

Abstract
Molecular dynamics (MD) simulations reveal molecular motions at atomic resolution. Recent advances in high-performance computing now enable microsecond-long simulations capable of sampling a wide range of biologically relevant events. But the disk space required to store an MD trajectory increases with simulation length and system size, complicating collaborative sharing and visualization. To overcome these limitations, we created PCAViz, an open-source toolkit for sharing and visualizing MD trajectories via the web browser. PCAViz includes two components: the PCAViz Compressor, which compresses and saves simulation data; and the PCAViz Interpreter, which decompresses the data in users’ browsers and feeds it to any of several browser-based molecular-visualization libraries (e.g., 3Dmol.js, NGL Viewer, etc.). An easy-to-install WordPress plugin enables “plug-and-play” trajectory visualization. PCAViz will appeal to a broad audience of researchers and educators. The source code is available at http://durrantlab.com/pcaviz/, and the WordPress plugin is available via the official WordPress Plugin Directory.
Introduction
Molecular dynamics (MD) simulations predict biomolecular dynamics by applying Newton’s laws of motion to atomic-resolution models. In brief, MD engines represent atoms and bonds as simple spheres connected by virtual springs.1 If the radii/partial charges of the spheres and the stiffness/length of the springs are properly parametrized, virtual Newtonian and actual quantum-mechanical forces are similar.2 The MD engine nudges virtual atoms per the approximated forces and advances the simulation mere femtoseconds. Repeating this process millions of times produces trajectories that capture protein motions.
Simulations reveal important information that static structures cannot. For example, they can resolve structural artifacts introduced by protein crystallography (e.g., crystal contacts that are not biologically relevant,3 potential steric clashes, etc.). Simulations can also demonstrate the relationships between protein structure and function,4 the effects of solvent pH on protein dynamics,5 and the paths through conformational space that a protein must traverse to reach different energetic states.6
Simulations also play a prominent role in structure-based, computer-aided drug discovery. Crystal structures capture single, static protein conformations. In contrast, MD simulations sample a more continuous range of conformations. As binding-pocket geometries transition between states, transient druggable subpockets sometimes form that are hidden to experiment. These cryptic pockets7,8 often play important roles in allostery and protein–protein interactions and so provide new drug-discovery opportunities for targeting otherwise challenging proteins.
JavaScript libraries such as 3DMol.js,9 NGL Viewer,10 and PV11 allow researchers and web developers to visualize static molecular structures in any modern web browser. Many of these libraries can also load small MD trajectories (e.g., several frames). But more extensive MD simulations are far larger, demanding too much bandwidth and memory for in-browser visualization. And yet, with advances in computer parallelization and software, the extensive microsecond-long simulations that are required to capture biological events such as ligand binding or protein folding (e.g., refs (12 and 13)) have become routine.
We here present PCAViz, an open-source Python/JavaScript toolkit for visualizing long MD trajectories in a web browser. Browser-based visualization is a powerful collaboration and teaching tool. It allows researchers to send URLs for convenient viewing rather than having to transfer large MD trajectories that are often many gigabytes in size. Educators can also share PCAViz URLs with their students in a classroom setting. Convenient 3D viewing of biomolecules on laptops, phones, and tablets often provides a clearer understanding of biological systems.
The PCAViz toolkit includes both a compression tool and an in-browser interpreter. The PCAViz Compressor is an easy-to-use, Python-based, command-line utility that extracts information from an MD trajectory using principal component analysis.14 It stores that information in the JSON format, a format specifically designed to ease data transfer between web servers and browsers. The PCAViz Interpreter is a JavaScript library that runs in the browser without requiring users to install any plugins. It accepts the JSON file from the server and converts it back into a format that can be visualized using any of several in-browser molecular visualization libraries.9,10,15,16 The PCAViz download includes examples showing how to integrate the Interpreter into existing web pages, and a PCAViz-powered WordPress plugin makes browser-based trajectory visualization particularly easy.
PCAViz will be a useful tool for the computational-biology community. It is available free of charge from http://durrantlab.com/pcaviz/ under the terms of the open-source GNU General Public License, version 2.
Results and Discussion
Atom-Position Accuracy
PCAViz uses several lossy techniques to reduce bandwidth, memory, and CPU demands. Consequently, the atomic positions of a PCAViz-processed trajectory will not exactly match those of the original simulation. But if one chooses the proper compression settings, the accuracy is acceptably high for in-browser visualization. In contrast, dedicated molecular-visualization programs such as VMD17 and PyMOL18 are better suited for more rigorous analyses of trajectory dynamics.
Understanding the trade-offs between atom-position accuracy and file size will help users pick the best PCAViz settings for their needs. The first potential source of atom-position inaccuracy is the PCA compression itself. The first principal component describes the predominant motions of the protein. But projecting a trajectory onto this component alone eliminates the more subtle motions that are only explained by subsequent components. After reverse transformation from PCA space back into Cartesian space, the atomic positions may differ substantially from those of the source trajectory (i.e., PCA compression is lossy). Using the top two components captures more detailed atomic motions, but the added data reduces compression, resulting in larger file sizes. At the other extreme, projecting a trajectory onto all principal components captures all simulated motions exactly, but the resulting data is not compressed at all.
The second potential source of atom-position inaccuracy arises due to rounding. To reduce the size of the JSON file containing the PCA data, our implementation rounds all principal-component vectors and coefficients to a user-defined number of decimal places. Rounding substantially reduces file sizes, but retaining too few decimal places introduces inaccuracies into the reverse-transformed atomic coordinates beyond those intrinsic to PCA itself.
To illustrate the impact of PCA compression and rounding, we used PCAViz to transform a simulation of La-related protein 1 (LARP1)19 into PCA space and then back into Cartesian space (Figure 1 and Table S1). The simulation consisted of 100 frames spaced 4.4 ns apart. We considered only non-hydrogen protein atoms (1326 atoms per frame). To remove global translational and rotation motions, each frame was aligned to a common reference by minimizing Cα RMSD using VMD.17 We applied PCAViz to this trajectory multiple times, each time retaining the top principal components required to account for 20%, 25%, 30%, 40%, 50%, 60%, 70%, 75%, 80%, 90%, and 100% of the positional variance captured by the aligned simulation, respectively. We also tested different rounding precisions, including rounding to the nearest tenth, hundredth, and thousandth. To measure atomic-position accuracy, in each case we calculated the non-hydrogen-atom RMSD between the original and processed frames (Figure 1A). We also recorded the file sizes associated with each setting (Figure 1B). Figure 2 illustrates the impact of these various settings on a specific frame taken from the simulation.
Figure 1.

Atom-position accuracy and file sizes. We compressed the benchmark LARP1 simulation using various positional-variance (X axis) and rounding-precision parameters (line styles). (A) To judge atom-position accuracy, we decompressed each simulation to recover the atomic Cartesian coordinates. We calculated the average RMSD between each frame of the original trajectory and the corresponding frame of the PCAViz-processed trajectory. (B) We also recorded the file size of each output JSON file produced using the same positional-variance/rounding-precision combinations.
Figure 2.
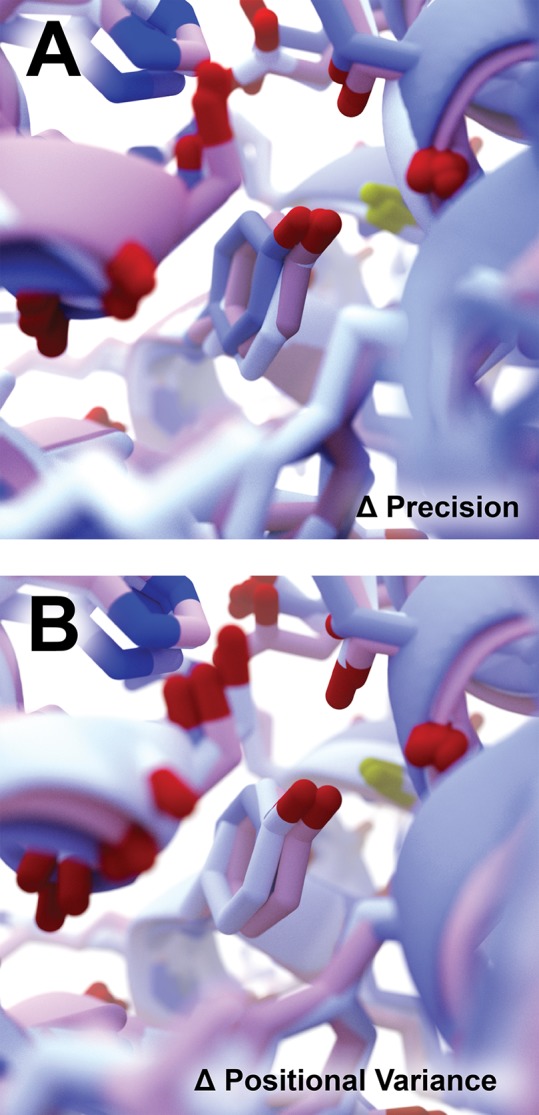
Atom-position accuracy depicted visually. We considered the 50th frame of our 100-frame LARP1 simulation. The original structure is shown in pink. PCAViz-processed structures are shown in blue and white. (A) Rounding PCA values to the nearest tenth and hundredth gave the blue and white structures, respectively. In both cases, principal components accounted for 100% of the positional variance. Rounding to the nearest hundredth and thousandth gave structures nearly identical to the original (pink). (B) Including sufficient principal components to account for 25% and 50% of the positional variance gave the blue and white structures, respectively. In both cases, the PCA values were rounded to the nearest hundredth. Accounting for 75% of the variance gave a structure nearly identical to the original (pink). These figures were generated using BlendMol.20
As is clear from Figure 1, accuracy improves as more principal components are retained. Similarly, accuracy improves with higher numeric precision (i.e., the number of digits retained after the decimal point). But these improvements come at the cost of file size. We note that when the output files are further compressed using GZIP, as is common when transmitting data over the Internet, the file sizes are further reduced (between ∼2.5 and ∼20 times in our tests).
As a second demonstration, we used PCAViz to transform a previously aligned trajectory of TEM-1 β-lactamase (TEM-1)21 into PCA space and then back into Cartesian space. The TEM-1 simulation consisted of 1000 frames spaced 50 ps apart. We considered only non-hydrogen protein atoms (2030 atoms per frame). The RMSDs between the original and PCAViz-processed trajectory frames for different variance and precision settings are given in Table S2, together with the sizes of the output JSON files. We saw the same trends with the TEM-1 simulation that we saw with the LARP1 simulation. Accuracy improved when we used higher positional-variance and numeric-precision cutoffs, at the cost of file size.
Comparison to Related Programs
We are not the first to use PCA to compress and expand MD trajectories. For example, the pyPcazip Python package22 uses a compression scheme similar to ours to enable PCA-based analysis via third-party packages such as MDAnalysis.14 But our primary goal is browser-based trajectory visualization, so PCAViz additionally:
-
1.
Stores the compressed PCA data as a JSON file to facilitate browser/server communication,
-
2.
Includes the information required to correctly represent atomic bonds in the browser,
-
3.
Allows users to stride the trajectory to further reduce file size, relying on interpolation between frames to approximate intermediate conformations,
-
4.
Rounds numeric values to further reduce file sizes,
-
5.
Includes a JavaScript decompression library (the PCAViz Interpreter) to decompress the trajectory files in users’ browsers.
Popular browser-based molecular visualization packages such as 3DMol.js,9 NGL Viewer,10 and PV11 can also display multiframe PDB files such as those produced by NMR. But visualizing a long-timescale MD trajectory using these tools is not practical given bandwidth and memory constraints. The PCAViz JavaScript interpreter effectively enhances these popular libraries. It downloads the compressed PCA data and decompresses it in the browser, delivering PDB-formatted frames to these packages as needed. The PCAViz download includes examples showing how to use PCAViz with 3DMol.js,9 NGL Viewer,10 and PV.11
Programs such as MDSrv23 and HTMoL24 similarly interface with browser-based molecular viewers to enable trajectory visualization. But MDSrv/HTMoL and PCAViz take fundamentally different approaches. MDSrv and HTMoL require users to separately set up a web server that streams trajectory frames to the browser on request, eliminating the need to approximate atomic positions. But few researchers and educators have the ability to set up a separate server dedicated exclusively to streaming simulation data. Additionally, on slow Internet connections the need to download each frame may lead to substantial lag times. In contrast, PCAViz downloads the PCA representation of the entire trajectory as a single file, using the same technology a standard Web site might use to download an image file. It then approximates the atomic positions of any frame in the browser itself, without requiring a separate trajectory-streaming server.
We have recently created other software tools that similarly enable browser-based trajectory visualization. For example, our BlendMol20 plugin for the popular 3D modeling program Blender allow users to import VMD- or PyMOL-visualized proteins as meshes. Our Pyrite25 plugin can then animate these meshes according to MD-captured motions. Additional plugins created by others allow Blender to export animations in file formats that are compatible with JavaScript libraries such as Babylon.js and three.js. Depending on the details of the workflow, this approach can produce photorealistic trajectory visualizations that are excellent for outreach and education. But the process is far from automated. Additionally, because the animations are applied to protein-shaped meshes rather than full atomistic models, accurately capturing atomic-resolution motions is more challenging. PCAViz overcomes these limitations.
Usage
The PCAViz download includes a detailed README file describing how to use the Python and JavaScript components of the toolkit, together with examples of use. The download also includes simple trajectory files for testing. To obtain a detailed description of the available PCAViz-Compressor command-line parameters, users need only pass the “--help” parameter to the Python script (i.e., python PCAViz.py --help). To see how to integrate the PCAViz Interpreter into an existing HTML page, users can examine the highly commented example HTML files included with the PCAViz download.
To encourage browser-based molecular visualization among educators and scientists without HTML and JavaScript expertise, we also developed a PCAViz WordPress plugin that automatically integrates the PCAViz Interpreter into any WordPress site. WordPress is an easy-to-use, open-source content management system that powers roughly 75 million websites around the world.26 WordPress shortcodes make it easy to add a PCAViz widget to any post or page. We have included several PCAViz JSON files with the WordPress plugin itself so educators without their own MD trajectories can teach their students about protein dynamics.
Conclusion
PCAViz is a useful tool for visualizing MD trajectories online that drastically reduces the amount of data that must be transmitted from the server to the browser. For example, consider PCAViz applied to our 100-frame, 1326-atom benchmark LARP1 simulation. When we retained only the components required to account for 90% of the variance and rounded all values to the nearest hundredth (see Figure 1 and Table S1), the resulting JSON file was only 497 K (101 K when further compressed with GZIP). In contrast, a PDB file containing the full trajectory was 10M. And yet the frames of the PCAViz-processed trajectory deviated from those of the original by only 0.62 Å on average (RMSD).
When performing detailed analyses of MD simulations, subangstrom accuracy may be necessary. In these cases, we recommend dedicated analysis programs such as VMD17 and PyMOL,18 or an online streaming solution such as MDSrv.23 But PCAViz is ideal when one wishes only to easily and effectively visualize MD simulations in the browser.
We expect PCAViz to appeal to a broad audience, including researchers who wish to collaboratively share their simulations, outreach coordinators who wish to communicate the power of computational biology, and students learning about protein motions in a classroom setting. We release it under the terms of the open-source GNU General Public License, version 2. A copy can be downloaded free of charge from http://durrantlab.com/pcaviz/. The WordPress plugin is available via the official WordPress Plugin Directory.
Materials and Methods
PCAViz Compressor (Python)
The PCAViz Compressor script is a command-line program written in Python that converts MD files from standard 3D-Cartesian formats (e.g., dcd, netcdf, xtc) to the compressed/simplified PCAViz JSON format. We have tested the PCAViz Compressor on several operating systems, using several different versions of the required Python libraries (Table 1). To obtain a copy of the compressor, users can (1) download (or clone) the entire PCAViz git repository at http://git.durrantlab.com/jdurrant/pcaviz, (2) download only the compressor-relevant files from the repository (pcaviz-compressor-python.zip), or (3) install the compressor via the pip package-management system (pip install pcaviz-durrantlab).
Table 1. PCAViz Compressor Compatibilitya.
| Operating System | Python | MD analysis | Scikit-learn | NumPy |
|---|---|---|---|---|
| macOS Mojave 10.14.4 | 3.6.7 | 0.19.2 | 0.20.3 | 1.16.3 |
| macOS Mojave 10.14.4 | 2.7.15 | 0.19.2 | 0.20.3 | 1.16.3 |
| Ubuntu 18.04.1 | 3.6.6 | 0.19.2 | 0.19.1 | 1.15.4 |
| Ubuntu 18.04.1 | 2.7.16 | 0.18.0 | 0.20.0 | 1.15.2 |
| Microsoft Windows 10 Home | 3.7.1 | 0.19.2 | 0.20.3 | 1.16.3 |
We have tested the PCAViz Compressor on several operating systems using different versions of Python, MDAnalysis, scikit-learn, and NumPy.
Loading the Trajectory
The PCAViz Compressor uses the MDAnalysis package14 to load simulation coordinate (trajectory) and topology files. MDAnalysis supports many popular coordinate-file formats (e.g., dcd, xtc, trr, out, trz, mdcrd, inpcrd, restrt, netcdf, nc) as well as many topology-file formats (e.g., psf, prmtop, parm7, top, and xml).
To preserve the global translational and rotational motions of the simulated system, PCAViz does not perform any least-squares fitting of trajectory frames to a common reference (e.g., the first frame). In many cases, users may wish to use a separate program (e.g., VMD17 or PyMOL18) to align the trajectory before PCAViz processing. Otherwise, global motions may dominate the positional variance of the system, degrading the representation of often more interesting internal motions.
Pruning the Trajectory
The compressor allows the user to remove atoms from the simulated system that will not be visualized in the browser. Removing unnecessary atoms simplifies the trajectory and accelerates subsequent steps. It also helps alleviate the memory and bandwidth constraints associated with in-browser viewing. For example, if the user does not wish to display solute–solvent interactions, water molecules can be removed. If the user is primarily interested in side-chain motions, hydrogen atoms may also be unnecessary. If the intent is to visualize a protein using the ribbon or new cartoon representations, retaining only backbone atoms is sufficient.
The user can also stride the simulation, keeping only every few frames. Striding drastically reduces the amount of data that must be stored, improving the performance of subsequent steps. Interpolating between the retained frames often reasonably reconstructs the dropped frames.
Principal Component Analysis
The PCAViz Compressor next uses principal component analysis (PCA),27 as implemented in the scikit-learn Python package,28 to approximate the motions of the remaining atoms. PCA involves identifying orthogonal vectors (principal components) that maximize the positional variation across the trajectory. The first component explains the most variation, with each consecutive component explaining less. The atomic coordinates of each frame can be expressed as a linear combination of the components, where each component is multiplied by a frame-specific coefficient. To reduce the dimensionality of the data while minimizing the loss of variation in a quantified way, one can approximate atomic coordinates by considering only the first n components. But, the resulting lossy compression comes at the cost of atom-position accuracy.
The user can specify the amount of positional variance that should be explained by the principal components. In a given PCA decomposition, each component accounts for a certain percentage of the total variance. The PCAViz Compressor identifies the smallest set of top components that—when considered together—account for the user-specified (cumulative) positional variance. Allowing users to specify the cumulative variance is more intuitive than requiring them to directly specify the number of principal components. For interested users, running the PCAViz Compressor with the “check_accuracy” flag outputs the number of top components retained. Tables S1 and S2 provide two illustrations of how the number of retained components varies according to the user-specified cumulative-variance cutoff. To obtain the best results, users should find a good balance between the variance cutoff and the desired atom-position accuracy. Accounting for more variance (by allowing PCAViz to include more components) improves accuracy at the cost of compression.
Controlling the JSON File Size
The compressor outputs a JSON file that contains the PCA components and frame-specific coefficients; the average atomic Cartesian coordinates (i.e., the origin in PCA space); the residue names and sequence numbers; the atom names; and the atomic coordinates of the first frame to enable in-browser bond-by-distance calculations. To optimize the JSON file for transfer over the web, the compressor stores this information as efficiently as possible. It deletes unnecessary spaces present in the file, rounds floating-point numbers to a user-defined precision, and represents all numeric values as integers (eliminating decimal points). These approaches substantially reduce the file size, enabling rapid transfer over the Internet.
Evaluating Accuracy
To evaluate the impact of compression on atom-position accuracy, users can optionally instruct the PCAViz Compressor to calculate the root-mean-square deviation (RMSD) between each pre- and postcompression frame. Examining the output CSV file containing these values provides a sense for how much accuracy is lost due to compression.
PCAViz Interpreter (JavaScript)
The PCAViz Interpreter displays the processed trajectories in any modern web browser. We have specifically tested it on Google Chrome, Firefox, and Safari. To obtain a copy of the interpreter, users can download the entire PCAViz git repository at http://git.durrantlab.com/jdurrant/pcaviz. The same Web site also includes a single ZIP file containing only the interpreter-relevant files (pcaviz-interpreter-javascript.zip).
Converting from PCA to Cartesian Space
The Interpreter first retrieves the JSON file from a web server and converts all the numeric values from integers back into decimal numbers. If the original trajectory was strided, the PCA coefficients associated with some trajectory frames may be missing. The JavaScript interpreter estimates the values of the missing coefficients by linearly interpolating between frames that do have defined coefficients.
Next, the JavaScript interpreter converts the PCA data back into 3D Cartesian coordinates, as required to visualize the trajectory using popular molecular-visualization JavaScript libraries such as 3DMol.js,9 NGL Viewer,10 and PV.11 For each frame, the original coordinates can be recovered by multiplying each principal component by the corresponding frame-specific coefficient, summing the scaled vectors, and adding the average atomic Cartesian coordinates.
Optimizing the Use of Available Resources
Converting from PCA space back to Cartesian space can be computationally expensive. The JavaScript interpreter provides three caching options to manage these calculations. The first, though CPU intensive, allows for quick start times and low memory usage. The coordinates of each frame are (re)calculated every time that frame is displayed. Previously calculated frame coordinates are discarded to keep memory usage low. Unfortunately, because this option is CPU intensive, trajectory playback on low-end machines is sometimes choppy.
The second option, though more memory intensive, allows for quick start times and lower CPU usage. The coordinates of each frame are calculated only once, when the frame is first displayed. The initial trajectory playback may be choppy as the CPU performs these initial calculations. But calculated coordinates are saved to memory, so the animation is smoother each time the trajectory is replayed (e.g., in loop playback mode).
The third option prioritizes smooth playback at the expense of both quick start times and memory usage. The coordinates of all frames are calculated and saved to memory before displaying any frame of the trajectory. As no new coordinates need be calculated during playback, playback tends to be much smoother once it starts.
Several other PCAViz options also aim to improve in-browser playback. The user can instruct the JavaScript interpreter to update atomic positions less frequently than the default 60 times per second (i.e., every 16.7 ms). Shortening the trajectory-playback duration can also reduce the number of frame coordinates that need be calculated in some cases. These adjustments may require the user to smooth the trajectory to improve visualization. If so, the PCAViz interpreter can also calculate a moving average of the frame coordinates, using a sample window of user-specified length.
Acknowledgments
This work was supported by a computing allocation from the University of Pittsburgh’s Center for Research Computing (allocation to J.D.D). We also acknowledge funding from the National Institutes of Health’s Building Infrastructure Leading to Diversity (BUILD) program (no. 8TL4GM118977-02).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.9b00703.
Tables S1 and S2, an extended description of PCAViz accuracy vs compression (PDF)
Author Contributions
† The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Durrant J. D.; McCammon J. A. Computer-Aided Drug-Discovery Techniques That Account for Receptor Flexibility. Curr. Opin. Pharmacol. 2010, 10, 770–774. 10.1016/j.coph.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Rhodes G.An Overview of Protein Crystallography. In Crystallography Made Crystal Clear; Academic Press: 2006; Chapter 2, pp 7–30. [Google Scholar]
- Hospital A.; Goni J. R.; Orozco M.; Gelpi J. L. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. W.; Brunetti L.; Brooks C. L. 3rd. Probing Ph-Dependent Dissociation of Hdea Dimers. J. Am. Chem. Soc. 2011, 133, 19393–19398. 10.1021/ja2060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan A.; Freddolino P. L.; Schulten K. Going Beyond Clustering in Md Trajectory Analysis: An Application to Villin Headpiece Folding. PLoS One 2010, 5, e9890. 10.1371/journal.pone.0009890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajda S.; Beglov D.; Wakefield A. E.; Egbert M.; Whitty A. Cryptic Binding Sites on Proteins: Definition, Detection, and Druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. 10.1016/j.cbpa.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglov D.; Hall D. R.; Wakefield A. E.; Luo L.; Allen K. N.; Kozakov D.; Whitty A.; Vajda S. Exploring the Structural Origins of Cryptic Sites on Proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3416–E3425. 10.1073/pnas.1711490115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego N.; Koes D. 3dmol.Js: Molecular Visualization with Webgl. Bioinformatics 2015, 31, 1322–1324. 10.1093/bioinformatics/btu829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose A. S.; Hildebrand P. W. Ngl Viewer: A Web Application for Molecular Visualization. Nucleic Acids Res. 2015, 43, W576–579. 10.1093/nar/gkv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M.Pv–Javascript Protein Viewer. https://biasmv.github.io/pv/ (accessed 7/23/2019).
- Durrant J. D.; Bush R. M.; Amaro R. E. Microsecond Molecular Dynamics Simulations of Influenza Neuraminidase Suggest a Mechanism for the Increased Virulence of Stalk-Deletion Mutants. J. Phys. Chem. B 2016, 120, 8590–8599. 10.1021/acs.jpcb.6b02655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan M.; Liu N.; Zhou W.; Li D.; Yang M.; Hou T. Structural Diversity of Ligand-Binding Androgen Receptors Revealed by Microsecond Long Molecular Dynamics Simulations and Enhanced Sampling. J. Chem. Theory Comput. 2016, 12, 4611–4619. 10.1021/acs.jctc.6b00424. [DOI] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson R. M. Jmol–a Paradigm Shift in Crystallographic Visualization. J. Appl. Crystallogr. 2010, 43, 1250–1260. 10.1107/S0021889810030256. [DOI] [Google Scholar]
- Hanson R. M.; Prilusky J.; Renjian Z.; Nakane T.; Sussman J. L. Jsmol and the Next-Generation Web-Based Representation of 3D Molecular Structure as Applied to Proteopedia. Isr. J. Chem. 2013, 53, 207–216. 10.1002/ijch.201300024. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsletter On Protein Crystallography 2002, 40, 82–92. [Google Scholar]
- Lahr R. M.; Mack S. M.; Heroux A.; Blagden S. P.; Bousquet-Antonelli C.; Deragon J. M.; Berman A. J. The La-related protein 1-Specific Domain Repurposes HEAT-like Repeats to Directly Bind a 5′TOP Sequence. Nucleic Acids Res. 2015, 43, 8077–8088. 10.1093/nar/gkv748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D. BlendMol: Advanced Macromolecular Visualization in Blender. Bioinformatics 2019, 35, 2323–2325. 10.1093/bioinformatics/bty968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec B.; Holtz K. M.; Wojciechowski C. L.; Kantrowitz E. R. Structure of the Wild-Type Tem-1 Beta-Lactamase at 1.55 a and the Mutant Enzyme Ser70Ala at 2.1 a Suggest the Mode of Noncovalent Catalysis for the Mutant Enzyme. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2005, 61, 1072–1079. 10.1107/S0907444905014356. [DOI] [PubMed] [Google Scholar]
- Shkurti A.; Goni R.; Andrio P.; Breitmoser E.; Bethune I.; Orozco M.; Laughton C. A. Pypcazip: A Pca-Based Toolkit for Compression and Analysis of Molecular Simulation Data. Data. SoftwareX. 2016, 5, 44. 10.1016/j.softx.2016.04.002. [DOI] [Google Scholar]
- Tiemann J. K. S.; Guixa-Gonzalez R.; Hildebrand P. W.; Rose A. S. Mdsrv: Viewing and Sharing Molecular Dynamics Simulations on the Web. Nat. Methods 2017, 14, 1123–1124. 10.1038/nmeth.4497. [DOI] [PubMed] [Google Scholar]
- Carrillo-Tripp M.; Alvarez-Rivera L.; Lara-Ramirez O. I.; Becerra-Toledo F. J.; Vega-Ramirez A.; Quijas-Valades E.; Gonzalez-Zavala E.; Gonzalez-Vazquez J. C.; Garcia-Vieyra J.; Santoyo-Rivera N. B.; Chapa-Vergara S. V.; Meneses-Viveros A. Htmol: Full-Stack Solution for Remote Access, Visualization, and Analysis of Molecular Dynamics Trajectory Data. J. Comput.-Aided Mol. Des. 2018, 32, 869–876. 10.1007/s10822-018-0141-y. [DOI] [PubMed] [Google Scholar]
- Rajendiran N.; Durrant J. D. Pyrite: A Blender Plugin for Visualizing Molecular Dynamics Simulations Using Industry-Standard Rendering Techniques. J. Comput. Chem. 2018, 39, 748–755. 10.1002/jcc.25155. [DOI] [PubMed] [Google Scholar]
- Munford M.How Wordpress Ate the Internet in 2016...And the World in 2017. Forbes 2016, Dec 22. [Google Scholar]
- Jolliffe I.Principal Component Analysis, 2nd ed.; Springer: New York, 2011; p 478. [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay É. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.