

Summary
Recent advances in genomics technologies have greatly accelerated the progress in both fundamental plant science and applied breeding research. Concurrently, high‐throughput plant phenotyping is becoming widely adopted in the plant community, promising to alleviate the phenotypic bottleneck. While these technological breakthroughs are significantly accelerating quantitative trait locus (QTL) and causal gene identification, challenges to enable even more sophisticated analyses remain. In particular, care needs to be taken to standardize, describe and conduct experiments robustly while relying on plant physiology expertise. In this article, we review the state of the art regarding genome assembly and the future potential of pangenomics in plant research. We also describe the necessity of standardizing and describing phenotypic studies using the Minimum Information About a Plant Phenotyping Experiment (MIAPPE) standard to enable the reuse and integration of phenotypic data. In addition, we show how deep phenotypic data might yield novel trait−trait correlations and review how to link phenotypic data to genomic data. Finally, we provide perspectives on the golden future of machine learning and their potential in linking phenotypes to genomic features.
Keywords: plant genomes, plant bioinformatics, plant genome annotation, phenotyping
Significance statement
This review addresses the state of the art on plant genome sequencing, phenotyping and how to bridge these. Genome sequencing sees a new revolution and phenotyping is becoming extremely important.
Introduction
The last decade has seen a considerable increase in published plant genomes, enabled by advances in sequencing technologies. The initial post‐Sanger sequencing advancement came in the form of high‐throughput short‐read technologies, frequently termed second‐generation sequencing (see glossary Table 1). Although the maximum read length of about 600 bases was considerably shorter than that available from contemporary Sanger sequencing, the high throughput and low relative cost ensured that this technology was quickly adapted. This was followed by the more recent long‐read technology (third‐generation sequencing) led by the PacBio platform. This overcame the read length problem inherent in second‐generation sequencing, enabling multi‐kilobase reads, but at a cost of read quality. Third‐generation sequencing was initially used to resequence well studied model species such as Arabidopsis thaliana, yeast and Drosophila (Berlin et al., 2015) before successfully sequencing new genomes, such as the small genome from Oropetium thomaeum (Van Buren et al., 2015).
Table 1.
Glossary table
| Term | Definition |
|---|---|
| Best linear unbiased predictions (BLUP) | A method used to estimate the ‘random’ effects of a mixed model. For a plant researcher this is of relevance when genotypes are considered a ‘random’ effect (reviewed in Piepho et al., 2008). |
| Chromosomal pseudomolecules | The largest sequences assembled and ordered by genome sequencing projects, each representing a single chromosome in the genome. These are not necessarily complete, i.e. they might contain stretches of ‘N's. |
| Contigs | Assembled sequences that contain no unknown (‘N’) bases. |
| Copy‐number variation (CNV) | An InDel that increases or decreases the number of copies of a specific DNA sequence. |
| De Bruijn graph method | A method of genome assembly particularly suited to datasets from short‐read sequencing platforms, due to its scalability to large numbers of reads. |
| De novo assembly | The method of assembling a genome from scratch when there is no reference sequence available. |
| Genome‐wide association studies (GWAS) | An observational study that tries to associate a genome‐wide set of variants (e.g. markers/polymorphisms) to determine whether a variant is associated with a particular trait. Usually requires many genotypes and relies on natural populations and/or panels with diverse cultivars as opposed to biparental populations. |
| Insertions/deletions (InDel) | A genomic variant in which one or more bases have been added and/or removed, resulting in a shorter or longer sequence than originally present. |
| Machine learning | The process of training computers to autonomously extract important information from a data set and identify patterns. Important subfields for a plant researcher include: (i) classification (e.g. is a plant diseased or healthy given an image); (ii) regression (e.g. predict plant biomass from several images); (iii) clustering (e.g. are there subtypes of plants in the experiment based on the measurement)? |
| Minimum information about plant phenotyping experiment (MIAPPE)’ | Presents guidelines and a checklist for describing plant phenotyping experiments so that they are understandable and reproducible. |
| Ontology | An ontology is extending controlled vocabularies (i.e. fixed lists of terms to be used) by relating these terms to each other. In the simplest case it could describe one term to always imply another term (e.g. if monocot, dicot and plant could represent a controlled vocabulary and the addition of monocot IS_A plant; dicot IS_A plant would start to add relationships towards an ontology). |
| Overlap‐layout consensus (OLC) method | A method of genome assembly particularly suited to datasets from long‐read sequencing platforms, originally developed for Sanger sequencing data. |
| Polish | A post‐assembly quality improvement procedure that aims to identify and correct small scale errors. |
| Quantitative trait locus (QTL) | A region of DNA containing one or more genes which are associated to the expression of a quantitative phenotypic trait. |
| Reduced representation libraries (RRL) | A protocol to create a sequencing library that aims to contain sequences only from selected subsets of the source genome. |
| Restriction site associated DNA sequencing (RAD‐seq) | A protocol using restriction enzymes to target specific sequences from a genome for including in a sequencing library. |
| Second‐generation sequencing/next‐generation sequencing | Usually sequencing by synthesis based, high‐throughput sequencing platforms that can sequence millions of DNA strands in parallel, but compared with Sanger sequencing have a higher error rate and limited read length, e.g. 50–600 bases, depending on the specific instrument used. Some platforms offer a paired‐end mode, whereby both ends of a DNA fragment are sequenced. |
| Single nucleotide polymorphism (SNP) | A genomic variant consisting of a single nucleotide substituted for an alternative nucleotide. |
| Third‐generation sequencing | Single‐molecule sequencing platforms that can create multi‐kilobase reads, but which have much higher error rates than Sanger or second‐generation sequencing platforms. |
| Variable importance prediction | A formalized method to predict the importance of variables in PLS type analyses. |
This trend continues as Oxford Nanopores, the latest long‐read technology, becomes more widely available. This technology (Jain et al., 2016) has already successfully been used to reconstruct the Arabidopsis genome (Michael et al., 2018) as well as the genome of a non‐model wild tomato species (Schmidt et al., 2017) and has the added advantage of not requiring large capital investment. Long reads can not only reveal small‐scale variation and presence−absence dynamics in genes, but also large‐scale variation, including rearrangements from for example transposon activity, and can lead to potentially novel insights into a plant species. Additionally, as pangenomic approaches based on multiple‐reference accessions becomes more common, de novo sequencing of many lines from each species can be expected (e.g. Brassica: Golicz et al., 2016, rice including wild relatives: Zhao et al., 2018). While genome research is certainly well established and advances in technologies allow for the delivery of data ever more quickly and efficiently, effective algorithms and storage capacity for genome data are becoming serious concerns (Stephens et al., 2015).
As with genomic developments, there are promising advances in plant phenotyping technology, such as the use of automated phenotyping machinery (Fiorani and Schurr, 2013) and advanced image analyses (Pound et al., 2014, 2017; Tsaftaris et al., 2016). This has resulted in unprecedented insights into plant physiology, architecture, and performance. Compared with genomic research, data output produced by established systems in plant phenotyping is still manageable (Coppens et al., 2017), although expanding the use of advanced imaging platforms such as hyperspectral cameras by the wider community will likely result in similar storage capacity concerns. As phenotyping equipment costs are still prohibitive for many plant laboratories, new lower cost phenotyping procedures, including the deployment of inexpensive sensors and set ups (Paulus et al., 2014) as well as machine‐learning techniques for low‐cost devices, are being developed and researched (Atanbori et al., 2018).
Analyses that combine advanced phenotyping and genomic datasets offer great potential for the discovery of novel insights, such as in GWAS (Millet et al., 2016, Borevitz et al., ibid) or genomic prediction technologies, even within the scope of a single project. Furthermore, machine‐learning and other data science techniques can extract novel insights from meta analyses of multiple datasets. However, there are several obstacles that need to be addressed before this can become widely applicable. This review outlines the current state of the art in genomics, plant phenotyping, and standardization. It explains how these data can be integrated using data science and machine‐learning techniques, and discusses current challenges that are being addressed by the plant science community.
From sequences to genomes
De novo sequencing and assembly of plants is often difficult and tedious (Claros et al., 2012). This is largely due to the high repeat content of many plant genomes, with repetitive elements derived from a wide range of sources, including transposons and tandem gene duplications. The situation can be further complicated by the fact that many plants are autopolyploid, or have undergone recent whole genome duplications (Vogel et al., 2018). This has often necessitated analyzing diploid relatives (e.g. wild strawberry, Shulaev et al., 2011) or using double‐monoploid lines (e.g. Potato Genome sequencing consortium, 2011) rather than a commercially relevant crop line. Even more problematic, plants may be derived from the hybridization of different but related species, giving rise to allopolyploid species such as rapeseed (Chaloub et al., 2014), tobacco (Sierro et al., 2014) or petunia (Bombarely et al., 2016), whose genomes are often tackled by first analyzing the extant parental genomes. This approach has also been applied to sequencing the D parent of the allohexaploid wheat (Luo et al., 2017).
While polyploidy forms an obvious problem, repeats and the complications that they cause have been known but not systematically analyzed. Jiao and Schneeberger (2017) investigated this issue in detail by comparing approximately 100 diverse plant and vertebrate genomes. The authors were able to demonstrate higher incidences of repeats in plant genomes, suggesting that some plant genome assemblies will require more advanced approaches to span repetitive regions. Another difficulty of plant genomes is often their sheer size, making costly long‐read sequencing technologies prohibitively expensive, both in terms of sequencing and computation. This was especially challenging for the complex 17 Gbp wheat genome, which consists of three subgenomes (International Wheat Genome Sequencing Consortium, 2014). Therefore, the initial assembly relied on sequencing sorted chromosomes, a difficult wet‐lab technique. Conversely, a whole‐genome shotgun assembly using long‐read data required 880 000 central processing unit (CPU) h to compute, taking more than 6 months, despite being run on a compute cluster (Zimin et al., 2017). Finally, many plants are self‐incompatible (Fujii et al., 2016) and consequently can be highly heterozygous, adding complexity to the assembly process.
Despite these hurdles, many standard pipelines and tools that can potentially assemble a reasonable quality genome, are available (Figure 1). The primary factor determining the choice of assembly pipeline is the type of reads in the dataset, as short and long reads are generally assembled using very different approaches. In the case of short‐read data from the Illumina platform, reads are typically quality controlled using for example FASTQC followed by adapter/quality trimming (Bolger et al., 2014b). After read trimming, the assembly process can be performed using a variety of short‐read assemblers such as ABySS (Simpson et al., 2009), DISCOVAR (de novo) (Weisenfeld et al., 2014), Velvet (Zerbino and Birney, 2008) or SOAPdenovo (Luo et al., 2012). SOAPdenovo is especially popular as it is easy to install, relatively easy to use and reasonably fast. Alternatively, commercial software such as the CLC assembler can be used with small computational resources and offers a graphical user interface, whereas the commercial NRGene suite enables the analysis of complex genomes using short‐read data (Avni et al., 2017; Luo et al., 2017).
Figure 1.
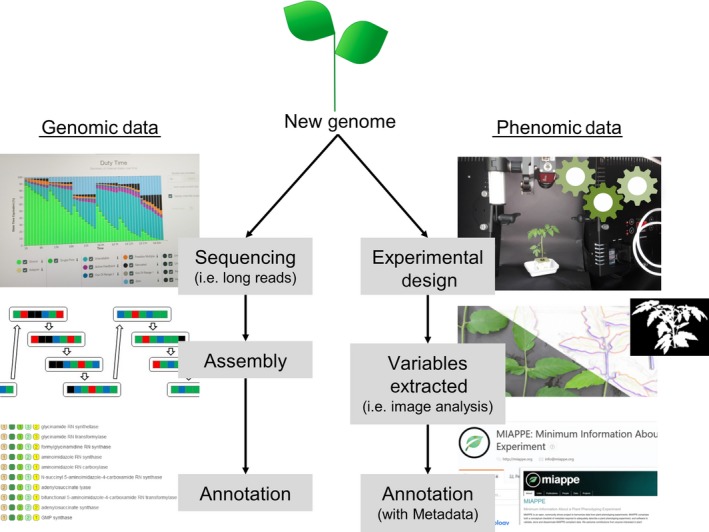
Preparatory analyses for genomics and phenomics data for new genomes.
Examples of long‐read assemblers include Miniasm (Li, 2016), Canu (Koren et al., 2017), SMART denovo or its successor, wtdbg, and Falcon. In some cases, steps of different assemblers can be ‘mixed and matched’ for speed and efficiency. For instance, it can be beneficial to use the error correction steps of Canu coupled to SMART denovo (Schmidt et al., 2017) or wtdbg (Koren: https://genomeinformatics.github.io/na12878update/).
The relative costs and high error rate of long‐read technologies negate some of their benefits. Error rate was particularly problematic for long reads from early versions of the third‐generation Oxford Nanopore platform, which offered a read correctness of below 70% (Rang et al., 2018). Its error rate has improved substantially in subsequent versions, but the technology still has difficulty resolving specific base patterns, such as long homopolymers. As a result, recent versions have been assessed to give a maximum read correctness of 88% (Wick et al., 2018), although this is potentially lower in plants (Schmidt et al., 2017). As these errors are systematic, they cannot be fully corrected by additional coverage. Therefore, even after post‐assembly read polishing, assemblies are currently capped at 99.9% accuracy when using Oxford data alone (Wick et al., 2018). In theory, PacBio reads should have much fewer systematic errors, and therefore should converge on the correct result, given sufficient coverage. Nonetheless, there are indications that real‐world assemblies may still suffer from some residual accuracy problems (Watson, 2018).
Given their complementary attributes, it is common to combine error‐prone long reads with highly accurate short reads to form a potentially superior hybrid assembly (Figure 2). Multi‐step hybrid approaches are necessary because established assembly algorithms, namely the Overlap‐Layout‐Consensus (OLC) method, used with long reads, and the De Bruijn Graph (DBG) method, used with short reads, are only suitable for their respective kinds of read dataset. An early approach to hybrid assembly was to first assemble the short reads, then scaffold the resulting contigs guided by the long reads (Figure 2). This process can be performed by a post‐assembler tool, such as PBJelly and SSPACE‐LongRead, or by integration directly into an assembler, such as SPAdes. The MaSuRCA (Zimin et al., 2013) approach is similar and works by first conservatively assembling the short reads into longer ‘super‐reads’ and then assembling the super‐reads in combination with the longer reads in an OLC approach. These short‐read‐first approaches work relatively well when only limited amounts of long‐read data are available.
Figure 2.
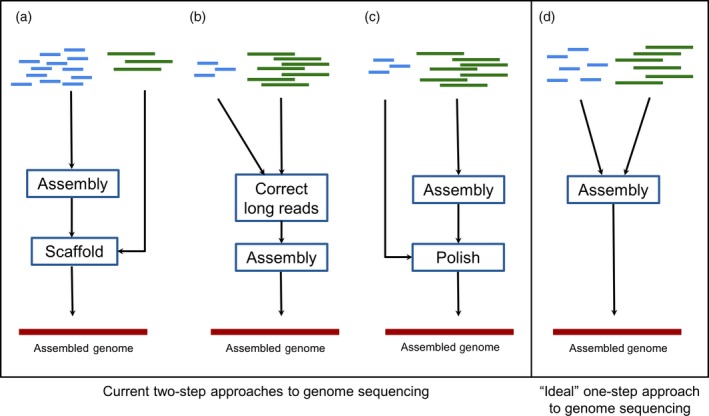
Approaches to genome sequencing.
Currently, when approaching genome sequencing, the method used depends on the read lengths available: (a) When more short reads are available, they are first assembled into contigs, which are then scaffolded, guided by the long reads. When more long reads are available, two assembly options exist. Either (b) short reads are used to first correct the long reads, which are then assembled or (c) the long reads are first assembled after which the short reads are used to ‘polish’ the assembly. As these approaches lose information at each step, a method (d) that could combine long and short reads in a single step (theoretically leading to an improved genome assembly) would be optimal.
However, when sufficient long‐read data are available, a long‐read assembly approach will generally give a better result. Short reads can be used before assembly to correct the individual long reads (Figure 2b) or after assembly to correct the contigs (Figure 2c), a process commonly referred to as ‘polishing’ the assembly. For pre‐assembly read correction, the simplest approach is to map individual short reads onto long reads and use the short‐read consensus to correct the long reads. This approach is implemented in tools such as Proovread (Hackl et al., 2014) and/or LSC (Au et al., 2012). As it is difficult to unambiguously align individual short reads against long reads, an alternative strategy involves an initial assembly of the short, accurate reads into contigs (HALC, Bao and Lan, 2017) or assembly graphs (LoRDEC, Salmela and Rivals, 2014) to correct the long reads. A recent comparison of long‐read correction tools found that HALC performed best on data sets from ‘complex’ genomes, such as that of humans or rice (Mahmoud et al., 2017).
Post‐assembly polishing using short reads can be performed using Pilon, while Racon supports polishing with either short or long reads. Although polishing with accurate short reads can dramatically improve assembly accuracy, in practice, this often applies only to unique genome regions.
Although these multistep hybrid approaches often outperform assemblers, which use short or long reads alone, they are inherently wasteful. Information is lost at each step in the analysis, and therefore results in a suboptimal assembly. A single step hybrid approach, which would allow for the seamless integration and combined analysis of short and long reads, could in principle yield an improved assembly (Figure 2d).
In addition, especially for some plant genomes, many short reads cannot be accurately mapped to one location due to transposon‐derived repeats and homologous genes with a high degree of identity, making the long‐read assembly errors unrecoverable by short reads.
The final endpoint of a genome assembly is ordering and orienting the assembled sequences to form chromosomal pseudomolecules. This can be guided by marker sequences from an independently determined genetic map. Alignment of these marker sequences against the assembly allows the approximate chromosomal position and potentially orientation of each scaffold to be determined. This last step is often not reached, as it is either not needed for the planned analyses or high resolution genetic maps are not available. However, in the context of combining genotypes with phenotypes, the exact chromosomal position of genes is essential for their correlation with known QTLs. Hi‐C, a new technology providing contact frequencies between sequences, has revolutionized the assembly to chromosomes. For plants it was notably applied to the 5 Gb barley and 12 Gb wild emmer genome (Avni et al., 2017; Mascher et al., 2017) and has allowed chromosome‐scale assemblies without a genetic map for example for raspberry (Van Buren et al., 2018).
One reference, multiple references and the pangenome
Short‐read sequencing technologies, in conjunction with annotated reference genomes, can be readily applied to a variety of biological questions, including detection (Zhang et al., 2017) and analysis of gene expression (Ezer et al., 2017), DNA methylation (Zhong et al., 2013), identification of transcription factor binding sites and the detection of causal regions and mutations in mutant screens (James et al., 2013; Klap et al., 2017) or populations (Thoen et al., 2017). However, the importance of next‐generation sequencing beyond the context of a single reference accession has long been recognized (Varshney et al., 2009). As sequencing became more accessible in terms of cost and availability, plant projects frequently sequenced multiple accessions or species in order to investigate natural diversity. This was initially applied to plant species with relatively small genomes such as rice or Arabidopsis, but has since been extended to field crops such as tomato (Lin et al., 2014).
Traditionally, the dominant analysis approach for such projects involved mapping reads from novel accessions to the reference genome to determine small‐scale variation, especially single‐nucleotide polymorphisms (SNPs) and, less commonly, insertions and deletions (InDels) including copy‐number variations (CNVs). A reduced representation of a genome is potentially the cheapest way to gain SNP and marker information in order to enable genome‐wide association studies (GWAS) and genomic selection studies (Bhat et al., 2016). The key idea was to reduce the sequencing cost per sample by only sequencing corresponding parts of genomes, albeit at the cost of a more complex library preparation, using restriction enzymes to selectively cut the DNA, therefore focusing the sequencing around the restriction sites. Multiple approaches have been developed, including reduced representation libraries (RRL; Van Tassell et al., 2008), restriction site associated DNA sequencing (RAD‐Seq; Baird et al., 2008) and genotyping‐by‐sequencing (GBS; Elshire et al., 2011). New variations of these techniques continue to be developed (He et al., 2014; Scheben et al., 2017).
Whole genome resequencing ranging from skim sequencing (approximately 1× coverage or below) to medium coverage resequencing (in the range of 20–40×) is increasingly common for small to mid‐size genomes (Scheben et al., 2017), but remains so far prohibitively expensive for large genomes such as wheat. Using this resequencing approach, sequences from the whole genome are used, thereby offering more comprehensive SNP detection than reduced representation approaches. This whole genome resequencing approach, which was successfully used in humans, often performs less well when applied to plants. This is due to the standard read mapping approaches, which were mostly tuned for human data sets and only tolerate minor variations from the reference sequence (Li and Durbin, 2009; Langmead and Salzberg, 2012). These are therefore ill suited to the high rates of variation found even within a single plant species. The frequent use of related wild species as breeding material further amplifies this problem, due to a broadening of the genomic pool. Other typical plant genomic characteristics, including large gene families, ancient whole genome duplications, polyploidy and a high amount of transposon derived repeats, further exacerbate the challenge.
While techniques based on mapping reads to reference genomes are well suited to GWAS and genomic selection, they are inadequate in identifying new genome variants, such as novel genes not present in the reference. In maize, it was estimated that an early genomic reference did not capture about a quarter of the low‐copy gene fraction from all inbred lines (Gore et al., 2009). Despite the estimated completeness of this reference being just 91%, mapping rates of above 95% for whole genome resequencing were obtained, illustrating that some reads were incorrectly mapped to repeat regions or paralogous genes (Bukowski et al., 2018). This represents a major issue as, in order to improve existing elite accessions using transgenic and new breeding technologies, finding novel genes or gene variants is necessary (Scheben and Edwards, 2018).
An alternative strategy to deal with this issue is to map the reads to the reference plant genome using relatively strict alignment criteria, followed by assembly of the ‘left‐over’ reads that could not be mapped. Using this two‐step approach, it is expected that the non‐mapping reads will assemble into novel genetic regions present in the particular strain under study. This strategy has been applied in the model plant Arabidopsis (Schneeberger et al., 2011), and more recently to crops cabbage (Golicz et al., 2016) and wheat (Montenegro et al., 2017). However, the resulting novel sequences are typically short and fragmented, as many of the reads belonging to these regions would have been inadvertently mapped to similar regions present on the reference, even if relatively strict alignment criteria are used.
A radically different approach is to ignore the existing reference entirely, instead jointly assembling read data from multiple genomes and tracking read origin (Iqabl et al., 2012, Muggli et al., 2017). This computationally elegant method allows the nodes and/or edges of the graph to be tagged with information, indicating which read dataset(s) support them. Given these tags, it is easy to determine the nodes/edges that are either shared by or unique to specific datasets. Chains of such nodes/edges can then be used to infer longer shared or unique sequences. Despite its elegance, this approach is only used occasionally in the eukaryotic field due to the computational resources needed.
Another alternative is the creation of multiple de novo assemblies that, from the wet‐lab perspective, has been made feasible by recent advances in long‐read sequencing technologies. However, the bioinformatics infrastructure required for such an endeavor presents a major barrier. A single gigabase scale assembly can require 10 000+ CPU h per iteration for Canu (Koren et al., 2017, Schmidt et al., 2017), but new sequence analysis algorithms (Bolger et al., 2017b,c) and assembly tools such as wtdbg (see above) promise to bring these computational costs down.
Multiple de novo genomes from a single species contain a more complete genetic repertoire than a single haploid reference. This approach can be extended to a set of related species such as a crop and its wild relatives. A recent study used more than 60 diverse rice (Oryza sativa) accessions together with a wild relative (Oryza rufipogon) to assemble multiple genomes, revealing gene loss and gain (Zhao et al., 2018). In a similar approach, 54 Brachypodium lines were all assembled de novo (Gordon et al., 2017). While illustrating the power of a multiple‐reference genome approach, these projects required multiple time‐intensive analysis pipelines, and several ad‐hoc developments.
A fundamental barrier to the wider adoption of this approach is that the vast majority of existing analysis tools and pipelines do not work with multiple‐reference genomes. The naïve creation of an in silico polyploid, formed by aggregating multiple‐reference genomes, is inadequate in many scenarios. It is necessary to have a clear conceptual difference between the sequences from a single line/species, which are generally considered in aggregate, and sequences from different lines/species, which are considered as alternatives. Furthermore, this in silico polyploid approach is highly inefficient when working with a large number of highly related genomes, as each is represented independently.
The creation of the pangenome promises to address the conceptual and computational limitations of the in silico polyploid approach. At its most basic, the pangenome must retain the distinction between multiple sequences from one origin genotype and sequences from different genotypes and, more critically, the analysis tools using the pangenome reference must act appropriately based on this origin information and the specific analysis being performed. For computational reasons, the pangenome is likely to be represented as a graph structure, as described above, rather than a large collection of independent linear sequences. This allows regions that are shared between many genomes to be represented once, saving both storage space and computational resources during alignment.
Despite the challenges of their creation, pangenomes promise to be an extremely powerful resource for analysis of genomic sequences. However, existing pangenomic aligners, such as BWBBLE (Huang et al., 2013) can handle only limited variation beyond what is already known. This limitation is not critical for genomes (e.g. human) in which genetic diversity is limited and in which the reference is very comprehensive. However, for optimal use with crop species and their wild relatives, pangenomic tools will also need to support highly divergent, novel sequences as well as large‐scale variations. One approach has been made by the Variant Graph Team (Variant Graph Team, 2018) that allows the representation of pangenomes in graphs or to map reads to these and also to visualize them.
In summary, using multiple‐reference genomes, it is possible to find new genes or new regulatory cis elements. This would not be possible with only one reference. Especially in the case of regulatory elements, line‐specific transposon insertions bringing their own regulatory elements might play an important role (Chuong et al., 2017).
Standardized genome annotations
To find and functionally annotate causal genes that underlie a QTL region, it is first necessary to identify these genes in the underlying DNA sequences (Figures 1 and 3, left panel). While gene finding can still be considered as an art, tools such as MAKER‐P (Campbell et al., 2014) and BRAKER2 (Hoff et al., 2016) have simplified this task considerably. In situations in which sufficient RNA‐Seq expression data are available, programs such as StringTie (Pertea et al., 2015) can be used to transform these data into a first draft gene space. This expression‐driven gene calling improves with the use of full‐length cDNA sequencing, made possible by long‐read technologies. However, expression‐driven gene annotation can only detect genes for which a data set exists and in which these genes are expressed. This necessitates that samples are subjected to a wide range of conditions to activate expression of the full gene space.
Figure 3.

Combining genomic and phenomic data.
The GWAS image was taken from Voiniciuc et al. (2016).
In comparison to gene finding, a comprehensive transposon detection method for plant genomes is still in a more experimental phase. There are currently no established pipelines that capture all transposon types in a single step. This does not pose a major problem when working with a new genome for which a well curated repeat library exists from closely related species. In such cases, a simple homology search against repeat libraries provided by repeat databases such as RepBase (Bao et al., 2015) or PGSB‐REdat (Spannagl et al., 2017) will be sufficient to provide a draft transposon annotation. Suitable matching tools are either RepeatMasker (Smit et al., 2016) or vmatch (http://www.vmatch.de/), which greatly improves running times for large genomes (Avni et al., 2017; Mascher et al., 2017). For novel species without curated repeat libraries, the transposon annotation is more cumbersome as de novo detection of species‐specific transposons needs to be performed first (Lerat, 2010). Here, packages like REPET (Flutre et al., 2011) perform well for smaller genomes. Transposons, formerly considered as junk DNA, are now believed to be a major contributor to genotype diversity. Their role in phenotype diversity has been shown for many well studied single examples (e.g. Butelli et al., 2012; Lutz et al., 2015) and also in some genome‐wide approaches (e.g. Bolger et al., 2014a,c; Makarevitch et al., 2015). Given the emerging importance of transposons in the study of stress and developmental responses, their consistent annotation and analysis is crucial and will be likely to provide many interesting insights and, when pangenomes are available, allow tracking of transposon evolution in a species.
Once genes and transposons have been structurally annotated, the next step is to ascribe each gene a biological function, in a process known as ‘functional annotation’. While using one‐off textual annotations can be beneficial when inspecting small QTL regions for potential candidates, using a priori biological knowledge is no longer feasible for large‐scale analyses. Therefore, a full genome annotation will usually first rely on an automatic functional annotation based on domain analyses and sequence similarity searches. In order to provide consistency, most tools that automatically annotate genomes frequently employ formalized ontologies such as Gene Ontology (GO) or MapMan ontology (Jaiswal and Usadel, 2017). The use of these (or other well defined) ontologies enables consistency of the annotation terms between different genomes.
There are many tools available that automatically annotate genes using ontologies such as the Mercator automated annotation tool (Lohse et al., 2014), BLAST2GO (Conesa and Gotz, 2008), KEGG Automatic Annotation Server (KAAS) (Moriya et al., 2007), and TRAPID (Van Bel et al., 2013) (reviewed in Bolger et al., 2017a). The overarching goal of these tools is the rapid automatic annotation of genes to a high standard, approaching that of manual annotation.
Phenotypes and their standardization
An important goal of plant genomics and other ’omics approaches is to better understand and predict plant phenotypes. Despite the challenges involved in plant genomics research, the generation and analysis of genomic data are largely outpacing the production and interpretation of phenotyping data (Furbank and Tester, 2011; Cobb et al., 2013). The reason for this ‘phenotyping bottleneck’ is that plants are highly plastic; one genotype may exhibit many different phenotypes depending on environmental conditions. Considerable efforts have been invested into the automation of plant phenotyping (Fiorani and Schurr, 2013; Fahlgren et al., 2015; Shakoor et al., 2017), which has dramatically improved the consistency and throughput of plant phenotyping.
However, even more than in genomics or other ’omics disciplines, plant phenotyping is a multi‐dimensional challenge, especially for crop species. This is because complex, commercially important targets, such as ‘yield improvement’, result from a variety of physiological, morphological, anatomical, and chemical aspects of plant performance. Therefore, many phenotyping efforts aim to understand one or more of these components such as photosynthesis, root architecture, or above ground biomass and subsequently build on crop models to scale to yield (Parent and Tardieu, 2014).
Given the developmental changes observed over time from seedling to mature plant, emphasis of most newly developed phenotyping techniques is on non‐destructive approaches, such as (three‐dimensional) imaging with red, green and blue (RGB) cameras (Figure 1), thermal and hyperspectral imaging, and/or fluorescence measurements of photosynthesis. Analyses of physiological processes such as enzyme activities (Gibon and Rolin, 2012), transpiration or carbon flux (carbon gain through photosynthesis, carbon loss through leaf, stem and root respiration) are far more challenging, as automation in these fields is not straightforward. Automated sampling of leaf material using robots will represent an important advance (Alenyà et al., 2012).
A single genotype has the potential to display a range of different phenotypes depending on the environmental conditions to which it is subjected. One challenge for researchers is to consider and address logistical issues that arise when coordinating physiological studies. For example large, in‐depth studies (such as for GWAS and QTL analyses) require considerable experimental space and resources necessary for the growth and analysis of a wide array (>200) of plant genotypes. Proper attention must be given to environmental conditions, ensuring that they are consistent across all replicates. Constant environmental conditions allow for a better assessment of physiological responses and most analyses are typically carried out with plants that are grown individually in pots, either in a growth room with small plants such as Arabidopsis, or in the glasshouse with larger, but agriculturally more relevant species such as Triticum or Zea. Small pot sizes ensure that there is enough space for many replicates as well as easy handling in automated phenotyping stations, but may also limit plant growth (Poorter et al., 2012a). Environmental conditions are generally under tight control in growth rooms and, to a lesser extent, in glasshouses. Nevertheless, both growth room and glasshouse environments are significantly more stable than the fluctuating environmental conditions that plants experience when subjected to field conditions. Consequently, genotypes that perform well in controlled environments may not necessarily be the ones that perform best in the field. Care has therefore to be taken when choosing and testing relevant conditions, i.e. light and temperature (Poorter et al., 2016). This is especially true when plants are tested under suboptimal conditions, such as a low nutrient or water supply (Ingestad, 1987). Limiting pot size, or improper timing of induced stresses could make the entire phenotypic analysis irrelevant (Passioura, 2012). Finally, when plant performance in the field is the ultimate aim, one has to keep in mind that a genotype that thrives well when grown individually in a pot may not necessarily be the genotype that will perform best under conditions in which plants are grown at high densities, such as in agriculture (Tollenaar and Wu, 1999).
Given the important role that the environment plays in plant growth and development, a comprehensive report of environmental conditions during experiments is of paramount importance, both for experiments carried out under controlled conditions as well as in the field (Poorter et al., 2012b). This enables the comparison of outputs of various experiments and to develop the ideotypes for different environmental scenarios (Chenu et al., 2011). Additionally, improved data sharing and standardization in reporting, particularly with regard to phenotype responses, is especially important in the agricultural sciences (Zamir, 2013). Making historic phenotypic data publicly available would allow plant researchers to share results, compare phenotypes, and analyze data that has been deposited in the past in order to identify new, and sometimes rare, alleles that improve productivity. Finally, the low‐barrier accessibility of data would invite computer scientists and computational biologists to develop or improve current algorithms for phenotypic data analysis (Minervini et al., 2016) and support the integration of scientific fields.
Although this is not always easy given the wide array of plant traits that are measured and the specific developmental time points during which those data are collected, efforts on data standardization are rapidly improving (Figure 1) (Krajewski et al., 2015; Ćwiek‐Kupczyńska et al., 2016). This will undoubtedly facilitate broader application of techniques such as genome‐wide association studies (GWAS; Millet et al., 2016) using high‐throughput field phenotyping (Pauli et al., 2016) (Figure 3). However, it is also important to keep in mind that there is not only a need for advancing phenotypic analysis and data integration, but also for better insights into the application of knowledge obtained under controlled conditions for the improvement of plant performance in the field (Junker et al., 2014; Poorter et al., 2016).
Phenotypic data storage
One key challenge in the plant sciences is the definition of appropriate data management procedures and infrastructures to preserve research data as a valuable scientific asset. This task has been centralized for genomic and expression data for all fields of the life sciences with the Short Read Archive (in the USA) and European Nucleotide Archive (in Europe). Phenotypic data, due to its high divergence, cannot easily be tackled by a highly streamlined and generalized platform. However, in line with the value of original data, funding agencies (Mons et al., 2017) and scientific journals are increasingly requesting scientists to publish research data under the FAIR (findable, accessible, interoperable, and reusable) data principles (Wilkinson et al., 2016). To make data reusable and interoperable in the plant phenotyping community, MIAPPE recommendations (i.e. required Minimal Information about Plant Phenotyping Experiments) are being developed to ensure a proper description of all necessary metadata, including the environment (Krajewski et al., 2015; Ćwiek‐Kupczyńska et al., 2016).
Nonetheless, complex, heterogeneous, or unstructured research data frequently remain publicly unavailable, often due to the lack of infrastructure needed to handle these data. In other cases, the data are published but remain obscured within the supplementary materials. While such data are human interpretable, the lack of standardized formatting and data semantics makes automated approaches difficult and error prone.
To provide a generalized resource with an emphasis on phenotypic data, the FAIR‐aware e!DAL software library was developed. Its aim was to lower the technical barriers and minimize the effort of researchers to make data publicly available (Arend et al., 2014). By contrast with popular data publication platforms such as Figshare (Singh, 2011) or DRYAD (White et al., 2008), e!DAL enables access to large volume research data stored in‐house by assigning Digital Object Identifiers (DOIs). While Figshare and DRYAD offer a comprehensive functionality, they are only free up to a relatively low data volume. This makes them an ideal solution for sharing condensed tables or reduced figures but these resources quickly become expensive and time consuming for larger phenotypic datasets. While there are other generic data repository infrastructure libraries available, for example Fedora (Lagoze et al., 2006), they do not provide a ready‐to‐use implementation as within e!DAL. Based on e!DAL, the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) Gatersleben and the German Plant Phenotyping Network jointly initiated the Plant Genomics and Phenomics Research Data Repository (PGP) (Arend et al., 2016a), which provides amongst others the first full MIAPPE compliant (Ćwiek‐Kupczyńska et al., 2016) phenotypic datasets (Arend et al., 2016b; Chen et al., 2018). The PGP repository currently provides 150 data records linked by DOIs and annotated by technical metadata. This comprises more than 1.2 million files with a volume of over two terabytes and is coupled to the ELIXIR European bioinformatics infrastructure, which allows a single sign‐on service. Furthermore, another unique feature of e!DAL‐PGP is the integrated peer‐review process, which guarantees a certain data quality for every released dataset. The intuitive submission process supports researchers in describing and sharing their phenotypic data to exploit the full scientific potential of their data.
The MIAPPE compliant form of data storage promises to overcome standardization issues especially for experimental factors, as discussed in the previous section. Therefore, these datasets will be immediately useful for experimental reproduction or offer a secondary use. Once enough data have accumulated they can be mined from different databases or e!DAL installations using for example DOIs and potentially identify relevant datasets by MIAPPE tags, offering a true multi‐player international data structure. This allows large‐scale data producers to share their data without a centralized resource by relying on existing infrastructure. Afterwards, the collective dataset might be subjected to machine‐learning approaches, discussed below.
However, to profit from existing phenotypic data straight away, it is potentially useful to simplify the environment to a single factor, such as water availability (see above and Poorter et al., 2012b) and the unit of measurement to a simple (ontological) term (e.g. ‘days to flowering’). A similar approach focusing on phenotypes is chosen by the AraPheno database, which collects several hundred phenotypes for the model plant Arabidopsis (Seren et al., 2017), many of which are derived from one large‐scale study by a multiauthor group (Atwell et al., 2010).
Due to the knowledge about the underlying populations, the data can be transmitted into a standardized GWAS pipeline in AraGWAS, which relies on standardized statistics and will therefore offer more comparable results (Togninalli et al., 2018).
Finally, it can be useful to store and summarize data even more simply, i.e. to only keep data relating to QTL for a specific species (Nijveen et al., 2017) or a group of species (Ni et al., 2009), as this provides, at the very least, a way to compare between different analyses and means to confirm results when a plant researcher or breeder conducts a similar analysis.
Bridging genotypes and phenotypes
Associating genotypes and phenotypes has become much more simple, as statistics have matured and state‐of‐the‐art tools that can be used on a user's desktop to associate data, for example in GWAS‐type settings (Figure 3), have been developed. These tools range from the efficient mixed model (EMMA) type family, through FAST‐LMM (Lippert et al., 2011) to TASSEL (Bradbury et al., 2007), to name but a few that are reviewed in this issue.
Additionally, user‐friendly online tools such easyGWAS (Grimm et al., 2017) or GWAPP (Seren et al., 2012) exist. These tools only require phenotypic data if using Arabidopsis. This is because these tools analyze the phenotypic data against an internally stored set of genomic data from a reference panel.
However, high‐throughput phenotyping of multiple traits allows not only association of traits with genotypes, but also association of traits with each other (e.g. Poorter et al., 2014, Figure 3). Once again, this works best within the same experimental setting, as under these conditions the environment and management is by definition ‘identical’. However, it is clear that novel insights would require pooling of multiple datasets or very large datasets that comprise many different phenotypic values; this has been done in AraPheno/AraGWAS (see above).
Another large advantage of an approach that relates phenotypes to phenotypes is that comparatively few variables are concerned, making statistical overfitting a minor problem. This advantage is because phenotypic data (on large populations) does not suffer from ‘p≫n’, i.e. the number of variables (phenotypes, p) is usually not larger than the number of samples (n). As an example, the Atwell study (2010) recorded 107 diverse phenotypic values in between 90 and 180 accessions or more. Therefore, many techniques from the extensively studied field of gene network reconstruction (reviewed by Emamjomeh et al., 2017) work well if not better when applied to phenotypes, given a large enough population. Indeed, for plant gene network analyses, gene expression data are often simply correlated, without putting too much detail into environmental or perturbation conditions. The only consideration is that expression data sets should represent a range of different conditions and not favor certain perturbations over others. This could be done either by hand, for example in CSBDB.DB (Steinhauser et al., 2004), or automatically, for example in ATTED‐II (Obayashi et al., 2018).
However, while a simple correlation analysis between phenotypes is a good start for an analysis and therefore supported in AraGWAS (Togninalli et al., 2018) and Phenome‐networks, more sophisticated approaches can be used. Indeed multiple different statistical and machine‐learning approaches are already being used currently.
Firstly, as a way to bridge for example well refined molecular measurements such as metabolic profiles to physiological parameters, one can use partial least square (PLS). This technique allows the determination of relationships between outcome variables and predictor variables. Gago et al. (2017) used this approach to relate canopy and stomatal conductance from a vineyard to a metabolite matrix. Typically, PLS results are then analyzed using variable importance prediction to determine important predictors (i.e. metabolites in this case). Gago et al. (2017) found for example phenylpropanoids and myo‐inositol to be predictive for both conductance values.
Alternatively, machine learning can be employed to predict important factors such as biomass. As an example, Maddison et al. (2017) used classical machine‐learning techniques (feature selection coupled to support vector regression) to predict biomass outcomes from non‐structural carbohydrates in Miscanthus, extending earlier observations by Sulpice and colleagues in Arabidopsis (2013).
However, these approaches implied that certain variables are considered a priori as more important than others. While this is clearly the case for Miscanthus biomass, deep phenotypic data allow the uncovering of novel associations not observed previously between the individual variables.
The QTL + phenotype supervised orientation (QPSO) approach, developed in the van Eeuwijk laboratory (Wang and van Eeuwijk, 2014; Wang et al., 2015), aims to generate directed networks between phenotypic traits by using known sparse QTL to orient the network, extending earlier work on gene network reconstruction and cleverly combining different data domains.
However, when only phenotypes are concerned, one can consider (full) partial correlation analysis that removes the influence of other variables on a variable pair or Bayesian network reconstruction. Common to all these methods is that they try to find relationships between two entities that are not dependent on the other variables. As an example, consider abscisic acid (ABA) that influences both stomatal conductance (Wilkinson and Davies, 2002) and primary root growth (Rowe et al., 2016) in response to drought stress. Assuming an overly simplified model in which stomatal conductance and primary root growth were only dependent on the ABA concentration, all three items would be correlated. However, controlling statistically for ABA would reveal that stomatal conductance and primary root growth were unrelated.
In any case, none of these takes hidden (not measured) but potentially important and causal variables into account. In addition, while these methods do not link traits or physiological variables with the underlying genomic basis (except for QPSO), they do provide structural insights about trait interrelationships. This understanding can be used to modify a target trait by genetically modifying another trait whose genetic basis is already understood. However, it has to be noted that all modelling insights are restricted to the data at hand, meaning many missing variables will make this more difficult.
Phenotypic prediction using phenotype ontologies
Another valid abstraction approach is to couple phenotypes to genes or genomic regions, leveraging a meaningful phenotypic ontology (Zamir, 2013, Hoehndorf et al., 2015; Deans et al., 2015; Coppens et al., 2017, Figure 3 top left). This strategy has been employed for many years for animals and humans, reaching from phenotypically described and formalized mouse data to integrated environments and reasoning, bridging data from different species (Robinson and Webber, 2014; Mungall et al., 2017; Rodríguez‐García et al., 2017). These data being animal−human centric are centered around disease associations, however the plant community has (at least for Arabidopsis) a massive resource for single knockouts using T‐DNA lines (O'Malley and Ecker, 2010; Kleinboelting et al., 2017). As a result, many ontologically defined phenotypic annotations are already available for knockouts and other transgenics in The Arabidopsis Information Resource (TAIR) and other databases (Lloyd and Meinke, 2012; Akiyama et al., 2014).
Therefore, data on phenotypes resulting from knockouts could be integrated with GWAS studies using the phenotype ontology data integration framework developed by the animal community (Hoehndorf et al., 2015). Therefore, typical candidate approaches, in which genes underlying a QTL region are investigated manually, could be extended by selecting candidate genes, based on their phenotypes and/or based on where in a phenotype network they reside. Indeed, the Planteome project tries to assess and integrate some of these data already with clever use of biomedical ontologies (Cooper et al., 2018).
The blessings and curses of machine learning
Over the past few years ‘deep’ machine‐learning methods, and particularly artificial neural network‐based approaches, have led to revolutionary results, particularly in image analysis. For example, this has greatly spurred the identification of plant features such as root tips and their localization in an image (Pound et al., 2017) to count leaves (Ubbens et al., 2018) or to derive vegetation indices from RGB images (Khan et al., 2018). In addition, this has led to the development of methods to detect plant diseases (Mahlein, 2016; Mohanty et al., 2016; Fuentes et al., 2017) and plant stress phenotyping (Ghosal et al., 2018). The latter application of deep learning to plant abiotic and biotic stress phenotyping has recently been reviewed by Singh et al. (2018).
The underlying frameworks are constantly driven forward by Google, Facebook, and other companies offering readily usable frameworks such as Tensorflow (https://www.tensorflow.org/) or Caffe2 (https://caffe2.ai/). In addition these big data centered companies develop dedicated hardware that promises to greatly accelerate training and analysis tasks. Therefore it is not surprising that plant image data is analyzed using a plethora of machine‐learning approaches (Pound et al., 2014; Tsaftaris et al., 2016). However convolutional neural networks have the potential to greatly advance the field of plant image analysis (Pound et al., 2017; Ubbens and Stavness, 2017; Figure 1).
One challenge with image analysis is that large‐scale datasets with data and ground truth outcomes are required. The former can be made readily available through plant phenotyping platforms, but finding the ground truth for a limited number of training datasets currently relies mostly on human experts. However, as this is costly and time consuming, smart solutions, such as those relying on citizen science (Giuffrida et al., 2018) are needed. A recent clever proof‐of‐concept study, which used the Amazon ‘mechanical turk platform’ (anonymous users are paid for small tasks), performed better than for‐credit students (Zhou et al., 2018). Without such data, algorithms can be compared based on standard datasets, such as those supplied by the International Plant Phenotypic Network (Minervini et al., 2014). This dataset is suitable for tasks such as plant detection and localization in images, as well as leaf detection, localization, and counting in images. This reliance on training datasets is necessary because there is, as of yet, no application of unsupervised reinforcement learning methods for image analysis purposes.
Other applications of machine learning, such as prediction of plant performance or the integration of heterogeneous datasets, are even less developed as researchers are currently embracing more traditional and/or data science driven methods for these applications. As an example, Chen et al. (2018) used regression and random forests, but not deep learning to predict plant biomass from plant images, whereas Coppens et al. (2017) reviews data integration.
Plant phenotypic data promise to be interesting vistas for machine‐learning approaches (Figure 3 top). Indeed, early studies have suggested that machine‐learning approaches for phenotype predictions stemming from a sufficiently genotyped population could be meaningful, especially in the p≫n setting in which more predictors from genomics data than plant samples are available (Crossa et al., 2017). As an example, Grinberg et al. (2018) tried to predict phenotypes using classical genomic Best Linear Unbiased Prediction (BLUP) as well as several machine‐learning techniques. The latter clearly outperformed BLUP for yeast with very controlled environments, whereas for wheat and rice, BLUP performed particularly well when there was population structure (Grinberg et al., 2018).
The non‐model/minor crop plant perspective
As has been shown above, both genomic and phenomic datasets are becoming more and more mature and cost efficient. At this time, it is the model plant Arabidopsis, rather than crop plants, that contains the most extensive datasets and that may enable ontology‐driven phenotype prediction. This is largely due to a number of points: (i) the availability of the machine‐readable ontology term‐enriched phenotypic datasets for well defined genes; (ii) the largest wealth of functional data for gene annotation, which is related to the former point; (iii) the use of standardized populations from the 1001 genome consortium, facilitating abstraction at the phenotypic level; and (iv) standardization, driven for example by TAIR. Also, for genetic and genomic studies, it is necessary to note the importance of accurate phenotyping. The most advanced (in terms of crop plants) is most likely maize that, despite its tremendous genetic variety, is tackled in a well planned and standardized way, driven both (pan)genomically (Gore et al., 2009; Hirsch et al., 2016) and phenomically (see e.g. AlKhalifah et al., 2018; for a well described dataset) and supported by user friendly tools, providing access to these resources such as TASSEL (Bradbury et al., 2007). However, while standardization is gaining traction and big datasets are becoming more available for major crops, minor crops remain less supported. Additionally, when studying this genotype−phenotype interaction, it is important to have access to detailed phenotypic data. In many cases, the selection and evaluation of phenotypes have been poorly developed in the experimental design of genetic and genomics studies (Houle et al., 2010). Therefore, efforts to identify gold standard experimental procedures and scoring protocols may contribute to the harmonization of phenotypic data and therefore to the improvement of data accessibility (Shrestha et al., 2012). In addition, the existence of biases is another new, important challenge in attaining knowledge from new high‐throughput techniques.
That said, for non‐model species general ‘cyberinfrastructures’ can also be used (Merchant et al., 2016) and specialized information systems, such as those for grapevine (Adam‐Blondon et al., 2016) or the Rosaceae community (Jung et al., 2017) have been developed. Indeed, while necessarily less data are available for non‐model (minor) crop plant communities (e.g. an apple researcher); they can learn from the lessons and mistakes made with big crops and within the International Plant Phenotyping Consortium.
Finally, it can be expected that even data from the model plant Arabidopsis will be transferable to dicots (and thus many horticultural minor crops) or at least related crops (i.e. Brassicaceae) on a large‐scale basis going beyond simple gene annotation.
Conclusions
The impact that the genomics revolution has made on plant science is undeniable and innovative pangenomic approaches that allow the integration of data of related species are beginning to take hold in the plant field. We are therefore in the middle of a genomics data explosion. We are also at an exciting time point, witnessing the next revolution in phenomics (Tardieu et al., 2017), and we begin to see how machine learning‐ and data science‐driven approaches are trickling into the area of bridging genomics and phenomics data. These developments are making plant science a truly modern science, inspired by artificial intelligence, robotics systems, and classical plant physiology. A whole new ‘breed’ of quantitative and computer science‐oriented plant scientists (Friesner et al., 2017) is therefore required to truly modernize the discipline.
Conflict of Interest
The authors declare no conflict of interest.
Box 1. Summary.
Plant genome sequencing has evolved to soon become a commodity approach for small genomes.
Phenotypic data standardization recommendations are provided by MIAPPE.
Many tools and databases facilitate bridging genotypes and phenotypes.
Box 2. Open questions.
Algorithms working on multiple genomes of a species are still in development.
It is still an open question how to best combine short and long reads into assemblies.
More rigorous phenotype ontologies and machine‐learning approaches are likely to improve our understanding of plants.
Acknowledgements
The authors want to acknowledge funding by the German Ministry of Education and Research FKZ 031B0293A and FKZ 031A536A‐C. In addition, we acknowledge partial funding by the German Ministry of Education and Research for the German Plant Phenotyping network 031A053 and the Plant Primary database FKZ 0315961 projects and the NRW Strategieprojekt BioSC (no. 313/323‐400‐002 13) and the European Union's Horizon 2020 Research and Innovation Programme (GoodBerry, grant agreement number 679303; EPPN2020, grant agreement number 731013; and BREEDCAFS, grant agreement number 727934).
References
- Adam‐Blondon, A.F. , Alaux, M. , Pommier, C. et al (2016) Towards an open grapevine information system. Hortic. Res. 3, 16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama, K. , Kurotani, A. , Lida, K. , Kuromori, T. , Shinozaki, K. and Sakurai, T. (2014) RARGE II: an integrated phenotype database of Arabidopsis mutant traits using a controlled vocabulary. Plant Cell Physiol. 55(1), e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenyà, G. , Dellen, B. , Foix, S. and Torras, C. (2012) Leaf segmentation from ToF data for robotized plant probing. IEEE Robot. Autom. Mag. 20, 50–59. [Google Scholar]
- AlKhalifah, N. , Campbell, D.A. , Falcon, C.M. et al (2018) Maize genomes to fields: 2014 and 2015 field season genotype, phenotype, environment, and inbred ear image datasets. BMC Res. Notes, 11(1), 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend, D. , Lange, M. , Chen, J. , Colmsee, C. , Flemming, S. , Hecht, D. and Scholz, U. (2014) e!DAL–a framework to store, share and publish research data. BMC Bioinformatics, 15, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend, D. , Junker, A. , Scholz, U. , Schuler, D. , Wylie, J. and Lange, M. (2016a) PGP repository: a plant phenomics and genomics data publication infrastructure. Database, 2016, baw033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend, D. , Lange, M. , Pape, J.‐M. , Weigelt‐Fischer, K. , Arana‐Ceballos, F. , Mücke, I. , Klukas, C. , Altmann, T. , Scholz, U. and Junker, A. (2016b) Quantitative monitoring of Arabidopsis thaliana growth and development using high‐throughput plant phenotyping. Sci. Data, 3, 160055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanbori, J. , Chen, F. , French, A.P. and Pridmore, T. (2018) Towards low‐cost image‐based plant phenotyping using reduced‐parameter CNN In: CVPPP 2018: Workshop on Computer Vision Problems in Plant Phenotyping. Newcastle upon Tyne, UK: http://eprints.nottingham.ac.uk/cgi/export/eprint/54696/Refer/nott-eprint-54696.refer [accessed on 28 December 2018] [Google Scholar]
- Atwell, S. , Huang, Y.S. , Vilhjálmsson, B.J. et al (2010) Genome‐wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature, 465(7298), 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au, K.F. , Underwood, J.G. , Lee, J.G. and Wong, W.H. (2012) Improving PacBio long read accuracy by short read alignment. PLoS ONE, 7, e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni, R. , Nave, M. , Barad, O. et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science, 357(6346), 93–97. [DOI] [PubMed] [Google Scholar]
- Baird, N.A. , Etter, P.D. , Atwood, T.S. , Currey, M.C. , Shiver, A.L. , Lewis, Z.A. , Selker, E.U. , Cresko, W.A. and Johnson, E.A. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3(10), e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, E. and Lan, L. (2017) HALC: high throughput algorithm for long read error correction. BMC Bioinformatics, 18, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin, K. , Koren, S. , Chin, C.S. , Drake, J.P. , Landolin, J.M. and Phillippy, A.M. (2015) Assembling large genomes with single‐molecule sequencing and locality‐sensitive hashing. Nat. Biotechnol. 33(6), 623–630. [DOI] [PubMed] [Google Scholar]
- Bhat, J.A. , Ali, S. , Salgotra, R.K. et al (2016) Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 7, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. , Scossa, F. , Bolger, M.E. et al (2014a) The genome of the stress‐tolerant wild tomato species Solanum pennellii. Nat. Genet. 46, 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014b) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, M.E. , Weisshaar, B. , Scholz, U. , Stein, N. , Usadel, B. and Mayer, K.F. (2014c) Plant genome sequencing ‐ applications for crop improvement. Curr. Opin. Biotechnol. 26, 31–37. [DOI] [PubMed] [Google Scholar]
- Bolger, M.E. , Arsova, B. and Usadel, B. (2017a) Plant genome and transcriptome annotations: from misconceptions to simple solutions. Brief. Bioinform. 19(3), 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Denton, A.K. , Bolger, M.E. and Usadel, B. (2017b) LOGAN: a framework for LOssless Graph‐based ANalysis of high throughput sequence data. BioRxiv, 175976 10.1101/175976 [DOI] [Google Scholar]
- Bolger, M. , Schwacke, R. , Gundlach, H. et al (2017c) From plant genomes to phenotypes. J. Biotechnol. 261, 46–52. [DOI] [PubMed] [Google Scholar]
- Bombarely, A. , Moser, M. , Amrad, A. et al (2016) Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants, 2(6), 16074. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Bukowski, R. , Guo, X. , Lu, Y. et al (2018) Construction of the third‐generation Zea mays haplotype map. Gigascience, 7(4), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli, E. , Licciardello, C. , Zhang, Y. , Liu, J. , Mackay, S. , Bailey, P. , Reforgiato‐Recupero, G. and Martin, C. (2012) Retrotransposons control fruit‐specific, cold‐dependent accumulation of anthocyanins in blood oranges. Plant Cell, 24, 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M.S. , Law, M. , Holt, C. et al (2014) MAKER‐P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. et al (2014) Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Chen, D. , Shi, R. , Pape, J.M. et al (2018) Predicting plant biomass accumulation from image‐derived parameters. Gigascience, 7(2), 10.1093/gigascience/giy001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenu, K. , Cooper, M. , Hammer, G.L. , Mathews, K.L. , Dreccer, M.F. and Chapman, S.C. (2011) Environment characterization as an aid to wheat improvement: interpreting genotype–environment interactions by modelling water‐deficit patterns in North‐Eastern Australia. J. Exp. Bot. 62(6), 1743–1755. [DOI] [PubMed] [Google Scholar]
- Chuong, E.B. , Elde, N.C. and Feschotte, C. (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18(2), 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claros, M.G. , Bautista, R. , Guerrero‐Fernandez, D. , Benzerki, H. , Seoane, P. and Fernandez‐Pozo, N. (2012) Why assembling plant genome sequences is so challenging. Biology, 1, 439–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb, J.N. , Declerck, G. , Greenberg, A. , Clark, R. and McCouch, S. (2013) Next‐generation phenotyping: requirements and strategies for enhancing our understanding of genotype‐phenotype relationships and its relevance to crop improvement. Theor. Appl. Genet. 126, 867–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. and Gotz, S. (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics, 2008, 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, L. , Meier, A. , Laporte, M.A. et al (2018) The Planteome database: an integrated resource for reference ontologies, plant genomics and phenomics. Nucleic Acids Res. 46(D1), D1168–D1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppens, F. , Wujts, N. , Inze, D. and Dhont, S. (2017) Unlocking the potential of plant phenotyping data through integration and data‐driven approaches. Curr. Opin. Syst. Biol. 4, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossa, J. , Pérez‐Rodríguez, P. , Cuevas, J. et al (2017) Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci. 22(11), 961–975. [DOI] [PubMed] [Google Scholar]
- Ćwiek‐Kupczyńska, H. , Altmann, T. , Arend, D. et al (2016) Measures for interoperability of phenotypic data: minimum information requirements and formatting. Plant Methods, 12, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans, A.R. , Lewis, S.E. , Huala, E. et al (2015) Finding our way through phenotypes. PLoS Biol. 13(1), e1002033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire, R.J. , Glaubitz, J.C. , Sun, Q. , Poland, J.A. , Kawamoto, K. , Buckler, E.S. and Mitchell, S.E. (2011) A robust, simple genotyping‐by‐sequencing (GBS) approach for high diversity species. PLoS ONE, 6(5), e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamjomeh, A. , Saboori Robat, E. , Zahiri, J. , Solouki, M. and Khosrav, P. (2017) Plant Biotechnol. Rep. 11, 71. [Google Scholar]
- Ezer, D. , Jung, J.H. , Lan, H. et al (2017) The evening complex coordinates environmental and endogenous signals in Arabidopsis. Nat. Plants, 3, 17087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren, N. , Gehan, M.A. and Baxter, I. (2015) Lights, camera, action: high‐throughput plant phenotyping is ready for a close‐up. Curr. Opin. Plant Biol. 24, 93–99. [DOI] [PubMed] [Google Scholar]
- Fiorani, F. and Schurr, U. (2013) Future scenarios for plant phenotyping. Annu. Rev. Plant Biol. 64, 267–291. [DOI] [PubMed] [Google Scholar]
- Flutre, T. , Duprat, E. , Feuillet, C. and Quesneville, H. (2011) Considering transposable element diversification in de novo annotation approaches. PLoS One, 6, e16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner, J. , Assmann, S.M. , Bastow, R. et al (2017) The next generation of training for Arabidopsis researchers: bioinformatics and quantitative biology. Plant Physiol. 175(4), 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes, A. , Yoon, S. , Kim, S.C. and Park, D.S. (2017) A robust deep‐learning‐based detector for real‐time tomato plant diseases and pests recognition. Sensors (Basel), 17(9), E2022 10.3390/s17092022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, S. , Kubo, K. and Takayama, S. (2016) Non‐self‐ and self‐recognition models in plant self‐incompatibility. Nat. Plants, 2, 16130. [DOI] [PubMed] [Google Scholar]
- Furbank, R.T. and Tester, M. (2011) Phenomics–technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 16(12), 635–644. [DOI] [PubMed] [Google Scholar]
- Gago, J. , Fernie, A.R. , Nikoloski, Z. et al (2017) Integrative field scale phenotyping for investigating metabolic components of water stress within a vineyard. Plant Methods, 13, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal, S. , Blystone, D. , Singh, A.K. , Ganapathysubramanian, B. , Singh, A. and Sarkar, S. (2018) An explainable deep machine vision framework for plant stress phenotyping. Proc. Natl Acad. Sci. USA, 115(18), 4613–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibon, Y. and Rolin, D. (2012) Aspects of experimental design for plant metabolomics experiments and guidelines for growth of plant material. Methods Mol. Biol. 860, 13–30. [DOI] [PubMed] [Google Scholar]
- Giuffrida, M.V. , Chen, F. , Scharr, H. and Tsaftaris, S.A. (2018) Citizen crowds and experts: observer variability in image‐based plant phenotyping. Plant Methods, 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A. , Bayer, P.E. , Barker, G.C. et al (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 7, 13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, S.P. , Contreras‐Moreira, B. , Woods, D.P. et al (2017) Extensive gene content variation in the Brachypodium distachyon pan‐genome correlates with population structure. Nat. Commun., 8, 2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore, M.A. , Chia, J.M. , Elshire, R.J. et al (2009) A first‐generation haplotype map of maize. Science, 326(5956), 1115–1117. [DOI] [PubMed] [Google Scholar]
- Grimm, D.G. , Roqueiro, D. , Salomé, P.A. et al (2017) easyGWAS: a cloud‐based platform for comparing the results of genome‐wide association studies. Plant Cell, 29, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg, N.F. , Orhobor, O.I. and King, R.D. (2018) An evaluation of machine‐learning for predicting phenotype: studies in yeast, rice and wheat. BioRxiv. 10.1101/105528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackl, T. , Hedrich, R. , Schultz, J. and Förster, F. (2014) Proovread: large‐scale high‐accuracy PacBio correction through iterative short read consensus. Bioinformatics, 30, 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, J. , Zhao, X. , Laroche, A. , Lu, Z.X. , Liu, H. and Li, Z. (2014) Genotyping‐by‐sequencing (GBS), an ultimate marker‐assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 5, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, C.N. , Hirsch, C.D. , Brohammer, A.B. et al (2016) Draft assembly of elite inbred line PH207 provides insights into genomic and transcriptome diversity in maize. Plant Cell, 28, 2700–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf, R. , Schofield, P.N. and Gkoutos, G.V. (2015) The role of ontologies in biological and biomedical research: a functional perspective. Brief. Bioinform. 16(6), 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K.J. , Lange, S. , Lomsadze, A. , Borodovsky, M. and Stanke, M. (2016) BRAKER1: unsupervised RNA‐seq‐based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32, 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle, D. , Goviandaraju, D.R. and Omholt, S. (2010) Phenomics: the next challenge. Nat. Rev. Genet. 11, 855–866. [DOI] [PubMed] [Google Scholar]
- Huang, L. , Popic, V. and Batzoglou, S. (2013) Short read alignment with populations of genomes. Bioinformatics, 29, i361–i370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingestad, T. (1987) New concepts on soil fertility and plant nutrition as illustrated by research on forest trees and stands. Geoderma, 40(3–4), 237–252. [Google Scholar]
- International Wheat Genome Sequencing Consortium . (2014) A chromosome‐based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science, 345, 1251788. [DOI] [PubMed] [Google Scholar]
- Iqbal, Z. , Caccamo, M. , Turner, I. , Flicek, P. and McVean, G. (2012) De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet., 44, 226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, M. , Olsen, H.E. , Paten, B. and Akeson, M. (2016) The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, P. and Usadel, B. (2016) Plant pathway databases. Methods Mol. Biol. 1374, 71–87. [DOI] [PubMed] [Google Scholar]
- James, G.V. , Patel, V. , Nordström, K.J. , Klasen, J.R. , Salomé, P.A. , Weigel, D. and Schneeberger, K. (2013) User guide for mapping‐by‐sequencing in Arabidopsis. Genome Biol. 14(6), R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, W.B. and Schneeberger, K. (2017) The impact of third generation genomic technologies on plant genome assembly. Curr. Opin. Plant Biol. 36, 64–70. [DOI] [PubMed] [Google Scholar]
- Jung, S. , Lee, T. , Cheng, CH. , Humann, J. , Yu, J. , Ficklin, S.P. and Main, D. (2017) Extension modules for storage, visualization and querying of genomic, genetic and breeding data in Tripal databases. Database, bax092 10.1093/database/bax092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker, A. , Muraya, M.M. , Weigelt‐Fischer, K. et al (2014) Optimizing experimental procedures for quantitative evaluation of crop plant performance in high throughput phenotyping systems. Front. Plant Sci. 5, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, Z. , Rahimi‐Eichi, V. , Haefele, S. , Garnett, T. and Miklavcic, S.J. (2018) Estimation of vegetation indices for high‐throughput phenotyping of wheat using aerial imaging. Plant Methods, 14, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klap, C. , Yeshayahou, E. , Bolger, A.M. , Arazi, T. , Gupta, S.K. , Shabtai, S. , Usadel, B. , Salts, Y. and Barg, R. (2017) Tomato facultative parthenocarpy results from SlAGAMOUS‐LIKE 6 loss of function. Plant Biotechnol. J. 15(5), 634–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinboelting, N. , Huep, G. and Weisshaar, B. (2017) Enhancing the GABI‐Kat Arabidopsis thaliana T‐DNA insertion mutant database by incorporating Araport11 annotation. Plant Cell Physiol. 58(1), e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski, P. , Chen, D. , Cwiek, H. et al (2015) Towards recommendations for metadata and data handling in plant phenotyping. J. Exp. Bot. 66, 5417–5427. [DOI] [PubMed] [Google Scholar]
- Lagoze, C. , Payette, S. , Shin, E. and Wilper, C. (2006) Fedora: an architecture for complex objects and their relationships. Int. J. Digit. Libr. 6(2), 124–138. [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–U354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat, E. (2010) Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity (Edinb), 104, 520–533. [DOI] [PubMed] [Google Scholar]
- Li, H. (2016) Minimap and Miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 32(14), 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. , Zhu, G. , Zhang, J. et al (2014) Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–12266. [DOI] [PubMed] [Google Scholar]
- Lippert, C. , Listgarten, J. , Liu, Y. , Kadie, C.M. , Davidson, R.I. and Heckerman, D. (2011) FaST linear mixed models for genome‐wide association studies. Nat. Methods, 8, 833–835. [DOI] [PubMed] [Google Scholar]
- Lloyd, J. and Meinke, D. (2012) A comprehensive dataset of genes with a loss‐of‐function mutant phenotype in Arabidopsis. Plant Physiol. 158(3), 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse, M. , Nagel, A. , Herter, T. et al (2014) Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant, Cell Environ., 37, 1250–1258. [DOI] [PubMed] [Google Scholar]
- Luo, R. , Liu, B. , Xie, Y. et al (2012) SOAPdenovo2: an empirically improved memory‐efficient short‐read de novo assembler. Gigascience, 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M.C. , Gu, Y.Q. , Puiu, D. et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii . Nature, 551, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz, U. , Pose, D. , Pfeifer, M. , Hagmann, J. , Wang, C. , Weigel, D. , Mayer, K.F. , Schmid, M. and Schwechheimer, C. (2015) Modulation of ambient temperature‐dependent flowering in Arabidopsis thaliana by natural variation of FLOWERING LOCUS M. PLoS Genet. 11, e1005588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison, A.L. , Camargo‐Rodriguez, A. , Scott, I.M. et al (2017) Predicting future biomass yield in Miscanthus using the carbohydrate metabolic profile as a biomarker. Glob. Change Biol. Bioenergy, 9(7), 1264–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlein, A.K. (2016) Plant disease detection by imaging sensors–parallels and specific demands for precision agriculture and plant phenotyping. Plant Dis. 100(2), 241–251. [DOI] [PubMed] [Google Scholar]
- Mahmoud, M. , Zywicki, M. , Twardowski, T. and Karlowski, W.M. (2017) Efficiency of PacBio long read correction by 2nd generation Illumina sequencing. Genomics. pii: S0888‐7543(17)30166‐0. 10.1016/j.ygeno.2017.12.011 [DOI] [PubMed] [Google Scholar]
- Makarevitch, I. , Waters, A.J. , West, P.T. , Stitzer, M. , Hirsch, C.N. , Ross‐Ibarra, J. and Springer, N.M. (2015) Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 11(1), e1004915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Gundlach, H. , Himmelbach, A. et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature, 544, 427–433. [DOI] [PubMed] [Google Scholar]
- Merchant, N. , Lyons, E. , Goff, S. , Vaughn, M. , Ware, D. , Micklos, D. and Antin, P. (2016) The iPlant collaborative: cyberinfrastructure for enabling data to discovery for the life sciences. PLoS Biol. 14, e1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael, T.P. , Jupe, F. , Bemm, F. et al (2018) High contiguity Arabidopsis thaliana genome assembly with a single Nanopore flow cell. Nat. Commun. 9(1), 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet, E.J. , Welcker, C. , Kruijer, W. et al (2016) Genome‐wide analysis of yield in Europe: allelic effects vary with drought and heat scenarios. Plant Physiol. 172, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minervini, M. , Abdelsamea, M.M. and Tsaftaris, S.A. (2014) Image‐based plant phenotyping with incremental learning and active contours. Ecol. Inform. 23, 35–48. [Google Scholar]
- Minervini, M. , Fischbach, A. , Scharr, H. and Tsaftaris, S.A. (2016) Finely‐grained annotated datasets for image‐based plant phenotyping. Pattern Recogn. Lett. 81, 80–89. 10.1016/j.patrec.2015.10.013 [DOI] [Google Scholar]
- Mohanty, S.P. , Hughes, D.P. and Salathé, M. (2016) Using deep learning for image‐based plant disease detection. Front. Plant Sci. 7, 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons, B. , Neylon, C. , Velterop, J. , Dumontier, M. , da Silva Santos, L.O.B. and Wilkinson, M.D. (2017) Cloudy, increasingly FAIR; revisiting the FAIR data guiding principles for the European open science cloud. Information Services and Use, 37(1), 49–56. [Google Scholar]
- Montenegro, J.D. , Golicz, A.A. , Bayer, P.E. et al (2017) The pangenome of hexaploid bread wheat. Plant J, 90, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Moriya, Y. , Itoh, M. , Okuda, S. , Yoshizawa, A.C. and Kanehisa, M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muggli, M.D. , Bowe, A. , Noyes, N.R. , Morley, P.S. , Belk, K.E. , Raymond, R. , Gagie, T. , Puglisi, S.J. and Boucher, C. (2017) Succinct colored de Bruijn graphs. Bioinformatics, 33, 3181–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall, C.J. , McMurry, J.A. , Köhler, S. et al (2017) The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 45(D1), D712–D722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, J. , Pujar, A. , Youens‐Clark, K. et al (2009) Gramene QTL database: development, content and applications. Database, 2009, bap005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijveen, H. , Ligterink, W. , Keurentjes, J.J. et al (2017) AraQTL ‐ workbench and archive for systems genetics in Arabidopsis thaliana . Plant J. 89, 1225–1235. [DOI] [PubMed] [Google Scholar]
- Obayashi, T. , Aoki, Y. , Tadaka, S. , Kagaya, Y. and Kinoshita, K. (2018) ATTED‐II in 2018: a plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant Cell Physiol. 59(2), 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley, R.C. and Ecker, J.R. (2010) Linking genotype to phenotype using the Arabidopsis unimutant collection. Plant J. 61(6), 928–940. [DOI] [PubMed] [Google Scholar]
- Parent, B. and Tardieu, F. (2014) Can current crop models be used in the phenotyping era for predicting the genetic variability of yield of plants subjected to drought or high temperature? J. Exp. Bot. 65(21), 6179–6189. [DOI] [PubMed] [Google Scholar]
- Passioura, J.B. (2012) Phenotyping for drought tolerance in grain crops: when is it useful to breeders? Funct. Plant Biol. 39(11), 851–859. [DOI] [PubMed] [Google Scholar]
- Pauli, D. , Chapman, S.C. , Bart, R. , Topp, C.N. , Lawrence‐Dill, C.J. , Poland, J. and Gore, M.A. (2016) The quest for understanding phenotypic variation via integrated approaches in the field environment. Plant Physiol. 172(2), 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus, S. , Behmann, J. , Mahlein, A.K. , Plümer, L. and Kuhlmann, H. (2014) Low‐cost 3D systems: suitable tools for plant phenotyping. Sensors (Basel), 14(2), 3001–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol., 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho, H.P. , Möhring, J. , Melchinger, A.E. and Büchse, A. (2008) BLUP for phenotypic selection in plant breeding and variety testing. Euphytica, 161, 229–228. [Google Scholar]
- Poorter, H. , Bühler, J. , van Dusschoten, D. , Climent, J. and Postma, J.A. (2012a) Pot size matters: a meta‐analysis of the effects of rooting volume on plant growth. Funct. Plant Biol. 39(11), 839–850. [DOI] [PubMed] [Google Scholar]
- Poorter, H. , Fiorani, F. , Stitt, M. et al (2012b) The art of growing plants for experimental purposes: a practical guide for the plant biologist. Funct. Plant Biol. 39(11), 821–838. [DOI] [PubMed] [Google Scholar]
- Poorter, H. , Lambers, H. and Evans, J.R. (2014) Trait correlation networks: a whole plant perspective on the recently criticized leaf economic spectrum. New Phytol. 201, 378–382. [DOI] [PubMed] [Google Scholar]
- Poorter, H. , Fiorani, F. , Pieruschka, R. , Wojciechowski, T. , van der Putten, W.H. , Kleyer, M. , Schurr, U. and Postma, J. (2016) Pampered inside, pestered outside? Differences and similarities between plants growing in controlled conditions and in the field. New Phytol. 212, 838–855. [DOI] [PubMed] [Google Scholar]
- Potato Genome Sequencing Consortium . (2011) Genome sequence and analysis of the tuber crop potato. Nature, 475(7355), 189–195. [DOI] [PubMed] [Google Scholar]
- Pound, M.P. , French, A.P. , Murchie, E.H. and Pridmore, T.P. (2014) Automated recovery of three‐dimensional models of plant shoots from multiple color images. Plant Physiol. 166, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pound, M.P. , Atkinson, J.A. , Townsend, A.J. et al (2017) Deep machine learning provides state‐of‐the‐art performance in image‐based plant phenotyping. Gigascience, 6(10), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang, F.J. , Kloosterman, W.P. and de Ridder, J. (2018) From squiggle to basepair: computational approaches for improving Nanopore sequencing read accuracy. Genome Biol. 19(1), 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P.N. and Webber, C. (2014) Phenotype ontologies and cross‐species analysis for translational research. PLoS Genet. 10(4), e1004268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐García, M.Á. , Gkoutos, G.V. , Schofield, P.N. and Hoehndorf, R. (2017) Integrating phenotype ontologies with PhenomeNET. J. Biomed. Semantics, 8(1), 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, J.H. , Topping, J.F. , Liu, J.L. and Lindsey, K. (2016) Abscisic acid regulates root growth under osmotic stress conditions via an interacting hormonal network with cytokinin, ethylene and auxin. New Phytol. 211(1), 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmela, L. and Rivals, E. (2014) LoRDEC: accurate and efficient long read error correction. Bioinformatics, 30, 3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheben, A. and Edwards, D. (2018) Towards a more predictable plant breeding pipeline with CRISPR/Cas‐induced allelic series to optimize quantitative and qualitative traits. Curr. Opin. Plant Biol. 45, 218–225. in press. S1369‐5266(18)30023‐2. 10.1016/j.pbi.2018.04.013 [DOI] [PubMed] [Google Scholar]
- Scheben, A. , Batley, J. and Edwards, D. (2017) Genotyping‐by‐sequencing approaches to characterize crop genomes: choosing the right tool for the right application. Plant Biotechnol. J. 15(2), 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, M.H. , Vogel, A. , Denton, A.K. et al. (2017) De Novo Assembly of a New Solanum pennellii Accession Using Nanopore Sequencing. Plant Cell, 29, 2336–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger, K. , Ossowski, S. , Ott, F. et al. (2011) Reference‐guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci U S A, 108, 10249–10254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seren, Ü. , Vilhjálmsson, B.J. , Horton, M.W. , Meng, D. , Forai, P. , Huang, Y.S. , Long, Q. , Segura, V. and Nordborg, M. (2012) GWAPP: a web application for genome‐wide association mapping in Arabidopsis. Plant Cell, 24, 4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seren, U. , Grimm, D. , Fitz, J. , Weigel, D. , Nordborg, M. , Borgwardt, K. and Korte, A. (2017) AraPheno: a public database for Arabidopsis thaliana phenotypes. Nucleic Acids Res. 45, D1054–D1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakoor, N. , Lee, S. and Mockler, T.C. (2017) High throughput phenotyping to accelerate crop breeding and monitoring of diseases in the field. Curr. Opin. Plant Biol., 38, 184–192. [DOI] [PubMed] [Google Scholar]
- Shrestha, R. , Matteis, L. , Skofic, M. , Portugal, A. , McLaren, G. , Hyman, G. and Arnaud, E. (2012) Bridging the phenotypic and genetic data useful for integrated breeding through a data annotation using the Crop Ontology developed by the crop communities of practice. Front. Physiol. 3, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulaev, V. , Sargent, D.J. , Crowhurst, R.N. et al (2011) The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro, N. , Battey, J.N. , Ouadi, S. , Bakaher, N. , Bovet, L. , Willig, A. , Goepfert, S. , Peitsch, M.C. and Ivanov, N.V. (2014) The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, J.T. , Wong, K. , Jackman, S.D. , Schein, J.E. , Jones, S.J. and Birol, I. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, J. (2011) FigShare. J. Pharmacol. Pharmacother. 2(2), 138–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A.K. , Ganapathysubramanian, B. , Sarkar, S. and Singh, A. (2018) Deep learning for plant stress phenotyping: trends and future perspectives. Trends Plant Sci. 23(10), 883–898. [DOI] [PubMed] [Google Scholar]
- Smit, A.F.A , Hubley, R. and Green, P. (2016) RepeatMasker Open‐4.0. 2013–2015. http://www.repeatmasker.org.
- Spannagl, M. , Nussbaumer, T. , Bader, K. , Gundlach, H. and Mayer, K.F. (2017) PGSB/MIPS PlantsDB database framework for the integration and analysis of plant genome data. Methods Mol. Biol. 1533, 33–44. [DOI] [PubMed] [Google Scholar]
- Steinhauser, D. , Usadel, B. , Luedemann, A. , Thimm, O. and Kopka, J. (2004) CSB.DB: a comprehensive systems‐biology database. Bioinformatics, 20, 3647–3651. [DOI] [PubMed] [Google Scholar]
- Stephens, Z.D. , Lee, S.Y. , Faghri, F. , Campbell, R.H. , Zhai, C. , Efron, M.J. , Iyer, R. , Schatz, M.C. , Sinha, S. and Robinson, G.E. (2015) Big data: astronomical or genomical? PLoS Biol. 13(7), e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice, R. , Nikoloski, Z. , Tschoep, H. et al (2013) Impact of the carbon and nitrogen supply on relationships and connectivity between metabolism and biomass in a broad panel of Arabidopsis accessions. Plant Physiol. 162(1), 347–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieu, F. , Cabrera‐Bosquet, L. , Pridmore, T. and Bennett, M. (2017) Plant phenomics, from sensors to knowledge. Curr. Biol. 27(15), R770–R783. [DOI] [PubMed] [Google Scholar]
- Thoen, M.P. , Davila Olivas, N.H. , Kloth, K.J. et al (2017) Genetic architecture of plant stress resistance: multi‐trait genome‐wide association mapping. New Phytol. 213(3), 1346–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togninalli, M. , Seren, Ü. , Meng, D. , Fitz, J. , Nordborg, M. , Weigel, D. , Borgwardt, K. , Korte, A. and Grimm, D.G. (2018) The AraGWAS Catalog: a curated and standardized Arabidopsis thaliana GWAS catalog. Nucleic Acids Res. 46(D1), D1150–D1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollenaar, M. and Wu, J. (1999) Yield improvement in temperate maize is attributable to greater stress tolerance. Crop Sci. 39, 1597–1604. [Google Scholar]
- Tsaftaris, S.A. , Minervini, M. and Scharr, H. (2016) Machine learning for plant phenotyping needs image processing. Trends Plant Sci. 21(12), 989–991. [DOI] [PubMed] [Google Scholar]
- Ubbens, J.R. and Stavness, I. (2017) Deep plant phenomics: a deep learning platform for complex plant phenotyping tasks. Front. Plant Sci. 8, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubbens, J. , Cieslak, M. , Prusinkiewicz, P. and Stavness, I. (2018) The use of plant models in deep learning: an application to leaf counting in rosette plants. Plant Methods, 18(14), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bel, M. , Proost, S. , Van Neste, C. , Deforce, D. , Van de Peer, Y. and Vandepoele, K. (2013) TRAPID: an efficient online tool for the functional and comparative analysis of de novo RNA‐Seq transcriptomes. Genome Biol. 14, R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buren, R. , Bryant, D. , Edger, P.P. et al (2015) Single‐molecule sequencing of the desiccation‐tolerant grass Oropetium thomaeum . Nature, 527(7579), 508–511. [DOI] [PubMed] [Google Scholar]
- Van Buren, R. , Wai, C.M. , Colle, M. et al (2018) A near complete, chromosome‐scale assembly of the black raspberry (Rubus occidentalis) genome. Gigascience, 7(8). 10.1093/gigascience/giy094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tassell, C.P. , Smith, T.P. , Matukumalli, L.K. , Taylor, J.F. , Schnabel, R.D. , Lawley, C.T. , Haudenschild, C.D. , Moore, S.S. , Warren, W.C. and Sonstegard, T.S. (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods, 5(3), 247–252. [DOI] [PubMed] [Google Scholar]
- Variant Graph Team . (2018) Tools for working with genome variation graphs. https://github.com/vgteam/vg
- Varshney, R.K. , Nayak, S.N. , May, G.D. and Jackson, S.A. (2009) Next‐generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530. [DOI] [PubMed] [Google Scholar]
- Vogel, A. , Schwacke, R. , Denton, A.K. et al (2018) Footprints of parasitism in the genome of the parasitic flowering plant Cuscuta campestris. Nat. Commun., 9, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voiniciuc, C. , Zimmermann, E. , Schmidt, M.H. , Günl, M. , Fu, L. , North, H.M. and Usadel, B. (2016) Extensive natural variation in Arabidopsis seed mucilage structure. Front. Plant Sci. 7, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. and van Eeuwijk, F.A. (2014) A new method to infer causal phenotype networks using QTL and phenotypic information. PLoS ONE, 9(8), e103997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Paulo, J. , Kruijer, W. , Boer, M. , Jansen, H. , Tikunov, Y. , Usadel, B. , van Heusden, S. , Bovy, A. and van Eeuwijk, F. (2015) Genotype‐phenotype modeling considering intermediate level of biological variation: a case study involving sensory traits, metabolites and QTLs in ripe tomatoes. Mol. BioSyst. 11(11), 3101–3110. [DOI] [PubMed] [Google Scholar]
- Watson, M. (2018) Mind the gaps – ignoring errors in long read assemblies critically affects protein prediction. BioRxiv. 10.1101/285049. [DOI] [PubMed] [Google Scholar]
- Weisenfeld, N.I. , Yin, S. , Sharpe, T. et al (2014) Comprehensive variation discovery in single human genomes. Nat. Genet. 46, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, H. , Carrier, S. , Thompson, A. , Greenberg, J. and Scherle, R. (2008) The Dryad Data Repository: A Singapore Framework Metadata Architecture in a DSpace Environment. Dublin Core Conference. http://dcpapers.dublincore.org/pubs/article/view/928
- Wick, R. , Judd, L.M. and Holt, K.E. (2018) Comparison of Oxford Nanopore basecalling tools. https://zenodo.org/record/1188469 [DOI] [PMC free article] [PubMed]
- Wilkinson, S. and Davies, W.J. (2002) ABA‐based chemical signalling: the co‐ordination of responses to stress in plants. Plant, Cell Environ. 25(2), 195–210. [DOI] [PubMed] [Google Scholar]
- Wilkinson, M.D. , Dumontier, M. , Aalbersberg, I.J. et al (2016) The FAIR guiding principles for scientific data management and stewardship. Sci. Data, 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir, D. (2013) Where have all the crop phenotypes gone? PLoS Biol. 11, e1001595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino, D.R. and Birney, E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. , Calixto, C.P.G. , Marquez, Y. et al (2017) A high quality Arabidopsis transcriptome for accurate transcript‐level analysis of alternative splicing. Nucleic Acids Res. 45(9), 5061–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Feng, Q. , Lu, H. et al (2018) Pan‐genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278–284. [DOI] [PubMed] [Google Scholar]
- Zhong, S. , Fei, Z. , Chen, Y.R. et al (2013) Single‐base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159. [DOI] [PubMed] [Google Scholar]
- Zhou, N. , Siegel, Z.D. , Zarecor, S. et al (2018) Crowdsourcing image analysis. BioRxiv. 10.1101/265918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin, A.V. , Marçais, G. , Puiu, D. , Roberts, M. , Salzberg, S.L. and Yorke, J.A. (2013) The MaSuRCA genome assembler. Bioinformatics, 29, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin, A.V. , Puiu, D. , Hall, R. , Kingan, S. , Clavijo, B.J. and Salzberg, S.L. (2017) The first near‐complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience, 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]