

Summary
Inflammatory macrophages play a critical role in gut and extra-gut inflammatory disorders, which may be promoted through the dysbiosis of gut microbiota. However, it is poorly understood how gut microbiota affect inflammatory macrophages. Here, we found that increased Escherichia coli (E. coli) in inflamed colon may induce inflammatory macrophages in gut and extra-gut tissues. These E. coli are different from other commensal and pathogenic E. coli in genomic components and also in ability to induce inflammatory responses. Dominant E. coli from colitic tissues induce gut inflammatory macrophages through a regulating network consisted of IL-18, IFN-γ, IL-12, and IL-22 in gut tissues. These E. coli also directly activate macrophages. Cytosolic inflammasome components PCKδ, NLRC4, caspase8, and caspase1/11 are involved in E. coli-mediated activation in both gut epithelial cells and macrophages. These disclose a novel mechanism for how dysbiosis of gut microbiota in colitis cause inflammatory macrophages related to multiple diseases.
Subject Areas: Biological Sciences, Pathophysiology, Immunology, Microbiology
Graphical Abstract
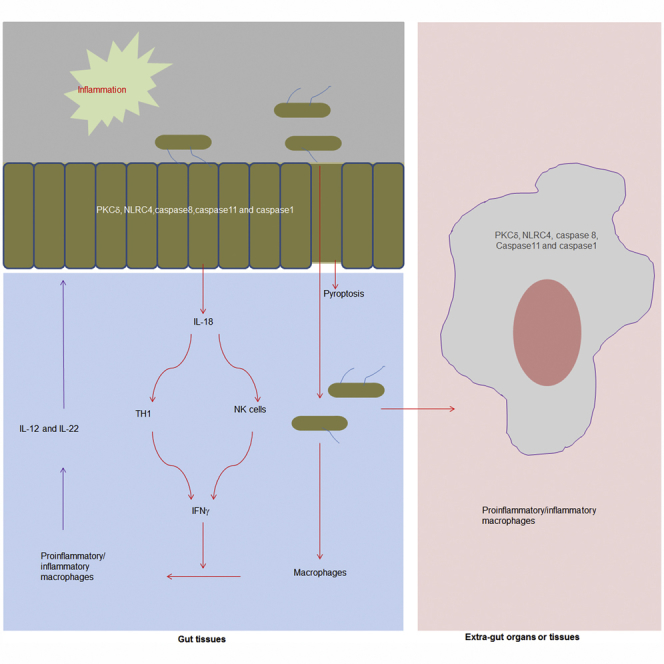
Highlights
-
•
Increased commensal E. coli in colitis induce inflammatory macrophages
-
•
Colitic E. coli are different from other commensal and pathogenic E. coli
-
•
Gut inflammatory macrophages by E. coli need IL-18, IFN-γ, IL-12, and IL-22
-
•
PCKδ, NLRC4, caspase8, and caspase1/11 are required for E. coli-mediated activation
Biological Sciences; Pathophysiology; Immunology; Microbiology
Introduction
The dysbiosis (aberrant gut microbiota composition and function) of gut microbiota may promote gut and extra-gut autoimmune and inflammatory disorders such as inflammatory bowel disease (IBD), obesity, atherosclerosis, carcinogenesis, etc (Blander et al., 2017). Although the mechanisms involved are not well understood, the inflammatory macrophages have a causal association with these diseases (Sekirov et al., 2010, Wynn et al., 2013). Thus it is critical to understand how gut microbiota regulate these macrophages.
Tissue-resident macrophages represent a highly heterogeneous cell population able to sense and quickly adapt to environmental cues such as gut tissue macrophages, which play either protective or tolerogenic roles. In steady state conditions, the gut lamina propria (LP) macrophages display an anergic phenotype and are essential for intestinal homeostasis (De Schepper et al., 2018); but under inflammatory settings such as DSS-mediated colitis, the conditioning of murine Ly6C+ blood monocytes is impaired, and they give rise to inflammatory macrophages (Zigmond et al., 2012). These inflammatory macrophages produce large amounts of mediators such as TNFα, IL6, IL-1β, reactive oxygen intermediaries, and nitric oxide to cause diseases (MacDonald et al., 2011). Thus, the transformation of suppressive macrophages back into proinflammatory phenotype or inflammatory macrophages into anti-inflammatory cells has a major impact on the progression and resolution of the inflammation-associated diseases. It is unclear how the transformation of these macrophages is induced and maintained in these diseases. Alterations in the microbiome population and/or changes in gut permeability may promote microbial translocation into the distal tissues and/or organs. Danger signals derived from the microbiome can trigger the inflammatory cascade and activate macrophages to transform into inflammatory macrophages. However, what danger signal(s) of gut microbiota induce inflammatory macrophages remains poorly understood.
Certain members of the microbiota have been linked to inflammatory responses and intestinal pathology in mouse models such as that the members of the Enterobacteriaceae family, Klebsiella pneumoniae and Proteus mirabilis (Garrett et al., 2010). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis (Garrett et al., 2010). E. coli, another member of Enterobacteriaceae family, is present in very less proportion in gut contents under normal physiological conditions (Schieber et al., 2015). However, a high abundance of commensal E. coli (facultative anaerobic Proteobacteria in phylum and Enterobacteriaceae in genus) is commonly observed during inflammation in the colon (Winter and Baumler, 2014), including chemically induced colitis, antibiotic-treated mice, infection with enteric pathogens, and genetically induced colitis (Winter and Baumler, 2014). Microbial communities in patients with inflammatory bowel diseases also exhibit an increased prevalence of E. coli (Winter and Baumler, 2014). However, the physiological and pathological function(s) of these E. coli are poorly understood. One isolated E. coli strain from antibiotic-treated mice may cause lethal inflammasome activation (Ayres et al., 2012), whereas another strain E. coli may protect mice against muscle wasting and loss of fat during enteric Salmonella typhimurium or respiratory Burkholderia thailandensis infections (Schieber et al., 2015). Here, we found that a high abundance of commensal E.coli in inflamed colon not only indirectly induce inflammatory macrophages through gut epithelial cells but also directly activate extra-gut macrophages through cytosolic inflammasome complexes consisted of PCKδ (phosphoenolpyruvate carboxykinase δ), NLRC4 (NLR family CARD domain-containing protein 4), caspase8, and caspase1/11. These inflamed tissues derived E. coli do not cause acute disease symptoms.
Results
E. coli O160:H7 Isolated from Inflamed Colon Promotes Sensitivity to DSS-mediated Colitis
To characterize inflammation-mediated E. coli, we employed chemically induced colitis (dextran sulfate sodium [DSS]-mediated colitis), in which there is a relative luminal abundance of Proteobacteria phylum (Enterobacteriaceae genus, E. coli species) (Schieber et al., 2015). Consistent with this report, the increased gut Proteobacteria phylum, Enterobacteriaceae genus, and E. coli was detected in the colonic contents and tissues of DSS-treated mice (Figures 1A and 1B and https://www.ncbi.nlm.nih.gov/sra/PRJNA512937). Using culturing techniques, serotyping, and genetic and molecular characterization, we identified a dominant E. coli strain from these inflamed colon tissues, named as E.coli O160:H7 strain (Figures S1A–S1C, 1C, and 1D, Table S1A and http://www.ncbi.nlm.nih.gov/bioproject/513139). E. coli O160: H7 strain was also present in the microbiota of unmanipulated mice but was not abundant, suggesting it is not able to compete efficiently for intestinal colonization. We next sequenced the genome of E. coli O160:H7 isolate and aligned the reads to reference E. coli genomes (Table S1B). The composition of E. coli O160:H7 gene clusters was different from other pathogenic E. coli O157:H7 and E. coli CFT073 and also unpathogenic E. coli str.k12 substr.MG1655 (Figures S1B and S1C). The fliC gene, encoding flagellin (H-antigen), was similar to that of E. coli O157:H7 isolates (Figure S1D). But, type III secretion system (T3SS) of E. coli O160:H7 was different from pathogenic E. coli O157.H7 such that T3SS of E. coli O160:H7 contained hxlB, irp1, HMWP1, pqqL, hokA, fhaB, fdoG, fdfH, ttuB, bax, PTS-Dga, EIID, dgaD, glmS, GFPT, ABC-2, and CPSE.A, which were not detected in E. coli O157:H7 (Table S1C). Notably, we did not find virulence-related membrane protein genes such as enterotoxin, EspB, EspA, SepZ, SepD, Hcp-like protein, protein TerZ, protein TerA, protein TerF, prohead protease, and antirepressor protein in E. coli O160:H7 isolate, which were encoded by E. coli O157:H7 (Table S1C). T3SS of E.coli O160:H7 was different from other unpathogenic E.coli str.k12.substr.MG1655 and pathogenic E.coli CFT073 (Table S1C). E.coli O160:H7 also encoded type IV secretion system (T4SS) (Table S1D) and other factors, including those for adhesion such as fim gene cluster (fimA, fimB, fimC, fimD, fimE, fimF, fimH, fimG, fimI, etc) and pil gene cluster (pilD, pilT), papC, and papD, and internalization gene such as csg etc. (Table S1D). However, other disease-associated factors such as Afa/Dr adhesins, traA (encoding pilin), and malX (marker for pathogenicity-associated island from strain CFT073), which were found in patients (Mansan-Almeida et al., 2013), was not detected in E.coli O160:H7 (Table S1D). E.coli O160:H7 also had multiple drug-resistant genes such as oprM, emhC, ttgC, cusC, adeK, smeF, mtrE, cmeC, gesC, acrA, mexA, adeI, smeD, mtrC, and cmeA (http://www.ncbi.nlm.nih.gov/bioproject/513139). Taken together, the gene composition of genome in E.coli O160:H7 is different from other identified pathogenic and unpathogenic E. coli.
Figure 1.

Characteristics of E. coli O160:H7 Isolated from Inflamed Colon Tissues
(A and B) 16s rDNA analyses of colon contents in DSS-treated wt (male, n = 5) and un-molested control mice (male, n = 5). The samples were clustered at phylum levels using the sample phylum count matrices and composition of colon bacteria (phylum levels) in control (A) and DSS-treated (B) mice. Mice were fed a 2.5% DSS solution in drinking water for 7 days.
(C) Fluorescent in situ hybridization (FISH) of E. coli in colon tissues of DSS-treated and un-molested mice (one representative, n = 6). Red, E. coli; Green, mucus; Blue, nuclei.
(D) Percent of E. coli O160:H7 clones in colitic tissues. The bacteria from colon tissues of DSS-treated and un-molested mice were in vitro cultured and then CFU of bacteria were sequenced through V1-V9 regions (n = 6).
(E and F) Survival rate (E), body weight, and disease activity index (F) in DSS-treated mice infused by E. coli O160:H7, heat-killed dead (killed) E. coli O160:H7, and E. coli IAI39 (isolated from mice by us) (n = 12). Mice were treated using pan-antibiotics for one week before infusing E. coli. Data in F are represented as mean ± SD.
(G) Length of colon were monitored at day 7 after DSS. Data are represented as mean ± SD.
(H) H&E staining and histological scores of colon tissues in DSS-treated mice infused by E. coli O160:H7 killed E. coli O160:H7. Scale bars = 40 μm. Data are represented as mean ± SD.
Two-side student's t-test in D; Kruskal Wallis test in E; analysis of variance test in F; ANOVA plus post-Bonferroni analysis in G and H; *p<0.05, **p<0.01, and ***p<0.001; NS, no significance. Data in D and H are represented as mean ± SEM. Data in F and G are represented as mean ± SD. Data in E–H are a representative of three independent experiments. See also Figure S1, Table S1, and https://www.ncbi.nlm.nih.gov/sra/PRJNA512937.
We next examine whether E. coli O160:H7 may cause pathological responses in gut tissues. Although E. coli O160:H7 were infused into wt mice, pan-antibiotic–treated wt mice and germ-free (GF) mice, no remarkable symptoms of acute gut diseases such as diarrhea, colonic bleeding, and reduced body weight were observed, consistent with the genome sequencing data that virulence-related genes are not detected in E. coli O160:H7. However, oral administration of E. coli O160:H7 promoted sensitivity to DSS-mediated colitis (Figures 1E–lH). This E.coli O160:H7 isolate was much more effective than un-dominant E. coli IAI39 strain isolated from same mice in promoting sensitivity to DSS-mediated colitis (Figures S2A–S2E). E. coli O160:H7was also different from other identified pathogenic E. coli. It was weaker than E. coli CFT073 but stronger than unpathogenic E. coli such as E. coli Str. k12.Substr.MG1655 and E. coli Nissle 1917 (Figures S2F–S2K). Oral administration of these E. coli resulted in high levels of colonization (Figures S2D and S2I)). Notably, E. coli O160:H7 isolate and other pathogenic and unpathogenic E.coli had a similar ability in inducing TLR4-mediated NF-κB activity (Figure S2L), implying that difference of O160:H7 with other gram-negative E.coli in promoting sensitivity to DSS-mediated colitis may not depend on LPS. Taken together, E.coli O160:H7 from inflamed colonic tissues promotes sensitivity to DSS-mediated colitis, but it is different from other pathogenic and unpathogenic E. coli.
E. coli O160:H7 Induces Inflammatory Macrophages in Gut and Extra-gut Tissues
To elucidate how E. coli O160:H7 promotes sensitivity to DSS-mediated colitis, we first examined the composition of gut immune cells in DSS-treated mice. There had remarkably increased F4/80+CD11B+, F4/80+CD11C+, and F4/80+TNFα+ macrophages in the colon lamina propria (LP) (Figure 2A) and higher levels of inflammatory cytokines in the colonic tissues of mice (Figure 2B), because CD11C and TNFα generally are markers of inflammatory macrophages (Bain et al., 2013), suggesting that there may exist increased inflammatory macrophages in the gut tissues of DSS-treated mice. For gut macrophage subsets, previous multiple studies (Bain et al., 2013, Mortha et al., 2014, Shouval et al., 2014, Tamoutounour et al., 2012) suggest that CX3CR1+CD11b+CD103−F4/80+Ly6C+MHCII+ cells belong to proinflammatory/inflammatory macrophages (P2 stage), whereas CX3CR1+ CD11b+CD103−F4/80+Ly6C−MHCII+cells as anti-inflammatory macrophages (P3 and P4 stage) (Figure S3A). We further investigated the gut macrophage subpopulations using this classification, which was used through this manuscript. The proportion of CD45+CX3CR1+CD11b+CD103−F4/80+ MHCII+Ly6C+ inflammatory macrophages remarkably increased in the DSS-treated mice (Figure 2C), suggesting that E. coli from inflamed colon may induce the inflammatory macrophages. To determine that E. coli O160:H7 may induce inflammatory macrophages, we employed E. coli-infused mice including broad-spectrum antibiotics AVNM (ampicillin, vancomycin, neomycin, and metronidazole)-treated mice, GF mice, and untreated normal mice. In E. coli-infused non-antibiotics wt mice, proinflammatory macrophages did not remarkably increase as compared with mice uninfused with E. coli (unshown). But, the colonization of E. coli O160:H7 isolate could cause significantly increased inflammatory macrophages in AVNM-treated mice and GF mice, in which there existed the dysbiosis of gut microbiota (Figures 2D–2H). There was also significantly increased inflammatory macrophages in extra-gut organs and tissues with higher levels of inflammatory cytokines in peripheral blood in E. coli-infused mice, in which E. coli were detected (Figures 2I–2K). Thus, although it does not cause remarkable bowel disease symptoms, E. coli from the inflamed colon may induce inflammatory macrophages not only in colon tissues but also in extra-gut organs and tissues under the dysbiosis of gut microbiota.
Figure 2.

E. coli O160:H7 Induces the Accumulation of Inflammatory Macrophages in Colon Tissues
(A) Flow cytometry of F4/80+CD11b+, F4/80+CD11c+, and F4/80+TNFα+ cells in DSS-treated and unmolested mice (n = 6).
(B) QRT-PCR of TNFα, IL-6, IL-1β, INOS, arginase-1, and IL-10 in the colon tissues of DSS-treated and unmolested mice (n = 6).
(C) Flow cytometry of MHCII+Ly6C+ inflammatory macrophages (CD45+CX3CR1+CD11b+CD103−F4/80+ MHCII+Ly6C+) in the colon LP of DSS-treated and unmolested mice (n = 6). % cells and total Ly6c + MHCII+ cell number per colon were analyzed (right).
(D) Flow cytometry of inflammatory macrophages in the colon LP of mice with or without different E. coli infusion (n = 3). % cells and total Ly6c + MHCII+ cell number per colon were analyzed (right).
(E) CFU of bacteria in colon tissues of mice infused different E. coli.
(F) Flow cytometry of inflammatory macrophages in the colon LP of E. coli colonized GF mice (n = 6). GF/pseudo, pseudomonas-colonized mice; GF/E.coli, E. coli O160-colonized mice; GF/killed E.coli, dead E. coli O160 infused mice. % cells and total Ly6c + MHCII+ cell number per colon were analyzed (right).
(G) Flow cytometry and immunostaining of F4/80+TNFα+ cells in the colon tissues of GF mice with or without E. coli infusion (n = 6). % cells and total F4/80+TNFα+ cell number per colon were analyzed (right). Scale bars = 40 μm.
(H) CFU of bacteria in intestine and colon tissues of E. coli infused GF mice.
(I) Flow cytometry of F4/80+TNFα+ cells in the spleen and liver of mice with or without E. coli infusion (n = 6).
(J) ELISA of IL-18, IL-1β, and TNFα in the sera of mice with or without E. coli infusion (n = 6).
(K) CFU of bacteria in intestine and colon tissues of mice infused E. coli.
Mice in A–C, untreated using pan-antibiotics. Mice in D and I–K, treated using pan-antibiotics for one week before infusing bacteria. For CFU in bacteria infused mice, 109 bacteria were orally infused and then CFU were counted after 7 days.
Scale bars = 40 μm in G. ANOVA plus post-Bonferroni analysis in D and F; Two-side Student's t-test in A– C, G, I, and J; *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A–K are represented as mean ± SD. Data in A–K are a representative of two or three independent experiments. See also Figure S3A.
IL-18, IFNγ, IL-12, and IL-22 Are Required in E. coli-mediated Gut Inflammatory Macrophages
We next want to understand how E. coli O160:H7 isolate induces inflammatory macrophages in colon tissues. Intestinal mononuclear phagocytes do not or only slightly produce inflammatory responses when stimulated with TLR (toll-like receptor) ligands, commensal, or pathogenic bacteria (Franchi et al., 2012). However, IFNγ may promote the generation of inflammatory macrophages (Hu and Ivashkiv, 2009). Remarkably increased IFNγ was detected not only in DSS-mediated colitis but also in E.coli O160:H7 colonized colon tissues as compared with their control mice, whereas other anti-inflammatory cytokines such as IL-4 did not significantly change (Figure 3A). There also existed a drastic expansion of interferon γ (IFNγ)-producing CD4+ T helper cells (Th1) cells (CD4+IFNγ+Th1 cells) and NKp46+ IFNγ+ cells in the LP tissues of DSS-mediated colitis and E. coli O160:H7-colonized mice (Figures 3B–3D). Because Ly6C+ monocytes can give rise to a CCR7-expressing CX3CR1int Ly6Clo cell population capable of migrating into lymph node and priming T cells toward Th1 under inflammatory conditions, increased CD4+Ki67+ T and CD4+IFNγ+cells were also detected in the PP (Figure 3E). The colonization of E. coli O160:H7 in IFNγ−/− mice did not cause accumulated inflammatory macrophages in the colonic LP (Figures 3F and 3G). Thus, our results demonstrate that gut IFN-γ plays a critical role in E. coli O160:H7-mediated inflammatory macrophages in colonic tissues.
Figure 3.

E. coli O160:H7-mediated Inflammatory Macrophages Depends on IFN in Gut Tissues
(A) QRT-PCR of IFNγ and IL4 in the colon tissues of DSS-treated (WT/DSS) and unmolested mice (WT/Vehicle) or GF mice with (GF/E. coli) or without E.coli (Vehicle) infused GF (n = 6).
(B) Flow cytometry of CD4+IFNγ+ and CD4−NKp46+cells in colon tissues of DSS-treated (WT/DSS) and unmolested (WT/Vehicle) mice (n = 6).
(C) Flow cytometry of CD45+CD4+IFNγ+ cells in the colon LP of E. coli infused GF mice (n = 6). GF/killed E.coli, dead E.coli infused GF mice.
(D) Immunostaining of CD4+IFNγ+ cells in colon tissues of E. coli infused GF mice (n = 6).
(E) Flow cytometry of CD4+Ki67+ and CD4+IFNγ+ cells in the payer's patch of DSS-treated and un-treated mice (n = 6).
(F) Flow cytometry of inflammatory macrophages in the colon LP of IFNγ−/− mice infused (E. coli) or uninfused (Vehicle) E.coli O160:H7 (n = 6). % cells and total Ly6c + MHCII+ cell number per colon were analyzed (right).
(G) CFU in intestine and colon tissues. GF mice were infused with 109E. coli and then CFU were counted after 7 days (n = 6).
Scale bars = 40 μm in D. Two-side student's t-test in A–C and E–G; *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A, B, C, E, F, and G are represented as mean ± SD. Data are a representative of two or three independent experiments. See also Figure S3.
IL-18 has been shown to play an important role in the induction of IFNγ production, increasing NK cell activity and T cell proliferation (Nielsen et al., 2016). There also are substantial evidences for the expression and secretion of IL-18 by the intestinal epithelium. Thus, we detected whether the accumulated IFNγ producing cells were related to IL-18 in the gut epithelial cells. Indeed, the increased IL-18 was detected in the gut epithelial cells of E. coli O160:H7-colonized mice and also DSS-induced colitic tissues (Figures 4A and 4B). More mature IL-18 was also detected in crypt supernatants after in vitro stimulation by E.coli O160:H7 (Figures 4C and 4D). The colonization of E. coli O160:H7 in IL-18 −/− mice did not cause the accumulation of inflammatory macrophages in the colonic LP (Figures 4E and 4F). In addition, both IL-22 and IL-12 also induce IL-18 expression in epithelial cells during intestinal infection (Munoz et al., 2015). Higher levels of IL-22 and IL-12 were detected in the gut tissues of E. coli-colonized mice (Figure S3B). IL-22 and IL-12 blocking reduced the expression of IL-18 and inhibited the accumulation of inflammatory macrophages (Figure S3C). Finally, we also compared the ability of E. coli O160:H7 with that of other E. coli strains in inducing mature IL-18. E. coli O160:H7 induced more IL-18 production than other unpathogenic bacteria but weaker as compared with pathogenic E. coli CFT073 in colon epithelial cells (Figure 4G). Taken together, gut epithelial cells derived IL-18 is involved in the increased inflammatory macrophages.
Figure 4.

E. coli O160:H7-mediated Inflammatory Macrophages depends on the Gut Epithelial Cells Derived IL-18
(A) ELISA of IL-18 in the colon tissues of mice with (DSS) or without DSS or GF mice infused with or without E. coli (n = 6, male). GF/killed E.coli, heat-killed E.coli infused mice.
(B) Immuno-staining of IL-18 in colon tissues of GF mice infused with E. coli (n = 6). GF/vehicle, only vehicle.
(C) ELISA of IL-18 in the supernatants of gut epithelial cells after exposed to E. coli O160:H7 or heat-killed E. coli O160:H7 (Killed E.coli) for 1 h.
(D) Immunoblotting of IL-18 in colon tissues, colon lamina propria, crypts, and crypt supernatants after exposed to E. coli O160:H7 or heat-killed E. coliO160:H7 (killed E.coli). Cytokeratin 19 (CK19) is a marker of epithelial cells; CD11b is a marker of macrophages; actin is a loading control.
(E) Flow cytometry of inflammatory macrophages and CD4+IFNγ+ cells in the colon LP of wt and IL-18−/− mice colonized E. coli O160:H7 (n = 6). % cells and total Ly6c + MHCII+ cell number per colon were analyzed (Lower).
(F) CFU/organ tissues in E. coli O160:H7 oral infused mice, which were treated with pan-antibiotics (AVNM) for 7 days.
(G) ELISA of IL-18 in the colon tissues of wt mice in response to different kinds of bacteria. E. coli 1917; O160, E.coli O160:H7, G1655, E. coli G1655; CFT073, E.coli CFT073.
Scale bars = 40 μm in B. Two-side Student's t-test in A (left), B and E; ANOVA plus post-Bonferroni analysis in A (right) and C; analysis of variance test in G. *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A–C and E–G are represented as mean ± SD. Data for all panels are a representative from two to three experiments.
PCKδ, NLRC4, caspase8, and caspase1/11 Are Required for E. coli O160:H7-Induced IL-18
Next question is how E. coli O160:H7 induces the expression of IL-18 in gut epithelial cells. The inactive 24 kDa precursor pro-IL-18 is constitutively expressed by gut epithelial cells and primed for release upon inflammasome activation. Gut epithelial cells have revealed an expression of an array of inflammasome components including NAIP, NLRP (NOD-like receptor protein) 1, NLRC4, NLRP6, AIM2, caspase1, caspase4/5 (human)/caspase11 (mouse), caspase8, ASC, and NLRP6/3 (von Moltke et al., 2013). Cytosolic pattern recognition receptors (PRRs) are often associated with the use of pore-forming toxins or injection of effecter molecules through specialized secretion systems of gram-negative bacteria (von Moltke et al., 2013), which are encoded by E. coli O160:H7. Because inner rod protein of type three secretion systems (TTSS) and functional flagellin (FliC) of gram-negative bacteria-mediated production of mature IL-18 mainly is through NLRC4/caspase1 signal pathway (Miao et al., 2010), we investigated the effects of NLRC4 and caspase1 on E.coli O160:H7-mediated mature IL-18. We found that NLRC4 and caspase1/11 was involved in E. coli O160:H7-mediated IL-18 release (Figures 5A–5C and S4A–S4C). More recent studies have revealed a requirement for caspase8 in activating caspase1 within the inflammasome complex (Man and Kanneganti, 2016). The caspase8 specific inhibitor did also affect E.coli O160:H7-mediated mature IL-18 (Figures 5D and 5E). The phosphorylation of NLRC4, which is activated by PKCδ in Salmonella infection, was necessary in macrophages (Qu et al., 2012). PKCδ inhibitor also impaired E.coli O160:H7-mediated mature IL-18 in gut epithelial cells (Figures 5D and 5E). Finally, immunoprecipitation further identified bioactive PCKδ, caspase1, caspase8, and ASC molecules being bound by NLRC4 in colon epithelial cells (Figure 5F), which are shown in macrophages infected with S. Typhimurium (Man et al., 2014). Interestingly, NLRC4 complexes also included caspase11 in colon epithelial cells (Figure 5F). It was also found that the noncanonical inflammasome also activates caspase11 in response to many gram-negative bacteria (Kayagaki et al., 2011). Critically, the colonization of E. coli O160:H7 in NLRC4 −/− or caspase1/11 −/− mice did not cause accumulated inflammatory macrophages in colonic LP (Figures 5G, 5H, and S4D). In addition, caspase11 and NLRC4 inflammasome activation in gut epithelial cells may lead to a lytic cell death, resembling pyroptosis (Rauch et al., 2017). There had increased PI+ cells (pyroptosis cells) in E.coli O160:H7 colonized GF mice, indicating that this strain of E. coli also induced pyroptosis of gut epithelial cells (Figure S5). As a result, this may promote the penetration of E. coli into extra-gut tissues. Taken together, we demonstrate that E. coli O160:H7 may induce the production of mature IL-18 through an inflammasome complex consisted of PKCδ, NLRC4, caspase8, and caspase1/11 in gut epithelial cells (Figure 5I).
Figure 5.

E. coli O160:H7 Induces IL-18 Through PCKδ/NLRC4/Caspase8/CASPASE11/1 Complexes in Colon Epithelial Cells
(A) ELISA of IL-18 in the supernatants of colon epithelial cells of wt, NLRC4−/−, or caspase1/11 −/− mice in response to E. coli O160:H7.
(B) Immunostaining of IL-18 in colon epithelial cells of wt, NLRC4−/−, and caspase1/11 −/− mice after infusing E. coli O160:H7 (n = 6). Scale bars = 40 μm.
(C) Immunoblotting in lamina propria, crypts, and crypt supernatants of colon tissues in wt and NLRC4 −/− mice after exposed to E. coli O160:H7 for 1 h.
(D) Immunoblotting in colon tissues, lamina propria, crypts, and crypt supernatants of wt mice after exposed to E. coli for 1 h with or without caspase8 and PKC inhibitors.
(E) ELISA of IL-18 in the supernatants of colon epithelial cells of wt mice after exposed to E. coli for 1 h with or without caspase8 and PKC inhibitors.
(F) Immunoprecipitation of NLRC4 after cross-linking in the colon epithelial cells of wt after exposed to E. coli O160:H7.
(G) Flow cytometry of inflammatory macrophages and CD4+IFNγ+ cells in the LP of colon tissues of NLRC4 −/− mice infused E. coli O160:H7 (n = 6).
(H) CFU/colon tissues in E. coli O160:H7 orally infused mice, which were treated with pan-antibiotics for 7 days.
(I) Model for E. coli O160:H7-mediated inflammatory macrophages. E. coli O160:H7 induces mature IL-18 in colon epithelial cells through PKCδ/NLRC4/Caspase8/caspase11/caspase1 complexes.
Scale bars = 40 μm in B. Two-side student's t-test in A, E, and G; *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A, E, G, and H are represented as mean ± SD; data in B are represented as mean ± SEM; data in all panels are a representative of two or three independent experiments. See also Figures S4 and S5.
E. coli O160:H7 Directly Induces IL-18 and IL-1β in Macrophages
We also observed effect(s) of E. coli O160:H7 isolate on the macrophages. E. coli O160:H7 directly activated macrophages to induce IL-1β and IL-18 in vitro (Figures 6A–6C). Furthermore, E. coli O160:H7-mediated production of IL-18 and IL-1β was also dependent on signal pathway consisted of PCKδ, NLRC4, caspase8, and caspase1/11 signal pathway in macrophages (Figures 6A–6E). Intravenous injection of E. coli O160:H7 into normal wt mice caused rapidly increased IL-18 and IL-1β in peripheral blood and accumulation of inflammatory macrophages in spleen and liver (Figures 6F and 6G). NLRC4−/− and caspase1/11 −/− and IL-18−/− impaired this ability of E. coli O160:H7 to induce the production of IL-1β and IL-18 (Figures 6F and 6G). These bacteria could effectively localize in these tissues and organs (Figure 6H). Meanwhile, we also found that E. coli O160:H7 was more effective in inducing mature IL-18 or IL-1β than other unpathogentic E. coli (Figures 6I and 6J). Thus, E. coli O160:H7 also directly induce production of IL-18 and IL-1β in macrophages through activating inflammasome complexes including PCKδ, NLRC4, caspase8, and caspase11/1.
Figure 6.

E. coli O160:H7 Directly Stimulates Macrophage to Produce IL-18 and IL-1 β
(A and B) ELISA of IL-18 and IL-1β in the supernatants of macrophages after exposed to E. coli O160:H7 at 3 h (A) and 24 h (B).
(C) Immunoblotting of pro-IL-1β, mature IL-1β, pro-caspase18, and mature IL-18 in the macrophages after exposed to E. coli for 1 h.
(D) IL-18 ELISA and immunoblotting after exposed to E. coli O160:H7 for 3 h in the presence of caspase8 and PKCδ inhibitor.
(E) Immunoblotting after exposed to E. coli O160:H7 with or without caspase8 and PKCδ inhibitors for 3 h.
(F) ELISA of IL-18, IL-1β, and TNFα in the peripheral blood of wt, NLRC4−/−, and caspase1/11−/− mice after injecting E. coli O160:H7 or heat-killed E. coli in tail vein at the indicated time (n = 6).
(G) Flow cytometry of F4/80+TNFα+ macrophages in spleen and liver of wt, NLRC4−/−, and caspase1/11 −/− mice after injecting E. coli or heat-killed E. coli in tail vein (n = 6).
(H) CFU of E. coli in the spleen, liver, and lung of wt, NLRC4−/−, and caspase1/11 −/− mice after injecting E. coli in tail vein.
(I) ELISA of IL-18 and IL-1β in the macrophages after exposed to DH5α, E.coli 1917, or E.coli O160:H7 for 3 h.
(J) Immunoblotting of in the macrophages after exposed to DH5α, E.coli nissle.1917 (1917); E. coli O160:H7 (O160).
ANOVA plus post-Bonferroni analysis in A, B, D, and F–I. *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A, B, D, F–I are represented as mean ± SD. Data are a representative of three independent experiments.
E. coli O55: HNT from Patients has Similar Function with E. coli O160:H7
We also investigate a dominant E. coli O55: HNT strain from colitic tissues of patients with inflammatory bowel disease. The increased E. coli could be detected in colitic tissues of patients with inflammatory bowel disease (Figures 7A and 7B), consistent with other data (Winter and Baumler, 2014). We found that the isolated E. coli O55: HNT from colitic tissues of patients with inflammatory bowel disease (Table S1E) had a similar function with mouse E. coli O160:H7 isolate. This strain E. coli O55: HNT also promoted sensitivity to DSS-mediated colitis (Figures 7C–7F) and induced inflammatory macrophages (Figure 7G). In vivo intravenously administration also caused increased inflammatory macrophages in the colonized tissues and organs (Figures 7H and 7I).
Figure 7.

Isolated E. coli 055:HNT from Colitic Tissues of Patients with Ulcerative Colitic Indirectly and Directly Induce Proinflammatory Macrophages
(A) H&E (Upper) and FISH (Lower) of healthy individuals and patients with ulcerative colitic (UC) and colon cancer (CC). Scale bars = 40 μm.
(B) Proportion of E.coli in colon tissues of healthy individuals (n = 17) and patients with ulcerative colitis (UC, n = 20) and colitic cancer (CC, n = 15), which were grown in LP plates.
(C and D) Survival rate (C), body weight, and disease index (D) were monitored in mice infused by E. coli 055:HNT after the start of DSS (n = 12).
(E) Length of colon were monitored in mice infused by E. coli 055:HNT after DSS treatment.
(F) H&E staining and histological scores of colon tissues in mice infused by E. coli 055: HNT after DSS treatment. Scale bars = 40 μm.
(G) Flow cytometry of inflammatory macrophages, F4/80+TNFα+ and CD4+IFNγ+ cells in mice infused by E. coli 055:HNT after DSS treatment (n = 5).
(H) Flow cytometry of F4/80+TNFα+ cells in the spleen and liver of wt mice after injecting E.coli 055:HNT for 24 h (n = 3).
(I) CFU of E. coli in the spleen and liver of mice after injecting different E. coli strains in tail vein for 24 h. 055, E. coli 055:HNT; O160, E.coli O160:H7, G1655, E. coli G1655; CFT073, E.coli CFT073.
Scale bars = 40 μm in A. ANOVA plus post-Bonferroni analysis in A and H; Two side Student's t-test in E– G; Wilcoxon's test in C; analysis of variance test in D; *p<0.05, **p<0.01, and ***p<0.001; NS, no significance; R. E, relative expression. Data in A and F are represented as mean ± SEM; data in D, E, G, H, and I are represented as mean ± SD. Data in C–I are a representative of two or three independent experiments. See also Figure S6.
We finally compared the effects of E. coli O55: HNT and E. coli O160:H7 strain with other identified pathogenic and unpathogenic E. coli on mortality and morbidity after oral administration and in vivo intravenous administration. Notably, E. coli O55: HNT was similar to E. coli O160:H7 strain but not to pathogenic E. coli such as E. coli CFT073 in mortality and morbidity. Although oral administration of E. coli CFT073 caused remarkable symptom of acute gut diseases, the mice administrated with E. coli O55: HNT did not exhibit detectable symptom (Figures S6A and S6B). Colon inflammation was observed only in E. coli CFT073 but not in E. coli O55: HNT or E. coli O160:H7 infused wt mice (Figures S6A and S6B). Although oral administration of E. coli in pan-antibiotics-treated mice or GF mice, E. coli O55: HNT or E. coli O160:H7 could cause symptom of acute gut diseases and colon inflammation but much slighter than E. coli CFT073 (Figures S6C and S6D). In in vivo intravenous administration mice, E. coli O55: HNT and E. coli O160:H7 could cause disease symptoms. However, these symptoms were remarkably slighter than pathogenic bacteria E. coli CFT073 (Figures S6E and S6F) although it is significantly severe than unpathogenic bacteria E. coli MG1655 (Figures S6E and S6F). Intravenous injection also exhibited tissue colonization pattern (Figure S6G). Taken together, there are remarkable differences in mortality and morbidity between E. coli O55:HNT and E. coli O160:H7 isolated from colitic tissues and other pathogenic E. coli.
Discussion
In this study, we found that a high abundance of commensal E. coli in inflamed colonic tissues are different from other unpathogenic commensal E. coli and also pathogenic E. coli in their genome, especially T3SS and virulent factors. These E. coli may induce inflammatory macrophages in the colon tissues and extra-gut tissues but not acute infection diseases. They stimulate gut epithelial cells to produce IL-18 through inflammasome complexes that consisted of PKC δ, NLRC4, caspase8, and caspase1/11. IL-18 derived from gut epithelial cells induces Th1- and NKp46+ IFNγ-producing cells, which are necessary for the generation of inflammatory macrophages. Meanwhile, higher levels of IL-12 and IL-22 in the colon tissues are also involved in E. coli-mediated inflammatory macrophages. The isolated E. coli not only induce gut inflammatory macrophages but also directly activate extra-gut macrophages to produce proinflammatory cytokines. There also have increased pyroptosis cells in the E. coli-colonized mice, which may potentially promote microbial translocation into the distal tissues and/or organs. These data disclose a new mechanism for how colitis associated gut microbiota to cause inflammatory macrophages in the gut and extra-gut tissues and organs. Since inflammatory macrophages are related to multiple systemic diseases such as inflammatory bowel disease (IBD), obesity, atherosclerosis, carcinogenesis, etc (Blander et al., 2017), our results imply that a high abundance of commensal E. coli in inflamed gut may play a role in the occurrence and development of these diseases. Thus, our data suggest a possible mechanism for the occurrence and development of chronic inflammation diseases, which are related to inflammatory macrophages.
Generally, gram-negative bacteria may activate inflammasomes through LPS-caspase11/1 and/or flagellin-NLRC4-caspase1 pathway to induce the production of mature IL-18 in macrophages and epithelial cells. However, several studies have exhibited difference of gram-negative bacteria in their ability to induce production of inflammatory cytokines. Pathogenic E. coli but not commensal bacteria can elicit substantial amounts of mature IL-1β by the NLRC4 pathway (Franchi et al., 2012, Lightfield et al., 2008). E. coli Nissle 1917 and commensal E. coli K12 also differentially affect the inflammasome in intestinal epithelial cells (Becker et al., 2014). We here also found that there exists a remarkable difference between inflamed colonic tissues derived E. coli and other unpathogenic and pathogenic E. coli in inducing inflammatory macrophages. Recently, E. coli strains from antibiotic-treated mice may cause lethal inflammasome activation through NLRC4 (Ayres et al., 2012), whereas another strain E. coli, which also activate NLRC4, may protect mice against muscle wasting and loss of fat during infections (Schieber et al., 2015). All of these may be derived from their genomic characteristics. Indeed, compared analyses of the genomes between inflamed colon tissues derived E. coli O160:H7 and other pathogenic and unpathogenic E. coli exhibit remarkable differences, especially in flagellin, rode-like proteins, and T3SS secreting system. Cytosolic PRRs (pattern recognition receptors) are critical for discriminating between pathogenic and nonpathogenic bacteria. Studies have found that cytosolic PRRs respond to patterns of pathogenesis that are often associated with virulent bacteria, such as the use of pore-forming toxins or injection of effector molecules through specialized secretion systems (von Moltke et al., 2013). The activation of NLRC4 inflammasome requires the presence of an intact type III (T3SS) or IV secretion system (T4SS) (Franchi et al., 2006). In addition, the release of T3SS PrgJ-like rod proteins into the cell cytosol can activate NLRC4. Thus, although the genetic factors of flagellin, rode-like protein, T3SS and/or IV secreting system change, these gram-negative E. coli may exhibit altered ability in inducing inflammatory cytokines and inflammation-associated diseases.
Our results suggest that gut epithelial cells exist in similar inflammasome complexes with macrophages to be involved in gram-negative bacteria (Qu et al., 2012). There exist multiple inflammasomes, which are broadly expressed in hematopoietic and non-hematopoietic cells, such as gut epithelial cells (Hu et al., 2010, Sellin et al., 2014), and can trigger numerous downstream responses including production of IL-1β, IL-18, and lytic cell death (Sellin et al., 2014). Despite the fact that the functional importance of inflammasomes within immune cells has been well established, the contribution of inflammasomes in non-hematopoietic cells remains comparatively understood. We here demonstrated that E. coli isolated from colitic tissues directly stimulate gut epithelial cells through inflammasome complexes that consisted of PKCδ, NLRC4, caspase8, and caspase11/1. Other studies also found the role of NAIP-NLRC4 (Rauch et al., 2017) and caspase4/11 (Hagar et al., 2013, Knodler et al., 2014) in gut epithelial cells. An inflammasome formed by NLRC4, ASC, and potentially caspase8 is also described in a model of enteric S. typhimurium infection (Rauch et al., 2017).
Although we demonstrate that inflamed E. coli directly and indirectly induce inflammatory macrophages through PKC δ, NLRC4, caspase8, and caspase1/11 complexes, the question is whether the inflamed E. coli-mediated activation of the inflammasomes in the gut and extra gut macrophages is a sufficient signal to trigger those chronic inflammatory diseases that remain unresolved. However, Kitamura et al. reported that transgenic mice expressing a constitutively active NLRC4 variant (H443P) develop an auto-inflammatory disease (Kitamura et al., 2014). Others also found that NAIP/NLRC4 inflammasome activation in MRP8+ cells is sufficient to cause systemic inflammatory diseases (Nichols et al., 2017).
Limitations of the Study
Although we analyzed the changes of cell population and subsets using flow cytometry, the exact changes of cell population and subsets, especially Ly6C+ inflammatory and anti-inflammatory macrophages in colon tissues, need to be solved through other technique(s) such as single cell analyses.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This research was supported by NSFC grants 91842302, 31470876, 91629102, ISF-NSFC program 31461143010, Tianjin science and technology commission (18JCZDJC35300), CAMS Innovation Fund for Medical Science (CIFMS2017-12M-2–005), a Ministry of Science and Technology grant (2016YFC1303604) and the State Key Laboratory of Medicinal Chemical Biology. We thanks Dr. Lu Gao in BGI, P. R. China for assistance in bacteria genome analyses.
Author Contributions
R.Y. designed the research and wrote the paper; H. Q., Y. L., X. S., J. W. and Yingquan. L. conducted in vivo experiments and immunoassay, participated in study design and performed the statistical analysis; Y. G conducted in vitro experiments, especially immunoblotting analyses; C. Z offered patient samples and conducted some in vivo experiments. H. Z, L. S conducted germ-free mouse experiments; Y. X. X.Y and Yanmei, X conducted bacteria typing; Y. Z. offered assistances for the animal experiments. All authors read and approve the final manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.046.
Data and Code Availability
Raw 16S rRNA gene sequence data for the feces microbiota: https://www.ncbi.nlm.nih.gov/sra/PRJNA512937; Raw genome components of E. coli O160:H7: http://www.ncbi.nlm.nih.gov/bioproject/513139.
Supplemental Information
References
- Ayres J.S., Trinidad N.J., Vance R.E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 2012;18:799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain C.C., Scott C.L., Uronen-Hansson H., Gudjonsson S., Jansson O., Grip O., Guilliams M., Malissen B., Agace W.W., Mowat A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. doi: 10.1038/mi.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker H.M., Apladas A., Scharl M., Fried M., Rogler G. Probiotic Escherichia coli Nissle 1917 and commensal E. coli K12 differentially affect the inflammasome in intestinal epithelial cells. Digestion. 2014;89:110–118. doi: 10.1159/000357521. [DOI] [PubMed] [Google Scholar]
- Blander J.M., Longman R.S., Iliev I.D., Sonnenberg G.F., Artis D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017;18:851–860. doi: 10.1038/ni.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schepper S., Verheijden S., Aguilera-Lizarraga J., Viola M.F., Boesmans W., Stakenborg N., Voytyuk I., Smidt I., Boeckx B., Dierckx de Casterle I. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175:400–415.e13. doi: 10.1016/j.cell.2018.07.048. [DOI] [PubMed] [Google Scholar]
- Franchi L., Amer A., Body-Malapel M., Kanneganti T.D., Ozoren N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- Franchi L., Kamada N., Nakamura Y., Burberry A., Kuffa P., Suzuki S., Shaw M.H., Kim Y.G., Nunez G. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett W.S., Gallini C.A., Yatsunenko T., Michaud M., DuBois A., Delaney M.L., Punit S., Karlsson M., Bry L., Glickman J.N. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar J.A., Powell D.A., Aachoui Y., Ernst R.K., Miao E.A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Elinav E., Huber S., Booth C.J., Strowig T., Jin C., Eisenbarth S.C., Flavell R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. U S A. 2010;107:21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Ivashkiv L.B. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kitamura A., Sasaki Y., Abe T., Kano H., Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 2014;211:2385–2396. doi: 10.1084/jem.20141091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knodler L.A., Crowley S.M., Sham H.P., Yang H., Wrande M., Ma C., Ernst R.K., Steele-Mortimer O., Celli J., Vallance B.A. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. 2014;16:249–256. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield K.L., Persson J., Brubaker S.W., Witte C.E., von Moltke J., Dunipace E.A., Henry T., Sun Y.H., Cado D., Dietrich W.F. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald T.T., Monteleone I., Fantini M.C., Monteleone G. Regulation of homeostasis and inflammation in the intestine. Gastroenterology. 2011;140:1768–1775. doi: 10.1053/j.gastro.2011.02.047. [DOI] [PubMed] [Google Scholar]
- Man S.M., Hopkins L.J., Nugent E., Cox S., Gluck I.M., Tourlomousis P., Wright J.A., Cicuta P., Monie T.P., Bryant C.E. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. U S A. 2014;111:7403–7408. doi: 10.1073/pnas.1402911111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S.M., Kanneganti T.D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016;16:7–21. doi: 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansan-Almeida R., Pereira A.L., Giugliano L.G. Diffusely adherent Escherichia coli strains isolated from children and adults constitute two different populations. BMC Microbiol. 2013;13:22. doi: 10.1186/1471-2180-13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao E.A., Mao D.P., Yudkovsky N., Bonneau R., Lorang C.G., Warren S.E., Leaf I.A., Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortha A., Chudnovskiy A., Hashimoto D., Bogunovic M., Spencer S.P., Belkaid Y., Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz M., Eidenschenk C., Ota N., Wong K., Lohmann U., Kuhl A.A., Wang X., Manzanillo P., Li Y., Rutz S. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity. 2015;42:321–331. doi: 10.1016/j.immuni.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Nichols R.D., von Moltke J., Vance R.E. NAIP/NLRC4 inflammasome activation in MRP8(+) cells is sufficient to cause systemic inflammatory disease. Nat. Commun. 2017;8:2209. doi: 10.1038/s41467-017-02266-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C.M., Wolf A.S., Goodier M.R., Riley E.M. Synergy between common gamma chain family cytokines and IL-18 potentiates innate and adaptive pathways of NK cell activation. Front Immunol. 2016;7:101. doi: 10.3389/fimmu.2016.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y., Misaghi S., Izrael-Tomasevic A., Newton K., Gilmour L.L., Lamkanfi M., Louie S., Kayagaki N., Liu J., Komuves L. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature. 2012;490:539–542. doi: 10.1038/nature11429. [DOI] [PubMed] [Google Scholar]
- Rauch I., Deets K.A., Ji D.X., von Moltke J., Tenthorey J.L., Lee A.Y., Philip N.H., Ayres J.S., Brodsky I.E., Gronert K., Vance R.E. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with Eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity. 2017;46:649–659. doi: 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber A.M., Lee Y.M., Chang M.W., Leblanc M., Collins B., Downes M., Evans R.M., Ayres J.S. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science. 2015;350:558–563. doi: 10.1126/science.aac6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I., Russell S.L., Antunes L.C., Finlay B.B. Gut microbiota in health and disease. Physiol. Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- Sellin M.E., Muller A.A., Felmy B., Dolowschiak T., Diard M., Tardivel A., Maslowski K.M., Hardt W.D. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014;16:237–248. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Shouval D.S., Biswas A., Goettel J.A., McCann K., Conaway E., Redhu N.S., Mascanfroni I.D., Al Adham Z., Lavoie S., Ibourk M. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamoutounour S., Henri S., Lelouard H., de Bovis B., de Haar C., van der Woude C.J., Woltman A.M., Reyal Y., Bonnet D., Sichien D. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012;42:3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- von Moltke J., Ayres J.S., Kofoed E.M., Chavarria-Smith J., Vance R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- Winter S.E., Baumler A.J. Why related bacterial species bloom simultaneously in the gut: principles underlying the 'Like will to like' concept. Cell Microbiol. 2014;16:179–184. doi: 10.1111/cmi.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn T.A., Chawla A., Pollard J.W. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E., Varol C., Farache J., Elmaliah E., Satpathy A.T., Friedlander G., Mack M., Shpigel N., Boneca I.G., Murphy K.M. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw 16S rRNA gene sequence data for the feces microbiota: https://www.ncbi.nlm.nih.gov/sra/PRJNA512937; Raw genome components of E. coli O160:H7: http://www.ncbi.nlm.nih.gov/bioproject/513139.