

Summary
This study evaluated 12‐week retreatment with sofosbuvir/velpatasvir/voxilaprevir in patients with chronic hepatitis C virus (HCV) infection who did not achieve sustained virologic response after previous treatment with a sofosbuvir‐ and velpatasvir‐containing regimen. All 31 patients maintained a sustained virologic response 12 weeks after the last sofosbuvir/velpatasvir/voxilaprevir dose.
Keywords: hepatitis C virus, direct‐acting antiviral agents, retreatment
Abbreviations
- FDC
fixed‐dose combination
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- RASs
resistance‐associated substitutions
- SOF
sofosbuvir
- VEL
velpatasvir
- VOX
voxilaprevir
1. INTRODUCTION
The fixed‐dose combination (FDC) of sofosbuvir (SOF) 400 mg, velpatasvir (VEL) 100 mg and voxilaprevir (VOX) 100 mg (SOF/VEL/VOX; Vosevi) is approved for DAA‐experienced patients in the United States, Canada, the European Union, Australia and other regions, and is now a guideline‐recommended salvage therapy.1, 2 The data supporting the use of SOF/VEL/VOX in NS5A inhibitor‐experienced patients are from GS‐US‐367‐1171 (POLARIS‐1), in which treatment with 12 weeks of SOF/VEL/VOX resulted in an overall sustained virologic response at posttreatment week 12 (SVR12) rate of 96% (253 of 263 patients).3 The most common prior NS5A inhibitors in POLARIS‐1 were ledipasvir (51%) and daclatasvir (27%); only 7% (19 patients) had received VEL. To gain more clinical experience with SOF/VEL/VOX in patients who were previously treated with SOF and VEL, we conducted an open‐label trial designed to assess the efficacy and safety of SOF/VEL/VOX for 12 weeks in patients who did not achieve a sustained virologic response following previous treatment with a SOF‐ and VEL‐containing regimen.
2. METHODS
Eligible patients previously received SOF/VEL/VOX for 8 weeks or SOF/VEL for 12 weeks in Studies GS‐US‐367‐1172 (POLARIS‐2), GS‐US‐367‐1173 (POLARIS‐3), or GS‐US‐367‐1170 (POLARIS‐4), or received a DAA‐based regimen in another Gilead‐sponsored study.3, 4 All patients completed the assigned regimen and attended all protocol‐mandated study visits. Patients were in general good health, with the exception of chronic HCV infection. Patients infected with any HCV genotype, with or without compensated cirrhosis were permitted to enrol. Patients with human immunodeficiency virus (HIV) or hepatitis B virus (HBV) infection, hepatocellular carcinoma or chronic liver disease of a non‐HCV aetiology at screening were excluded.
The institutional review board at each participating institution approved this study, and all patients provided written informed consent.
Samples to determine HCV RNA levels were collected from patients at screening; baseline/Day 1 (predose); Weeks 2, 4, 8, and 12 (or upon early termination); and posttreatment Weeks 4 and 12. The COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, v2.0 was used to quantify HCV RNA in this study. The lower limit of quantitation of this assay is 15 IU/ml.
The primary efficacy endpoint was the proportion of patients with SVR12, defined as HCV RNA less than the lower limit of quantitation (<15 IU/mL) 12 weeks after cessation of treatment.
Secondary efficacy endpoints included the proportion of patients with sustained virologic response at posttreatment week 4 (SVR4), the proportion of patients with HCV RNA less than the lower limit of quantitation while on treatment, HCV RNA (log10 IU/mL) and changes from baseline in HCV RNA (log10 IU/mL) through the end of treatment, and the proportion of patients with virologic failure.
For efficacy endpoints, the 2‐sided 95% exact confidence interval (CI) was calculated for the percentages of patients with SVR12, SVR4 and HCV RNA below the lower limit of quantitation at each post‐baseline visit using the Clopper‐Pearson method. Summary statistics were calculated for absolute values and changes from baseline in HCV RNA (log10 IU/mL) by visit through the end of treatment. No statistical comparisons were conducted.
Baseline deep sequencing analysis was performed for all patients (15% assay cut‐off). The HCV NS3, NS5A and NS5B coding regions were amplified using standard reverse transcriptase‐polymerase chain reaction (RT‐PCR) technology. Following amplification, RT‐PCR products were deep sequenced.
3. RESULTS
Of the 38 patients screened for the study, 31 patients were enrolled. The 7 patients not enrolled did not meet all inclusion and exclusion criteria (3 patients had cirrhosis but did not have liver imaging within 6 months to exclude hepatocellular carcinoma, 4 patients had exclusionary medical history or were not in general good health, 2 patients had laboratory parameters outside acceptable ranges, 1 patient had clinically relevant alcohol or drug abuse within 12 months prior to screening and 1 patient had not received a prior HCV treatment regimen required for enrolment). The 31 patients enrolled had HCV genotype 1a (48%), 1b (13%), 2 (7%), 3 (26%), 4 (3%) or 5 (3%), and approximately half (48%) had cirrhosis. All patients had previously experienced virologic relapse after completing a SOF‐ and VEL‐containing regimen (Figure 1A). Seventeen patients (55%) had previously received SOF/VEL/VOX for 8 weeks, all of whom had been treated in Study GS‐US‐342‐1172 (POLARIS‐2).4 Eleven patients (35%) had previously received SOF/VEL for 12 weeks, 2 patients treated in GS‐US‐367‐1172 (POLARIS‐2), 1 patient in GS‐US‐367‐1173 (POLARIS‐3) and 8 patients in GS‐US‐367‐1170 (POLARIS‐4).4 Three patients had previously received other SOF/VEL‐containing regimens in other studies (GS‐US‐337‐1468 [LEPTON], GS‐US‐337‐0122 and GS‐US‐367‐1168).5, 6, 7 At baseline, 32% of patients had NS5A resistance‐associated substitutions (RASs) and 26% had NS3 RASs. No patients had both NS5A and NS3 RASs.
Figure 1.
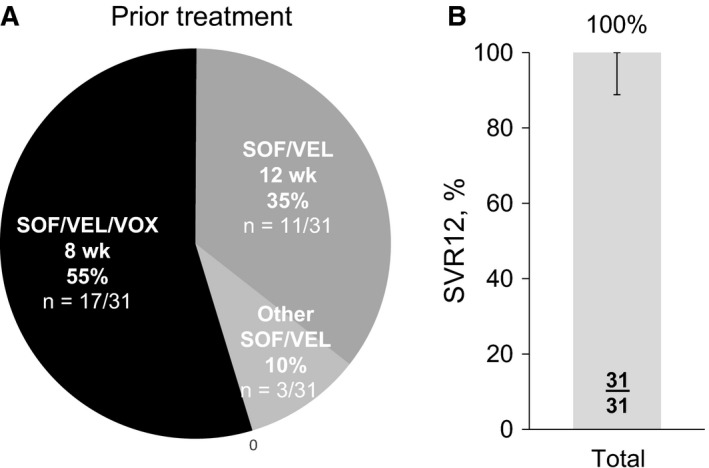
A, Prior HCV treatment and B, Rate of sustained virologic response 12 weeks after treatment with sofosbuvir‐velpatasvir‐voxilaprevir for 12 weeks
All 31 patients in this study achieved SVR12 after completing the 12 weeks of SOF/VEL/VOX treatment (100% [95% CI: 89%‐100%]) (Figure 1B). Potent and rapid suppression of HCV RNA while on treatment was observed. After 2 weeks of treatment, the mean (SD) change from baseline in HCV RNA level was −5.16 (0.560) log10 IU/mL, and this decrease was maintained during treatment from Weeks 4 through 12 (at Week 12, mean HCV RNA level was 1.15 log10 IU/mL, and changes from baseline ranged from −6.16 to −3.76 log10 IU/mL). During treatment, the proportions of patients with HCV RNA below the lower limit of quantitation were 52% at Week 2, 97% at Week 4 and 100% at Weeks 8 and 12.
No patient discontinued treatment with SOF/VEL/VOX due to an adverse event. One patient had a treatment interruption due to hospitalization. This 63‐year‐old male patient with cirrhosis and a history of a Dieulafoy lesion was admitted to the hospital with a Grade 3 serious adverse event of gastrointestinal haemorrhage on Day 12 of SOF/VEL/VOX treatment and interrupted treatment on Days 13 and 17. This patient also experienced Grade 3 adverse events of acute respiratory failure, cellulitis and asthenia (considered serious). No other patients experienced Grade 3 or higher adverse events, or serious adverse events.
Overall, 61% of patients experienced adverse events. The only adverse events reported in more than 10% of patients were fatigue or nausea (16% each), and headache (13%).
Two patients had Grade 3 laboratory abnormalities. The patient described above who was hospitalized with the gastrointestinal haemorrhage had Grade 3 decreased haemoglobin, Grade 3 decreased neutrophils and Grade 3 increased serum glucose. Another patient had Grade 3 increased serum glucose; both patients had a medical history of diabetes.
4. DISCUSSION
SOF/VEL for 12 weeks has demonstrated high efficacy in clinical trial and real‐world settings 8, 9, 10 and is a recommended first‐line regimen for DAA‐naïve patients regardless of genotype or cirrhosis status.1, 2 In Europe and in other regions, SOF/VEL/VOX for 8 weeks is available for use in DAA‐naïve patients without cirrhosis. In the POLARIS‐2 and POLARIS‐3 studies, few patients who failed treatment with 8 weeks of SOF/VEL/VOX had treatment‐emergent RASs (1 of 23 virologic failures),4 suggesting that retreatment with the same regimen for a longer duration may be successful.
A 12‐week course of SOF/VEL/VOX is an approved salvage regimen for NS5A inhibitor‐experienced patients, but due to the timing of the POLARIS‐1 study, limited data existed for patients who had failed a SOF‐ and VEL‐containing regimen. In this study, treatment with SOF/VEL/VOX for 12 weeks resulted in an SVR12 rate of 100% (31 of 31 patients; 95% CI: 89% to 100%) in patients with chronic HCV infection who did not achieve a sustained virologic response with prior exposure to SOF and VEL. Most patients had received prior treatment with SOF/VEL/VOX for 8 weeks (55%) or SOF/VEL for 12 weeks (35%).
Treatment with SOF/VEL/VOX for 12 weeks was well tolerated in this study. Adverse events were reported in 61% of patients, and most were Grade 1 or 2 in severity. The only adverse events reported for more than 10% of patients were fatigue or nausea (each 16%), and headache (13%). Of note, in previous studies of SOF/VEL/VOX,3 diarrhoea was reported in 19% of patients receiving 12 weeks of SOF/VEL/VOX. However, in the current study, diarrhoea was reported in only 7% of patients.
Overall, these results demonstrate the contribution of both the addition of the protease inhibitor VOX to the potent nucleotide/NS5A inhibitor combination of SOF/VEL, and the longer treatment period with SOF/VEL/VOX for retreatment of both noncirrhotic and cirrhotic patients who had been previously treated with SOF/VEL for 12 weeks or SOF/VEL/VOX for 8 weeks. The results also further support the use of SOF/VEL/VOX for 12 weeks as a salvage regimen for patients who have failed prior therapy with NS5A inhibitor‐containing regimens.
CONFLICTS OF INTEREST
Peter Ruane has served as a speaker, consultant, owns stock in and has received research funding from Gilead, has served as a consultant for ViiV, Merck and AbbVie, and has received research funding from ViiV, Merck, AbbVie and Allergan. Simone I. Strasser has received honoraria for advisory board participation or speaker fees from Gilead Sciences, Bayer, Bristol‐Myers Squibb (BMS), AbbVie, MSD, Ipsen, Eisai, Sirtex, Astellas and Novartis. Edward J. Gane receives research grants from Gilead Sciences, serves on the advisory boards of AbbVie, Boehringer Ingelheim, Gilead Sciences, Janssen, Novartis, Roche and Tibotec, and is a speaker for Gilead Sciences, Novartis, Roche and Tibotec. Robert H. Hyland, Jiang Shao, Hadas Dvory‐Sobol, Tram Tran, Luisa M. Stamm and Diana M. Brainard are employees of and hold stock in Gilead Sciences. Lisa Nyberg discloses conflicts with Gilead Sciences, AbbVie, Intercept and Allergan. Stephen Shafran has received funding for HCV clinical trials from AbbVie, BMS, Gilead Sciences, and Merck and honoraria from BMS, Gilead Sciences, and Merck.
ACKNOWLEDGEMENTS
Writing assistance was provided by Holly Dippold of Gilead Sciences.
Ruane P, Strasser SI, Gane EJ, et al. Sofosbuvir/Velpatasvir/Voxilaprevir for patients with HCV who previously received a Sofosbuvir/Velpatasvir‐containing regimen: Results from a retreatment study. J Viral Hepat. 2019;26:770–773. 10.1111/jvh.13067
REFERENCES
- 1. American Association for the Study of Liver Diseases (AASLD), Infectious Disease Society of America (IDSA) . HCV guidance: recommendations for testing, managing, and treating hepatitis C. Available at: https://hcvguidelines.org/. Accessed 16 August 2018.
- 2. European Association for the Study of the Liver (EASL) . EASL recommendations on treatment of hepatitis C 2018. J Hepatol, 2018; 69:461‐511. [DOI] [PubMed] [Google Scholar]
- 3. Bourliere M, Gordon SC, Flamm SL, et al. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N Engl J Med. 2017;376:2134‐2146. [DOI] [PubMed] [Google Scholar]
- 4. Jacobson IM, Lawitz E, Gane EJ, et al. Efficacy of 8 weeks of sofosbuvir, velpatasvir, and voxilaprevir in patients with chronic HCV infection: 2 phase 3 randomized trials. Gastroenterology. 2017;153:113‐122. [DOI] [PubMed] [Google Scholar]
- 5. Gane E, Schwabe C, Hyland RH, et al. Efficacy of the combination of sofosbuvir, velpatasvir, and the NS3/4A protease inhibitor GS‐9857 in treatment‐naïve or previously treated patients with hepatitis C virus genotype 1 or 3 infections. Gastroenterology. 2016;151:448‐456. [DOI] [PubMed] [Google Scholar]
- 6. Gane EJ, Hyland RH, An D, et al. Once daily sofosbuvir with GS‐5816 for 8 weeks with or without ribavirin in patients with HCV genotype 3 without cirrhosis result in high rates of SVR12: the ELECTRON2 study [Abstract 79]. Hepatology. 2014;60(Suppl):236A‐237A. [Google Scholar]
- 7. Lawitz E, Reau N, Hinestrosa F, et al. Efficacy of sofosbuvir, velpatasvir, and GS‐9857 in patients with genotype 1 hepatitis C virus infection in an open‐label, phase 2 trial. Gastroenterology. 2016;151:893‐901. [DOI] [PubMed] [Google Scholar]
- 8. Grebely J, Dalgard O, Conway B, et al. Sofosbuvir and velpatasvir for hepatitis C virus infection in people with recent injection drug use (SIMPLIFY): an open‐label, single‐arm, phase 4, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3:153‐161. [DOI] [PubMed] [Google Scholar]
- 9. Landis CS, Sulkowski MS, Reau N, et al. Safety and efficacy of velpatasvir and sofosbuvir‐based regimens for the treatment of HCV genotype 1‐6: results of the HCV‐TARGET study [Poster 1096]. The Liver Meeting 2017 ‐ The 68th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); 2017. 20‐24 October; Washington, D. C.
- 10. Mangia A, Piazzolla V, Losappio R, et al. High SVR rates in patients with and without cirrhosis treated in real life with sofosbuvir/velpatasvir (SOF/VEL) combination for 12 weeks without ribavirin (RBV). [Poster THU 323]. European Association for the Study of the Liver (EASL); 2018. 11‐15 April; Paris, France.