

Abstract
Transposable elements (TEs) are ubiquitous sequences in genomes of virtually all species. While TEs have been investigated for several decades, only recently we have the opportunity to study their genome‐wide population dynamics. Most of the studies so far have been restricted either to the analysis of the insertions annotated in the reference genome or to the analysis of a limited number of populations. Taking advantage of the European Drosophila population genomics consortium (DrosEU) sequencing data set, we have identified and measured the dynamics of TEs in a large sample of European Drosophila melanogaster natural populations. We showed that the mobilome landscape is population‐specific and highly diverse depending on the TE family. In contrast with previous studies based on SNP variants, no geographical structure was observed for TE abundance or TE divergence in European populations. We further identified de novo individual insertions using two available programs and, as expected, most of the insertions were present at low frequencies. Nevertheless, we identified a subset of TEs present at high frequencies and located in genomic regions with a high recombination rate. These TEs are candidates for being the target of positive selection, although neutral processes should be discarded before reaching any conclusion on the type of selection acting on them. Finally, parallel patterns of association between the frequency of TE insertions and several geographical and temporal variables were found between European and North American populations, suggesting that TEs can be potentially implicated in the adaptation of populations across continents.
1. INTRODUCTION
The rise of next‐generation sequencing technologies has allowed us to enter the era of population genomics. It is now possible to have access to genomes from various individuals representing different populations of the same species. This allows us to directly observe the intraspecies variability on a very large scale. These observed differences can correspond to various types of events, from small indels or point mutations (SNPs) to larger events encompassing several kilobases corresponding to structural variations (Alkan, Coe, & Eichler, 2011). These later events include chromosomal rearrangements, such as inversions and translocations, and large insertions/deletions leading to copy number variations (Escaramís, Docampo, & Rabionet, 2015). Several mechanisms have been shown to promote structural variations such as recombination errors, and errors in replication (Escaramís et al., 2015). Transposable elements (TEs) have also been shown to be one major cause of structural variations by their capacity of mobilizing DNA sequences within the genome (Korbel et al., 2007; Morgante, Depaoli, & Radovic, 2007), but also by generating insertion/deletion polymorphisms (Batzer & Deininger, 2002; Boulesteix, Weiss, & Biémont, 2006; Kalendar et al., 2011).
Transposable elements are middle repetitive sequences that are dispersed in the genomes where they have the capacity to move and replicate themselves. According to the species, they can represent various genomic proportions; in eukaryotes, it ranges from ~3% in yeast (Kim, Vanguri, Boeke, Gabriel, & Voytas, 1998) to more than 80% in maize and wheat (Mascher et al., 2017; Schnable et al., 2009). Several decades of studies have shown that TEs do not have the same activity in all genomes (González & Petrov, 2012; Guio & González, 2019). For example, in humans, only a few families are still functional and thus able to move and to promote changes in the genome (Mills, Bennett, Iskow, & Devine, 2007). On the other hand, Drosophila melanogaster harbours numerous active TE families, which is illustrated by a wide proportion of identical copies (Kaminker et al., 2002; Lerat, Rizzon, & Biémont, 2003) and a predominant role in the amount of spontaneous mutations observed in this species (Ashburner, Golic, & Hawley, 2005; Green, 1988). Indeed, there is experimental evidence showing that some of the D. melanogaster TE families are able to transpose (Kim et al., 1994; Leblanc et al., 2000).
The study of structural variants at the scale of populations provides information concerning the evolution of a species. For several years, simple repeats and TEs have been used as evolutionary markers in several species due to the insertion polymorphism they can generate. Recently, the genetic diversity represented by TE polymorphisms has been shown to reflect human evolution suggesting the possibility that there may be a connection between TE‐based genetic divergence and population‐specific phenotypic differences (Rishishwar, Tellez Villa, & Jordan, 2015). In Arabidopsis, the analysis of TE insertion polymorphism among more than 200 accessions revealed that most TE insertions are rare but they are associated with local extremes of gene expression and DNA methylation levels within the population (Stuart et al., 2016). Moreover, several common TE insertions were found to be associated with modified expression of nearby genes (Stuart et al., 2016), suggesting that TE polymorphisms are a rich source of genetic diversity likely to play an important role in facilitating epigenomic and transcriptional differences between individuals in this species. Similarly, TE polymorphisms represent more than half of large insertions and deletions in the rice genome and were estimated to generate about 14% of the genomic DNA differences between the indica and the japonica rice subspecies (Huang, Lu, Zhao, Liu, & Han, 2008).
Drosophila melanogaster is a perfect model to study TE polymorphism and their impact on the evolution of this species: The genome is quite small, the geographical distribution is wide, and it is relatively easy to sample natural populations. Drosophila melanogaster originated in sub‐Saharan Africa and colonized Europe over the last 13,000–43,000 years, and more recently North America and Australia (Baudry, Viginier, & Veuille, 2004; Duchen, Zivkovic, Hutter, Stephan, & Laurent, 2013; Kapopoulou et al., 2018), which indicates that different natural populations are likely to have evolved and adapted differently to distinct environments. Drosophila melanogaster has been thoroughly analysed for its TE content. It has been known since a long time that its genome contains around 15% of TEs (Dowsett & Young, 1982), and with the sequencing of the Drosophila genome in the 2000s, it was possible to get access to the copies of all the families facilitating their precise identification (Bargues & Lerat, 2017; Kaminker et al., 2002; Lerat, Burlet, Biémont, & Vieira, 2011; Quesneville et al., 2005). Moreover, numerous bioinformatic tools have been recently developed to detect TE polymorphism from either pool‐seq or individual high‐throughput sequencing data (see for reviews Ewing, 2015; Modolo & Lerat, 2014). While there are several studies that have analysed the abundance and population dynamics of the TE insertions annotated (present) in the reference genome (e.g., Barrón, Fiston‐Lavier, Petrov, & González, 2014; Petrov et al., 2011), few analysis so far have detected and analysed de novo TE insertions, that is, insertions present in a sample but not in the reference genome, in natural populations of Drosophila (Chakraborty et al., 2018; Cridland, Macdonald, Long, & Thornton, 2013; Disdero & Filée, 2017; Kofler, Betancourt, & Schlötterer, 2012; Linheiro & Bergman, 2012; Rahman et al., 2015; Zhuang, Wang, Theurkauf, & Weng, 2014). Moreover, the majority of genomes in which de novo insertions have been analysed correspond to North American strains from the DGRP and the DSRP panels (Chakraborty et al., 2018; Cridland et al., 2013; Disdero & Filée, 2017; Kofler et al., 2012; Linheiro & Bergman, 2012; Zhuang et al., 2014).
With the aim of studying population variation of the D. melanogaster genome, the European Drosophila Population Genomics Consortium (DrosEU) was recently founded (Kapun et al., 2018). Towards this aim, this collaborative consortium is extensively sampling and sequencing natural European populations on a continent‐wide scale, and across distinct timescales (Kapun et al., 2018). This framework thus offers the unique opportunity to evaluate the TE insertion polymorphism role in the evolutionary history of this species across space and time. A first preliminary analysis of the European populations collected by the consortium in 2014 (Figure 1) showed that the repetitive content, mostly TE insertions, varied between 16% and 21% with respect to the nuclear genome size. This analysis also revealed that some of the TE insertions showed significant correlation with geographical and temporal variables suggesting that they could be under selection (Kapun et al., 2018). However, an in‐depth analysis of the population dynamics of TEs in European populations was not pursued. In this work, we aimed at (a) assessing the dynamics and population structure of TE families in European natural populations; (b) identifying and characterizing individual TE insertions; and (c) testing for parallel patterns of selection between European and North American populations for reference TE insertions.
Figure 1.
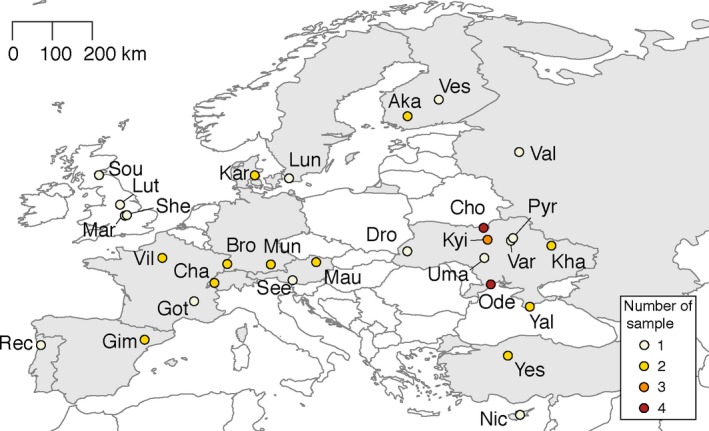
Geographical localization of the populations analysed in this work (Kapun et al., 2018). Acronyms are detailed in Supporting Information Table S1. Colours indicate the number of samples at each geographical location [Colour figure can be viewed at http://wileyonlinelibrary.com]
2. MATERIAL AND METHODS
2.1. Sequence data
We used available pool‐sequencing data for most (see below) of the 48 samples of the DrosEU consortium data set, each one of them containing at least 33 wild‐caught individuals (Figure 1, Supporting Information Table S1, Kapun et al., 2018). These samples were collected from 32 geographical locations at different time points across Europe. Data from North American populations were obtained from Bergland, Behrman, O'Brien, Schmidt, and Petrov (2014), Reinhardt, Kolaczkowski, Jones, Begun, and Kern (2014) and Machado et al. (2018).
2.2. Estimation of TE family abundance and average TE family divergence
For each pool‐sequencing data set, a sample of cleaned single‐end reads (pair R1 of the original library), representing 0.5X of the D. melanogaster genome (assuming 175 Mb), was created combining five samples of 0.1X previously generated (Kapun et al., 2018) with dnaPipeTE (Goubert et al., 2015). We removed from the analysis six samples (5, 7, 8, 34, 35 and 51, Supporting Information Table S1) that were previously found to be contaminated with D. simulans reads. Note that for 11 of the populations analysed, we had samples corresponding to two seasons: spring and fall.
For each pool, the sampled reads were mapped against a collection of consensus sequences for the TE families attributed to D. melanogaster, accessible online at flybase.org (the consensus sequences belonging to other species were removed from the original data set), using RepeatMasker (Smit, Hubley, & Green, 2013). RepeatMasker was used with the following parameters: ‐a ‐nolow ‐no_is. The parameter –a allows to calculate the Kimura 2 parameter (K2P) distance (CpG corrected) for each hit between a read and the TE consensus. Only hits covering a minimum of 80% of the sample reads were kept for further analysis in order to discard false positives.
A first matrix representing the genome percentage of each TE family per pool was generated using the total number of base pairs mapped to one TE family relative to the total number of bases in the 0.5X sample. A second matrix representing the average divergence (K2P) per TE family was also generated. Here, for each pool and each TE family, a weighted average was computed using for each hit, the number of bp mapped as weight of the divergence value.
2.2.1. Data filtering and analysis
All statistical analysis and figures were carried out using r version 3.4.2 (R Core Team, 2017). First, TE families with an average genome representation lower than 0.01% per pool were removed from the analysis. Then, analyses were repeated independently for LTR, DNA and LINE elements.
The heatmaps were generated using the function heatmap.2 for the package gplots (Warnes et al., 2015) with default parameters for clustering of rows (TE families) and columns (populations). Multivariate analyses were computed using the ade4 package (Dray & Dufour, 2007). To compute the link between the matrices of TE abundance and of TE divergence, we used the rv.rtest (Heo & Gabriel, 1997). The obtained correlation value was tested against a null distribution obtained by the permutation of the data in each matrix, with 999 replicates (Monte‐Carlo test). We performed a coinertia analysis (Thioulouse et al., 2018) on the two previous standardized PCA to reveal structures common to both data sets.
Linear correlations were tested between either the TE content (percentage of the genome per TE family) or the average divergence (K2P) and different geographical/environmental variables. Specifically, we used 19 bioclimatic, three environmental, and three geographical variables. The bioclimatic variables are derived from monthly climatic data in order to obtain a bioclimatic profile, that is, a range of climatic conditions reflecting annual trends, seasonality and extreme environmental features (http://worldclim.org/bioclim). For each variable and TE family, the p‐value was corrected for multiple testing using a false discovery rate of 0.05 (FDR, Benjamini & Hochberg, 1995). To avoid population overrepresentation, we considered a maximum of two samples per population (in the case where there are summer and fall samples for the same locality) or one sample per locality if multiple samples were collected in the same season. We thus removed samples 3, 18, 20, 21, 24, 37 and 39 (Supporting Information Table S1).
2.3. Detection of de novo TE insertions
In order to detect TE insertions that are either present in the reference genome (thereafter referred to as “reference insertions”) or present in a sample but not in the reference genome (de novo insertions), we used two different programs: popoolationte2 (Kofler, Gómez‐Sánchez, & Schlötterer, 2016) and tidal (Rahman et al., 2015). The popoolationte2 program computes population frequencies of both reference and de novo TE insertions in the analysed populations compared to the reference D. melanogaster genome (version 6.04) using the pooled sequencing data as input. Since popoolationte2 performs better for sequencing data having a very deep coverage (Kofler et al., 2016), we decided to group data from several locations corresponding to the same geographical regions (Supporting Information Table S1). We thus obtained from the 48 individual samples a total of 14 data sets with a sequencing coverage of at least 90X. Following the manual of popoolationte2, we generated a masked genome by converting the bases covered by the 5,416 copies annotated in the reference genome to N using the bedtool command maskFastaFromBed (Quinlan & Hall, 2010). The read sequences were aligned on the masked genome and the TE copies using bowtie2 (Langmead & Salzberg, 2012) with the ‐‐local option. Using the bam outfile, we successively run the popoolationte2 programs using default parameters (unless specified): ppileup (‐‐map‐qual 15), identifySignatures (‐‐min‐count 2), frequency, filterSignatures (‐‐max‐otherte‐count 2, ‐‐max‐structvar‐count 2) and pairupSignatures to obtain the list of the TE insertions and frequencies in each of the 14 data sets analysed. tidal was run on the same data set using default settings (Rahman et al., 2015). This program allows to detect TE insertions and depletions when compared to the reference genome by using a split‐read approach.
A perl script was written to determine insertions found in common between the two programs by comparing their start positions within a range of 150 bp. In the comparison between popoolationte2 and tidal, we removed from the insertions detected by popoolationte2 those corresponding to insertions found in the reference genomes to make sure that we were comparing only de novo insertions. Statistical analyses were performed using the r software version 3.2.3 (R Core Team, 2017).
2.4. Comparison of popoolationte2 and tidal results with t‐lex2
To determine whether the insertions detected by popoolationte2 and tidal represented real events, we compared the results of these two methods with the results obtained by t‐lex2 (Fiston‐Lavier, Barron, Petrov, & Gonzalez, 2015). t‐lex2 only identifies TE insertions that are present in the reference genome. Thus, we focused on 1,630 reference insertions previously identified to be located in euchromatic regions (Kapun et al., 2018). t‐lex2 has been previously used and experimentally validated in Drosophila with an estimated error rate of only 5% (Fiston‐Lavier et al., 2015).
Not all the 1,630 insertions were found by popoolationte2 and t‐lex2, which most probably indicates the absence of these specific insertions in the populations analysed. However, popoolationte2 consistently detected less reference insertions than t‐lex2, suggesting that it has a high rate of false negatives. On the other hand, we found that a large proportion of the insertions that were detected by both programs were the same, in all populations (Supporting Information Figure S1). Besides, popoolationte2 does not detect any of the insertions that t‐lex2 classifies as absent. Thus, we considered that popoolationte2 has a very low rate of false positives.
We categorized the insertions found by popoolationte2 and t‐lex2 according to their frequency in each population, as computed by popoolationte2 (Supporting Information Figures S2 and S3 for frequency distributions in each population). As expected, since we are considering insertions present in the reference genome, the common insertions were the most numerous in all populations, followed by the fixed insertions, while there was a small number of rare insertions (Supporting Information Figure S2a). To determine whether this representation could have a bias due to the type of insertions that popoolationte2 is able to detect, we looked at the same distribution but for the insertions detected only by t‐lex2 (Supporting Information Figure S2b). We found that in all populations, the most numerous insertions correspond to fixed insertions, with rare and common insertions being less numerous, which indicates that popoolationte2 could have a bias to detect reference insertions that are common.
Because tidal does not detect shared insertions between the reference genome and the genome under investigation but only indicates the absence of reference insertions, we compared the insertions found as absent by tidal with the insertions found as absent or insertions for which the frequency could not be estimated by t‐lex2 (Supporting Information Figure S4). For most populations, tidal usually found more absent insertions than t‐lex2 suggesting that tidal has a high false negative rate. However, the overlapping between both programs is very good since on average 89% of the insertions found to be absent by t‐lex2 are also found to be absent by tidal (Supporting Information Figure S4). Thus, tidal also has a low rate of false positives.
2.5. Experimental validation of de novo TE insertions
We focused on the 5,424 de novo insertions that were detected by both tidal and popoolationte2 in the 14 data sets analysed (Supporting Information Table S2). An arbitrary name was set starting with “te” for each one of the 5,424 de novo insertions. We first checked whether any of these 5,424 de novo insertions were present in the DGRP population using the data available in Rahman et al. (2015). Because tidal does not predict the exact insertion position but rather provides a range of nucleotides where the TE is inserted, we considered a insertion predicted in the DrosEU data set to be the same insertion predicted in the DGRP data set when the two insertions were annotated in ±10 bp, and both insertions belong to the same TE family. A total of 1,542 out of 5,424 de novo insertions were present in the DGRP data set in at least one strain. We then choose 37 de novo insertions to experimentally validate their presence in the D. melanogaster genome (Supporting Information Table S3). All these 37 insertions were not present in the Y chromosome and their canonical length was <6 kb to avoid technical problems in the PCR amplification (except for te1163, te2964 and te1569).
Genomic DNA was extracted from a pool of 10 female flies from each strain (Supporting Information Table S3). Primers were designed in the flanking region of the predicted insertion regions amplifying a minimum of 400 bp when the TE was not present (Supporting Information Table S4). PCR programs were set considering the canonical length for each TE insertion. PCR bands evidencing the presence of a particular TE insertion (in homozygous or heterozygous state) were Sanger‐sequenced using either forward or reverse primers to discard nonspecific PCR amplifications. Some PCR bands were cloned using TOPO‐TA Cloning Kit for Sequencing (Invitrogen) following the manufacturer's instructions and Sanger‐sequenced using either M13 forward or M13 reverse primers. While not all the polymorphic bands could be cloned, the ones that were cloned and sequenced revealed unspecific amplifications.
The 37 TEs that were experimentally validated were randomly chosen from the ones that are present in at least one European population at >10% frequency according to the popoolationte2 software. In addition, 19 of the 37 insertions were also present in the DGRP population at >10% frequency according to popoolationte2 (Rahman et al., 2015). For those TEs predicted in both continents (19), their presence was PCR‐validated in a subset of the DGRP strains, while the other 18 de novo TEs were validated in European isofemale strains. The strains used for validation were as follows: 48 inbred strains from the DGRP population, 15 isofemale strains collected in 2015 from Gimenells (Spain), 12 isofemale strains collected in 2015 from Karensminde (Denmark) or 15 isofemale strains collected in 2015 from Akaa/Vesanto (Finland; Supporting Information Table S3).
We confirmed the presence of 16 de novo insertions, while results were consistent with the absence for 19 insertions, and PCR bands were unspecific in the tested strains for the other two insertions. Note that the 16 experimentally validated TEs were predicted to be present in populations from North America and Europe, and showed a higher average frequency compared with the 18 TEs, only present in European populations, that were not validated (Supporting Information Table S5). Because the sequenced flies used for tidal and popoolationte2 predictions were collected in 2014 and the isofemale lines used for the PCR validation were collected in 2015, it is possible that some of the TEs that were not validated corresponded to false negatives. We expect this limitation to have a bigger effect when the population frequency of the TE is lower. Thus, we considered that 16 out of 18 de novo predicted insertions that were present at an average frequency of >10% in the two continents analysed were validated by PCR. This validation rate (16/18) is similar to the one reported by Rahman et al. (2015) (11/12), and the one reported by Zhang and Kelleher (2017) (28/28). However, we cannot discard that some of the nonvalidated insertions are false positives.
We also tested whether the TE frequency estimates based on popoolationte2 for three de novo insertions, te2319, te5038 and te0036, were accurate. We found that the three insertions were present in the two European populations tested at similar frequencies to the ones predicted by popoolationte2 software (Supporting Information Table S5). Finally, we analysed whether tidal predicted correctly the size of the insertion, the insertion site and the family identity of the 16 validated insertions. TE length was correctly predicted for eight of the 16 insertions while it differed for the other eight insertions (Supporting Information Table S5). All of the validated 16 de novo TEs are inserted in the region predicted by tidal software, except te1163 and te5288, that were inserted 366 bp and 4 bp away from the predicted insertion region, respectively. Finally, 15 out of 16 insertions belong to the predicted TE family. Only the BS element te1163 was wrongly predicted to belong to the HMS‐Beagle TE family.
2.6. Correlations of individual TE copies with geographical and temporal variables
To test whether the correlation with geographical and temporal variables found for a subset of reference TE insertions in the DrosEU populations was also present in North American populations, we used 17 pool‐sequenced samples of D. melanogaster collected in nine different geographical locations at different seasons across North America (Bergland et al., 2014; Reinhardt et al., 2014; Machado et al., 2018; Supporting Information Table S6). We followed the same procedure as described in Kapun et al. (2018). Briefly, we used the population frequencies of 1,615 TE insertions annotated in the D. melanogaster reference genome version 6.04 estimated using t‐lex2 (Fiston‐Lavier et al., 2015) provided by Rech et al. (2018). We excluded those TEs with the interquartile range (IQR) <10. Then, we assessed correlations with population frequencies between TEs and latitude, longitude, altitude and season using generalized linear models (ANCOVA) with a binomial error structure in R, and applying Moran's I test to account for residual spatio‐temporal autocorrelation (Kühn & Dormann, 2012; Moran, 1950). We adjusted the p‐values using the Bonferroni method to correct for multiple testing. To be more conservative, we only considered as significant those TEs with p‐values <0.001, and that were located in regions of high recombination (>0 cM/Mb) according to the recombination rate estimates reported in Comeron, Ratnappan, and Bailin (2012) and Fiston‐Lavier, Singh, Lipatov, and Petrov (2010). Chi‐square test with Yate's correction was used to determine TE family enrichment among the significant TEs.
3. RESULTS
3.1. Transposable element family abundance in European populations does not reflect the geographical population structure
To determine whether the variability in TE content reflected the population geographical structure, we analysed the TE family abundance in 42 samples from 28 natural populations collected across Europe (Kapun et al., 2018, see Material and Methods). We calculated the number of base pairs per population occupied by TEs from the three different TE classes: LTRs, LINEs and DNA elements (Supporting Information File S1). We observed variation in the amount of TEs per population (Figure 2). However, no geographical structure was observed using the total TE abundance (Supporting Information Figure S5) or the individual TE presence/absence patterns (Supporting Information Figure S6a,b, see below).
Figure 2.
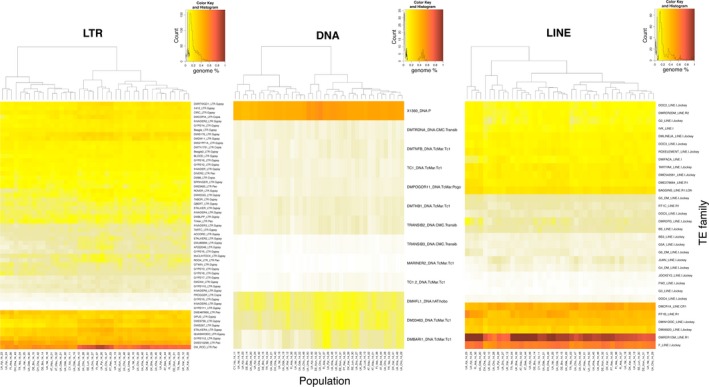
Heatmap representing the genome percentage estimate of each TE family per sample. Abundance was estimated by mapping 0.5X of unassembled reads of each sample against the D. melanogaster TE library with RepeatMasker. The colour scale, from yellow to red, is proportional to the TE family abundance. Cladograms indicate the level of similarity between populations based on their relative TE abundance per family [Colour figure can be viewed at http://wileyonlinelibrary.com]
The clustering of the populations was very similar for LTR and DNA elements, but less discriminant for LINE elements (Figure 2). The most abundant LTR elements in all populations belong to the Pao (e.g., ROO and DME010298 (Batumi)), and the Gypsy (e.g., GYPSY12, and OPUS) superfamilies (Figure 2). 1,360 was the most abundant DNA element in all the populations (Figure 2). Finally, the most abundant LINEs were from the Jockey (e.g., F‐element, and DM06920 (HeT‐A)), R1 (e.g., DMRER1DM), and CR1 (DMCR1A) superfamilies (Figure 2). On the other hand, some TE families were very rare in most of the populations analysed, such as the LTR elements INVADER5, GYPSY9 and FROGGER, the DNA element MARINER2 or the LINE element G3 (Figure 2). Thus, while there is variation in the TE content among European natural populations, some particular families are abundant or rare in most populations (Figure 2).
We tested whether the abundance of the different TE families correlated with 25 geographical/environmental variables mainly derived from monthly climatic data (Supporting Information Table S7, see Material and Methods). We found a negative correlation between DM6920 (jockey), DME542581 (jockey) and INVADER5 abundance with altitude, and between DMHFL1 (hobo) abundance and diurnal range mean of monthly maximum and minimum temperature, and the maximum temperature of the warmest month (Table 1, Supporting Information Table S7). Thus, the abundance of only a few TE families is negatively associated with some geographical (altitude) and environmental variables (temperature).
Table 1.
Significant correlations between transposable elements family abundance and different geographical and environmental variables
| ID | Altitude | Diurnal range mean of monthly max and min temp | Max temperature of warmest month | |||
|---|---|---|---|---|---|---|
| p Valuea | Correlationb | p Value | Correlation | p Value | Correlation | |
| DM06920_LINE.I.Jockey | 4,65E−03 | −6,40E−01 | 6,76E−01 | −2,89E−01 | 9,39E−01 | 3,39E−02 |
| DME542581_LINE.I.Jockey | 1,44E−02 | −5,85E−01 | 6,76E−01 | −2,56E−01 | 9,13E−01 | 4,23E−02 |
| DMHFL1_DNA.hAT.hobo | 7,62E−01 | −8,14E−02 | 2,06E−02 | −5,96E−01 | 1,65E−02 | −6,03E−01 |
| INVADER5_LTR.Gypsy | 2,45E−02 | −5,52E−01 | 3,69E−01 | −4,37E−01 | 7,95E−01 | −1,31E−01 |
Significant p‐values are in bold.
Pearson correlation coefficient.
3.2. Transposable element family divergence varied among European populations
The level of divergence between TE copies from the same family and their consensus sequence reflects the average age of the TE family. In order to assess the dynamics of the transposition among European populations, we calculated the divergence between the consensus sequences and the reads mapping to that particular family for each population (Supporting Information File S2). We observed that, for the three TE classes, two groups of populations were separated based on this criterium (Figure 3). This result suggested that some natural populations harbour older TE copies, including samples from United Kingdom, Austria, Turkey, Ukraine and Switzerland. However, this pattern is not explained by geographical population structure (Supporting Information Figure S7). We also found that some families are old, in all populations, such as the LTR elements INVADER5, FROGGER, INVADER6, GYPSY5, DMDM11 (gypsy), the DNA element DMTHB1, and the LINE elements JOCKEY2, TARTYAK, DME542581 (jockey) and DM06920 (jockey). Other TE families had a different dynamic depending on the population analysed such as the LTR element DME9736 (gypsy), the DNA element DMTNFB (Tc1) or the LINE element BS3. We also observed that DNA elements are in general younger than the other two classes (Figure 3). Finally, we did not find any significant correlations between the TE family divergence and any of the geographical and environmental variables analysed (Supporting Information Table S8).
Figure 3.
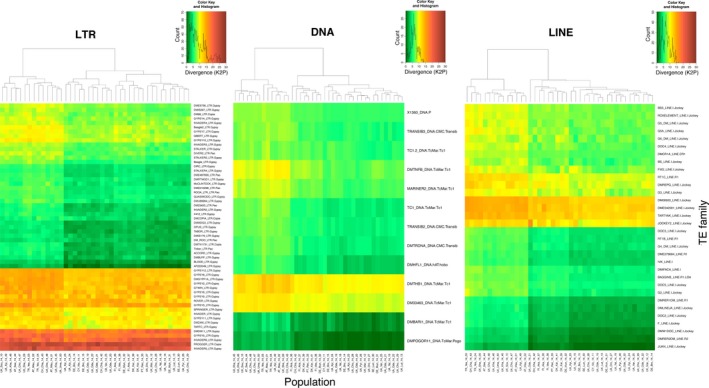
Heatmap representing the average Kimura substitution level (CpG adjusted, K2P) of each TE family per sample. Divergence was estimated by mapping 0.5X of unassembled reads of each sample against the Drosophila melanogaster TE library with RepeatMasker. The colour scale, from green to red, is proportional to the K2P distance. Cladograms indicate the level of similarity between populations based on the TE divergence per family [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.3. The TE content in European populations is negatively correlated with the TE family divergence
Whether the amount of TEs from a given family is linked to the age of the TEs is still an open question. We calculated the link between the matrix of these two variables using the multivariate analysis rv.rtest and found a significant association between TE abundance and TE divergence (correlation coefficient = 0.256, p‐value: 0.003). We then investigated the coinertia between the two standardized PCAs previously performed for the TE abundance and TE divergence (Supporting Information Figure S8). No geographical structure was detected; however, a split between two groups of populations that are closer in space when using the TE divergence as a variable could be observed (Supporting Information Figure S8, axis 1, vertical line). To better understand the link between the two PCAs, we computed the correlations between the PCA axes. We found a strong negative correlation between Axis 1 of the PCA on TE abundance and Axis 2 of the PCA on TE divergence (r = −0.9187). In order to identify the TE families that could be driving this correlation, we calculated the correlation between TE abundance and the Axis 2 coefficients from the PCA on TE divergence. We found that the majority of the TE families displays negative correlations (Supporting Information Figure S9). For all families of DNA elements, the correlations were negative (13/13: range from −0.82 to −0.06); for LINE elements, 15/32 showed negative correlations and 17/32 showed positive correlations (range from −0.84 to 0.88); for LTR, 41/58 showed a negative correlation and 17/58 a positive one (range from −0.91 to 0.83). Thus, for 67% of the families analysed abundance is negatively correlated with the TE family divergence in European populations.
3.4. Identification of de novo individual TE insertions in European populations
Besides analysing the abundance and divergence of TE families, we also identified de novo individual TE insertions in the European populations. Because all the programs that have been designed to identify de novo TE insertions have biases in the TEs they are able to identify, we used two different programs: popoolationte2 and tidal (see Material and Methods; Kofler et al. (2016), Rahman et al. (2015)). The comparison of the results of these two programs with those of t‐lex2 suggested that both methods have a low rate of false positives and a high rate of false negatives (see Material and Methods).
The popoolationte2 program detected a large number of TE insertions in all data sets (from 4,277 in Switzerland to 11,649 in Ukraine, Supporting Information Table S9), and the total number of insertions was correlated with the sequencing coverage (Spearman correlation test ρ = 0.63, p‐value=0.01918). For each population, we classified the insertions based on their frequencies, as computed by popoolationte2, discriminating among rare (0 < frequency ≤ 10%), common (10 < frequency ≤ 95%) and fixed (frequency > 95%) insertions (Supporting Information Figures S10 and S11 for TE frequency distribution in each population). We then tested the correlation between the number of rare, common and fixed insertions and the sequencing coverage in each population. The correlation mentioned above is explained by the rare insertions (Spearman correlation test ρ = 0.83, p‐value = 0.0003346), whereas the number of common and fixed insertions did not correlate with the coverage (p‐values = 0.1458 and 0.8557 for common and fixed insertions, respectively). This indicates that the number of rare insertions is not directly comparable between populations, unless they have similar sequencing coverage. If we do compare populations with similar coverage, for example, Finland and Austria (150X, Supporting Information Table S1), we found that the number of rare de novo insertions varies among populations, with Austria displaying more than twice the amount of rare insertions present in Finland (Supporting Information Figure S10).
We also used tidal to detect de novo insertions in the European populations. tidal is designed to detect de novo insertions and absent reference insertions when compared to the reference genome. The number of detected new insertions varies across populations, as was observed with popoolationte2 (Supporting Information Table S9). However, the number of de novo insertions is smaller compared to the number of insertions detected by popoolationte2, which could be explained by the different methodologies (e.g., split‐reads vs. discordant reads) used by the two programs (Supporting Information Table S9). As observed for popoolationte2, there is a positive correlation between the number of insertions found by tidal and the sequence coverage (Spearman correlation test ρ = 0.86, p‐value = 6.438e−05). However, in this case both the number of rare insertions (Spearman correlation test ρ = 0.85, p‐value = 0.0001785) and the number of common insertions (Spearman correlation test ρ = 0.65, p‐values = 0.01371) showed a significant correlation with coverage, while the fixed insertions displayed no correlation (p‐value = 0.291). Thus, only the numbers of fixed insertions can be directly compared among populations.
Overall, our results showed that while the ability of popoolationte2 to detect rare insertions depends on the coverage of the sample analysed, tidal underestimates both the number of rare and common insertions when the coverage is low.
3.5. popoolationte2 and tidal detect different subsets of de novo TE insertions
As mentioned above, tidal and popoolationte2 have both the ability to detect de novo insertions. Thus, we compared the results obtained by both programs to determine how many of the individual insertions were found by both of them (Supporting Information Figure S12). The overlapping proportion was not as good as previously seen for the reference insertions when comparing popoolationte2 with t‐lex2 (Supporting Information Figure S1). On average, 8.3% of the new insertions found by popoolationte2 and 26% of those found by tidal are the same (based on the chromosomal location and on the TE family name). This overlapping represents, however, a large number of insertions in each population (from 142 in the populations from Turkey to 1,538 in the populations from Ukraine, Supporting Information Figure S12). Although some of the insertions that were detected by only one of the two softwares might be real insertions, to be conservative, we decided to analyse only the 5,424 insertions that were detected both by popoolationte2 and tidal (Supporting Information Table S2). Thus, we are only analysing a subset of all the insertions that might be present in these populations. Experimental validation of a subset of these insertions suggested that a significant proportion of the insertions found by both popoolationte2 and tidal are likely to be real insertions, although we cannot discard that some of the low‐frequency insertions are false positives (see Material and Methods). The TE frequency estimates based on popoolationte2 method also appear to be accurate (Supporting Information Table S5, see Material and Methods). Finally, tidal predicts correctly the insertion site and the family identity of de novo TE insertions, while the size prediction is not so accurate (Supporting Information Table S5, see Material and Methods).
3.6. Characterization of the individual TE insertions found by popoolationte2 and tidal
We categorized the 5,424 insertions detected by popoolationte2 and tidal according to their frequency in the populations as computed by popoolationte2 (Figure 4 and Supporting Information Figure S13 for frequency distributions in each population). We observed that in most populations, these new insertions represented rare insertions, except in Denmark, Spain, Finland and Switzerland where they rather corresponded to common insertions. However, almost no fixed insertions are detected in common by both programs (Figure 4).
Figure 4.
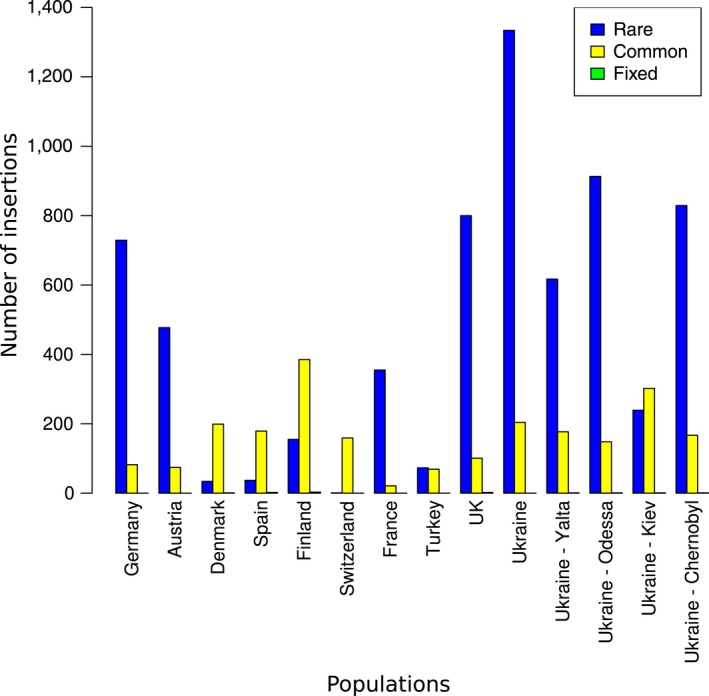
Frequency distribution of de novo insertions identified by both popoolationte2 and tidal. For each population, the different insertions were categorized according to their population frequency in “rare,” “common” and “fixed” insertions [Colour figure can be viewed at http://wileyonlinelibrary.com]
We also determined the TE families that are the most represented among the new insertions in the different populations (Figure 5 and Supporting Information Table S10). Two families are largely overrepresented. They correspond to the LTR retrotransposon ROO and the LINE JOCKEY (DMLINEJA_LINE.I.Jockey). Both elements are indeed known to present numerous potentially active copies in the reference genome (Kaminker et al., 2002; Lerat et al., 2003; Rahman et al, 2015). The DNA transposon POGO is also well represented, especially in the populations of Denmark, Switzerland and Turkey. Although these three families appear to be active in all the populations, the number of copies varies among populations. Note that these results are consistent with the divergence for these families: All of them are young insertions (Figure 3).
Figure 5.
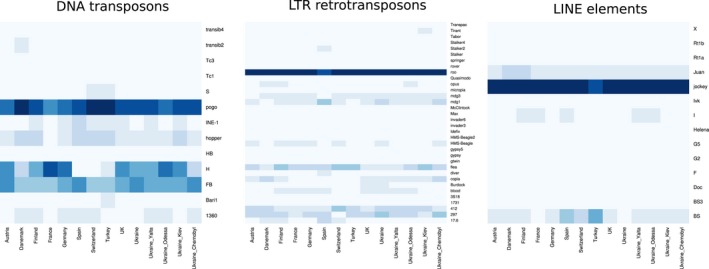
The most represented TE families among the de novo insertions. The heatmaps represent the proportion of each TE family found among de novo insertions in each population, according to each TE class. The representation is normalized by population; thus, the TE families that are the most abundant in that particular population can be identified. The abundance of a particular TE family is higher as the blue colour intensifies [Colour figure can be viewed at http://wileyonlinelibrary.com]
We also analysed the genomic context of the individual insertions identified (Supporting Information Table S2). We observed that 38.59% of the new insertions are intergenic (39.59%, 42.02% and 30.15% when considering separately LTRs, LINEs and DNA transposons, respectively), 49.10% occurred in introns or in 5’UTR‐introns (47.30%, 47.96% and 56.65% for LTRs, LINEs and DNA transposons, respectively), 4.29% occurred in exons (4.62%, 3.46% and 4.16% for LTRs, LINEs and DNA transposons, respectively), while the remaining 8.02% of the insertions (8.49%, 6.56% and 9.04% for LTRs, LINEs and DNA transposons, respectively) were distributed in the other genomic compartments. The distribution of the new insertions is in agreement with the expected distribution, with an overrepresentation of TEs in intergenic regions and introns, and a depletion in exons and other genetic compartments, for all TE classes (χ 2 = 12, df = 9, p‐value = 0.2133).
Finally, the majority of the insertions, 3,758 out of 5,424, were located in regions with high recombination rates (Supporting Information Table S2), suggesting that the data set of insertions analysed is biased as it has been previously reported that TE insertions are more abundant in low recombination regions (e.g., Cridland et al., 2013; Kofler et al., 2012). Note that 283 out of the 3,758 insertions were common (10% < frequency ≤ 95%) in at least two populations, and thus could be evolving under positive selection. However, the increase in frequency due to neutral processes should be discarded before concluding that these insertions are adaptive.
3.7. Several TEs showed parallel correlation patterns between their frequencies and geographical and temporal variables in Europe and North America
In a previous work, we found significant correlations between the frequency of 57 reference TE insertions, located in regions with high recombination, and several geographical and temporal variables (Kapun et al., 2018). We thus tested whether the previously reported correlations can be observed in North American populations (Kapun et al., 2018). We focused on the 115 TE insertions that showed frequency variability among North American samples (IQR > 10). Of these 115 insertions, 48 showed significant associations with geographical or temporal variables after correction for multiple testing (Adjusted p‐value <0.001). None of the insertions showed significant signals of residual spatio‐temporal autocorrelation among samples (Moran's I >0.05 for all tests; Supporting Information Table S11). We focused on the 34 out of the 48 insertions with significant correlations that were present in high recombination regions (see Material and Methods). For these 34 TEs, we observed significant correlations of 10 TE frequencies with latitude, five with altitude, four with season and two with longitude, (Supporting Information Table S11). The frequencies of the other 17 insertions were significantly correlated with more than one of the above‐mentioned variables (Supporting Information Table S11). These significant TEs were scattered along the main five chromosome arms and did not show enrichment for any particular family (chi‐square p‐values after Yate's correction >0.05).
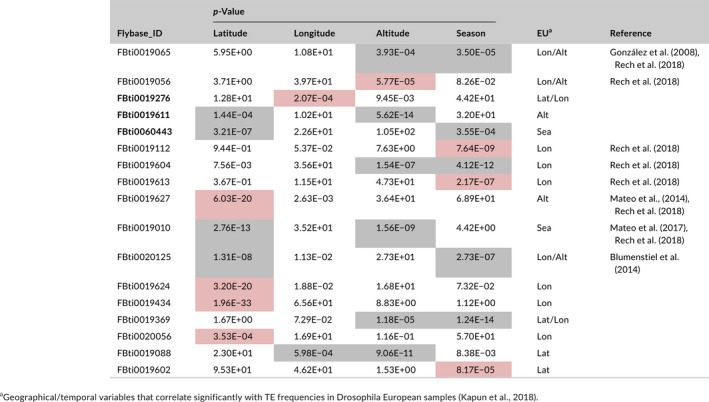
We found that 17 TEs showed significant correlations with either geographical or temporal variables in both North American and European samples (Table 2). This overlap between the two sets of populations was significant (hypergeometric p‐value = 0.009), suggesting that some TE insertions could be under the action of positive selection in both continents. Indeed, although some of these 17 shared TEs correlated with more than one variable in both continents, five of them correlated significantly with the same variable (Table 2). In the European sample analysis, 14 of the 57 significant TEs were previously identified as candidate adaptive insertions using different approaches (Blumenstiel, Chen, He, & Bergman, 2014; González, Lenkov, Lipatov, Macpherson, & Petrov, 2008; Mateo, Ullastres, & González, 2014; Mateo, Rech, & González, 2018; Rech et al., 2018). Among the 17 significant TEs exhibiting significant correlations both in Europe and North American samples, eight TEs were previously identified as candidate adaptive TEs (14 out of 57 and eight out of 17, hypergeometric p‐value = 0.01). This seems to indicate that there is an enrichment for those TEs with additional evidences of selection among the 17 significant TEs shared between Europe and North America. In addition, there were three TEs, FBti0019276, FBti0019611 and FBti0060443 which showed significant correlations with the same variable in both continents and that have never been reported to exhibit any signature of positive selection (Table 2).
Table 2.
Transposable elements (TEs) showing parallel correlation patterns in North American and European populations. p‐values are Bonferroni adjusted (<0.001). In red, TEs that correlate with one variable. In grey, TEs that correlate with more than one variable. In bold, TEs that correlate with the same variable in both continents.
4. DISCUSSION
The extend of variation in the TE content of D. melanogaster has been extensively studied, suggesting a very dynamic mobilome (Biémont et al., 1994; Charlesworth & Langley, 1989; Kofler et al., 2012; Vieira & Biémont, 2004). However, the majority of these studies have only reported data concerning either a handful of TE families, or were restricted to the analysis of reference TE insertions in a single or a few population (e.g., González et al., 2008; Kofler et al., 2012; Cridland et al, 2013). In order to perform a comprehensive analysis of the dynamics and population structure of TEs in D. melanogaster, we constructed heatmaps based either on the relative abundance of each TE family (Figure 2) or on the average divergence between sampled reads and TE consensus sequences (Figure 3) for 42 samples collected in 28 European populations. Considering the most abundant TE families (≥0.01% of the genome in average), we showed that the abundance of TE families is highly variable among European D. melanogaster natural populations. Using divergence between reads and the consensus sequence as a proxy of the age of the different TE families, we confirmed that most of the TE copies detected in D. melanogaster are relatively young (Lerat et al., 2011, 2003). Our population‐wide survey shows clear signatures of recent transposition among the youngest families (Figure 3, darker green areas); however, these bursts do not affect all populations equally, with LTR elements, because of their abundance, as major contributors to the observed variation. It also shows that the activity of specific families seems to be population‐specific and thus appears to depend on the genetic background, as previously described by Adrion, Song, Schrider, Hahn, and Schaack, (2017) in North American lines.
The presence/absence of TE insertions has been successfully used as a neutral marker to describe population genetic structure (Esnault et al., 2008; Goubert et al., 2017). We have used both TE abundance and TE divergence to investigate the population genetic structure in European populations. Clustering by relative abundance and divergence do not provide evidence for geographical structuring based on the global TE dynamics (Figures 2 and 3). Supporting these results, PCAs using either TE families’ relative abundance or divergence (Supporting Information Figures S5 and S6) did not reveal population clustering based on geography. Both metrics, abundance and divergence, are population‐ and TE‐specific and likely reflect the recent activity of TEs, further preventing to identify geographical structure.
Similar results have been reported in D. simulans, where population geographical structure was found for gene expression but not for TE activity (Lerat, Fablet, Modolo, Lopez‐Maestre, & Vieira, 2017). Our picture of the TE dynamics in European D. melanogaster supports a scenario where the expected balance between the endogenous rate of transposition of each TE family and selection against TE insertions generates segregating insertions whose fate is predominantly determined by population‐specific factors such as demography, rather than local adaptation. Interestingly, we found some significant correlations between the abundance of a few TE families and some environmental variables, which indicate that some TEs can be potentially implicated in adaptive processes as discussed below (Table 1).
We wondered if the TE abundance and TE divergence used to describe the TE dynamics were linked. The correlation calculated between the two matrices clearly shows a strong link between the two variables (Supporting Information Figure S9). The large majority of the TE content seems to be negatively correlated with the divergence, which indicates that high copy number TE families present in general low levels of divergence. This is expected since copies from TE families that have recently transposed did not have time to accumulate mutations and thus showed low levels of divergence. Conversely, the accumulation of mutations with time prevents us from detecting older copies of a given TE family. In addition, the deletion rate in the genome of D. melanogaster has been reported to be very high (Petrov, 2002), reducing further the number of copies of the older TE families. However, we do observe positive correlation between abundance and divergence for some TE families, which may indicate that some TE families could insert in genomic locations where they are protected from deletion, such as piRNA cluster regions.
Besides analysing global patterns of TE abundance and divergence across European populations, we have also detected de novo individual TE insertions using two different programs: popoolationte2 and tidal (Kofler et al., 2016; Rahman et al., 2015). We found that the overlap of the predictions between the two methods was 8% for the insertions predicted by popoolationte2 and 26% for those predicted by tidal. Although some of the insertions predicted by only one of the methods might be real insertions, combining the predictions is likely to yield numerous false positives (Rishishwar, Mariño‐Ramírez, & Jordan, 2016). Thus, we were conservative and we only further analyse the insertions predicted by the two methods. Our analysis showed that our data set was biased towards insertions present in high recombination regions, and as such, it could be a good data set to identify putatively adaptive insertions (Cridland et al., 2013; Kofler et al., 2012). Indeed, 283 insertions were present at frequencies ranging from 10% to 95% in at least two of the populations analysed representing a good data set for future functional validation. Because some of these insertions could have increased in frequency neutrally, further experiments are needed before reaching any conclusion about their adaptive role.
Finally, we found a significant correlation between the frequency of 17 insertions and several geographical/temporal variables both in European and North American populations (Table 2). Eight of these 17 insertions have been previously identified as candidate adaptive TEs, while the remaining nine are described for the first time in this work. For example, one of these new insertions FBti0019276 is located inside a gene coding for the transcriptional activator of Adh, a gene widely accepted to be involved in D. melanogaster adaptation to ethanol‐rich habitats (e.g., rotting fruit; Fry, 2014; Fry, Donlon, & Saweikis, 2008; Kreitman, Shorrocks, & Dytham, 1992; Thomson, Jacobson, & Laurie, 1991; see also Siddiq, Loehlin, Montooth, & Thornton, 2017). Indeed, parallel clines in allele frequencies for genes of the ethanol detoxification pathway have been extensively described (Berry & Kreitman, 1993; David et al., 1986; Fry et al., 2008; Oakeshott et al., 1982).
Although we did not find a full overlap in the correlations between the significant TEs and the geographical/temporal variables in the two continents analysed (i.e., the same TEs correlating exactly with the same variables), this does not necessarily mean that the significant TEs found in each continent are not likely to be adaptive. Indeed, it has been reported that the same trait/phenotype (and thus its underlying genotype) can correlate with different variables in different geographical locations (Pool & Aquadro, 2007). Moreover, there are well‐known cases where association between genotype (with additional evidence of being the causative locus of a given phenotype) and phenotype in one continent is not replicated in other continents (Schmidt, Matzkin, Ippolito, & Eanes, 2005). This could happen because the selective pressures could be different along the clines or due to the polygenic nature of the traits under selection than can be also operating in different genetic backgrounds or simply because the genetic targets are different (i.e., phenotypic convergence adaptation). Overall, the fact that the set of 17 significant TEs shared between both continents is enriched with TEs that have been previously identified as candidates to be adaptive, suggesting that some of these 17 TEs might have a potential role in adaptation (Table 2).
We also observed that the abundance of some particular TE families correlates significantly with temperature and altitude. This is not particularly surprising since it is well known that TE activity is influenced by temperature (Chakrani, Capy, & David, 1992; Giraud & Capy, 1996; Ratner, Zabanov, Kolesnikova, & Vasilyeva, 1992; Vieira, Aubry, Lepetit, & Biémont, 1998), and that the temperature‐dependent regulation of different TE families is strongly affected by the genetic background of the host (Jakšić, Kofler, & Schlötterer, 2017). In addition, Kreiner and Wright (2018) have recently found that, in maize, TE abundance is significantly correlated with altitude. Even though they did not find any evidence of adaptation due to TE abundance (but due to genome size), they concluded that adaptation to altitude could be determined by the TE abundance through its effects on genome size.
Other works have shown some links between TE abundance and environmental conditions (Belyayev et al., 2010; Kalendar, Tanskanen, Immonen, Nevo, & Schulman, 2000), pointing towards a positive correlation between TE abundance and stress. However, the growing body of data regarding this issue shows that TEs can be either activated or repressed under stress conditions, and thus, the association between TEs and stress is context‐dependent (see Horváth, Merenciano, & González, 2017 for a detailed review). In the light of these results, it is not unreasonable to think that TE abundance could have an indirect implication in environmental adaptation.
Our work highlights how genome‐wide analysis of natural populations uncovers the considerable amount of genetic variability present in nature. The population‐specific dynamics of TE insertions described in this work implies that analysing the abundance and activity of TEs in a few populations does not provide a realistic picture of the contribution of TEs to genome evolution and genome function. Large consortiums such as the DrosEU consortium, which coordinates the sampling and sequencing of D. melanogaster natural populations, provide us with the unique opportunity to perform population genomic analysis at a continent‐wide level that should help reveal all the genetic variability present in nature.
AUTHOR CONTRIBUTIONS
J.G. and C.V. conceived and designed the study; E.L., C.G., S.G.‐R., M.M. and A.‐B.D. performed the analysis and interpreted the results; J.G., C.V., E.L., C.G. and S.G.‐R., wrote the manuscript; and all authors edited various versions of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Anna Ullastres, Laura Aguilera, Maria Bogaerts and Gabriel E. Rech for technical help. We also thank members of the DrosEU consortium for sharing isofemale lines from their laboratory collections.
Lerat E, Goubert C, Guirao‐Rico S, et al. Population‐specific dynamics and selection patterns of transposable element insertions in European natural populations. Mol Ecol. 2019;28:1506–1522. 10.1111/mec.14963
Funding information
J.G. is funded by the ERC (H2020‐ERC‐2014‐CoG‐647900), and C.V. is funded by the ANR Exhyb (14‐CE19‐0016).
Contributor Information
Cristina Vieira, Email: cristina.vieira@univ-lyon1.fr.
Josefa González, Email: josefa.gonzalez@ibe.upf-csic.es.
DATA ACCESSIBILITY
Supporting Information File S1. Relative TE family abundance (in per cent of the total number of bp sampled) per sample ftp://pbil.univ-lyon1.fr/pub/divers/goubert/DrosoEU/TEbp_001pc
Supporting Information File S2. Averaged divergence between reads and TE family consensus sequence (K2P distance) per sample ftp://pbil.univ-lyon1.fr/pub/divers/goubert/DrosoEU/TEMdiv_001pc.
REFERENCES
- Adrion, J. R. , Song, M. J. , Schrider, D. R. , Hahn, M. W. , & Schaack, S. (2017). Genome‐Wide estimates of transposable element insertion and deletion rates in Drosophila melanogaster . Genome Biology and Evolution, 9(5), 1329–1340. 10.1093/gbe/evx050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan, C. , Coe, B. P. , & Eichler, E. E. (2011). Genome structural variation discovery and genotyping. Nature Reviews Genetics, 12(5), 363–376. 10.1038/nrg2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M. , Golic, K. , & Hawley, R. (2005). Drosophila: A laboratory handbook, 2nd ed (Cold Sprin). New‐York, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Bargues, N. , & Lerat, E. (2017). Evolutionary history of LTR‐retrotransposons among 20 Drosophila species. Mobile DNA, 8(1), 7 10.1186/s13100-017-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrón, M. G. , Fiston‐Lavier, A.‐S. , Petrov, D. A. , & González, J. (2014). Population genomics of transposable elements in Drosophila . Annual Review of Genetics, 48(1), 561–581. 10.1146/annurev-genet-120213-092359 [DOI] [PubMed] [Google Scholar]
- Batzer, M. A. , & Deininger, P. L. (2002). Alu repeats and human genomic diversity. Nature Reviews. Genetics, 3(5), 370–379. 10.1038/nrg798 [DOI] [PubMed] [Google Scholar]
- Baudry, E. , Viginier, B. , & Veuille, M. (2004). Non‐African Populations of Drosophila melanogaster Have a Unique Origin. Molecular Biology and Evolution, 21(8), 1482–1491. 10.1093/molbev/msh089 [DOI] [PubMed] [Google Scholar]
- Belyayev, A. , Kalendar, R. , Brodsky, L. , Eviatar, N. , Schulman, A. H. , & Raskina, O. (2010). Transposable elements in a marginal plant population: Temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Modile DNA, 1, 6 10.1186/1759-8753-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological). Wileyroyal Statistical Society, 57(1), 289–300. 10.2307/2346101 [DOI] [Google Scholar]
- Bergland, A. O. , Behrman, E. L. , O’Brien, K. R. , Schmidt, P. S. , & Petrov, D. A. (2014). Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genetics, 10(11), e1004775 10.1371/journal.pgen.1004775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry, A. , & Kreitman, M. (1993). Molecular analysis of an allozyme cline: Alcohol dehydrogenase in Drosophila melanogaster on the east coast of North America. Genetics, 134(3), 869–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biémont, C. , Lemeunier, F. , Guerreiro, M. P. G. , Brookfield, J. F. , Gautier, C. , Aulard, S. , & Pasyukova, E. G. (1994). Population dynamics of the copia, mdg1, mdg3, gypsy, and P transposable elements in a natural population of Drosophila melanogaster . Genetical Research, 63(3), 197 10.1017/S0016672300032353 [DOI] [PubMed] [Google Scholar]
- Blumenstiel, J. P. , Chen, X. , He, M. , & Bergman, C. M. (2014). An age‐of‐allele test of neutrality for transposable element insertions. Genetics, 196(2), 523–538. 10.1534/genetics.113.158147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulesteix, M. , Weiss, M. , & Biémont, C. (2006). Differences in genome size between closely related species: The Drosophila melanogaster species subgroup. Molecular Biology and Evolution, 23(1), 162–167. 10.1093/molbev/msj012 [DOI] [PubMed] [Google Scholar]
- Chakraborty, M. , VanKuren, N. W. , Zhao, R. , Zhang, X. , Kalsow, S. , & Emerson, J. J. (2018). Hidden genetic variation shapes the structure of functional elements in Drosophila. Nature Genetics, 50(1), 20–25. 10.1038/s41588-017-0010-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrani, F. , Capy, P. , & David, J. R. (1992). Developmental temperature and somatic excision rate of mariner transposable element in three natural populations of Drosophila simulans . Genetics Selection Evolution, 25, 121–132. 10.1186/1297-9686-25-2-121 [DOI] [Google Scholar]
- Charlesworth, B. , & Langley, C. H. (1989). The population genetics of Drosophila transposable elements. Annual Review of Genetics, 23(1), 251–287. 10.1146/annurev.ge.23.120189.001343 [DOI] [PubMed] [Google Scholar]
- Comeron, J. M. , Ratnappan, R. , & Bailin, S. (2012). The many landscapes of recombination in Drosophila melanogaster . PLoS Genetics, 8(10), e1002905 10.1371/journal.pgen.1002905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland, J. M. , Macdonald, S. J. , Long, A. D. , & Thornton, K. R. (2013). Abundance and distribution of transposable elements in two drosophila QTL mapping resources. Molecular Biology and Evolution, 30(10), 2311–2327. 10.1093/molbev/mst129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, J. R. , David, J. , Merçot, H. , Capy, P. , McEvey, S. , & Van Herrewege, J. (1986). Alcohol tolerance and ADH gene frequencies in European and African populations of Drosophila melanogaster . Genetics Selection Evolution, 18, 405–416. 10.1186/1297-9686-18-4-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disdero, E. , & Filée, J. (2017). LoRTE: Detecting transposon‐induced genomic variants using low coverage PacBio long read sequences. Mobile DNA, 8, 5 10.1186/s13100-017-0088-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsett, A. P. , & Young, M. W. (1982). Differing levels of dispersed repetitive DNA among closely related species of Drosophila. Proceedings of the National Academy of Sciences of the United States of America, 79(15), 4570–4574. 10.1073/pnas.79.15.4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray, S. , & Dufour, A.‐B. (2007). The ade4 package: Implementing the duality diagram for ecologists. Journal of Statistical Software, 22(4), 1506–20. 10.18637/jss.v022.i04 [DOI] [Google Scholar]
- Duchen, P. , Zivkovic, D. , Hutter, S. , Stephan, W. , & Laurent, S. (2013). Demographic inference reveals African and European admixture in the North American Drosophila melanogaster population. Genetics, 193(1), 291–301. 10.1534/genetics.112.145912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escaramís, G. , Docampo, E. , & Rabionet, R. (2015). A decade of structural variants: Description, history and methods to detect structural variation. Briefings in Functional Genomics, 14(5), 305–314. 10.1093/bfgp/elv014 [DOI] [PubMed] [Google Scholar]
- Esnault, C. , Boulesteix, M. , Duchemin, J. B. , Koffi, A. A. , Chandre, F. , Dabiré, R. , … Biémont, C. (2008). High genetic differentiation between the M and S molecular forms of Anopheles gambiae in Africa. PLoS ONE, 3(4), 1506–7. 10.1371/journal.pone.0001968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing, A. D. (2015). Transposable element detection from whole genome sequence data. Mobile DNA, 6(1), 24 10.1186/s13100-015-0055-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiston‐Lavier, A. S. , Barron, M. G. , Petrov, D. A. , & Gonzalez, J. (2015). t‐lex2: Genotyping, frequency estimation and re‐annotation of transposable elements using single or pooled next‐generation sequencing data. Nucleic Acids Research, 43(4), e22 10.1093/nar/gku1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiston‐Lavier, A.‐S. , Singh, N. D. , Lipatov, M. , & Petrov, D. A. (2010). Drosophila melanogaster recombination rate calculator. Gene, 463(1–2), 18–20. 10.1016/j.gene.2010.04.015 [DOI] [PubMed] [Google Scholar]
- Fry, J. D. (2014). Mechanisms of naturally evolved ethanol resistance in Drosophila melanogaster . The Journal of Experimental Biology, 217(Pt 22), 3996–4003. 10.1242/jeb.110510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J. D. , Donlon, K. , & Saweikis, M. (2008). A worldwide polymorphism in aldehyde dehydrogenase in Drosophila melanogaster: Evidence for selection mediated by dietary ethanol. Evolution; International Journal of Organic Evolution, 62(1), 66–75. 10.1111/j.1558-5646.2007.00288.x [DOI] [PubMed] [Google Scholar]
- Giraud, T. , & Capy, P. (1996). Somatic activity of the mariner transposable element in natural populations of Drosophila simulans . Procedings of the Royal Society of London (Biological sciences), 263(1376), 1481–1486. 10.1098/rspb.1996.0216 [DOI] [PubMed] [Google Scholar]
- González, J. , Lenkov, K. , Lipatov, M. , Macpherson, J. M. , & Petrov, D. A. (2008). High rate of recent transposable element‐induced adaptation in Drosophila melanogaster . PLoS Biology, 6(10), 2109–2129. 10.1371/journal.pbio.0060251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González, J. , & Petrov, D. A. (2012). Evolution of genome content: Population dynamics of transposable elements in flies and humans. Methods in Molecular Biology (N. J. Clifton), 855, 361–383. 10.1007/978-1-61779-582-4_13 [DOI] [PubMed] [Google Scholar]
- Goubert, C. , Henri, H. , Minard, G. , Valiente Moro, C. , Mavingui, P. , Vieira, C. , & Boulesteix, M. (2017). High‐throughput sequencing of transposable element insertions suggests adaptive evolution of the invasive Asian tiger mosquito towards temperate environments. Molecular Ecology, 26(15), 3968–3981. 10.1111/mec.14184 [DOI] [PubMed] [Google Scholar]
- Goubert, C. , Modolo, L. , Vieira, C. , Moro, C. V. , Mavingui, P. , & Boulesteix, M. (2015). De novo assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biology and Evolution, 7(4), 1192–1205. 10.1093/gbe/evv050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, M. (1988). Mobile DNA elements and spontaneous gene mutation. Banbury Reports, 41–50. [Google Scholar]
- Guio, L. , & González, J. (2019). New insights on the evolution of genome content: Population dynamics of transposable elements in flies and humans. Evolutionary Genomics: Statistical and Computational Methods. Methods in Molecular Biology. Springer. (in press). [DOI] [PubMed] [Google Scholar]
- Heo, M. , & Gabriel, K. R. (1997). A permutation test of association between configurations by means of the RV coefficient. Communications in Statistics ‐ Simulation and Computation, 27, 843–856. [Google Scholar]
- Horváth, V. , Merenciano, M. , & González, J. (2017). Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends in Genetincs, 33(11), 832–841. 10.1016/j.tig.2017.08.007 [DOI] [PubMed] [Google Scholar]
- Huang, X. , Lu, G. , Zhao, Q. , Liu, X. , & Han, B. (2008). Genome‐wide analysis of transposon insertion polymorphisms reveals intraspecific variation in cultivated rice. Plant Physiology, 148(1), 25–40. 10.1104/pp.108.121491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakšić, A. M. , Kofler, R. , & Schlötterer, C. (2017). Regulation of transposable elements: Interplay between TE‐encoded regulatory sequences and host‐specific trans‐acting factors in Drosophila melanogaster . Molecular Ecology, 26(19), 5149–5159. 10.1111/mec.14259 [DOI] [PubMed] [Google Scholar]
- Kalendar, R. , Flavell, A. J. , Ellis, T. H. N. , Sjakste, T. , Moisy, C. , & Schulman, A. H. (2011). Analysis of plant diversity with retrotransposon‐based molecular markers. Heredity, 106(4), 520–530. 10.1038/hdy.2010.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalendar, R. , Tanskanen, J. , Immonen, S. , Nevo, E. , & Schulman, A. H. (2000). Genome evolution of wild barley (Hordeum spontaneum) by BARE‐1 retrotransposon dynamics in response to Sharp microclimatic divergence. Proceedings of the National Academy of Sciences of the United States of America, 97(12), 6603–6607. 10.1073/pnas.110587497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker, J. S. , Bergman, C. M. , Kronmiller, B. , Carlson, J. , Svirskas, R. , Patel, S. , … Celniker, S. E. (2002). The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biology, 3(12), RESEARCH0084 10.1186/gb-2002-3-12-research0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapopoulou, A., Kapun, M., Pavlidis, P., … S. (2018). Early split between African and European populations of Drosophila melanogaster . bioRxiv, 34022 10.1101/340422 [DOI] [Google Scholar]
- Kapun, M. , Aduriz, M. G. B. , Staubach, F. , Vieira, J. , Obbard, D. , Goubert, C. , … Gonzalez, J. (2018). Genomic analysis of European Drosophila melanogaster populations on a dense spatial scale reveals longitudinal population structure and continent‐wide selection. Biorxiv, 313759 10.1101/313759 [DOI] [Google Scholar]
- Kim, A. , Terzian, C. , Santamaria, P. , Pélisson, A. , Purd’homme, N. , & Bucheton, A. (1994). Retroviruses in invertebrates: The gypsy retrotransposon is apparently an infectious retrovirus of Drosophila melanogaster . Proceedings of the National Academy of Sciences of the United States of America, 91(4), 1285–1289. 10.1073/pnas.91.4.1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. M. , Vanguri, S. , Boeke, J. D. , Gabriel, A. , & Voytas, D. F. (1998). Transposable elements and genome organization: A comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Research, 8(5), 464–478. [DOI] [PubMed] [Google Scholar]
- Kofler, R. , Betancourt, A. J. , & Schlötterer, C. (2012). Sequencing of pooled DNA samples (Pool‐Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster . PLoS Genetics, 8(1), e1002487 10.1371/journal.pgen.1002487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, R. , Gómez‐Sánchez, D. , & Schlötterer, C. (2016). popoolationte2: Comparative population genomics of transposable elements using Pool‐seq. Molecular Biology and Evolution, 33(10), 2759–2764. 10.1093/molbev/msw137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel, J. O. , Urban, A. E. , Affourtit, J. P. , Godwin, B. , Grubert, F. , Simons, J. F. , … Snyder, M. (2007). Paired‐end mapping reveals extensive structural variation in the human genome. Science, 318(5849), 420–426. 10.1126/science.1149504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner, J. M. , & Wright, S. I. (2018). A less selfish view of genome size evolution in maize. PLOS Genetics, 14(5), e1007249 10.1371/journal.pgen.1007249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitman, M. , Shorrocks, B. , & Dytham, C. (1992). Genes and ecology: Two alternative perspectives using Drosophila In Berry R. J., Crawford T. J., & Hewitt G. M. (Eds.), Genes and ecology (pp. 281–312). Hoboken, NJ: Blackwell Scientific. [Google Scholar]
- Kühn, I. , & Dormann, C. F. (2012). Less than eight (and a half) misconceptions of spatial analysis. Journal of Biogeography, 39, 995–998. 10.1111/j.1365-2699.2012.02707.x [DOI] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc, P. , Desset, S. , Giorgi, F. , Taddei, A. R. , Fausto, A. M. , Mazzini, M. , … Vaury, C. (2000). Life cycle of an endogenous retrovirus, ZAM, Drosophila melanogaster . Journal of Virology, 74(22), 10658–10669. 10.1128/JVI.74.22.10658-10669.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat, E. , Burlet, N. , Biémont, C. , & Vieira, C. (2011). Comparative analysis of transposable elements in the melanogaster subgroup sequenced genomes. Gene, 473(2), 100–109. 10.1016/j.gene.2010.11.009 [DOI] [PubMed] [Google Scholar]
- Lerat, E. , Fablet, M. , Modolo, L. , Lopez‐Maestre, H. , & Vieira, C. (2017). TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Research, 45(4), e17 10.1093/nar/gkw953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat, E. , Rizzon, C. , & Biémont, C. (2003). Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Research, 13(8), 1889–1896. 10.1101/gr.827603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linheiro, R. S. , & Bergman, C. M. (2012). Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster . PLoS ONE, 7(2), 10.1371/journal.pone.0030008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, H. , Bergland, A. O. , Taylor, R. , Tilk, S. , Behrman, E. , Dyer, K. , … Petrov, D. A. (2018). Broad geographic sampling reveals predictable and pervasive seasonal adaptation in Drosophila. bioRxiv, 337543 10.1101/337543 [DOI] [Google Scholar]
- Mascher, M. , Gundlach, H. , Himmelbach, A. , Beier, S. , Twardziok, S. O. , Wicker, T. , … Stein, N. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nature, 544(7651), 427–433. 10.1038/nature22043 [DOI] [PubMed] [Google Scholar]
- Mateo, L. , Rech, G. E. , & González, J. (2018). Genome‐wide patterns of local adaptation in Drosophila melanogaster: Adding intra European variability to the map. Scientific Reports, 8, 16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo, L. , Ullastres, A. , & González, J. (2014). A transposable element insertion confers xenobiotic resistance in Drosophila. PLoS Genetics, 10(8), e1004560 10.1371/journal.pgen.1004560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills, R. E. , Bennett, E. A. , Iskow, R. C. , & Devine, S. E. (2007). Which transposable elements are active in the human genome? Trends in Genetics, 23(4), 183–191. 10.1016/j.tig.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Modolo, L. , & Lerat, E. (2014). Identification and analysis of transposable elements in genomic sequences In Poptsova M. (Ed.), Genome analysis: current procedures and applications (Vol. 9, pp. 165–181). Norfolk, UK: Caister Academic Press. [Google Scholar]
- Moran, P. A. P. (1950). Notes on Continuous Stochastic Phenomena. Biometrika, 37, 17 10.1093/biomet/37.1-2.17 [DOI] [PubMed] [Google Scholar]
- Morgante, M. , Depaoli, E. , & Radovic, S. (2007). Transposable elements and the plant pan‐genomes. Current Opinion in Plant Biology, 10(2), 149–155. 10.1016/j.pbi.2007.02.001 [DOI] [PubMed] [Google Scholar]
- Oakeshott, J. G. , Gibson, J. B. , Anderson, P. R. , Knibb, W. R. , Anderson, D. G. , & Chambers, G. K. (1982). Alcohol dehydrogenase and glycerol‐36‐phosphate dehydrogenase clines in Drosophila melanogaster on different continents. Evolution, 36(1), 86–96. 10.1111/j.1558-5646.1982.tb05013.x [DOI] [PubMed] [Google Scholar]
- Petrov, D. A. (2002). DNA loss and evolution of genome size in Drosophila. Genetica, 115(1), 81–91. 10.1023/A:101607621516 [DOI] [PubMed] [Google Scholar]
- Petrov, D. A. , Fiston‐Lavier, A.‐S. , Lipatov, M. , Lenkov, K. , González, J. , Gonzalez, J. , & González, J. (2011). Population genomics of transposable elements in Drosophila melanogaster . Molecular Biology and Evolution, 28(5), 1633–1644. 10.1093/molbev/msq337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool, J. E. , & Aquadro, C. F. (2007). The genetic basis of adaptive pigmentation variation in Drosophila melanogaster . Molecular Ecology, 16(14), 2844–2851. 10.1111/j.1365-294X.2007.03324.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesneville, H. , Bergman, C. M. , Andrieu, O. , Autard, D. , Nouaud, D. , Ashburner, M. , & Anxolabehere, D. (2005). Combined evidence annotation of transposable elements in genome sequences. PLoS Computational Biology, 1(2), 0166–0175. 10.1371/journal.pcbi.0010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). bedtools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A language and environment for statistical computing. Retrieved from https://www.r-project.org
- Rahman, R. , Chirn, G. , Kanodia, A. , Sytnikova, Y. A. , Brembs, B. , Bergman, C. M. , Lau, N. C. (2015). Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Research, 43(22), 10655–10672. 10.1093/nar/gkv1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner, V. A. , Zabanov, S. A. , Kolesnikova, O. V. , & Vasilyeva, L. A. (1992). Induction of the mobile genetic element Dm‐412 transpositions in the Drosophila genome by heat shock treatment. Proceedings of the National Academy of Sciences of the United States of America, 89(12), 5650–5654. 10.1073/pnas.89.12.5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rech, G. E. , Bogaerts-Marquez, M. , Barron, M. G. , Merenciano, M. , Villanueva-Canas, J. L. , Horvath, V. , … Gonzalez, J. (2018). Stress response, behavior, and development are shaped by transposable element-induced mutations in Drosophila. Retrieved from 10.1101/380618 [DOI] [PMC free article] [PubMed]
- Reinhardt, J. A. , Kolaczkowski, B. , Jones, C. D. , Begun, D. J. , & Kern, A. D. (2014). Parallel geographic variation in Drosophila melanogaster . Genetics, 197(1), 361–373. 10.1534/genetics.114.161463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishishwar, L. , Mariño‐Ramírez, L. , & Jordan, I. K. (2016). Benchmarking computational tools for polymorphic transposable element detection. Briefings in Bioinformatics, 18(6), 908–918. 10.1093/bib/bbw072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishishwar, L. , Tellez Villa, C. E. , & Jordan, I. K. (2015). Transposable element polymorphisms recapitulate human evolution. Mobile DNA, 6, 21 10.1186/s13100-015-0052-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, P. S. , Matzkin, L. , Ippolito, M. , & Eanes, W. F. (2005). Geographic variation in diapause incidence, life‐history traits, and climatic adaptation in Drosophila melanogaster . Evolution; International Journal of Organic Evolution, 59(8), 1721–1732. 10.1111/j.0014-3820.2005.tb01821.x [DOI] [PubMed] [Google Scholar]
- Schnable, P. S. , Ware, D. , Fulton, R. S. , Stein, J. C. , Wei, F. , Pasternak, S. , … Wilson, R. K. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science (New York, N.Y.), 326(5956), 1112–1115. 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Siddiq, M. A. , Loehlin, D. W. , Montooth, K. L. , & Thornton, J. W. (2017). Experimental test and refutation of a classic case of molecular adaptation in Drosophila melanogaster . Nature Ecology and Evolution, 1, 0025 10.1038/s41559-016-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A. , Hubley, R. , & Green, P. (2013. –2015). RepeatMasker Open‐4.0.
- Stuart, T. , Eichten, S. R. , Cahn, J. , Karpievitch, Y. V. , Borevitz, J. O. , & Lister, R. (2016). Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. eLife, 5, e20777 10.7554/eLife.20777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thioulouse, J. , Dray, S. , Dufour, A.‐B. , Siberchicot, A. , Jombart, T. , & Pavoine, S. (2018). Multivariate analysis of ecological data in ade4. New York, NY: Springer‐Verlag New York. [Google Scholar]
- Thomson, M. S. , Jacobson, J. W. , & Laurie, C. C. (1991). Comparison of alcohol dehydrogenase expression in Drosophila melanogaster and D. simulans . Molecular Biology and Evolution, 8(1), 31–48. 10.1093/oxfordjournals.molbev.a040630 [DOI] [PubMed] [Google Scholar]
- Vieira, C. , Aubry, P. , Lepetit, D. , & Biémont, C. (1998). A temperature cline in copy number for 412 but not roo/B104 retrotransposons in populations of Drosophila simulans . Proceedings. Biological Sciences, 265(1402), 1161–1165. 10.1098/rspb.1998.0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, C. , & Biémont, C. (2004). Transposable element dynamics in two sibling species: Drosophila melanogaster and Drosophila simulans . Genetica, 120(1–3), 115–123. 10.1023/B:GENE.0000017635.34955.b5 [DOI] [PubMed] [Google Scholar]
- Warnes, G. R. , Bolker, B. , Bonebakker, L. , Gentleman, R. , Liaw, W. H. A. , Lumley, T. , … Venables, B. (2015). gplots: Various R programming tools for plotting data. R package version 2.17.0. Retrieved from http://CRAN.R-project.org/package=gplots
- Zhang, S. , & Kelleher, E. S. (2017). Targeted identification of TE insertions in a Drosophila genome through hemi-specific PCR. Mobile DNA, 8(1), 10 10.1186/s13100-017-0092-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, J. , Wang, J. , Theurkauf, W. , & Weng, Z. (2014). TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Research, 42(11), 6826–6838. 10.1093/nar/gku323 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting Information File S1. Relative TE family abundance (in per cent of the total number of bp sampled) per sample ftp://pbil.univ-lyon1.fr/pub/divers/goubert/DrosoEU/TEbp_001pc
Supporting Information File S2. Averaged divergence between reads and TE family consensus sequence (K2P distance) per sample ftp://pbil.univ-lyon1.fr/pub/divers/goubert/DrosoEU/TEMdiv_001pc.