A > T | KRAS Q61L |
| #27 | c.436G > A | KRAS A146T | c.436G > A | KRAS A146T | c.436G > A | KRAS A146T |
| #28 | c.35G > A | NRAS G12D | c.35G > A | NRAS G12D | c.35G > A | NRAS G12D |
| #29 | c.38G > T | NRAS G13 V | c.38G > T | NRAS G13 V | c.38G > T | NRAS G13 V |
| #30 | c.181C > A | NRAS Q61K | c.181C > A | NRAS Q61K | c.181C > A | NRAS Q61K |
| #31 | c.181C > A | NRAS Q61K | c.181C > A | NRAS Q61K | c.181C > A | NRAS Q61K |
| #32 | c.182A > G | NRAS Q61R | c.182A > G | NRAS Q61R | c.182A > G | NRAS Q61R |
| #33 | NA | NA | c.1799 T > A | BRAF V600E | c.1799 T > A | BRAF V600E |
| #34 | NA | NA | c.1799 T > A | BRAF V600E | c.1799 T > A | BRAF V600E |
| #35 | NA | NA | c.1799 T > A | BRAF V600E | c.1799 T > A | BRAF V600E |
| #36–50 | Wild-type | Wild-type | Wild-type | Wild-type | Wild-type | Wild-type |
NA not applicable
KRAS and NRAS mutations detected by BNA-clamp PCR with Sanger sequencing
We also analyzed the 50 samples for KRAS and NRAS mutations using BNA-clamp PCR (Fig. 1). Of 50 samples, amplification plots of 26 samples reached to threshold, but those of five samples did not reached (Supplemental Table 1). In these five samples, we detected either KRAS and/or NRAS mutations by Sanger sequencing using PCR product of BNA-clamp PCR. Therefore, at least one mutation in KRAS or NRAS was identified in 31 samples (Table 1). Twenty six mutations were found in KRAS (17 at codon 12, 5 at codon 13, 2 at codon 61, and 2 at codon 146). Six mutations were identified in NRAS (2 at codon 12, 1 at codon 13, and 3 at codon 61). In one sample (sample #3), two mutations (KRAS at codon 146 and NRAS at codon 12) were identified (Table 1).
Comparison of identified mutations by PCR-rSSO and BNA-clamp PCR with Sanger sequencing
We next compared the KRAS and NRAS mutation status identified by the two different methods. There was 94% (47/50) concordance between PCR-rSSO and BNA-clamp PCR with Sanger sequencing. Positive percent agreement was 94% (29/31) and negative percent agreement was 95% (18/19). In three samples (sample #1-#3 in Table 1), there were discordant results. In sample #1, KRAS c.180_181delTCinsAA (p.Q61K) was detected by BNA-clamp PCR, but not by PCR-rSSO (Table 1). In sample #2, NRAS c.34G > T (p.G12C) was detected by PCR-rSSO, but not by BNA-clamp PCR. In sample #3, BNA-clamp PCR with Sanger sequencing identified two nucleotide changes, KRAS c.436G > A (p.A146T) and NRAS c.35G > A (p.G12D), whereas PCR-rSSO identified only one nucleotide change, KRAS c.436G > A.
Validation by NGS
To validate the results from the two methods, we subjected the three discordant samples (samples #1-#3) as well as 47 concordant samples (#4-#50) to NGS covering hotspot mutations of 50 cancer-associated genes. NGS yielded the sufficient sequencing reads mapped on the target regions (mean: 97%) and an average base coverage depth on targeted reference region (mean: 10,849-fold) (Table 2).
Table 2.
Quality and coverage depth of next generation sequencing
| Sample | Mapped reads | On target | Mean depth | Uniformity |
|---|---|---|---|---|
| #1 | 3,896,308 | 98% | 17,984 | 98% |
| #2 | 2,686,903 | 98% | 12,262 | 96% |
| #3 | 1,202,870 | 98% | 5057 | 80% |
| #4 | 2,101,697 | 98% | 9467 | 92% |
| #5 | 2,069,593 | 94% | 8935 | 99% |
| #6 | 2,623,488 | 98% | 11,775 | 90% |
| #7 | 2,950,289 | 97% | 13,056 | 99% |
| #8 | 1,834,917 | 98% | 8227 | 87% |
| #9 | 3,270,772 | 98% | 14,836 | 96% |
| #10 | 3,957,906 | 99% | 18,101 | 100% |
| #11 | 2,980,026 | 98% | 13,450 | 100% |
| #12 | 1,518,770 | 98% | 6864 | 86% |
| #13 | 2,569,739 | 91% | 10,930 | 100% |
| #14 | 3,089,053 | 99% | 14,112 | 100% |
| #15 | 3,471,557 | 99% | 15,934 | 100% |
| #16 | 2,385,272 | 98% | 10,832 | 90% |
| #17 | 2,990,670 | 99% | 14,066 | 100% |
| #18 | 2,803,954 | 98% | 12,871 | 100% |
| #19 | 1,818,393 | 97% | 8086 | 99% |
| #20 | 649,510 | 97% | 2794 | 92% |
| #21 | 3,236,156 | 99% | 14,950 | 99% |
| #22 | 2,215,854 | 98% | 9819 | 100% |
| #23 | 2,600,289 | 99% | 11,942 | 98% |
| #24 | 2,487,267 | 97% | 11,077 | 96% |
| #25 | 2,127,006 | 98% | 9555 | 85% |
| #26 | 1,227,270 | 68% | 3341 | 75% |
| #27 | 2,386,360 | 95% | 10,366 | 99% |
| #28 | 3,042,162 | 98% | 13,665 | 95% |
| #29 | 3,321,952 | 98% | 14,756 | 96% |
| #30 | 2,905,123 | 98% | 12,934 | 92% |
| #31 | 2,437,114 | 98% | 10,958 | 90% |
| #32 | 3,031,339 | 97% | 13,566 | 98% |
| #33 | 2,819,335 | 98% | 12,766 | 93% |
| #34 | 2,335,610 | 98% | 10,532 | 82% |
| #35 | 3,136,401 | 97% | 14,138 | 98% |
| #36 | 2,583,625 | 97% | 11,885 | 99% |
| #37 | 1,987,561 | 96% | 8886 | 97% |
| #38 | 1,161,909 | 95% | 5181 | 98% |
| #39 | 1,592,153 | 98% | 7337 | 97% |
| #40 | 1,421,966 | 98% | 6490 | 91% |
| #41 | 1,716,683 | 98% | 7854 | 93% |
| #42 | 2,121,085 | 99% | 9920 | 97% |
| #43 | 1,970,851 | 98% | 8928 | 99% |
| #44 | 2,510,670 | 99% | 11,608 | 99% |
| #45 | 1,398,962 | 98% | 6231 | 99% |
| #46 | 2,872,517 | 97% | 13,063 | 98% |
| #47 | 1,760,828 | 97% | 7928 | 97% |
| #48 | 2,259,832 | 97% | 9915 | 90% |
| #49 | 1,948,999 | 98% | 8921 | 99% |
| #50 | 3,134,126 | 98% | 14,287 | 96% |
Mapped reads: number of sequencing reads that were mapped to the human genome
On target: percentage of mapped reads that were aligned over the target region
Mean depth: Average base coverage depth over all bases targeted in the reference
Uniformity: percentage of target bases covered by at least 0.2x the average base read depth
NGS detected KRAS c.180_181delTCinsAA (p.Q61K) in sample #1, KRAS c.205G > A (p.D69N) in sample #2 and KRAS c.436G > A (p.A146T) and NRAS c.35G > A (p.G12D) mutations in sample #3 (Table 3). Although NRAS c.34G > T (p.G12C) was identified by PCR-rSSO in sample #2, this mutation was not identified by NGS. Both BNA-clamp PCR and PCR-rSSO methods did not detect NRAS p.D69N in sample #2, because this variant was not covered by either method (Table 3). Furthermore, remaining 47 samples were concordant among PCR-rSSO, BNA-clamp PCR with Sanger sequencing and NGS. Overall, the NGS results were concordant with those of BNA-clamp PCR with Sanger sequencing. These results indicate that KRAS and NRAS mutations were accurately detected by BNA-clamp PCR with Sanger sequencing.
Table 3.
Mutations in discordant samples identified by next generation sequencing using a panel of 50 cancer-associated genes
| Sample No. | Position | Reference | Variant | Gene | Nucleotide changes | Deduced amino acid changes | VAF (%) | coverage |
|---|---|---|---|---|---|---|---|---|
| #1 | chr12:25380277 | GA | TT | KRAS | c.180_181delTCinsAA | Q61K | 23.5 | 1982 |
| #2 | chr1:115256506 | C | T | NRAS | c.205G > A | D69N | 17.5 | 1995 |
| chr14:105246470 | C | T | AKT1 | c.130G > A | D44N | 5.5 | 2000 | |
| chr17:7578431 | G | A | TP53 | c.499C > T | Q167* | 21.1 | 1970 | |
| chr19:1223030 | C | T | STK11 | c.967C > T | P323S | 5.1 | 2000 | |
| chr19:1223054 | C | T | STK11 | c.991C > T | R331W | 5.1 | 1997 | |
| #3 | chr12:25378562 | C | T | KRAS | c.436G > A | A146T | 28.3 | 1161 |
| chr1:115258747 | C | T | NRAS | c.35G > A | G12D | 9.1 | 1998 | |
| chr3:37067240 | T | A | MLH1 | c.1151 T > A | V384D | 21.6 | 1998 | |
| chr5:112173917 | C | T | APC | c.2626C > T | R876* | 16.1 | 2000 | |
| chr5:112175589 | C | T | APC | c.4298C > T | P1433L | 7.4 | 2000 | |
| chr13:49033902 | T | C | RB1 | c.2039 T > C | I680T | 6.5 | 1628 | |
| chr17:7577551 | C | A | TP53 | c.730G > T | G244C | 12.6 | 2000 | |
| chr17:7578479 | G | A | TP53 | c.451C > T | P151S | 18.0 | 2000 | |
| chr19:1223125 | C | G | STK11 | c.1062C > G | F354 L | 41.2 | 818 |
Bold text indicates mutations covered by BNA-clamp and PCR-rSSO
VAF variant allele frequency
To examine the reasons for the discordant results, we checked the NGS results and BNA-clamp PCR with Sanger sequencing. In sample #1, harboring KRAS p.Q61K, there were multi-nucleotide variants (c.180_181delTCinsAA) across codon 60 and 61 (Fig. 2 and Table 3). The nucleotide change at codon 60 (GGT > GGA) led to a synonymous mutation (KRAS p.G60G). Because PCR-rSSO detects only perfectly-matched single nucleotide variants, it did not detect the multi-nucleotide variants [29]. In sample #2, NRAS c.34G > T (p.G12C) was not identified by either BNA-clamp PCR or NGS (Fig. 2 and Additional file 2: Figure S2).
Fig. 2.
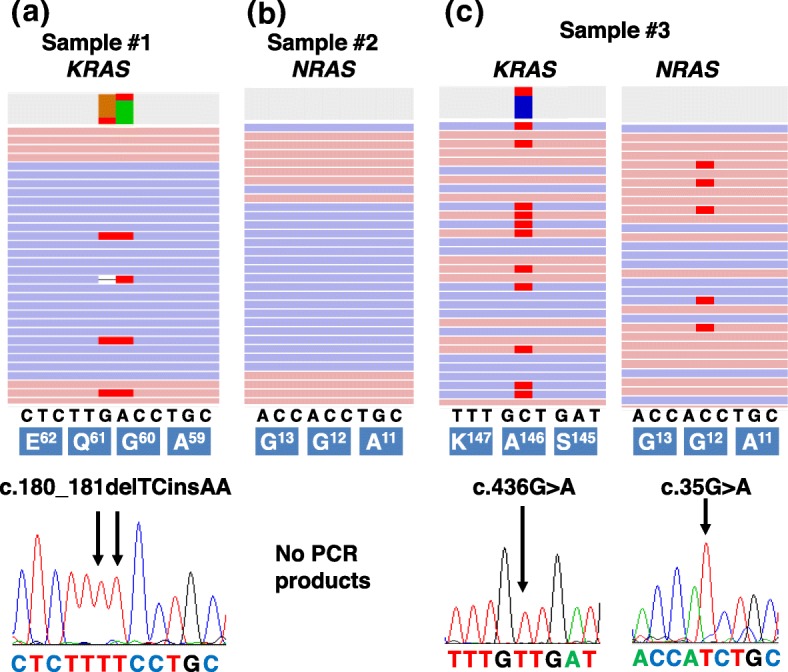
Discordant results were validated by NGS and Sanger sequencing. a-c Representative images of read alignments (BAM files) of sample #1 (a), #2 (b) and #3 (c) were visualized with Ion Reporter Genome Viewer (upper images). PCR products produced by BNA-clamp PCR were purified and used as templates for Sanger sequencing. Sequencing chromatograms show the mutations in each sample (lower image). Arrows indicate the position of the mutations. a In sample #1, KRAS p.Q61K was detected by NGS. At this site, multi-nucleotide variants (c.180_181delTCinsAA) existed in codons 60 and 61. b In sample #2, there was no apparent variant at codon 12 of NRAS. We did not observe an amplification plot signal by real-time PCR and obtained no visible PCR product for subsequent Sanger sequencing analysis. c Two different mutations (NRAS p.G12D and KRAS p.A146T) were observed in sample #3
BRAF mutations
BRAF mutations occur in approximately 10% of colorectal cancers and are associated with resistance to anti-EGFR therapy [9]. PCR-rSSO using the MEBGEN™ RASKET kit did not cover BRAF mutations. However, BRAF mutations were detected by BNA-clamp PCR and NGS in 3 of the 50 (6%) samples (sample #33–35), which were wild-type for KRAS and NRAS (Table 1).
Turnaround time
We assessed the turnaround time of PCR-rSSO and BNA-clamp PCR methods. It takes approximately 4.5 h with PCR-rSSO, whereas 2 h with BNA-clamp PCR. Because PCR-rSSO method uses DNA-probe for hybridization to detect mutated DNA, it takes more long time for hybridize reaction. Contrary, BNA-clamp method contains mixed BNA-clamping probe and primers in reaction reagent and needs one-step real-time PCR reaction for detecting mutation. If PCR product of BNA-clamp PCR is analyzed by Sanger sequencing, it additionally takes about 3 h. Combined with BNA-clamp PCR and Sanger sequencing, it takes a total of 5 h.
Discussion
In this study, we compared mutation detection in KRAS and NRAS genes between PCR-rSSO and BNA-clamp PCR with Sanger sequencing. Overall, the concordance rate was 94% (47/50 samples) between the two methods. However, there were three discordant results which were further analyzed by NGS with high-depth coverage. The NGS results were consistent with the results of BNA-clamp PCR with Sanger sequencing. Our results demonstrated the BNA-clamp PCR method with Sanger sequencing have high accuracy for the detection of KRAS, NRAS and BRAF mutations in colorectal cancer.
According to manufacturer’s instructions, the limit of detections of both BNA-clamp PCR and PCR-rSSO methods were 1–5%. We confirmed the sensitivity by diluted experiment of BNA-clamp PCR (Additional file 3: Figure S3). Although the performance of sensitivity is comparable, BNA-clamp PCR with Sanger sequencing has several advantages. First, it requires only standard clinical laboratory equipment (e.g. a real-time PCR and capillary sequencer). We could qualitatively evaluate the presence of mutations by real-time PCR. The BNA probe binds to wild-type template DNA and inhibits its PCR amplification, whereas mutated alleles are selectively amplified during real-time PCR (Additional file 1: Figure S1). Second, BNA-clamp PCR can analyze multi-nucleotide variants within KRAS, NRAS and BRAF genes. Third, BNA-clamp PCR is a simple method with a short turnaround time. PCR-rSSO takes 4.5 h per run: PCR reaction (2.3 h), hybridization (1.4 h) and detection of fluorescence with dedicated equipment (0.8 h). BNA-clamp PCR takes only 2 h per run: preparation of PCR master mix (0.5 h) and real time PCR reaction (1.5 h). Even when PCR products of BNA-clamp PCR were investigate by the Sanger sequencing, it takes about 30 min longer compared to PCR-rSSO. Fourth, the running cost is lower than PCR-rSSO; it costs approximately 50 USD per BNA-clamp PCR reaction compared with approximately 178 USD per PCR-rSSO reaction. It costs additionally 20 USD when Sanger sequencing is conducted.
We examined the three discordant results between PCR-rSSO with Sanger sequencing and BNA-clamp methods. The reason for discordance was explained in only sample #1. The PCR-rSSO method detects perfectly-matched mutated alleles in tumor samples; therefore, it missed the KRAS multi-nucleotide variant, c.180_181delTCinsAA, and reported this site as wild type (Table 1). A previous report also showed PCR-rSSO could not detect KRAS p.G12C because of a multi-nucleotide variant at codon 11 (c.33_34delTGinsCT) [29]. In sample #2, PCR-rSSO reported an NRAS c.34G > T (p.G12C) mutation but both BNA-clamp PCR and deep sequencing did not. To exclude the possibility that the variant calling filtered out the NRAS mutation, we further visualized BAM data using Ion Reporter™ Genomic Viewer. However, we could not confirm the corresponding mutated reads in NRAS (Additional file 2: Figure S2). In sample #3, both BNA-clamp PCR and NGS detected the NRAS (c.35G > A) p.G12D mutation at 9.1% variant allele fraction. Although the reagent kit included a perfectly-matched probe corresponding to NRAS c.35G > A (p.G12D), PCR-rSSO did not detect this mutation.
The reasons for these two discordant results remain unclear, but one possible explanation is tumor heterogeneity. We prepared sections from patient FFPE tissues and each method analyzed different sections (Fig. 1). PCR-rSSO was conducted by a commercial laboratory on tissue without microdissection. In contrast, BNA-clamp PCR with Sanger sequencing and NGS used microdissected tumor tissue samples (Arcturus; Thermo Fisher Scientific, MA, USA) [19, 38]. Alternatively, there may be differences in quality control between laboratories. Our laboratory (GAC-Genome Analysis Center) sent specimens to the College of American Pathologists (CAP) for proficiency testing and achieved a 100% match.
BNA-clamp PCR has limitations. Although we could identify the presence of mutations in samples by real-time PCR, Sanger sequencing is needed to determine the nucleotide changes. In addition, the kit contains nine primer/probe mixtures, which are designed to interrogate KRAS, NRAS and BRAF exons. When a small number of samples (less than eight) is to be tested, they can be analyzed in one reaction in a 96-well format. When more samples need to be analyzed, several real-time PCRs are needed. Therefore, small to medium numbers of samples are suitable for analysis by BNA-clamp PCR. In one sample (#3), the amplification plot did not reached to threshold line by BNA-clam PCR, though variant allele fraction of KRAS A146T and NRAS G12D by the NGS were 28.3 and 9.1%, respectively. Although the precise reason is not unclear, there are several possibilities. One is the quality of the DNA from FFPE tissue. Fragmented FFPE DNA may be not effectively amplified by BNA-clamp PCR in this sample. Second possibility may be the difference of DNA polymerase enzyme used for reaction. In general, high-activity and fidelity DNA polymerase is used in NGS library construction. The PCR amplification efficiency may be different between BNA-clamp PCR and NGS. According to these possibilities, NGS could detect the mutation nevertheless less quality of DNA was used as long as targeted regions was successfully amplified and NGS library was constructed.
Overall, we confirmed the clinical utility of BNA-clamp PCR for detecting KRAS, NRAS and BRAF mutations in colorectal cancers. This less time-consuming and less laborious method can enable precision medicine to be offered to patients with metastatic colorectal cancers, such as anti-EGFR therapy. Furthermore, circulating tumor DNA (ctDNA) was shed into the blood stream and body fluids, called as liquid biopsy. The detection of ctDNA is useful for monitoring tumor recurrence, predicting treatment effect and detecting drug-resistant mutation in patients with colorectal cancer. BNA-clamp PCR would be one of the candidate methods for detecting rare mutations in liquid biopsy in a clinical laboratory.
Conclusions
In this study, we estimated the performance of BNA-clamp PCR with Sanger sequencing method to detect KRAS, NRAS and BRAF mutations in colorectal cancers and compared the results from PCR-rSSO and NGS. BNA-clamp PCR accurately detected KRAS, NRAS and BRAF mutations in patients with colorectal cancer. Genetic testing by BNA-clamp PCR with Sanger sequencing has potential to be used in routine clinical practice for the selection of appropriate patients for anti-EGFR therapy.
Supplementary information
Additional file 1: Figure S1. Principle of BNA-clamp PCR. Forward and reverse primers amplify the targeted mutation. A BNA probe binds to the wild-type allele but not to the mutated allele. The BNA probe selectively inhibits PCR amplification of the wild-type allele. F, fluorescence; Q, quencher.
Additional file 2: Figure S2. Sequence reads were visualized by Ion Reporter Genome Viewer. Representative images of read alignments (BAM files) of sample #2 were visualized with Ion Reporter Genome Viewer. There are no mutated reads corresponding to NRAS p.G12C (c.34G > T: chr1:115,258,748) in the next generation sequencing data.
Additional file 3: Figure S3. Amplification plot of dilution experiment by BNA-clamp PCR method. Wild-type control DNA was spiked in the Tru-Q 7 (1.3% Tier) Reference Standard. BNA-clamp PCR was performed using serial dilution DNA. (A) DNA containing KRAS mutation at codon 12/13 (dilution range: 1–33% variant allele fraction) and (B) BRAF mutation at codon 600 (dilution range: 0.4–12% variant allele fraction).
Additional file 4: Table S1. Threshold cycle values of real-time PCR using BNA-clamp PCR method.
Acknowledgements
We thank all medical and ancillary staff of the hospital and the patients for consenting to participate. We thank Jeremy Allen, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Abbreviations
- BAM
Binary SAM
- BNA
Bridged nucleic acid
- BNA-clamp PCR
bridged nucleic acid-clamp real-time PCR
- BRAF
v-raf murine sarcoma viral oncogene homolog B1
- CAP
College of American Pathologists
- EGFR
Epidermal growth factor receptor
- FFPE
Formalin-fixed paraffin embedded
- KRAS
Kirsten rat sarcoma viral oncogene homolog
- NGS
Next generation sequencing
- NRAS
Neuroblastoma RAS viral oncogene homolog
- PCR-rSSO
PCR–reverse sequence-specific oligonucleotide probe
- SA-PE
Phycoerythrin-labeled streptavidin
- UNG
Uracil-N-glycosylase
Authors’ contributions
YN and YH drafted the work and wrote the manuscript. YN, YH and KA performed experiments, acquired and interpreted data. TO participated in pathological examination and acquisition of data. YH, HM, and MO designed of the work and supervised this study. MO was involved in the final editing. All authors have read and approved the final manuscript.
Funding
This study was supported by a Grant-in-Aid for Genome Research Project from Yamanashi Prefecture (to M.O. and Y.H.), The Japan Society for the Promotion of Science (JSPS) KAKENHI Early-Career Scientists (Grant Number JP18K16292 to Y.H.), a Research Grant for Young Scholars (to Y.H.), The YASUDA Medical Foundation (to Y.H.) and The Uehara Memorial Foundation (to Y.H.). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Written informed consent was obtained from all participants and this study was approved by the Institutional Review Board of clinical research and genome research committee at Yamanashi Central Hospital. The study complied with Declaration of Helsinki principles.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuki Nagakubo and Yosuke Hirotsu contributed equally to this work.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12920-019-0610-8.
References
- 1.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 2.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from Cetuximab in advanced colorectal Cancer. N Engl J Med. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 3.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-refractory metastatic colorectal Cancer. N Engl J Med. 2004;351(4):337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 6.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 7.Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015;26(1):13–21. doi: 10.1093/annonc/mdu378. [DOI] [PubMed] [Google Scholar]
- 8.De Roock W, Claes B, Bernasconi D, De SJ, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 9.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50(5):307–312. doi: 10.1002/gcc.20854. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torigoe H, Hari Y, Sekiguchi M, Obika S, Imanishi T. 2′-O,4′-C-methylene bridged nucleic acid modification promotes pyrimidine motif triplex DNA formation at physiological pH: thermodynamic and kinetic studies. J Biol Chem. 2001;276(4):2354–2360. doi: 10.1074/jbc.M007783200. [DOI] [PubMed] [Google Scholar]
- 12.Rahman S, Seki S, Obika S, Yoshikawa H, Miyashita K, Imanishi T. Design, synthesis, and properties of 2′,4′-BNA (NC): a bridged nucleic acid analogue. J Am Chem Soc. 2008;130(14):4886–4896. doi: 10.1021/ja710342q. [DOI] [PubMed] [Google Scholar]
- 13.Shivarov V, Ivanova M, Naumova E. Rapid detection of DNMT3A R882 mutations in hematologic malignancies using a novel bead-based suspension assay with BNA (NC) probes. PLoS One. 2014;9(6):e99769. doi: 10.1371/journal.pone.0099769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morishita S, Takahashi K, Araki M, Hironaka Y, Sunami Y, Edahiro Y, Tsutsui M, Ohsaka A, Tsuneda S, Komatsu N. Melting curve analysis after T allele enrichment (MelcaTle) as a highly sensitive and reliable method for detecting the JAK2V617F mutation. PLoS One. 2015;10(3):e0122003. doi: 10.1371/journal.pone.0122003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iida T, Mizuno Y, Kaizaki Y. Real-time PCR-based method for the rapid detection of extended RAS mutations using bridged nucleic acids in colorectal cancer. Clin Chim Acta. 2019;489:164–168. doi: 10.1016/j.cca.2017.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Mizuki N, Ohno S, Sugimura K, Seki T, Kikuti Y, Ando A, Ota M, Tsuji K, Inoko H. PCR-RFLP is as sensitive and reliable as PCR-SSO in HLA class II genotyping. Tissue Antigens. 1992;40(2):100–103. doi: 10.1111/j.1399-0039.1992.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 17.Iijima Y, Hirotsu Y, Amemiya K, Ooka Y, Mochizuki H, Oyama T, Nakagomi T, Uchida Y, Kobayashi Y, Tsutsui T, et al. Very early response of circulating tumour-derived DNA in plasma predicts efficacy of nivolumab treatment in patients with non-small cell lung cancer. Eur J Cancer. 2017;86:349–357. doi: 10.1016/j.ejca.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Sakamoto I, Hirotsu Y, Nakagomi H, Ouchi H, Ikegami A, Teramoto K, Amemiya K, Mochizuki H, Omata M. BRCA1 and BRCA2 mutations in Japanese patients with ovarian, fallopian tube, and primary peritoneal cancer. Cancer. 2016;122(1):84–90. doi: 10.1002/cncr.29707. [DOI] [PubMed] [Google Scholar]
- 19.Amemiya K, Hirotsu Y, Goto T, Nakagomi H, Mochizuki H, Oyama T, Omata M. Touch imprint cytology with massively parallel sequencing (TIC-seq): a simple and rapid method to snapshot genetic alterations in tumors. Cancer Med. 2016;5(12):3426–3436. doi: 10.1002/cam4.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goto T, Hirotsu Y, Nakagomi T, Shikata D, Yokoyama Y, Amemiya K, Tsutsui T, Kakizaki Y, Oyama T, Mochizuki H, et al. Detection of tumor-derived DNA dispersed in the airway improves the diagnostic accuracy of bronchoscopy for lung cancer. Oncotarget. 2017;8(45):79404–79413. doi: 10.18632/oncotarget.18159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goto T, Hirotsu Y, Amemiya K, Nakagomi T, Shikata D, Yokoyama Y, Okimoto K, Oyama T, Mochizuki H, Omata M. Distribution of circulating tumor DNA in lung cancer: analysis of the primary lung and bone marrow along with the pulmonary venous and peripheral blood. Oncotarget. 2017;8(35):59268–59281. doi: 10.18632/oncotarget.19538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goto T, Hirotsu Y, Mochizuki H, Nakagomi T, Oyama T, Amemiya K, Omata M. Stepwise addition of genetic changes correlated with histological change from "well-differentiated" to "sarcomatoid" phenotypes: a case report. BMC Cancer. 2017;17(1):65. doi: 10.1186/s12885-017-3059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goto T, Hirotsu Y, Mochizuki H, Nakagomi T, Shikata D, Yokoyama Y, Oyama T, Amemiya K, Okimoto K, Omata M. Mutational analysis of multiple lung cancers: discrimination between primary and metastatic lung cancers by genomic profile. Oncotarget. 2017;8:31133–31143. doi: 10.18632/oncotarget.16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirotsu Y, Kojima Y, Okimoto K, Amemiya K, Mochizuki H, Omata M. Comparison between two amplicon-based sequencing panels of different scales in the detection of somatic mutations associated with gastric cancer. BMC Genomics. 2016;17(1):833. doi: 10.1186/s12864-016-3166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Mochizuki H, Omata M. Detection of BRCA1 and BRCA2 germline mutations in Japanese population using next-generation sequencing. Mol Genet Genomic Med. 2015;3(2):121–129. doi: 10.1002/mgg3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirotsu Y, Zheng TH, Amemiya K, Mochizuki H, Guleng B, Omata M. Targeted and exome sequencing identified somatic mutations in hepatocellular carcinoma. Hepatol Res. 2016;46(11):1145–1151. doi: 10.1111/hepr.12663. [DOI] [PubMed] [Google Scholar]
- 27.Takaoka Shinya, Hirotsu Yosuke, Ohyama Hiroshi, Mochizuki Hitoshi, Amemiya Kenji, Oyama Toshio, Ashizawa Hiroshi, Yoshimura Dai, Nakagomi Keiko, Hosoda Kenji, Suzuki Yoji, Kojima Yuichiro, Omata Masao. Molecular subtype switching in early-stage gastric cancers with multiple occurrences. Journal of Gastroenterology. 2019;54(8):674–686. doi: 10.1007/s00535-019-01547-z. [DOI] [PubMed] [Google Scholar]
- 28.Amemiya K, Hirotsu Y, Oyama T, Omata M. Relationship between formalin reagent and success rate of targeted sequencing analysis using formalin fixed paraffin embedded tissues. Clin Chim Acta. 2019;488:129–134. doi: 10.1016/j.cca.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Yoshino T, Muro K, Yamaguchi K, Nishina T, Denda T, Kudo T, Okamoto W, Taniguchi H, Akagi K, Kajiwara T, et al. Clinical validation of a multiplex kit for RAS mutations in colorectal Cancer: results of the RASKET (RAS KEy testing) prospective, Multicenter Study. EBioMedicine. 2015;2(4):317–323. doi: 10.1016/j.ebiom.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taniguchi H, Okamoto W, Muro K, Akagi K, Hara H, Nishina T, Kajiwara T, Denda T, Hironaka S, Kudo T, et al. Clinical validation of newly developed multiplex kit using Luminex xMAP Technology for Detecting Simultaneous RAS and BRAF mutations in colorectal Cancer: results of the RASKET-B study. Neoplasia. 2018;20(12):1219–1226. doi: 10.1016/j.neo.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kojima Y, Hirotsu Y, Omata W, Sugimori M, Takaoka S, Ashizawa H, Nakagomi K, Yoshimura D, Hosoda K, Suzuki Y, et al. Influence of NUDT15 variants on hematological pictures of patients with inflammatory bowel disease treated with thiopurines. World J Gastroenterol. 2018;24(4):511–518. doi: 10.3748/wjg.v24.i4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirotsu Y, Nakagomi H, Amemiya K, Oyama T, Inoue M, Mochizuki H, Omata M. Intrinsic HER2 V777L mutation mediates resistance to trastuzumab in a breast cancer patient. Med Oncol. 2017;34(1):3. doi: 10.1007/s12032-016-0857-2. [DOI] [PubMed] [Google Scholar]
- 33.Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Oyama T, Mochizuki H, Omata M. Multigene panel analysis identified germline mutations of DNA repair genes in breast and ovarian cancer. Mol Genet Genomic Med. 2015;3(5):459–466. doi: 10.1002/mgg3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirotsu Y, Ooka Y, Sakamoto I, Nakagomi H, Omata M. Simultaneous detection of genetic and copy number alterations in BRCA1/2 genes. Oncotarget. 2017;8(70):114463–114473. doi: 10.18632/oncotarget.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takano A, Hirotsu Y, Amemiya K, Nakagomi H, Oishi N, Oyama T, Mochizuki H, Omata M. Genetic basis of a common tumor origin in the development of pancreatic mixed acinar-neuroendocrine-ductal carcinoma: a case report. Oncol Lett. 2017;14(4):4428–4432. doi: 10.3892/ol.2017.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyashita Y, Hirotsu Y, Tsutsui T, Higashi S, Sogami Y, Kakizaki Y, Goto T, Amemiya K, Oyama T, Omata M. Analysis of significantly mutated genes as a clinical tool for the diagnosis in a case of lung cancer. Respir Med Case Rep. 2017;20:171–175. doi: 10.1016/j.rmcr.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakagomi T, Goto T, Hirotsu Y, Shikata D, Yokoyama Y, Higuchi R, Amemiya K, Okimoto K, Oyama T, Mochizuki H, et al. New therapeutic targets for pulmonary sarcomatoid carcinomas based on their genomic and phylogenetic profiles. Oncotarget. 2018;9(12):10635–10649. doi: 10.18632/oncotarget.24365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amemiya K, Hirotsu Y, Oyama T, Omata M: Simple and rapid method to obtain high-quality tumor DNA from clinical-pathological specimens using touch imprint cytology. J Vis Exp. 2018(133). 10.3791/56943. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Principle of BNA-clamp PCR. Forward and reverse primers amplify the targeted mutation. A BNA probe binds to the wild-type allele but not to the mutated allele. The BNA probe selectively inhibits PCR amplification of the wild-type allele. F, fluorescence; Q, quencher.
Additional file 2: Figure S2. Sequence reads were visualized by Ion Reporter Genome Viewer. Representative images of read alignments (BAM files) of sample #2 were visualized with Ion Reporter Genome Viewer. There are no mutated reads corresponding to NRAS p.G12C (c.34G > T: chr1:115,258,748) in the next generation sequencing data.
Additional file 3: Figure S3. Amplification plot of dilution experiment by BNA-clamp PCR method. Wild-type control DNA was spiked in the Tru-Q 7 (1.3% Tier) Reference Standard. BNA-clamp PCR was performed using serial dilution DNA. (A) DNA containing KRAS mutation at codon 12/13 (dilution range: 1–33% variant allele fraction) and (B) BRAF mutation at codon 600 (dilution range: 0.4–12% variant allele fraction).
Additional file 4: Table S1. Threshold cycle values of real-time PCR using BNA-clamp PCR method.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.