

Abstract
Hyperuricemia (HU) is a cause of gout. Clinical studies show a link between HU and cardiovascular disease. However, the role of soluble serum urate (SU) on atherosclerosis development remains elusive. We aimed to use a new HU mouse model [Uricase/Uox knockout (KO)] to further investigate the relationship between HU and atherosclerosis. A mouse model by perivascular collar placement of induced carotid atherosclerosis was established in male Uox‐ KO mice. The Uox‐ KO mice had elevated SU levels and enhanced levels of atherosclerosis inflammatory response proteins. In contrast, Uox‐ KO mice with carotid atherosclerosis showed severe neointimal changes in histology staining consistent with increases in intimal area and increases in proliferating cell nuclear antigen (PCNA)‐ and F4/80‐positive cells. Allopurinol reduced neointimal areas induced by the perivascular collar in hyperuricemic mice, accompanied by decreased expression of PCNA‐ and F4/80‐positive cells. Urate‐lowering treatment alleviated atherosclerosis inflammatory response factors and reactive oxygen species (ROS) intensities in both collar placement Uox‐ KO mice and urate‐stimulated human umbilical vein endothelial cells (HUVECs). In vitro results using HUVECs showed ROS was induced by urate and ROS induction was abrogated using antioxidants. These data demonstrate that urate per se does not trigger atherosclerosis intima lesions in male mice. Urate worsens carotid neointimal lesions induced by the perivascular collar and urate‐lowering therapy partially abrogates the effects. The current study warrants clinical studies on the possible benefits of urate‐lowering therapy in atherosclerosis patients with HU.
Keywords: allopurinol, animal model, atherosclerosis, reactive oxygen species, urate
Clinical studies show a link between hyperuricemia (HU) and cardiovascular disease. However, the role of soluble urate on atherosclerosis development remains unclear. A mouse model of induced carotid atherosclerosis was established in a spontaneous HU mouse with the uricase gene inactivated. This study demonstrates that urate worsens carotid neointimal lesions induced by the perivascular collar and that urate‐lowering therapy partially abrogates the effects.

Abbreviations
- BUN
blood urea nitrogen
- CKD
chronic kidney disease
- CVD
cardiovascular disease
- HDL‐C
high‐density lipoprotein cholesterol
- HU
hyperuricemia
- HUVECs
human umbilical vein endothelial cells
- ICAM‐1
intercellular adhesion molecule‐1
- KO
knockout
- LDL‐C
low‐density lipoprotein cholesterol
- MCP‐1
monocyte chemoattractant protein‐1
- NAC
N‐acetylcysteine
- PCNA
proliferating cell nuclear antigen
- ROS
reactive oxygen species
- SU
serum urate
- TC
total cholesterol
- TG
triglyceride
- ULT
urate‐lowering treatment
- Uox
uricase/urate oxidase
- VCAM‐1
vascular cell adhesion molecule‐1
- WT
wild type
- XO
xanthine oxidase
Introduction
Cardiovascular disease (CVD) is the leading cause of death world‐wide 1. Atherosclerosis is one of the major CVDs characterized by focal intimal thickening and ultimately luminal obstruction induced by fibro‐proliferation and an inflammatory process mediated by cytokine production and vascular regulatory mechanisms 2. Neointimal formation happens at the early stage of atherosclerosis which is a complex process initiated by the damage of endothelial cells and exposure of vascular smooth muscle cells to circulating blood elements 3. Hyperuricemia (HU) causes gout and has been implicated in hypertension and atherosclerosis in humans 4. Epidemiological studies have associated HU with atherosclerotic vascular diseases 5, and the level of serum urate (SU) can predict cardiovascular outcomes and mortalities in both sexes 6. HU is also a risk factor for hypertension, which is a potent contributor of atherosclerosis 7. A meta‐analysis of 18 prospective cohort studies, including data from more than 55 000 patients, showed an increased risk of incident hypertension in subjects with HU, and the overall risk increased by 13% per 1 mg·dL−1 increase in SU 8. More recently, a retrospective cohort study demonstrated that HU plays a significant role in the progression from prehypertension to hypertension with a 35% increasing ratio of hypertension risk in men 9. These findings have prompted a growing research interest on the possible benefits of urate‐lowering treatment (ULT) in cardiovascular diseases. However, it has not been definitively established whether urate is merely a marker or a causal agent of CVD, or whether ULT affects outcomes.
Our previous data demonstrated elevated blood pressure in female but not male mice suggesting distinct mechanism accounting for the vascular phenotypes between males and females 10. Given HU is more important for males in term of prevalence, we wanted to ask: (a) does HU in males induce endothelial injury at micro level if not high blood pressure at macro level; (b) if not, does HU worsen existing neointimal lesions induced using established atherosclerosis model. In this study, males were employed to address these questions.
Results
Soluble urate does not induce atherosclerotic phenotypes
Serum urate levels in male uricase/urate oxidase (Uox)‐KO mice were almost three‐fold increased compared to wild‐type (WT) counterparts (563.9 μmol·L−1 ± 16.9 vs 176.3 μmol·L−1 ± 6.7, P < 0.001; Table 1). Compared with WT controls, blood urea nitrogen (BUN) and serum creatinine were significantly elevated in Uox‐KO mice (Table 1). No apparent changes occurred in the fasting glucose level of male Uox‐KO mice compared with WT mice (Table 1). Neither did lipid profiles change [total cholesterol (TC), high‐density lipoprotein cholesterol (HDL‐C) and low‐density lipoprotein cholesterol (LDL‐C); Table 1].
Table 1.
Blood biochemistry in WT and Uox‐KO mice. Biochemical indicators in WT and Uox‐KO mice (n = 8 per group, males, 16 weeks of age). Data were shown as mean ± SEM. Differences between groups were analyzed by the student's t test and one‐way analysis of variance followed by Newman–Keuls multiple comparison test
| WT | KO | P value | WT‐collared | KO‐collared | P value | KO‐collared + allopurinol | P value (compared to KO‐collared) | |
|---|---|---|---|---|---|---|---|---|
| Uric acid (μmol·L−1) | 176.3 ± 6.7 | 563.9 ± 16.9 | < 0.001 | 187.5 ± 4.9 | 573.0 ± 16.4 | < 0.001 | 325.8 ± 9.2 | < 0.001 |
| BUN (mmol·L−1) | 4.47 ± 0.16 | 10.04 ± 0.35 | < 0.001 | 5.19 ± 0.28 | 10.42 ± 0.42 | < 0.001 | 7.91 ± 0.63 | 0.005 |
| Creatinine (μmol·L−1) | 16.53 ± 1.67 | 23.63 ± 2.18 | 0.04 | 15.36 ± 1.83 | 30.5 ± 0.97 | < 0.001 | 21.77 ± 2.47 | 0.0047 |
| Fasting glucose (mmol·L−1) | 6.0 ± 0.2 | 6.3 ± 0.2 | 0.25 | 6.1 ± 0.2 | 6.2 ± 0.2 | 0.71 | 6.4 ± 0.3 | 0.6 |
| TC (mmol·L−1) | 3.36 ± 0.12 | 3.67 ± 0.13 | 0.1 | 3.59 ± 0.08 | 3.85 ± 0.12 | 0.08 | 3.62 ± 0.14 | 0.23 |
| TGs (mmol·L−1) | 1.49 ± 0.16 | 1.13 ± 0.03 | 0.03 | 1.35 ± 0.07 | 1.23 ± 0.03 | 0.13 | 1.27 ± 0.06 | 0.62 |
| HDL cholesterol (mmol·L−1) | 1.61 ± 0.13 | 1.74 ± 0.05 | 0.35 | 1.60 ± 0.05 | 1.80 ± 0.08 | 0.06 | 1.59 ± 0.13 | 0.21 |
| LDL cholesterol (mmol·L−1) | 0.49 ± 0.07 | 0.55 ± 0.04 | 0.44 | 0.48 ± 0.06 | 0.55 ± 0.06 | 0.43 | 0.50 ± 0.03 | 0.44 |
To evaluate the effects of urate on the cardiovascular system, blood pressure, endothelium‐dependent vasodilatation and cardiac function were tested. Systolic blood pressure (SBP), diastolic blood pressure (DBP) and mean blood pressure (MBP) exhibited no difference between male Uox‐KO mice and WT controls (Fig. 1A). Neither did aortae dilation nor cardiovascular function change (Fig. 1B; Table 2). This does not support a direct role of urate in the control of blood pressure, vasodilation and cardiac function in males. As shown in Fig. 1C,D pathogenic proteins in atherosclerosis were up‐regulated in both plasma and carotid lesions – monocyte chemoattractant protein (MCP‐1), intercellular adhesion molecule‐1 (ICAM‐1), and vascular cell adhesion molecule‐1 (VCAM‐1; P < 0.05). However, Uox‐KO mice did not present an atherosclerotic phenotype, showing no changes in intimal areas, proliferation (proxied by PCNA‐positive cells) and inflammation (proxied by F4/80‐positive cells) in Fig. 1E,F.
Figure 1.

Soluble urate does not induce atherosclerosis phenotypes. (A) Blood pressure was measured by the CODA programmable non‐invasive tail‐cuff sphygmomanometer. Mice underwent an acclimation period of seven consecutive days to the sphygmomanometer before experiments. SBP, DBP and MBP in non‐fasting 16‐week‐old WT and Uox‐KO mice (males, n = 10, 7). (B) Isolated aortic rings from 16‐week‐old WT and Uox‐KO mice (males, n = 3) were precontracted with 0.1 μmol·L−1 noradrenaline after an equilibration period of 60 min. Dilation at each acetylcholine concentration (0.001, 0.01, 0.1, 1.0, 10 μmol·L−1) was measured and expressed as the percentage response to noradrenaline. (C) Protein levels of MCP‐1, ICAM‐1, and VCAM‐1 in serum of 16‐week‐old WT and Uox‐KO mice (males, n = 6) detected by ELISA. (D) Protein levels of MCP‐1, ICAM‐1, and VCAM‐1 in carotid arteries of 16‐week‐old WT and Uox‐KO mice determined by three independent western blotting experiments (males, n = 3). (E, F) Carotid arteries were removed and fixed in formalin followed by paraffin‐embedding of 5 μm serial sections. Pathological assessment in WT and Uox‐KO mice (males, n = 6) quantified by intimal area, PCNA‐ and F4/80‐ positive cell counts. Scale bars = 50 μm. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs WT (student's t‐test).
Table 2.
The results of transthoracic ultrasound. No significant difference was observed between male Uox‐KO and WT counterparts (n = 6, 16 weeks of age). LVAWd: end‐diastolic left ventricular anterior wall thickness; LVAWs: end‐systolic left ventricular anterior wall thickness; LVIDd: end‐diastolic left ventricular inner diameter; LVIDs: end‐systolic left ventricular inner diameter; LVPWd: end‐dastolic left ventricular posterior wall thickness; LVPWs: end‐systolic left ventricular posterior wall thickness; EF: ejection fraction; FS: fractional shortening; LV Mass AW: left ventricle mass. Differences between groups were analyzed by the student's t test
| WT (n = 6) | KO (n = 6) | P | |||
|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | ||
| LVAW; d (mm) | 0.88 | 0.05 | 0.84 | 0.05 | 0.57 |
| LVAW; s (mm) | 1.20 | 0.09 | 1.14 | 0.08 | 0.60 |
| LVID; d (mm) | 3.43 | 0.07 | 3.72 | 0.13 | 0.08 |
| LVID; s (mm) | 2.48 | 0.14 | 2.73 | 0.15 | 0.26 |
| LVPW; d (mm) | 0.81 | 0.04 | 0.85 | 0.07 | 0.66 |
| LVPW; s (mm) | 1.05 | 0.07 | 1.11 | 0.09 | 0.62 |
| EF (%) | 54.38 | 4.40 | 52.04 | 4.93 | 0.73 |
| FS (%) | 27.77 | 2.81 | 26.52 | 3.01 | 0.77 |
| LV mass AW (mg) | 98.18 | 4.74 | 111.40 | 5.21 | 0.09 |
| LV mass AW (corrected; mg) | 78.54 | 3.79 | 89.12 | 4.16 | 0.09 |
| LV Vol; d (μL) | 48.47 | 2.43 | 59.50 | 4.91 | 0.07 |
| LV Vol; s (μL) | 22.61 | 3.11 | 28.59 | 3.89 | 0.26 |
| Stroke volume (μL) | 25.86 | 1.09 | 30.92 | 3.98 | 0.25 |
| Cardiac output (mL·min−1) | 9.71 | 0.74 | 10.17 | 1.15 | 0.74 |
| Heart rate (b.p.m.) | 375 | 22 | 342 | 35 | 0.50 |
Hyperuricemia accelerates carotid neointimal lesions with collar placement
We next addressed the questions: is the effect of urate evident only once a stress is given, and does ULT affect the atherosclerosis pathogenesis? This perivascular carotid collar model along with western‐type diet represents a useful complementary model in which neointimal lesions show up at the early‐stage of atherosclerosis. With collar induction HU makes neointimal lesions worse, indicated by the increases in intimal area, more PCNA‐ and F4/80‐ positive cells in the carotid artery compared with WT collared mice (Fig. 2C,D). Administration of allopurinol (100 mg·kg−1), a xanthine oxidase (XO) inhibitor and urate‐lowering drug, significantly alleviated the intimal phenotypes including intimal area, PCNA‐positive cell counts and F4/80‐positive cell counts. Allopurinol also alleviated both SU and renal functions indicated by BUN and creatinine in Uox‐KO collared mice compared to WT collared mice (Table 1). Consistently, allopurinol decreased expression of MCP‐1, as well as ICAM‐1 and VCAM‐1, in both plasma and carotid tissues determined by ELISA and RT‐PCR, respectively (Fig. 2A,B). The down‐regulation of MCP‐1, ICAM‐1 and VCAM‐1 in carotid tissue indicated that inflammation was improved by ULT. Echocardiographic analysis of heart rate, diastolic left ventricular internal diameter and ejection fraction exhibited no difference between collared Uox‐KO mice and WT controls (data not shown).
Figure 2.

HU accelerates carotid neointimal lesions with collar placement. A perivascular carotid collar placement model was generated with non‐occlusive silastic collars in 10‐week‐old mice. Four weeks before surgery, the animals received HF, HC western‐type diets [21% (wt/wt) fat and 0.15% cholesterol]. Mice were gavaged for a 10‐week allopurinol treatment (100 mg·kg−1). Shown are effects of collar induction and allopurinol treatment on MCP‐1, ICAM‐1, and VCAM‐1 levels in (A) plasma (n = 6) and (B) carotid tissues (males, n = 6), and carotid morphology, proliferation and inflammation represented by intimal area, PCNA‐ and F4/80‐ positive cell counts in (C, D) (males, n = 6), respectively. Scale bars = 50 μm. Error bars represent SEM. *P < 0.05, ***P < 0.001 vs WT control and † P < 0.05 vs untreated Uox‐KO mice (student's t test and one‐way analysis of variance followed by Newman–Keuls multiple comparison test).
Hyperuricemia elevates ROS in carotid artery in vivo
Given reactive oxygen species (ROS) plays a putative role in the pathogenesis of atherosclerosis, we tested the ROS intensity by fluorescent dye dihydroethidium (DHE) staining in the carotid artery of WT and Uox‐KO mice. Uox‐KO mice had elevated ROS levels compared with WT, with or without collar placement (Fig 3A, 3B). ROS intensities were reduced in Uox‐KO mice with 100 mg·kg−1 allopurinol treatment for 10 weeks vs WT controls in the presence or absence of collar placement (Fig 3A,B), implying that urate imposes additional oxidative stress which may accelerate atherosclerosis development.
Figure 3.
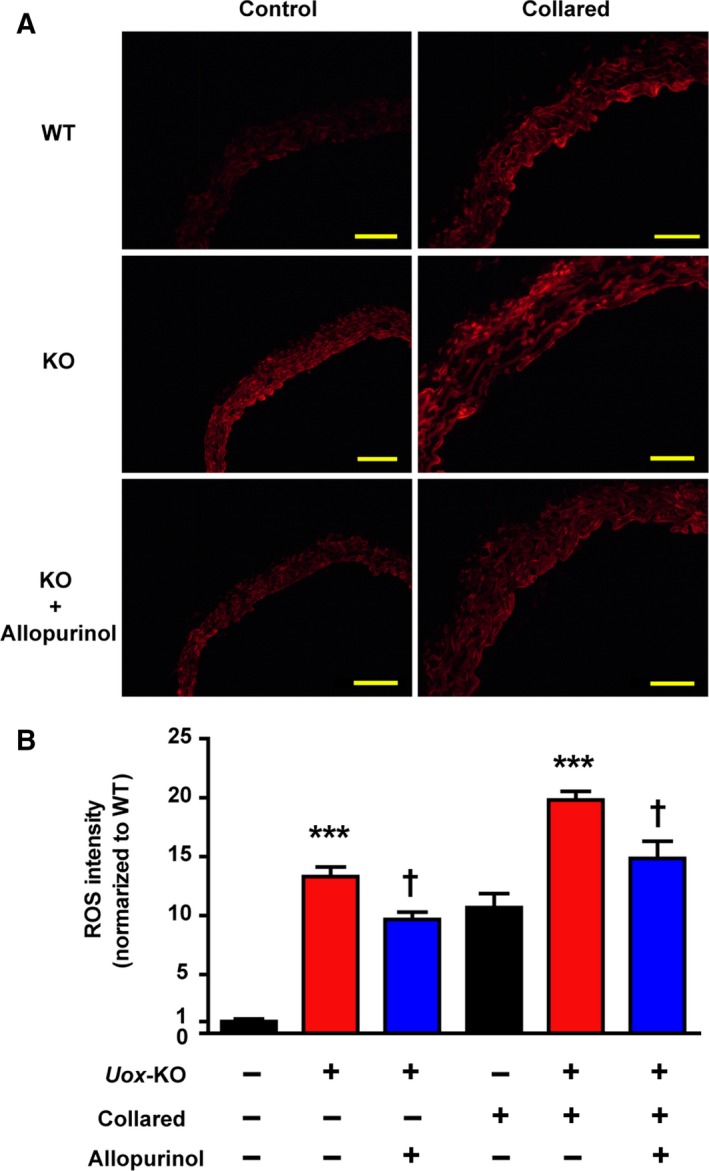
HU elevates ROS in carotid artery in vivo. Carotid arteries were incubated with 2 μmol·L−1 DHE fluorescence probe for 30 min at 37 °C in the dark to measure ROS levels. (A) Representative images of fluorescent dye DHE staining from carotid artery of WT and Uox‐KO mice with or without collar placement are shown (males, n = 6). (B) Effects of 8‐week allopurinol treatment (100 mg·kg−1) on ROS were quantified. Scale bars = 50 μm. Error bars represent SEM. ***P < 0.001 vs WT control. † P < 0.05 vs untreated Uox‐KO mice (student's t test and one‐way analysis of variance followed by Newman–Keuls multiple comparison test).
Soluble urate induces ROS enhancement in vitro
Human umbilical vein endothelial cell (HUVEC) (105/well) viability was measured when co‐incubated with soluble urate (200, 400, 600 and 800 μmol·L−1) at 24, 48 and 72 h. As shown in Fig. 4A–C, soluble urate decreased cell viability in a dose‐dependent and time‐dependent manner and elevated ROS levels – for example with 800 μmol·L−1 urate, ROS intensity was almost 10‐fold increased. ROS levels were extenuated by pre‐incubation for 12 h with probenecid (1 mmol·L−1), an organic anion transport inhibitor, or N‐acetylcysteine (NAC; 10 μmol·L−1), a ROS scavenger, or with both (Fig. 4D). Relative mRNA expression of MCP‐1, ICAM‐1 and VCAM‐1 in soluble urate (800 μmol·L−1, 48 h)‐treated HUVECs was significantly higher than controls and this was lessened by pre‐incubation with probenecid or NAC or both (Fig. 5).
Figure 4.

Soluble urate induces ROS enhancement in vitro. (A) HUVECs (105/well) viability measurement co‐incubated with soluble urate (200, 400, and 800 μmol·L−1) at 24, 48 and 72 h. (B, C) Representative images of fluorescent dye DHE staining from soluble urate stimulated HUVECs and quantified ROS levels. (D) DHE staining on soluble urate (800 μmol·L−1, 48 h) stimulated HUVECs after 12 h pre‐incubation with 1 mmol·L−1 probenecid or 10 μmol·L−1 N‐acetyl‐l‐cysteine (NAC) or both. Scale bars = 20 μm. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs control, † P < 0.05 vs UA group (student's t test and one‐way analysis of variance followed by Newman–Keuls multiple comparison test).
Figure 5.
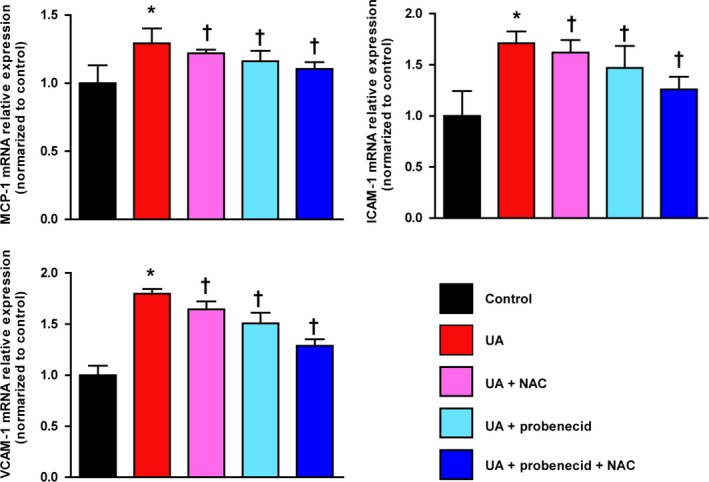
ULT alleviates atherosclerosis inflammatory response factors in vitro. Soluble urate ‐stimulated HUVECs (800 μmol·L−1, 48 h) after 12 h pre‐incubation with 1 mmol·L−1 probenecid or 10 μmol·L−1 NAC or both. Relative mRNA expression of MCP‐1, ICAM‐1, and VCAM‐1 in HUVECs was estimated. Error bars represent SEM. *P < 0.05 vs control, † P < 0.05 vs UA group (student's t test and one‐way analysis of variance followed by Newman–Keuls multiple comparison test).
Discussion
The presence of hepatic Uox is the reason that rodents have lower SU compared to humans 11. When Uox is knocked out, mice develop spontaneous HU similar to the SU level of humans 10. Based on this HU mouse model, we further generated an atherosclerosis model with right carotid artery peri‐collar placement and a western‐type diet. Using this model we showed that, because Uox‐KO mice did not present an atherosclerotic phenotype despite of facts that inflammatory response and ROS indeed were induced in Uox‐KO mice. However, HU worsened the development of neointimal lesions through in vivo ROS enhancement.
Epidemiological studies have detected an association between HU and hypertension, while evidence for causation, that includes data from Mendelian randomization studies, is limited and inconclusive 12, 13, 14. Hypertension is a risk factor in the pathogenesis of atherosclerosis. Here, we describe the cardiovascular characteristics in Uox‐KO male mice, which exhibit no signs of heart dysfunction, heart morphology alteration or blood pressure change. This is consistent with data from another HU mouse model with liver Glut9 deficiency 15. These data therefore do not support a direct causal role of urate on blood pressure in mice.
To the best of our knowledge, this study is the first to assess conducted dilation responses of aortae in the spontaneous HU mouse and showed that urate cannot induce experimentally significant changes of endothelium‐dependent vasodilatation. Indeed, when expressed as a function of the dilation, the conducted dilation of aortae in male Uox‐KO and WT mice was relatively similar. Interestingly, the endothelial relaxation dysfunction reported in aortae from low‐density lipoprotein receptor/apolipoprotein E (LDL/ApoE) double KO mice was in regions with significant lesions, and not in other regions or in aortae from the ApoE single KO mice where lesions were minimal 16. Similarly, even in diabetic ApoE‐KO mice, endothelial dysfunction was only reported in plaque‐prone regions of the aortae, while plaque‐resistant segments maintained a normal acetylcholine response 17. These pieces of evidence are consistent with our observation of no vasodilation dysfunction in our Uox‐KO mice compared with WT mice.
Our results show that spontaneous HU without any stress does not induce obvious atherosclerosis phenotypes. However, HU alone was sufficient to induce oxidative stress and enhance levels of atherosclerosis associated inflammatory cytokines indicating that HU is only a promoting factor rather than an initiator in atherosclerosis. HU may contribute to an activated atherosclerotic pathological process. MCP‐1, ICAM‐1 and VCAM‐1, crucial pathogenic elements in atherosclerosis, are up‐regulated in atherosclerotic lesions and influence growth factor production and medial smooth muscle cell migration 18, 19. HU enhanced MCP‐1, ICAM‐1 and VCAM‐1 as shown in serum level and protein expression, suggesting that these three molecules are also involved in a HU‐driven atherosclerosis promoting effect that would exacerbate the pathogenesis of atherosclerosis. The augmented response of MCP‐1, ICAM‐1 and VCAM‐1 to a given concentration of urate would stimulate downstream inflammation and thereby induce atherosclerosis progressing to a greater extent than controls in vitro. Increasing of intimal area due to smooth muscle cell movement and reproduction is an essential component of atherosclerosis, which can be indicated by proliferating cell nuclear antigen (PCNA) 20. Macrophages accumulate in atherosclerotic plaques, playing crucial roles in atherosclerotic immune responses 21. Significant increases in intimal areas, PCNA‐ and F4/80‐ positive cells in the carotid artery indicates that HU contributes to atherosclerosis, though no plague were found. Despite a few hundred systematic reviews, meta‐analyses, and Mendelian randomization studies exploring 136 unique health outcomes, convincing evidence of a clear causal role of urate in disease pathogenesis only exists for gout and nephrolithiasis 22. Urate is involved in a diverse array of biological functions, while possibly contributing to the pathogenesis of cardiovascular phenotypes, rendering it a pathogenic but not causal role 11. HU did associate with an increased risk of cardiovascular death only in participants with gout and existing cardiovascular disease 23, consistent with our experimental in vivo data.
Reactive oxygen species are another major but non‐specific mediator in the formation of atherosclerosis‐inducing endothelial dysfunction. It can reduce the bioavailability of nitric oxide, a potential anti‐atherosclerotic factor 24. Urate can lead to ROS enhancement which facilitates atherosclerosis by oxidative stress 25, which is consistent with our in vivo and in vitro data. Changes in ROS exhibited a similar trend as the change of neointimal lesions in collar‐induced Uox‐KO mice, which were partially rescued by ULT. This suggests that ROS may also contribute to urate‐induced atherosclerosis‐promoting effects. The effect of allopurinol (via the active metabolite oxypurinol) that inhibits XO activity and suppresses urate biosynthesis 26, also reduces ROS production by inhibiting XO. Other non‐XO effects of allopurinol include LDL oxidation prevention, heat shock protein expression inhibition and decreasing early changes in inflammation such as leukocyte activation by reducing adherence, rolling and extravasation 27. Given our data shows HU was sufficient to induce both ROS production and atherosclerosis associated inflammatory cytokines, worsened ROS level, inflammatory molecules and neointimal lesions observed in collared Uox‐KO mice could be effects due to an ‘additional action’ imposed by HU. Thus, all the neointimal lesions and associated inflammatory factors as well as ROS production were alleviated when ULT was present. HUVECs are wildly used cell models to investigate the mechanisms of vascular phenotypes, especially atherosclerosis 28, 29. HUVECs have urate transporter 1 (URAT1) on their apical‐surface, and probenecid is an effective inhibitor of URAT1. Pretreating HUVECs with probenecid will stop urate from entering inside the cells and its intracellular biologic behaviours efficaciously at the dose of 1 mmol·L−1 30. In vitro, our study showed the same trend of ROS in vivo. ROS intensities were lowered with probenecid intervention. Combined with probenecid and NAC, the strongest ROS lowering effect was observed. Thus, the benefits of XO inhibitors such as allopurinol might rely on blocking the production of oxidants rather than on lowering urate 31. A random, double‐blind, crossover study also showed that the mechanism of improvement in endothelial function with high‐dose allopurinol lies in its ability to reduce vascular oxidative stress and not in urate reduction 32. Therefore, although our data exclude a direct causal role of urate per se on atherosclerosis in male Uox‐KO mice, further studies to address a potential role of XO activity on the cardiovascular function are warranted. Overall, this work reinforces the conclusion that urate accelerates pathogenesis of atherosclerosis and ROS lowering may bring the anti‐atherosclerotic effects.
It remains controversial as to whether asymptomatic HU should be treated for the purpose of improving cardiovascular outcomes 33. Stamp and Dalbeth 34 suggested that asymptomatic HU treatment needs to be cautiously considered, due to the limited data and the potential risks of treatment. Kok et al. 35 reported that allopurinol therapy in patients with gout does not yield beneficial cardiovascular outcomes. However, Kuwabara et al. 36 promoted the use of ULT for asymptomatic HU in a 5‐year Japanese cohort study with 13 201 subjects. ULT was associated to better outcomes in HU patients with cardiovascular diseases accompanied by benefits in endothelial dysfunction and systemic inflammation 37. A randomized controlled trial reported that allopurinol reduces central blood pressure and carotid intima‐media thickness progression at 1 year in patients with recent ischemic stroke and transient ischemic attack 38. A mouse study reported that allopurinol represents a potential novel strategy for preventing left ventricular remodelling and dysfunction after myocardial infarction 39. As well, significant anti‐atherosclerotic effects were seen in a HU study in ApoE‐KO mice 40. Consistently, our results suggest that ULT with allopurinol for hyperuricemic mice may improve neointimal lesions significantly.
Given multiple lines of evidence suggest patients with chronic kidney disease (CKD) are at an increased risk cardiovascular disease 41 and Uox‐KO mice develops renal failure 10, we could not rule out CKD is involved in the neointimal lesions in HU mice. Generally, CKD worsens classical risk factors including hypertension, dyslipidemia and malnutrition 42, 43, which are associated with increased thickness and stiffness of arteries independent of major confounders 44, 45. However, none of these phenotypes can be observed in the male Uox‐KO mice according to our previous report 10, which is distinct from 5/6 nephrectomy CKD mouse model with developing hypertension and atherogenesis 46. On the other hand, urate production is abundantly detected in microvascular endothelial cells and urate reduces endothelial nitric oxide 47, 48, 49, a crucial protective molecule from vascular injury. In addition, hyperuricemic rats have impaired endothelial function which can be reversed by lowering urate 50. Clinically, it has been shown that CKD stage 3A alone may not be a strong risk factor for cardiovascular events without HU, while dramatically elevated the risk accompanied by HU, indicating HU is a major determinant of increased CVD risk in CKD stage 3A 51. Taken together, our results tend to suggest a direct toxicity of urate to endothelial cells rather than secondary effects of CKD is not sufficient to induce atherosclerosis but may contribute to the neointimal lesions in perivascular carotid collar model.
It is important to note that there was only approximately 40% of survivors in the Uox‐KO birth cohort 10. Thus, the survived Uox‐KO mice with constant high urate levels may represent biased subjects and they normally develop significant metabolic and renal dysfunction, which actually mimics the human cases of gout and HU with severe complications. A transient increase in uricosuria during the first 2 weeks of life in Uox‐deficient mice might be responsible for the relative low survival rate 52. The current model is useful to study the asymptomatic HU from a metabolic perspective though it is not fully understood the reasons of poor survival rate. There is phenotypic heterogeneity existed between sexes in the Uox‐KO model. To avoid confounding factor due to gender or sex hormone difference, we only chose male mice in the current study. Further investigation in females would help to delineate a full picture of the effect of HU on cardiovascular diseases.
In conclusion, this is the first evidence to demonstrate that urate plays a contributing rather than a causal role in the carotid neointimal lesions, while ULT may bring additional benefits in this spontaneous HU male mouse model. Clinical and mechanism studies are warranted to investigate the ULT's anti‐atherosclerotic benefits in atherosclerosis patients with HU.
Materials and methods
Mouse model
The spontaneous HU mice were developed by knock‐out of the hepatically expressed Uox gene 10. Controls were their WT littermates on the C57BL/6J background. As previously described 53, we generated perivascular carotid collar placement mouse models with non‐occlusive silastic collars (length, 5 mm; internal diameter, 0.3 mm; external diameter, 0.6 mm) in 10‐week‐old males. Four weeks before surgery, the animals received high‐fat, high‐cholesterol (HF/HC) western‐type diets (21% (wt/wt) fat and 0.15% cholesterol). Mice were anesthetized by ketamine/xylazine and kept on continuous anesthesia during the surgery. Subsequently, a midline neck incision was made to surgically expose the right carotid artery, and the collar was positioned around the right carotid artery and held in place with a nylon sleeve. Both carotid sheaths were openned, and the common carotid arteries were dissected free from the surrounding connective tissue. The carotid arteries were then returned to their original position and the wound was sutured. After a 6‐week collar placement with western‐type diet, all mice were sacrificed for subsequent measurements. To evaluate the urate‐lowering effect, mice were gavaged with allopurinol at a dose of 100 mg·kg−1 from 6 to 16 weeks of age with collar induction at 10‐week‐old.
Mice were housed under specific pathogen free conditions at 22 °C under a 12‐h light/dark photoperiod with ad libitum access to rodent diet and sterile water 54. The Animal Research Ethics Committee of the Affiliated Hospital of Qingdao University approved this study.
Blood biochemistry
Mice were fasted overnight before blood collection from the outer canthus. All biochemical indicators including SU, BUN, serum creatinine, fasting glucose, lipid profiles [TC, triglyceride (TG), high‐ and low‐ density lipoprotein cholesterol (HDL‐C, LDL‐C) were measured by an automatic biochemical analyser (Toshiba, Tokyo, Japan).
Elisa
Plasma MCP‐1, ICAM‐1 and VCAM‐1 levels were quantified using an ELISA kit (R&D Systems, Minneapolis, MN, USA).
Quantitative real‐time RT‐PCR
Total RNA was isolated from the carotid artery or cell line using trizol reagent (Roche Pharmaceuticals, Basel, Switzerland) and then reverse transcribed using a Fast Quant RT kit (Takara‐Bio, Shiga, Japan), followed by amplification using primers for MCP‐1, ICAM‐1 and VCAM‐1 under the condition: 95 °C for 10 min and 40 cycles of 95 °C for 15 s, 58 °C for 20 s and 68 °C for 20 s. The threshold cycle (C t) was determined and used to calculate ΔC T values. The ΔΔC t ( ) was used to calculate relative mRNA expression, with each measurement performed in triplicate. Primer sequences are shown in Table 3.
Table 3.
Primers of quantitative real‐time RT‐PCR
| Primers | (5′‐sequences‐3′) |
|---|---|
| h‐MCP‐1_F | GATGCAATCAATGCCCCAGTC |
| h‐MCP‐1_R | TCCTTGGCCACAATGGTCTTG |
| h‐ICAM‐1_F | GGCTGGAGCTGTTTGAGAAC |
| h‐ICAM‐1_R | TCACACTGACTGAGGCCTTG |
| h‐VCAM‐1_F | TCCCTACCATTGAAGATACTGGAAA |
| h‐VCAM‐1_R | GCTGACCAAGACGGTTGTATCTC |
| h‐β‐actin_F | CGCAAAGACCTGTACGCCAAC |
| h‐β‐actin_R | CACGGAGTACTTGCGCTCAGG |
Western blot
Protein was extracted from the carotid artery. Fifty microgram protein lysates were loaded and transferred to nitrocellulose membranes. Blots were incubated with the primary antibodies (1 : 2000; Abcam, Cambridge, UK) against MCP‐1 (Catalog no. ab25124), ICAM‐1 (Catalog no. ab119871), VCAM‐1 (Catalog no. ab134047) and GAPDH (Catalog no. ab8245) at 4 °C overnight. After incubation with horseradish peroxidase‐conjugated goat anti‐rabbit secondary antibodies, visualization was performed with an enhanced chemiluminescence kit (Thermo Scientific, Waltham, MA, USA). Each measurement was performed in triplicate.
Echocardiographic analysis
Echocardiography was performed after anesthesia with isoflurane using a Vevo 2100 ultrasound system equipped with a MS400 probe (VisualSonics, Toronto, ON, Canada) for in vivo transthoracic ultrasound imaging. The heart was imaged in a two‐dimensional mode in the parasternal long‐axis view. An M‐mode cursor was positioned perpendicular to the interventricular septum and the posterior wall of the left ventricle at the level of the papillary muscles. Stroke volume was calculated as (LV Vol; d − LV Vol; s) and cardiac output as [(LV Vol; d − LV Vol; s) * HR]/1000, where, LV Vol d: end‐diastolic left ventricle volume; LV Vol s: end‐systolic left ventricle volume.
Blood pressure
Systolic and DBP blood pressure was measured by the CODA programmable noninvasive tail‐cuff sphygmomanometer (Kent Scientific, Torrington, CT, USA). Mice underwent an acclimation period of seven consecutive days to the sphygmomanometer before experiments. MBP was calculated as [DBP + 1/3 (SBP − DBP)].
Assessment of endothelium‐dependent vasodilatation
Vasorelaxation of isolated aortic ring segments were determined in oxygenated Kreb's solution. Aortic rings were precontracted with 0.1 μmol·L−1 noradrenaline after an equilibration period of 60 min. Dilation at each acetylcholine (0.001, 0.01, 0.1, 1.0, 10 μmol·L−1) concentration was measured and expressed as the percentage in response to noradrenaline.
Pathology analysis and immunohistochemistry
Carotid arteries were removed and fixed in formalin followed by paraffin embedding of 5 μm serial sections. Tissue serial sections were incubated with anti‐mouse PCNA (1 : 50; Santa Cruz, CA, USA, Catalog no. sc25280) and anti‐mouse F4/80 (macrophage‐specific marker, 1 : 50; Santa Cruz, Catalog no. sc52664) rabbit polyclonal antibodies. Images were captured by Nikon Eclipse TE2000‐S microscope (Nikon, Chiyoda, Japan) and analysed by image‐pro plus software (version 6.0; Houston, TX, USA).
Reactive oxygen species (ROS) measurement
Samples were incubated with 2 μmol·L−1 DHE fluorescence probe (Thermo Scientific) for 30 min at 37 °C in the dark to measure ROS levels. Fluorescence was determined using a Nikon 90i (Nikon) with excitation wavelength at 480 nm and emission wavelength at 610 nm.
Soluble urate
As previously described 55, soluble urate was prepared by dissolving uric acid (UA; Sigma, St. Louis, MO, USA) in warmed media containing 1 m NaOH. The solution was tested to be free of mycoplasma, endotoxin and filtered before use. Crystals were not detectable under polarizing microscopy, nor did they develop during cell incubation.
Cell culture
Human umbilical vein endothelial cells (Cell bank of Chinese Academy of Sciences) were cultured in human endothelial cell‐specific growth medium C‐22010 (PromoCell, Heidelberg, Germany) with 10% fetal bovine serum, 100 units·mL−1 penicillin and 100 μg·mL−1 streptomycin (Invitrogen, Waltham, MA, USA). The viability of HUVECs was measured by Cell Counting Kit‐8 (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's instruction. Cells (105/well) were plated in 96‐well plates and co‐incubated with soluble urate (200–800 μmol·L−1) after 1 mmol·L−1 probenecid (an organic anion transport inhibitor that blocks UA entry into cells) or 10 μmol·L−1 NAC (a ROS scavenger) intervention for 12 h.
Statistical analysis
All statistical analyses were performed using graphpad prism software (version 7; San Diego, CA, USA). Data were presented as the mean ± SEM. Differences between groups were analyzed by Student's t test or one‐way analysis of variance followed by Newman–Keuls multiple comparison test as appropriate. P < 0.05 was considered to be statistically significant.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
All authors approved the final version to be published. JL, MS, XW and CL design the study. JL, XY, ZL, XQ and XJ performed the experiments. JL, XW and CL analysed and interpreted the data. Prof. Li had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Acknowledgements
All authors have written and edited the article. This study is supported by the research project grants from National Key Research and Development Program (#2016YFC0903400 and #2016YFC0903403), National Science Foundation of China (#81520108007, #81770869, #31371272, #81500346, #81441401, #31471195), Science and Technology Development Project of Shandong Province (#2014GSF118013), Basic Application Research Plan of Qingdao (#15‐9‐1‐98‐jch).
Jie Lu, Mingshu Sun and Xinjiang Wu contributed equally to this article.
Contributor Information
Tony R. Merriman, Email: tony.merriman@otago.ac.nz.
Changgui Li, Email: lichanggui@medmail.com.cn.
References
- 1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY et al (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zmuda EJ, Powell CA & Hai T (2011) A method for murine islet isolation and subcapsular kidney transplantation. J Vis Exp 50, e2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tian DY, Jin XR, Zeng X & Wang Y (2017) Notch signaling in endothelial cells: is it the therapeutic target for vascular neointimal hyperplasia? Int J Mol Sci 18, e1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Odden MC, Amadu AR, Smit E, Lo L & Peralta CA (2014) Uric acid levels, kidney function, and cardiovascular mortality in US adults: National Health and Nutrition Examination Survey (NHANES) 1988‐1994 and 1999‐2002. Am J Kidney Dis 64, 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu AH, Gladden JD, Ahmed M, Ahmed A & Filippatos G (2016) Relation of serum uric acid to cardiovascular disease. Int J Cardiol 213, 4–7. [DOI] [PubMed] [Google Scholar]
- 6. Xia X, He F, Wu X, Peng F, Huang F & Yu X (2014) Relationship between serum uric acid and all‐cause and cardiovascular mortality in patients treated with peritoneal dialysis. Am J Kidney Dis 64, 257–264. [DOI] [PubMed] [Google Scholar]
- 7. Wu M & Chan C (2012) Learning transcriptional regulation on a genome scale: a theoretical analysis based on gene expression data. Brief Bioinform 13, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grayson PC, Kim SY, LaValley M & Choi HK (2011) Hyperuricemia and incident hypertension: a systematic review and meta‐analysis. Arthritis Care Res (Hoboken) 63, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Do OH, Low JT & Thorn P (2015) Lepr(db) mouse model of type 2 diabetes: pancreatic islet isolation and live‐cell 2‐photon imaging of intact islets. J Vis Exp 99, e52632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu J, Hou X, Yuan X, Cui L, Liu Z, Li X, Ma L, Cheng X, Xin Y, Wang C et al (2018) Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int 93, 69–80. [DOI] [PubMed] [Google Scholar]
- 11. Alvarez‐Lario B & Macarron‐Vicente J (2010) Uric acid and evolution. Rheumatology 49, 2010–2015. [DOI] [PubMed] [Google Scholar]
- 12. Sluijs I, Holmes MV, van der Schouw YT, Beulens JW, Asselbergs FW, Huerta JM, Palmer TM, Arriola L, Balkau B, Barricarte A et al (2015) A Mendelian randomization study of circulating uric acid and type 2 diabetes. Diabetes 64, 3028–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J et al (2008) SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 40, 437–442. [DOI] [PubMed] [Google Scholar]
- 14. Dehghan A, Kottgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC et al (2008) Association of three genetic loci with uric acid concentration and risk of gout: a genome‐wide association study. Lancet 372, 1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Preitner F, Pimentel A, Metref S, Berthonneche C, Sarre A, Moret C, Rotman S, Centeno G, Firsov D & Thorens B (2015) No development of hypertension in the hyperuricemic liver‐Glut9 knockout mouse. Kidney Int 87, 940–947. [DOI] [PubMed] [Google Scholar]
- 16. Beleznai T, Takano H, Hamill C, Yarova P, Douglas G, Channon K & Dora K (2011) Enhanced K(+)‐channel‐mediated endothelium‐dependent local and conducted dilation of small mesenteric arteries from ApoE(‐/‐) mice. Cardiovasc Res 92, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding H, Hashem M, Wiehler WB, Lau W, Martin J, Reid J & Triggle C (2005) Endothelial dysfunction in the streptozotocin‐induced diabetic apoE‐deficient mouse. Br J Pharmacol 146, 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsueh TP, Sheen JM, Pang JH, Bi KW, Huang CC, Wu HT & Huang ST (2016) The anti‐atherosclerotic effect of naringin is associated with reduced expressions of cell adhesion molecules and chemokines through NF‐kappab pathway. Molecules 21, e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dzau VJ, Braun‐Dullaeus RC & Sedding DG (2002) Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8, 1249–1256. [DOI] [PubMed] [Google Scholar]
- 20. Lu J, Hou X, Yuan X, Cui L, Liu Z, Li X, Ma L, Cheng X, Xin Y, Wang C et al (2017) Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int 93, 69–80. [DOI] [PubMed] [Google Scholar]
- 21. Li Z, Zhou Z, Hou X, Lu D, Yuan X, Lu J, Wang C, Han L, Cui L, Liu Z et al (2017) Replication of Gout/Urate concentrations GWAS susceptibility loci associated with gout in a Han Chinese population. Sci Rep 7, 4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu R, Han C, Wu D, Xia X, Gu J, Guan H, Shan Z & Teng W (2015) Prevalence of hyperuricemia and gout in mainland China from 2000 to 2014: a systematic review and meta‐analysis. Biomed Res Int 2015, 762820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muoio DM & Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta‐cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9, 193–205. [DOI] [PubMed] [Google Scholar]
- 24. Montezano AC & Touyz RM (2012) Reactive oxygen species and endothelial function–role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol 110, 87–94. [DOI] [PubMed] [Google Scholar]
- 25. Forstermann U, Xia N & Li H (2017) Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res 120, 713–735. [DOI] [PubMed] [Google Scholar]
- 26. Elion GB (1966) Enzymatic and metabolic studies with allopurinol. Ann Rheum Dis 25, 608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. George J & Struthers AD (2009) Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vasc Health Risk Manag 5, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esfahani M, Saidijam M, Najafi R, Goodarzi MT & Movahedian A (2018) The effect of salusin‐beta on expression of pro‐ and anti‐inflammatory cytokines in human umbilical vein endothelial cells (HUVECs). ARYA Atheroscler 14, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jung TW, Park HS, Choi GH, Kim D, Ahn SH, Kim DS, Lee T & Jeong JH (2018) Maresin 1 attenuates pro‐inflammatory reactions and ER stress in HUVECs via PPARalpha‐mediated pathway. Mol Cell Biochem 448, 335–347. [DOI] [PubMed] [Google Scholar]
- 30. Tassone EJ, Cimellaro A, Perticone M, Hribal ML, Sciacqua A, Andreozzi F, Sesti G & Perticone F (2018) Uric acid impairs insulin signaling by promoting Enpp1 binding to insulin receptor in human umbilical vein endothelial cells. Front Endocrinol 9, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson RJ, Sanchez‐Lozada LG, Mazzali M, Feig DI, Kanbay M & Sautin YY (2013) What are the key arguments against uric acid as a true risk factor for hypertension? Hypertension 61, 948–951. [DOI] [PubMed] [Google Scholar]
- 32. Civelek M & Lusis AJ (2014) Systems genetics approaches to understand complex traits. Nat Rev Genet 15, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abeles AM (2015) Hyperuricemia, gout, and cardiovascular disease: an update. Curr Rheumatol Rep 17, 13. [DOI] [PubMed] [Google Scholar]
- 34. Stamp L & Dalbeth N (2017) Urate‐lowering therapy for asymptomatic hyperuricaemia: a need for caution. Semin Arthritis Rheum 46, 457–464. [DOI] [PubMed] [Google Scholar]
- 35. Kok VC, Horng JT, Chang WS, Hong YF & Chang TH (2014) Allopurinol therapy in gout patients does not associate with beneficial cardiovascular outcomes: a population‐based matched‐cohort study. PLoS ONE 9, e99102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuwabara M, Niwa K, Hisatome I, Nakagawa T, Roncal‐Jimenez CA, Andres‐Hernando A, Bjornstad P, Jensen T, Sato Y, Milagres T et al (2017) Asymptomatic hyperuricemia without comorbidities predicts cardiometabolic diseases: five‐year Japanese cohort study. Hypertension 69, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hayashino Y, Okamura S, Tsujii S & Ishii H (2016) Association of serum uric acid levels with the risk of development or progression of albuminuria among Japanese patients with type 2 diabetes: a prospective cohort study [Diabetes Distress and Care Registry at Tenri (DDCRT 10)]. Acta Diabetol 53, 599–607. [DOI] [PubMed] [Google Scholar]
- 38. Higgins P, Walters MR, Murray HM, McArthur K, McConnachie A, Lees KR & Dawson J (2014) Allopurinol reduces brachial and central blood pressure, and carotid intima‐media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Heart 100, 1085–1092. [DOI] [PubMed] [Google Scholar]
- 39. Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, Fuchs M, Hilfiker‐Kleiner D, Hornig B, Drexler H et al (2004) Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation 110, 2175–2179. [DOI] [PubMed] [Google Scholar]
- 40. Wakuda H, Uchida S, Ikeda M, Tabuchi M, Akahoshi Y, Shinozuka K & Yamada S (2014) Is hyperuricemia a risk factor for arteriosclerosis? Uric acid and arteriosclerosis in apolipoprotein e‐deficient mice Biol Pharm Bull 37, 1866–1871. [DOI] [PubMed] [Google Scholar]
- 41. Olechnowicz‐Tietz S, Gluba A, Paradowska A, Banach M & Rysz J (2013) The risk of atherosclerosis in patients with chronic kidney disease. Int Urol Nephrol 45, 1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bermudez‐Lopez M, Arroyo D, Betriu A, Masana L, Fernandez E & Valdivielso JM (2017) New perspectives on CKD‐induced dyslipidemia. Exp Opin Ther Targets 21, 967–976. [DOI] [PubMed] [Google Scholar]
- 43. Rao P, Reddy GC & Kanagasabapathy AS (2008) Malnutrition‐inflammation‐atherosclerosis syndrome in chronic kidney disease. Indian J Clin Biochem 23, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson CT & Brewster LP (2013) Carotid artery intima‐media thickness and the renin‐angiotensin system. Hosp Pract 41, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maruyama K, Nakagawa N, Saito E, Matsuki M, Takehara N, Akasaka K, Sato N & Hasebe N (2016) Malnutrition, renal dysfunction and left ventricular hypertrophy synergistically increase the long‐term incidence of cardiovascular events. Hypertens Res 39, 633–639. [DOI] [PubMed] [Google Scholar]
- 46. Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K & Nielsen LB (2003) Chronic renal failure accelerates atherogenesis in apolipoprotein E‐deficient mice. J Am Soc Nephrol 14, 2466–2474. [DOI] [PubMed] [Google Scholar]
- 47. Johnson RJ, Nakagawa T, Sanchez‐Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY & Lanaspa MA (2013) Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 62, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ & Henderson GN (2008) Inactivation of nitric oxide by uric acid. Nucleosides, Nucleotides Nucleic Acids 27, 967–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang DH, Park SK, Lee IK & Johnson RJ (2005) Uric acid‐induced C‐reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 16, 3553–3562. [DOI] [PubMed] [Google Scholar]
- 50. Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S & Johnson RJ (2005) Hyperuricemia induces endothelial dysfunction. Kidney Int 67, 1739–1742. [DOI] [PubMed] [Google Scholar]
- 51. Ito S, Naritomi H, Ogihara T, Shimada K, Shimamoto K, Tanaka H & Yoshiike N (2012) Impact of serum uric acid on renal function and cardiovascular events in hypertensive patients treated with losartan. Hypertens Res 35, 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flannick J & Florez JC (2016) Type 2 diabetes: genetic data sharing to advance complex disease research. Nat Rev Genet 17, 535–549. [DOI] [PubMed] [Google Scholar]
- 53. Baetta R, Silva F, Comparato C, Uzzo M, Eberini I, Bellosta S, Donetti E & Corsini A (2007) Perivascular carotid collar placement induces neointima formation and outward arterial remodeling in mice independent of apolipoprotein E deficiency or Western‐type diet feeding. Atherosclerosis 195, e112–e124. [DOI] [PubMed] [Google Scholar]
- 54. Zhang X, Zhu X & Chen B (2010) Inhibition of collar‐induced carotid atherosclerosis by recombinant apoA‐I cysteine mutants in apoE‐deficient mice. J Lipid Res 51, 3434–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Eisenbacher JL, Schrezenmeier H, Jahrsdorfer B, Kaltenmeier C, Rojewski MT, Yildiz T, Beyer T, Erle A, Wiegmann DS, Grassl S et al (2014) S100A4 and uric acid promote mesenchymal stromal cell induction of IL‐10 + /IDO+ lymphocytes. J Immunol 192, 6102–6110. [DOI] [PubMed] [Google Scholar]