

Abstract
Plastid sequencing is an essential tool in the study of plant evolution. This high‐copy organelle is one of the most technically accessible regions of the genome, and its sequence conservation makes it a valuable region for comparative genome evolution, phylogenetic analysis and population studies. Here, we discuss recent innovations and approaches for de novo plastid assembly that harness genomic tools. We focus on technical developments including low‐cost sequence library preparation approaches for genome skimming, enrichment via hybrid baits and methylation‐sensitive capture, sequence platforms with higher read outputs and longer read lengths, and automated tools for assembly. These developments allow for a much more streamlined assembly than via conventional short‐range PCR. Although newer methods make complete plastid sequencing possible for any land plant or green alga, there are still challenges for producing finished plastomes particularly from herbarium material or from structurally divergent plastids such as those of parasitic plants.
Keywords: chloroplast, genomic, next‐generation sequencing, plant evolution, plastid
Introduction
DNA sequences of plastids have provided many important insights into plant ecology and evolution over the past three decades (Palmer 1987; Chase et al. 1993; Petit & Vendramin 2007; Hollingsworth et al. 2016). The continued popularity and utility of plastid sequencing is due to properties that make it the most accessible genome to the plant molecular biologist. The highly conserved gene order, near absence of recombination and low levels of nucleotide substitution (Box 1), make the plastid the ideal target for universal primers that amplify homologous loci in phylogenetically divergent species (Palmer 1987; Taberlet et al. 1991; Clegg et al. 1994; Shaw et al. 2005). In addition, the high‐copy number of plastids per cell means that genomic DNA extracts are naturally enriched for plastids (Bendich 1987) and thus an easier target than low‐copy nuclear genes for sequencing, particularly from small or degraded samples (Staats et al. 2013). Although attention is shifting from the sole‐reliance on plastid genes, to exploiting DNA variation in the nuclear genome (Hollingsworth et al. 2011; Lemmon & Lemmon 2013; Mandel et al. 2014; Weitemier et al. 2014), many research fields such as phylogenetics and phylogeography will continue to use plastid sequences for both technical and biological reasons.
Box 1. Typical and atypical plastid genome structures.
Land plant plastomes are typically considered to be 120–160 Kb in the length, nonrecombinant, circular, maternally inherited, strongly AT‐biased and with highly conserved gene order. While these general observations hold for many species, there are notable exceptions to each of these generalities, for example presence of recombination (Maréchal & Brisson 2010; Ness et al. 2016), noncircular plastids (Lilly et al. 2001), biparental plastid inheritance (Metzlaff et al. 1981), giant plastomes (e.g. chlorophyte green alga Floydiella terrestris, 521 Kb plastome sequence, Brouard et al. 2010) and miniaturized plastids <100 Kb (Wicke et al. 2013).
Most plastids are organized into a long single copy section (LSC) and a short single copy section (SSC), typically flanked by two inverted repeats (IRs) ~20–25 Kb long (Kolodner & Tewari 1979). These IRs are the most prominent structural feature of the plastome and appear to be maintained by concerted evolution and thus are near identical in their sequences. However, it is important to note that some groups have lost part of one, all of one or both inverted repeats (Palmer et al. 1987).
Plastomes are generally repeat poor and do not contain long repeats outside of the IR. For example, Camellia plastids contain just 156 repeats over 30 bp in length, with the longest repeat 82 bp long (Huang et al. 2014). Seldom are repeats longer than current sequence read length, with rare exceptions (e.g. longest repeat in Hordeum vulgare is 540 bp, Saski et al. 2007). Short repeats are also present and may be used as a variable marker in population studies. A/T mononucleotide repeats are the most abundant form of repeat, with 700 such repeats over 8 units in length in the alga Chlorella vulgaris (Wheeler et al. 2014).
Many biological properties of the plastid make them ideal for ecological and evolutionary studies. For example, predominantly uniparental inheritance makes plastid sequences informative for population genetic studies investigating seed flow (Ennos 1994; Petit et al. 2005), and low effective population sizes and thus short coalescent times make it ideal for phylogeography (Petit & Vendramin 2007). More generally, the plastid contains a core set of genes for photosynthesis, protein synthesis and ribosome production, and thus, plastid studies provide insights into key biochemical pathways and cellular functions (Kleffmann et al. 2004; Naumann et al. 2016). Plastid sequencing can also reveal the cyanobacterial origins of plastids and the genomic changes associated with endosymbiosis (McFadden 2001). As such, sequencing plastid loci has been instrumental in improving our understanding of phylogenetic relationships (Palmer 1987; Chase et al. 1993; Jansen et al. 2007), phylogeographic patterns (Soltis et al. 1997; Petit & Vendramin 2007), species discrimination (Hollingsworth et al. 2009; Nock et al. 2011), hybridization (Palme et al. 2004), photosynthesis (Leister 2003) and genome evolution (Wicke et al. 2011, 2016).
In each of these research fields, the move from the analysis of single‐gene regions that can be amplified by PCR, to complete plastid genomes (plastomes), is important to provide higher resolution and address previously unanswered questions (Hollingsworth et al. 2016). For example, recent studies have shown that (near) complete plastid DNA sequences improve phylogenetic support in analyses of recent rapid radiations (Parks et al. 2009; Barrett et al. 2014) and increase the ability to discriminate species with DNA barcoding (Ruhsam et al. 2015). Complete plastid sequences facilitate the study of mechanisms of gene loss and genome evolution in lineages where the plastid is subject to an altered selection regime, such as parasitic, carnivorous and mycoheterotrophic plants (Box 1, Barrett & Davis 2012; Wicke et al. 2013, 2014). Complete plastid genomes are necessary for detecting intracellular gene transfer between plastids, mitochondria and the nucleus (Iorizzo et al. 2012; Straub et al. 2013; Ma et al. 2015; : Wysocki et al. 2015). Sequencing all plastid genes also allows the discovery of the most variable loci for phylogenetic and population genetic inference (e.g. plastid microsatellites, Provan et al. 2001), for use over different spatial and temporal scales (Parks et al. 2009; Doorduin et al. 2011; Zhang et al. 2011). Overall, the widespread interest in plastid genome sequencing, in conjunction with improved sequencing techniques (discussed below), has led to a surge of published plastomes, with over 1000 available in GenBank, representing the full taxonomic scope of green plants and a more sparse sampling of other plastid bearing lineages (Donaher et al. 2009; Janouškovec et al. 2015; Smith & Keeling 2015).
Plastid genome sequencing, like many areas of molecular biology, has been influenced by numerous technical innovations in DNA sequencing. The first complete plastid sequence was produced by sequencing overlapping clones from restriction endonuclease fragments of Nicotiana tabacum (Shinozaki et al. 1986; Fig. 1a). This approach was superseded by PCR amplification and Sanger Sequencing (Taberlet et al. 1991). Now next‐generation sequencing of total genomic DNA is emerging as a direct and cost‐effective way to assemble the complete plastid sequence for any plant species (Nock et al. 2011). However, the rapid development of many sequencing and bioinformatic approaches to recover the plastome sequence can lead to some confusion in choosing the most effective option. Here, we give examples and explain the underlying principles of the most popular approaches and provide recommendations for how to sequence the plastome of nonmodel species with minimal cost and effort. In particular, we consider strategies for when only a single plastid sequence is required, through to scalable approaches for retrieving plastid sequences for many individuals and species. With each of these approaches, we consider the end‐goal to be a complete plastid sequence free of sequencing gaps and errors. We start by outlining the potential approaches to retrieve the plastome (via enrichment and nonenriched samples, Box 2), before considering the suitability of different sequencing technologies and assembly approaches.
Figure 1.
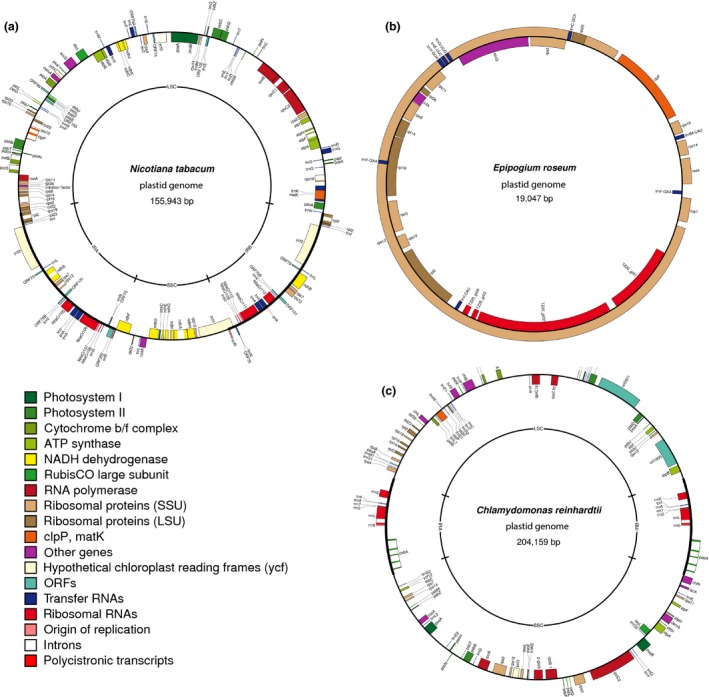
Structural diversity in plastids. (a) Nicotiana has a typical land plant plastid of 156 Kb and tripartite structure, (b) the mycoheterotrophic nonphotosynthetic orchid Epipogium roseum has the smallest plastid genome to date at 19 Kb, and with greatly reduced gene content, (c) green algae, here represented by Chlamydomonas reinhardtii, demonstrate dramatic plastid genome variation and include species with giant plastomes over 500 Kb in length. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Box 2. Selecting an enrichment approach.
A key decision for plastid assembly is whether to enrich samples for plastids through organelle isolation, PCR, hybrid baits or methylation enrichment, or to proceed with direct sequencing of a nonenriched genomic DNA extracts. Plastid enrichment introduces a time‐consuming (and potentially expensive) laboratory procedure, but has the benefit of focusing the downstream sequencing on the desired genomic regions, aiding analysis and reducing sequencing costs. In contrast, sequencing a total gDNA extract and subsequently identifying and assembling plastid reads may be faster and more cost‐effective in terms of laboratory procedures. However, it can be wasteful in terms of unnecessarily sequencing other genomic regions, reducing the multiplexing potential and also introduces an additional set of bioinformatic challenges. Typically, the choice of whether or not to enrich for plastids depends on a mixture of biological and technical aspects.
First, consideration must be given to the plastid biology of the group of interest and the availability of a related reference genome. Species with structurally rearranged plastids, such as many holoparasitic plants, will typically be assembled de novo from nonenriched gDNA or isolated plastids, as enrichment strategies relying on PCR primers or baits may not be suitable due to reduced sequence conservation. Similar strategies are often employed for generating a new reference plastome from a given clade, with different strategies such as lower‐coverage sequencing or enrichment for additional individuals (e.g. Curci et al. 2015).
The choice to enrich also depends on the nuclear genome size of the species of interest. Land plant genomes vary 2400‐fold in size, from 61 Mb in the carnivorous plant Genlisea tuberosa (Lentibulariaceae) to 149 Gb in Paris japonica (Melanthiaceae). The larger the nuclear genome size, the smaller the number of reads in a gDNA sample that will match the plastome and thus the lower plastid sequencing coverage (Fig. 2). As such, enrichment becomes increasingly important for species with large genome sizes, which include many economically important species (e.g. many pines, orchids and wheat).
Finally, the library preparation approach may simply be dictated by the type of expertise within a research group, with groups with wet‐laboratory technical expertise (or limited access to NGS) more likely to use enrichment, and groups with bioinformatic expertise more likely to directly sequence unenriched gDNA. However, the increasing availability of NGS, and bioinformatics tools for plastid assembly, means gDNA sequencing is likely to become the sample type of choice.
Library preparation strategies
Direct sequencing of genomic DNA
A genomic DNA (gDNA) sample contains a mix of nuclear and organellar DNA (plastid and mitochondrion). Thus, in many cases, the plastid can be assembled directly from a gDNA next‐generation sequencing (NGS) library, without prior enrichment or isolation of plastid DNA (Nock et al. 2011). In particular, it is becoming popular to perform a ‘genome skim’ (Straub et al. 2012), where gDNA is sequenced at low nuclear genome coverage (~0.1–10×), and this often provides sufficient data for complete plastid assembly (Coissac et al. 2016). This approach circumvents the need for optimizing species‐specific enrichment protocols (see below) and thus has dramatically streamlined plastome sequencing. This is perhaps the ‘gold standard’ for plastome assembly, often being relatively quick and cheap, and usually leading to high‐quality complete sequence assemblies. While genome skims have proven successful even for degraded herbarium material (Staats et al. 2013), special attention may be required during assembly (Box 3).
Box 3. Strategies for technically challenging plastome assemblies.
Structurally rearranged plastomes of mycoheterotrophs and parasitic plants, as well as degraded herbarium DNA samples, present particular challenges for current plastome assembly workflows. However, new tools are promising for overcoming many of the current limitations.
Herbarium samples present the joint challenge of low levels of recoverable DNA, in conjunction with high sample degradation. Low DNA yields are best overcome by optimizing DNA extraction (Savolainen et al. 1995), and the use of low‐input DNA library preparation kits (e.g. NuGEN Ovation Ultralow Library System), or via target enrichment with hybrid baits. Current consensus is that even the most degraded herbarium samples contain DNA potentially suitable for genomic analysis (Staats et al. 2013; Bakker et al. 2016). DNA degradation may impact the quality of NGS library preparations, because the shearing of poor quality template DNA will result in nonuniform bands. Downstream, assembly of plastomes from herbarium material may be fragmented or incomplete, while a minority may fail entirely (Bakker et al. 2016). In practice, it seems that some sample failure is inevitable and may be a limitation that cannot be overcome via new sequence technologies and pipelines. However, even partial plastomes are sufficient for many applications such as phylogenetic reconstruction.
Structurally atypical plastids, such as those of parasitic plants and mycoheterotrophs, often contain rearrangements, pseudogenes and gene deletions. This makes these plastids difficult to assemble using pipelines based on sequence conservation to plastid sequence databases. They may also pose difficulties for de novo plastid assembly pipelines due to low plastid copy number, or unusual GC‐content. As such, assembling a circularized plastome typically required bioinformatic refinement or additional laboratory work (e.g. Naumann et al. 2016). Solutions to streamline this process lie both in the generation of sequence data and in improved assembly pipelines. For example, sequence technologies generating reads many Kb in length will greatly facilitate de novo assembly in these groups, resulting in less need to connect scaffolds of unknown order. Improved de novo pipelines using read extension will make it possible to assemble accurate circularized plastome sequences, as has recently been shown with holoparasitic Cytinus hypocistic (Roquet et al. 2016). Overall it seems technological solutions will improve assembly in groups that have traditionally been a challenge for complete plastome sequencing.
There is a large choice of suitable NGS library types for these gDNA samples, including commonly used PCR‐based libraries (such as Illumina TruSeq, Illumina Nextera and Illumina compatible libraries such as NEB Ultra) and increasingly popular PCR‐free libraries that are less‐error prone but require more input DNA (>1 μg). While library preparation costs vary greatly, many service providers now charge less than $125 per sample. Some of these library preparations can be automated with robot liquid handlers to increase throughput (e.g. Illumina with the Neoprep). A gDNA library will contain a variable amount of plastid data depending on the nuclear genome size and tissue type, with <0.5% plastid reads in some gDNA samples of sugarcane (Hoang et al. 2015) to over 20% in milkweeds [Fig. 2, Table S1 (Supporting information), Straub et al. 2012]. Thus, the primary concern is designing a multiplex pooling strategy that sequences the desired number of samples with suitable plastid coverage, and choosing bioinformatic analyses that can correctly assign and assemble plastid sequence reads (discussed later).
Figure 2.
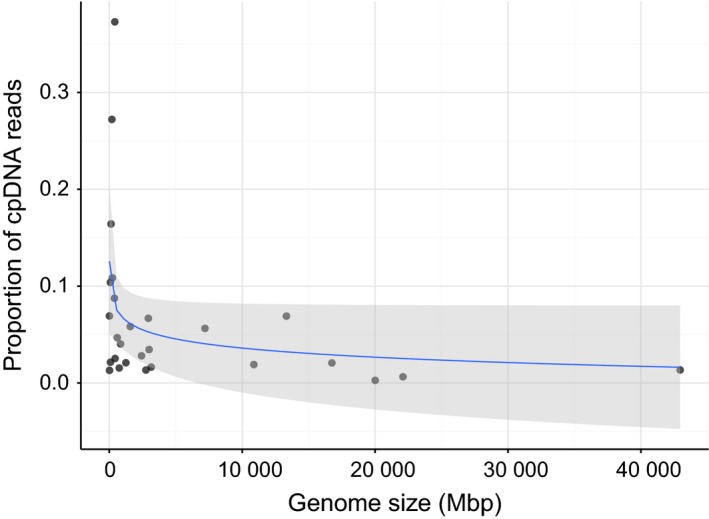
Representation of plastid reads in gDNA sequence libraries of species with different genome sizes. The graph shows the proportion of sequence reads from a phylogenetically diverse range of 27 green plant species that map to a reference database of 100 plant plastomes. Grey shading indicates the 95% confidence interval of the fitted line. Full details are given in Appendix S1 (Supporting information). [Colour figure can be viewed at http://wileyonlinelibrary.com]
While there are major benefits to assembling plastids directly from unenriched gDNA extracts, some laboratories may prefer to enrich their samples and focus sequencing effort only on the plastid (Box 2). We next explore these options.
Enrichment via plastid isolation
Sequencing plastid isolates is an intuitive route to focus sequencing coverage only on the plastid genome. Intact plastids can be isolated from fresh leaves via a sucrose density gradient, using either a home‐made protocol (e.g. Miflin & Beevers 1974) or a proprietary kit (e.g. Sigma Chloroplast Isolation Kit). It is also possible to isolate organellar DNA by high salt precipitation, or by degrading nuclear DNA in a gDNA extraction with DNase I treatment, although these two approaches can give low yields or contamination with mitochondrial DNA (Shi et al. 2012). The isolated plastids are typically recovered at a low yield and may require further amplification before sequencing. The main benefit of this approach is that de novo assembly of the enriched DNA sample is simple and will likely lead to a complete assembly even with a small number of sequence reads. This was the case in a chloroplast extraction optimization study by Shi et al. (2012), where 5–10 μg of isolated plastid DNA was subject to short‐read sequencing, with 50 Mb of data giving 100× coverage and a complete assembly.
Despite the benefits, there are substantial limitations to plastid isolation approaches, not least the requirement of large quantities of plant tissue which may exceed 5 g of fresh leaves. Further issues are that isolation protocols typically require species‐specific optimization which may hamper large‐scale comparative studies. The sample may also contain nuclear DNA contamination and thus require bioinformatic filtering. As such, the technical challenge associated with retrieving a high‐yield of intact plastids means that genome skimming or other approaches (discussed below) are increasingly popular alternatives for plastid sequencing.
Enrichment via methylation‐sensitive capture
Plant organelles demonstrate numerous characteristics that distinguish them from the nuclear genome. One rarely exploited feature is that eukaryotic nuclear genomes possess methylated CpG sites, a form of methylation associated with gene expression, while prokaryote‐derived organelles have dramatically lower total methylation (Feng et al. 2010). Yigit et al. (2014) showed that gDNA could be partitioned into a high‐methylated‐CpG nuclear fraction, and a fraction of low‐methylated‐CpG elements. The methyl‐poor fraction was enriched for plastids by 3.2‐ to 11.2‐fold, depending on the species in question. Subsequent to enrichment, the NGS library for each sample is prepared using standard protocols. Methylation‐sensitive capture is promising as it does not require a priori knowledge of the sequence of interest, and is inexpensive (~$30/sample). However, it requires careful testing before being widely adopted. For example, it is already apparent that this route will not be viable for degraded DNA samples such as herbarium material, where short gDNA fragments may lack a methylated site (here a CpG island), and thus will not be partitioned correctly.
Enrichment via hybrid bait capture
An alternative way to enrich for plastid DNA is through the use of oligonucleotide probes designed to capture complete plastids. In sequence capture, short probes (‘baits’) are used to isolate complementary sequences from a genomic DNA extract. Post‐capture library pools are then sequenced with NGS. For example, Stull et al. (2013) designed a collection of 55 000 baits for eudicots, with each bait intended to capture 120 bp sequences, with a 50‐bp overlap. Their approach worked for enriching a broad range of angiosperm gDNA extracts for plastid DNA. Sequence capture is extremely promising, especially as it would be suitable for a wide‐range of plant material including degraded herbarium samples, and thus deserves further development. In particular, it will be valuable to find less expensive alternatives to the rather expensive commercial enrichment kits (e.g. SureSelect Reagent Kit $1120/16 samples). One example would be MYcroarray, which has been successfully used in a plastid study by Comer et al. (2015). In addition to cost, another drawback is the potential to enrich for nuclear‐encoded plastid genes or plastid genes transferred to mitochondria (Box 3). Given the initial expenditure usually associated with this approach, and the subsequent high level of multiplexing required to fill a lane of sequencing, it is best suited to large‐scale analyses of plastomes and is a promising route for whole‐plastid DNA barcoding.
Enrichment via PCR
PCR is an effective way to enrich a gDNA extract for plastid DNA. The small size (c.150 Kb) and conserved sequence of plastids make it feasible to amplify the complete plastid genome either with short‐range PCR and Sanger Sequencing, or long‐range PCR and NGS. A set of universal primers has been developed to amplify the entire angiosperm plastome in 138 PCRs, with amplicons 0.8–1.5 Kb in length (Dong et al. 2013; however, see Prince 2015 for critique of the primers). These amplicons are easy to assemble as they have been designed to overlap by c.100 bp. Short‐range PCR has been successfully used to assemble a wide‐range of representative taxa across the angiosperms. There are also clade‐specific primer sets available for short‐range amplification of plastid DNA (e.g. for monocots, Scarcelli et al. 2011). Short‐range PCR represents one of the easiest ways to obtain (near) complete plastids for research laboratories with limited access to NGS or without bioinformatics expertise. However, it does present major limitations. First, it does not scale‐well. Unlike assembly from NGS reads from genomic DNA, which can be highly automated, the Sanger approach requires manual laboratory handling and scoring of sequence chromatograms. Moreover, this approach is only suited to ‘typical’ plastids (see Box 1), and even so the assembly of some regions, such as the boundaries of the inverted repeat, repeat‐rich regions or rapidly evolving genes such as matK and ycf1, may require the design of species‐specific primers. As such, short‐range PCR is better suited to applications requiring partial plastids (e.g. population genetic studies such as Whittall et al. 2010), rather than complete assemblies (e.g. studies of plastid genome evolution).
The second PCR‐based approach is long‐range PCR and NGS. Yang et al. (2014) and Uribe‐Convers et al. (2014) have developed suites of universal primers for the long‐range amplification of plastomes in amplicons of 4–23 Kb in length. These large amplicons are then sequenced on an NGS platform. The reduced number of primers relative to the short‐range PCR approach makes this method less time‐consuming in the laboratory, and the longer amplicon size allows all primers to be anchored in low variability regions of the genome. The tagging of different amplicons also allows the multiplexing of many individuals in a single lane of NGS. However, as a PCR‐based approach, it shares limitations outlined above in terms of amplifying known genome regions, and the failure of a single PCR will result in a large gap in the assembled sequence. The large amplicon size also requires high molecular weight DNA, which can be a limitation when working with degraded DNA samples such as herbarium material (e.g. Staats et al. 2013). Except in cases where PCR and ligation of barcoded adapters are automated (e.g. Uribe‐Convers et al. 2016), long‐range PCR approaches have all the challenges associated with NGS library preparation (expensive and time‐consuming) but without the benefits of direct assembly from gDNA.
Sequencing strategies
Once a library preparation approach has been chosen, the next choice is picking a sequencing strategy to match. The goal of producing high‐quality complete plastids is increasingly feasible with NGS data. In general, read lengths of current NGS platforms (e.g. 100 bp or longer, paired‐end sequences) have overcome the threshold of repeats in the plastome and thus are sufficient for de novo assemblies (Malé et al. 2014). This is a great improvement from early short NGS reads, such as the 36‐bp reads used to assemble the plastome of Pinus in up to 183 contigs (Cronn et al. 2008). As such, priority should be given to the use of long reads and/or the use of paired‐end data (Straub et al. 2012).
When designing a plastid sequencing study, 30× should be considered the minimum planned plastid sequence coverage, with >100× usually desirable. There appears to be no benefit of having very high coverage (over ~200x, A. D. Twyford, Unpublished). As a ballpark figure for sequencing gDNA, 500 Mb of sequence data should be sufficient to assemble the plastid for a typical leaf gDNA extract from a species with a small genome. For example, the ratio of plastid to nuclear genome coverage in gDNA libraries of Mimulus guttatus (Phrymaceae, 440 Mb genome size, Fig. 3) is approximately 67:1, which implies that ~3.1% of reads are derived from the plastid. If we sequenced 500 Mb of data, it would result in ~100× coverage of the plastid. Given the small plastid genome size, it is the ideal sample type to run on lower output machines such as the Illumina MiniSeq or MiSeq (Twyford 2016), or other platforms such as Ion Torrent PGM. This would particularly be the case for enriched libraries. For larger numbers of genome skims, it would be more cost‐effective to use high‐output sequencers such as the Illumina HiSeq 4000 (750 Gb/run); see http://www.molecularecologist.com/next-gen-fieldguide-2016 for comparison of sequencing platforms. As sequencing output increases, the potential number of plastids that can be pooled in a single sequencing run is very large (many 100s). This is made possible by the growing number of available adapters (e.g. commercial 384‐plex adapter sets) and dual‐indexing strategies (Sickel et al. 2015).
Figure 3.
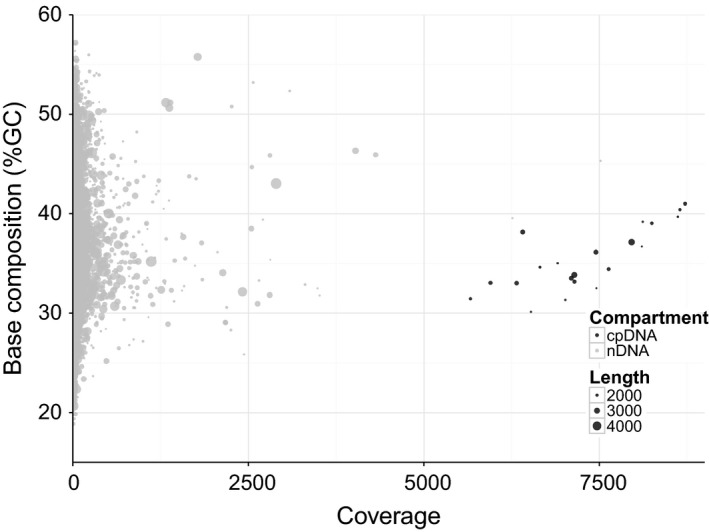
GC by coverage plot of a draft genome assembly. Short reads of the monkey flower Mimulus guttatus (population IM, SRR010318) were assembled with SPAdes genome assembler (v3.7.1) and annotated according to matches to the published Mimulus plastome (Vallejo‐Marín et al. 2016).
Long‐read third‐generation sequencing is extremely promising for the assembly of small genomes such as plastids. For example, Pacific Biosciences long‐read sequencers typically generates reads >15 Kb, with the longest reads up to 60 Kb in length. These long reads, in conjunction with lack of bias in AT‐rich regions, make it ideal for plastid assembly. The current high costs on a per‐read or per‐Mb basis means isolation of plastid DNA or enrichment is commonly done prior to sequencing (e.g. >100× coverage, Wu et al. 2014; Chen et al. 2015), although gDNA has also been sequenced (c.2000× plastid coverage, Stadermann et al. 2015). These long sequence reads are becoming increasingly cost‐effective as new platforms are released, such as the PacBio Sequel.
Assembly
Plastid sequence reads can be assembled to a reference genome or de novo. Reference‐guided assembly is most well suited to studies of related taxa where a reference genome exists. De novo assembly is preferable across phylogenetically divergent groups, species without available reference sequences, or groups with structural rearrangements or major gene loss or genome expansion (Fig. 1b,c, Box 3).
A common first stage in many de novo plastid assembly approaches is to separate plastid reads from nuclear and mitochondrial reads. Filtering before assembly can be an important way to reduce the complexity of a library, which greatly facilitates de novo assembly. Moreover, because the expected coverage of plastid reads is so much higher, assembling before filtering can lead to problems with error correction and de novo assembly algorithms that expect even coverage. Only in instances where researchers have a priori information that plastomes could have atypical gene content or copy number would it be necessary to conduct assembly before filtering plastid‐derived sequences, such as in the highly rearranged plastome of the parasitic plant Hydnora (Naumann et al. 2016).
There are two nonmutually exclusive approaches for isolating plastid reads; first, they can be separated based on similarity to known plastid sequence. For example, reads can be matched to a database of plastid sequences using blast or aligned to a related plastid using a short‐read aligner like Bowtie 2 (Langmead & Salzberg 2012). This may lead to some gaps in regions divergent from the reference sequence or database. Stringent read filtering by sequence similarity is not advised in lineages with atypical plastome structure or from lineages where no close reference sequence exists, as this can lead to incomplete assemblies. This filtering strategy also has the downside that it may incorrectly remove mitochondrial DNA that has been transferred to the plastid genome (Iorizzo et al. 2012; Straub et al. 2013; Ma et al. 2015; Wysocki et al. 2015). Additional plastid reads can be recovered using read extension approaches such as the GetOrganelle script (https://github.com/Kinggerm/GetOrganelle). Here, the first set of reads matching reference plastid(s) are used as seeds for successive rounds of extension, where additional reads that overlap the seeds are incorporated into the pool of plastid reads.
A second approach to recover plastid reads in gDNA is from distinct properties of the plastid, rather than similarity to known sequences. In particular, plastid reads are usually present with many‐fold higher coverage than nuclear DNA (though see Box 3). For example, a genome skimming study in Jerusalem artichoke (Helianthus tuberosus) found plastid DNA had over 1400 times higher coverage than the 9.4 Gbp nuclear genome (plastid DNA = 355×, nDNA = 0.25×), and an approximately 20‐fold greater coverage than the mitochondrial genome (Bock et al. 2014). Second, most land plant plastids have a distinct GC‐content to the nuclear genome (plastid DNA ~37%, Civáň et al. 2014; nDNA ~41% Li & Du 2014), although this distinction is not always clear (Smith et al. 2011; Šmarda et al. 2012). Taking these properties together, plots of GC‐content against read depth can be effective for distinguishing plastid reads (Fig. 3). This approach can be combined with best‐matching sequences in annotated databases to provide an effective filtering strategy (Kumar et al. 2013). An advantage of using coverage and GC‐content rather than similarity is that it may remove potentially problematic regions where plastid genes have been translocated to the nuclear genome and share sequence properties with nuclear rather than plastid DNA (Oliver et al. 1990). Similarly, by breaking raw reads into pieces of length k (so‐called kmers), we can count the frequency with which each kmer occurs using software such as BFcounter (Melsted & Pritchard 2011). The resulting count distribution, known as the kmer frequency, can be used to extract reads from high‐copy DNA such as the plastome.
Suitable de novo assemblers for these filtered plastid reads include ABySS, CLC Genomic Workbench, Edena, Euler‐sr, Geneious de novo, MIRA, Newbler, SOAPdenovo, SPAdes, SSAKE or Velvet (reviewed in Ekblom & Wolf 2014). In many cases, assembly performance and run‐time may be improved by down sampling the number of reads. A de novo assembly with a large kmer value, typically with minimal optimization of assembly parameters, will often yield good results assembling the plastid into a small number of large contigs. Further refinements are required to join scaffolds such as those broken by the inverted repeats (discussed below).
Instead of filtering nonplastid reads prior to assembly, there are a growing number of programs (mitobim, org.asm, fast‐plast) that merge filtering with assembly. The approach is to use known plastid (or mitochondrial) sequence as seeds to identify or ‘bait’ plastid reads and approximate coverage. From these seeds, assembly proceeds by finding reads that overlap the reads already incorporated. The organelle asembler (org.asm, http://pythonhosted.org/ORG.asm) uses baiting followed by cycles of stack filling, extension, cleaning and gap filling to assemble circular plastomes. It is reported to return 70% of plastids as complete (Coissac et al. 2016). The assembly software fast‐plast (https://github.com/mrmckain) is similar in its use of seed‐based baiting, but also uses a conventional assembler and is designed to correctly orientate the inverted repeat. These assemblers are the most direct means to produce circularized assemblies and can be highly automated for large sample sizes. However, the lack of published comparisons with other assemblers means careful examination should be given to the assembly quality particularly in repetitive regions. It is also unclear how well they perform in structurally atypical plastids such as those found in parasitic plants or whether it always accurately assembles the full plastome including both inverted repeats (A. D. Twyford & R. W. Ness, Unpublished).
The most common outcome of de novo plastid assembly is a small number of long contigs with breaks corresponding to the large single copy (LSC), small single copy (SSC) and inverted‐repeat (IR) regions. This is because many assemblers struggle to cope with the pair of near identical IRs, and as such collapse both IRs and display double the read depth for this region. These contigs can subsequently be stitched together, bearing in mind that plastids exist in two different states within a cell with alternate SSC orientation (Walker et al. 2015). Care should be given to check reads bridging the IR‐boundary give an accurate sequence assembly. For studies where precise IR boundaries are important, any remaining uncertainty can be examined using PCR primers that span IR boundaries. This plastid finishing step is however increasingly unnecessary with methods using read extension or approaches using long sequence reads.
While plastome assembly can be a routine and easy task from DNA extracted from fresh tissue of autotrophic land plants, this is not always the case. One relatively common yet often unexpected issue is intracellular gene transfer (Iorizzo et al. 2012; Straub et al. 2013; Ma et al. 2015; : Wysocki et al. 2015). It is now apparent that plastids can exchange DNA with the nucleus and mitochondria. Foreign DNA in plastids (and plastid DNA in the mitochondria and nucleus) can often be distinguished from unique properties of the plastome, described above, such as copy number. This must be accounted for to complete a plastome sequence.
Conclusions
Plastome sequencing is at an exciting turning point. Large‐scale NGS library preparation, increasing read lengths and sequencing throughput, and automated assembly pipelines, make the prospect of plastid sequences for all lineages of land plants and algae a real possibility. These plastid sequences can increasingly be harnessed to their full potential with improved downstream processing including automatic annotation (Huang & Cronk 2015) and many integrated pipelines suited to large data sets (such as The Plastome Database, http://verdant.iplantcollaborative.org/plastidDB/). These data have great potential for increasing our understanding of plant biology and genome evolution and will set the context for future exploration of plastid gene expression (Sanitá Lima et al. 2016), as well as complementary investigations of the nuclear genome.
A.D.T. and R.W.N. wrote the manuscript and prepared the figures.
Supporting information
Appendix S1 Proportion of plastid reads in gDNA sequence libraries of species with different genome sizes.
Acknowledgements
We thank the Editor and two Reviewers for useful comments on the manuscript. We also thank Claude dePamphilis, Julia Naumann and Wen‐Bin Yu for useful discussions about plastid biology. Collaborative research by AT and RN is funded by NERC International Opportunities Fund Grant NE/N006739/1. Research by AT is supported by NERC Fellowship NE/L011336/1.
References
- Bakker FT, Lei D, Yu J et al (2016) Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Biological Journal of the Linnean Society, 117, 33–43. [Google Scholar]
- Barrett CF, Davis JI (2012) The plastid genome of the mycoheterotrophic Corallorhiza striata (Orchidaceae) is in the relatively early stages of degradation. American Journal of Botany, 99, 1513–1523. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Specht CD, Leebens‐Mack J et al (2014) Resolving ancient radiations: can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Annals of Botany, 113, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich AJ (1987) Why do chloroplasts and mitochondria contain so many copies of their genome? BioEssays, 6, 279–282. [DOI] [PubMed] [Google Scholar]
- Bock DG, Kane NC, Ebert DP, Rieseberg LH (2014) Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phytologist, 201, 1021–1030. [DOI] [PubMed] [Google Scholar]
- Brouard J‐S, Otis C, Lemieux C, Turmel M (2010) The exceptionally large chloroplast genome of the green alga Floydiella terrestris illuminates the evolutionary history of the Chlorophyceae. Genome Biology and Evolution, 2, 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MW, Soltis DE, Olmstead RG et al (1993) Phylogenetics of seed plants: an analysis of nucleotide sequences from the plastid gene rbcL. Annals of the Missouri Botanical Garden, 80, 528–580. [Google Scholar]
- Chen X, Li Q, Li Y, Qian J, Han J (2015) Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Frontiers in Plant Science, 6, doi: 10.3389/fpls.2015.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Foster PG, Embley MT, Séneca A, Cox CJ (2014) Analyses of Charophyte chloroplast genomes help characterize the ancestral chloroplast genome of land plants. Genome Biology and Evolution, 6, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg MT, Gaut BS, Learn GH, Morton BR (1994) Rates and patterns of chloroplast DNA evolution. Proceedings of the National Academy of Sciences, 91, 6795–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coissac E, Hollingsworth PM, Lavergne S, Taberlet P (2016) From barcodes to genomes: extending the concept of DNA barcoding. Molecular Ecology, 25, 1423–1428. [DOI] [PubMed] [Google Scholar]
- Comer JR, Zomlefer WB, Barrett CF et al (2015) Resolving relationships within the palm subfamily Arecoideae (Arecaceae) using plastid sequences derived from next‐generation sequencing. American Journal of Botany, 102, 888–899. [DOI] [PubMed] [Google Scholar]
- Cronn R, Liston A, Parks M et al (2008) Multiplex sequencing of plant chloroplast genomes using Solexa sequencing‐by‐synthesis technology. Nucleic Acids Research, 36, e122, doi: 10.1093/nar/gkn502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curci PL, De Paola D, Danzi D, Vendramin GG, Sonnante G (2015) Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE, 10, e0120589. doi: 10.1371/journal.pone.0120589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaher N, Tanifuji G, Onodera NT et al (2009) The complete plastid genome sequence of the secondarily nonphotosynthetic alga Cryptomonas paramecium: reduction, compaction, and accelerated evolutionary rate. Genome Biology and Evolution, 1, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W, Xu C, Cheng T, Lin K, Zhou S (2013) Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biology and Evolution, 5, 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorduin L, Gravendeel B, Lammers Y et al (2011) The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Research, 18, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekblom R, Wolf JBW (2014) A field guide to whole‐genome sequencing, assembly and annotation. Evolutionary Applications, 7, 1026–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennos R (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity, 72, 250–259. [Google Scholar]
- Feng S, Cokus SJ, Zhang X et al (2010) Conservation and divergence of methylation patterning in plants and animals. Proceedings of the National Academy of Sciences, 107, 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang NV, Furtado A, McQualter RB, Henry RJ (2015) Next generation sequencing of total DNA from sugarcane provides no evidence for chloroplast heteroplasmy. New Negatives in Plant Science, 1–2, 33–45. [Google Scholar]
- Hollingsworth PM, Forrest LL, Spouge JL et al (2009) A DNA barcode for land plants. Proceedings of the National Academy of Sciences, 106, 12794–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth PM, Graham SW, Little DP (2011) Choosing and using a plant DNA Barcode. PLoS ONE, 6, e19254, doi: 10.1371/journal.pone.0019254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth P, De‐Zhu L, Van der Bank M, Twyford A (2016) Telling plant species apart with DNA: from barcodes to genomes. Philosophical Transactions of the Royal Society B, 371, 20150338, doi: 10.1098/rstb.2015.0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DI, Cronk QC (2015) Plann: a command‐line application for annotating plastome sequences. Applications in Plant Sciences, 3, doi: 10.3732/apps.1500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Shi C, Liu Y, Mao S‐Y, Gao L‐Z (2014) Thirteen Camellia chloroplast genome sequences determined by high‐throughput sequencing: genome structure and phylogenetic relationships. BMC Evolutionary Biology, 14, 151, doi: 10.1186/1471-2148-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorizzo M, Grzebelus D, Senalik D et al (2012) Against the traffic the first evidence for mitochondrial DNA transfer into the plastid genome. Mobile Genetic Elements, 2, 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janouškovec J, Tikhonenkov DV, Burki F et al (2015) Factors mediating plastid dependency and the origins of parasitism in apicomplexans and their close relatives. Proceedings of the National Academy of Sciences, 112, 10200–10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA et al (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome‐scale evolutionary patterns. Proceedings of the National Academy of Sciences, 104, 19369–19374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleffmann T, Russenberger D, von Zychlinski A et al (2004) The Arabidopsis thaliana chloroplast proteome reveals pathway abundance and novel protein functions. Current Biology, 14, 354–362. [DOI] [PubMed] [Google Scholar]
- Kolodner R, Tewari KK (1979) Inverted repeats in chloroplast DNA from higher plants. Proceedings of the National Academy of Sciences, 76, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M (2013) Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon‐annotated GC‐coverage plots. Frontiers in Genetics, 4, 237, doi: 10.3389/fgene.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leister D (2003) Chloroplast research in the genomic age. Trends in Genetics, 19, 47–56. [DOI] [PubMed] [Google Scholar]
- Lemmon EM, Lemmon AR (2013) High‐throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics, 44, 99–121. [Google Scholar]
- Li X‐Q, Du D (2014) Variation, evolution, and correlation analysis of C+G content and genome or chromosome size in different kingdoms and phyla. PLoS ONE, 9, e88339, doi: 10.1371/journal.pone.0088339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly JW, Havey MJ, Jackson SA, Jiang J (2001) Cytogenomic analyses reveal the structural plasticity of the chloroplast genome in higher plants. The Plant Cell, 13, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P‐F, Zhang Y‐X, Guo Z‐H, Li D‐Z (2015) Evidence for horizontal transfer of mitochondrial DNA to the plastid genome in a bamboo genus. Scientific Reports, 5, 11608, doi: 10.1038/srep11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malé P‐JG, Bardon L, Besnard G et al (2014) Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family. Molecular Ecology Resources, 14, 966–975. [DOI] [PubMed] [Google Scholar]
- Mandel JR, Dikow RB, Funk VA et al (2014) A target enrichment method for gathering phylogenetic information from hundreds of loci: an example from the Compositae. Applications in Plant Sciences, 2, 1300085, doi: 10.3732/apps.1300085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, Brisson N (2010) Recombination and the maintenance of plant organelle genome stability. New Phytologist, 186, 299–317. [DOI] [PubMed] [Google Scholar]
- McFadden GI (2001) Primary and secondary endosymbiosis and the origin of plastids. Journal of Phycology, 37, 951–959. [Google Scholar]
- Melsted P, Pritchard JK (2011) Efficient counting of k‐mers in DNA sequences using a bloom filter. BMC Bioinformatics, 12, 333, doi: 10.1186/1471-2105-12-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzlaff M, Börner T, Hagemann R (1981) Variations of chloroplast DNAs in the genus Pelargonium and their biparental inheritance. Theoretical and Applied Genetics, 60, 37–41. [DOI] [PubMed] [Google Scholar]
- Miflin BJ, Beevers H (1974) Isolation of intact plastids from a range of plant tissues. Plant Physiology, 53, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann J, Der JP, Wafula EK et al (2016) Detecting and characterizing the highly divergent plastid genome of the nonphotosynthetic parasitic plant Hydnora visseri (Hydnoraceae). Genome Biology and Evolution, 8, 345–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Kraemer SA, Colegrave N, Keightley PD (2016) Direct estimate of the spontaneous mutation rate uncovers the effects of drift and recombination in the Chlamydomonas reinhardtii plastid genome. Molecular Biology and Evolution, 33, 800–808. [DOI] [PubMed] [Google Scholar]
- Nock CJ, Waters DL, Edwards MA et al (2011) Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnology Journal, 9, 328–333. [DOI] [PubMed] [Google Scholar]
- Oliver JL, Marin A, Martínez‐Zapater JM (1990) Chloroplast genes transferred to the nuclear plant genome have adjusted to nuclear base composition and codon usage. Nucleic Acids Research, 18, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palme A, Su Q, Palsson S, Lascoux M (2004) Extensive sharing of chloroplast haplotypes among European birches indicates hybridization among Betula pendula, B. pubescens and B. nana . Molecular Ecology, 13, 167–178. [DOI] [PubMed] [Google Scholar]
- Palmer JD (1987) Chloroplast DNA evolution and biosystematic uses of chloroplast DNA variation. The American Naturalist, 130, S6–S29. [Google Scholar]
- Palmer JD, Osorio B, Aldrich J, Thompson WF (1987) Chloroplast DNA evolution among legumes: loss of a large inverted repeat occurred prior to other sequence rearrangements. Current Genetics, 11, 275–286. [Google Scholar]
- Parks M, Cronn R, Liston A (2009) Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biology, 7, 84, doi: 10.1186/1741-7007-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit RJ, Vendramin GG (2007) Plant phylogeography based on organelle genes: an introduction In: Phylogeography of Southern European Refugia (eds Weiss S Ferrand N.), pp. 23–97. Springer, AA Dordrecht, The Netherlands. [Google Scholar]
- Petit RJ, Duminil J, Fineschi S et al (2005) INVITED REVIEW: comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Molecular Ecology, 14, 689–701. [DOI] [PubMed] [Google Scholar]
- Prince LM (2015) Plastid primers for angiosperm phylogenetics and phylogeography. Applications in Plant Sciences, 3, doi: 10.3732/apps.1400085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provan J, Powell W, Hollingsworth PM (2001) Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends in Ecology & Evolution, 16, 142–147. [DOI] [PubMed] [Google Scholar]
- Roquet C, Coissac É, Cruaud C et al (2016) Understanding the evolution of holoparasitic plants: the complete plastid genome of the holoparasite Cytinus hypocistis (Cytinaceae). Annals of Botany, 118, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhsam M, Rai HS, Mathews S et al (2015) Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Molecular Ecology Resources, 15, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Sanitá Lima M, Woods LC, Cartwright MW, Smith DR (2016) The (in)complete organelle genome: exploring the use and nonuse of available technologies for characterizing mitochondrial and plastid chromosomes. Molecular Ecology Resources, 16, 1279–1286. [DOI] [PubMed] [Google Scholar]
- Saski C, Lee S‐B, Fjellheim S et al (2007) Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes. Theoretical and Applied Genetics, 115, 571–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen V, Cuénoud P, Spichiger R et al (1995) The use of herbarium specimens in DNA phylogenetics: evaluation and improvement. Plant Systematics and Evolution, 197, 87–98. [Google Scholar]
- Scarcelli N, Barnaud A, Eiserhardt W et al (2011) A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. PLoS ONE, 6, doi: 10.1371/journal.pone.0019954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J, Lickey EB, Beck JT et al (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany, 92, 142–166. [DOI] [PubMed] [Google Scholar]
- Shi C, Hu N, Huang H et al (2012) An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE, 7, doi: 10.1371/journal.pone.0031468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K, Ohme M, Tanaka M et al (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. The EMBO Journal, 9, 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickel W, Ankenbrand MJ, Grimmer G et al (2015) Increased efficiency in identifying mixed pollen samples by meta‐barcoding with a dual‐indexing approach. BMC Ecology, 15, 20, doi: 10.1186/s12898-015-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šmarda P, Bureš P, Šmerda J, Horová L (2012) Measurements of genomic GC content in plant genomes with flow cytometry: a test for reliability. New Phytologist, 193, 513–521. [DOI] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ (2015) Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proceedings of the National Academy of Sciences, 112, 10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Burki F, Yamada T et al (2011) The GC‐rich mitochondrial and plastid genomes of the green alga Coccomyxa give insight into the evolution of organelle DNA nucleotide landscape. PLoS ONE, 6, e23624–e23624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Gitzendanner MA, Strenge DD, Soltis PS (1997) Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Systematics and Evolution, 206, 353–373. [Google Scholar]
- Staats M, Erkens RHJ, van de Vossenberg B et al (2013) Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS ONE, 8, e69189, doi: 10.1371/journal.pone.0069189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadermann KB, Weisshaar B, Holtgräwe D (2015) SMRT sequencing only de novo assembly of the sugar beet (Beta vulgaris) chloroplast genome. BMC Bioinformatics, 16, 295, doi: 10.1186/s12859-015-0726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SC, Parks M, Weitemier K et al (2012) Navigating the tip of the genomic iceberg: next‐generation sequencing for plant systematics. American Journal of Botany, 99, 349–364. [DOI] [PubMed] [Google Scholar]
- Straub SC, Cronn RC, Edwards C, Fishbein M, Liston A (2013) Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biology and Evolution, 5, 1872–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull GW, Moore MJ, Mandala VS et al (2013) A targeted enrichment strategy for massively parallel sequencing of angiosperm plastid genomes. Applications in Plant Sciences, 1, doi: 10.3732/apps.1200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non‐coding regions of chloroplast DNA. Plant Molecular Biology, 17, 1105–1109. [DOI] [PubMed] [Google Scholar]
- Twyford AD (2016) Will benchtop sequencers resolve the sequencing trade‐off in plant genetics? Frontiers in Plant Science, 7, doi: 10.3389/fpls.2016.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe‐Convers S, Duke JR, Moore MJ, Tank DC (2014) A long PCR‐based approach for DNA enrichment prior to next‐generation sequencing for systematic studies. Applications in Plant Sciences, 2, 1300063, doi: 10.3732/apps.1300063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe‐Convers S, Settles ML, Tank DC (2016) A phylogenomic approach based on PCR target enrichment and high throughput sequencing: resolving the diversity within the South American Species of Bartsia L (Orobanchaceae). PLoS ONE, e0148203, doi: 10.1371/journal.pone.0148203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo‐Marín M, Cooley AM, Lee MY et al (2016) Strongly asymmetric hybridization barriers shape the origin of a new polyploid species and its hybrid ancestor. American Journal of Botany, 103, 1–17. [DOI] [PubMed] [Google Scholar]
- Walker JF, Jansen RK, Zanis MJ, Emery NC (2015) Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. American Journal of Botany, 11, 1751–1752. [DOI] [PubMed] [Google Scholar]
- Weitemier K, Straub SCK, Cronn RC et al (2014) hyb‐seq: combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences, 2, apps.1400042, doi: 10.3732/apps.1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler GL, Dorman HE, Buchanan A, Challagundla L, Wallace LE (2014) A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Applications in Plant Sciences, 2, doi: 10.3732/apps.1400059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittall JB, Syring J, Parks M et al (2010) Finding a (pine) needle in a haystack: chloroplast genome sequence divergence in rare and widespread pines. Molecular Ecology, 19, 100–114. [DOI] [PubMed] [Google Scholar]
- Wicke S, Schneeweiss GM, Müller KF, Quandt D (2011) The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology, 76, 273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Müller KF, de Pamphilis CW et al (2013) Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. The Plant Cell, 25, 3711–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Schäferhoff B, Müller KF (2014) Disproportional plastome‐wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Molecular Biology and Evolution, 31, 529–545. [DOI] [PubMed] [Google Scholar]
- Wicke S, Müller KF, dePamphilis CW et al (2016) Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proceedings of the National Academy of Sciences, 113, 9045–9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Gui S, Quan Z et al (2014) A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: insight into the plastid evolution of basal eudicots. BMC Plant Biology, 14, 289, doi: 10.1186/s12870-014-0289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki WP, Clark LG, Attigala L, Ruiz‐Sanchez E, Duvall MR (2015) Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evolutionary Biology, 15, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JB, Li DZ, Li HT (2014) Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Molecular Ecology Resources, 14, 1024–1031. [DOI] [PubMed] [Google Scholar]
- Yigit E, Hernandez DI, Trujillo JT, Dimalanta E, Bailey CD (2014) Genome and metagenome sequencing: Using the human methyl‐binding domain to partition genomic DNA derived from plant tissues. Applications in Plant Sciences, 2, doi: 10.3732/apps.1400064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐J, Ma P‐F, Li D‐Z (2011) High‐throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE, 6, e20596, doi: 10.1371/journal.pone.0020596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Proportion of plastid reads in gDNA sequence libraries of species with different genome sizes.