

Abstract
Neurofibromatosis type 1 (NF1) is a cancer predisposition syndrome with an incidence of 1:2,000. Patients with NF1 have an increased cancer risk and mortality, but there are no population‐based cohort studies specifically investigating the risk of childhood malignancies. We used the Finnish NF1 cohort to analyze the incidence, risk and prognosis of malignancies in NF1 patients <20 years of age. Persons born in 1987–2011 were included, and 524 persons were followed through the files of the Finnish Cancer Registry from birth up to age 20 years. This amounted to 8,376 person years. Fifty‐three patients had cancer <20 years of age, yielding a standardized incidence ratio (SIR) of 35.6. The most frequent location of pediatric cancers was the central nervous system (CNS); there were 45 cases and the SIR was 115.7. Exclusion of 22 optic pathway gliomas (OPGs) gave an SIR of 59.1 for the CNS and 21.6 for all cancers. There were nine malignant peripheral nerve sheath tumors (MPNSTs); their cumulative risk was 2.7% by age 20. No cases of leukemia were observed. NF1 patients showed considerable excess mortality with a standardized mortality ratio (SMR) of 73.1. The survival of NF1 patients with CNS tumors other than OPGs did not differ from that of non‐NF1 controls (HR 0.64, 95% CI 0.23 to 1.76). In conclusion, brain tumors in childhood and MPNSTs in adolescence are malignancies of major concern in patients with NF1. The risk for myeloid malignancies may not be as high as suggested in the literature.
Keywords: astrocytoma, glioma, leukemia, malignant peripheral nerve sheath tumor, plexiform neurofibroma
Short abstract
What's new?
Patients with neurofibromatosis type 1 (NF1) are known to have a high risk of various cancers. What has not been well‐studied, however, are the types of cancers that are most common among children with NF1, and how those cancers impact mortality. In this cohort study, the authors found that malignancies do cause increased mortality in patients under age 20. Brain tumors in childhood and malignant peripheral nerve sheath tumors (MPNSTs) in adolescence are of particular concern. On the other hand, the risk of myeloid malignancies may be lower than previously assumed.
Abbreviations
- CNS
central nervous system
- ICD‐10
International Statistical Classification of Diseases and Related Health Problems 10th Revision
- MPNST
malignant peripheral nerve sheath tumor
- NF1
neurofibromatosis type 1
- OPG
optic pathway glioma
- SIR
standardized incidence ratio
- SMR
standardized mortality ratio
Introduction
Neurofibromatosis type 1 (NF1; Mendelian Inheritance in Man, 162200) is a cancer predisposition syndrome with an incidence of 1:2,000 as reported in Finland,1 and a prevalence of 1:2,000–4,000.2, 3 The inheritance of NF1 follows an autosomal dominant trait, but in ~50% of the patients the syndrome is caused by a de novo mutation which may delay the diagnosis especially in children. The NF1 gene underlying the syndrome is located on chromosome 17 and encodes neurofibromin tumor suppressor protein. Neurofibromin functions as a Ras‐GTPase activating protein (RasGAP), and NF1 mutations lead to over‐activation of the Ras signaling pathway.4, 5
Patients with NF1 have a high risk for nervous system malignancies which are uncommon in the general population, such as malignant peripheral nerve sheath tumors (MPNSTs) and astrocytomas.6, 7 Brain and CNS tumors have been reported in approximately 20% of patients with NF1 and are usually detected in early childhood.8, 9, 10 Optic pathway gliomas (OPGs) account for about 70% of all CNS tumors in children with NF1 while the second most common brain tumor is brainstem glioma, representing about 17% of all CNS tumors.10, 11 OPG of NF1 patients is morphologically nearly always grade 1 pilocytic astrocytoma. The incidence of OPG in NF1 may be as high as 15–20%, and it frequently develops before the age of 7 years, but can present at any age.9 The course of NF1‐associated OPGs is more favorable than that of sporadic ones: in NF1 patients, OPG is most often asymptomatic and less than half require treatment.8, 12, 13 Similarly, the brain stem glioma has a more benign course in NF1 patients compared to the children without NF1.10 In addition to optic nerve and brain stem tumors, low‐grade pilocytic astrocytomas and high‐grade gliomas occur also in other locations of the CNS in NF1.11 However, the survival of the NF1 patients with non‐OPG brain tumors is poorly understood13 (for review see Ref. 14). The incidence and survival related to childhood brain tumors among patients with NF1 have not been previously studied in population‐based cohorts.
Most OPGs are recorded as benign tumors, D33 in the International classification of diseases (ICD‐10), and thus are not registered as cancers in all countries. Despite a possibly benign morphology, intracranial tumors can be coded as cancers with the topographic codes C70‐C72 in ICD‐10; this is the case, for example, in Finland and the United States. All indolent OPGs are not registered, but tumors which have been treated or biopsied are more likely to be covered by the Finnish Cancer Registry.15 Thus, the present study does not attempt to report incidence, survival or mortality associated with OPGs in the study population.
MPNST is an aggressive tumor which accounts for 38–45% of the cancer deaths of all NF1 patients.6, 16 MPNST in the general population is a rare tumor of the elderly, while the highest risk of MPNST in NF1 coincides with young adulthood.6, 17 MPNST is one of the hallmark complications of NF1 and arises often from a congenital plexiform neurofibroma. Patients with NF1 microdeletion, i.e., a large deletion of the NF1 gene and its flanking regions, are especially susceptible to MPNSTs.18 Treatment of MPNST in children with NF1 has not been systematically studied, but the mainstay of treatment is radical operation while the role of adjuvant therapy is still unclear. NF1 is an unfavorable prognostic factor of MPNST.19 Our knowledge on MPNST in children with NF1 comes mainly from case series studies,20, 21, 22, 23 but an MPNST risk of 0.5% for NF1 patients under 20 years of age has been estimated in one population‐based cohort.24
Children with NF1 are frequently mentioned in the literature as having an increased risk of malignant myeloid disorders, particularly juvenile myelomonocytic leukemia.25 The NF1 gene functions as a tumor suppressor in early myelopoiesis and this provides the basis for this connection.26 However, good quality population‐based epidemiologic data on the incidence of myeloid disorders associated with NF1 are sparse. Also, pheochromocytoma is one of the tumors typically associated with NF16 and it has been described in a 16‐year‐old teenager with NF127 but there are no reports on its incidence in adolescence.
The current knowledge of the epidemiology of NF1‐related malignancies in children relies mainly on hospital register studies, while population‐based studies on the incidence, survival and mortality are lacking. Our recent population‐based study on NF1 and cancer6 described the cancer risk in NF1 in various age groups and showed a highly increased cancer incidence and mortality in childhood. The purpose of the present study is to analyze the risks of various malignancies in childhood in more detail.
Data and statistical methods
The study adhered to the Declaration of Helsinki principles and was approved by the Ethics Committee of the Hospital District of Southwest Finland, the Ministry of Social Affairs and Health and the National Institute for Health and Welfare.
The Finnish NF1 Registry is population‐based and encompasses all NF1 patients who have visited the 5 university and 15 central hospitals in Finland during the ascertainment period of 1987–2011.1 Currently, the cohort includes a total of 1,476 patients (770 females, 706 males) whose NF1 diagnosis has been verified from patient records according to the diagnostic criteria of the NIH. The NF1 registry was cross‐linked with national registries using the personal identity code as a key. Dates of death and emigration were obtained from the Population Register Center of Finland (https://vrk.fi/en). The Finnish Cancer Registry (https://cancerregistry.fi) was searched for cancer diagnoses of the NF1 patients. Population reference rates of cancer incidence and mortality were generated by the Cancer Registry.
All intracranial tumors can be registered as cancers with ICD‐10 topographic codes C70‐C72, regardless of morphology. However, underreporting of tumors which are not histologically verified, e.g., non‐malignant tumors of the central nervous system has been noted in the Finnish Cancer Registry. Specifically, 78.9% of all benign brain tumors were registered in the Finnish Cancer Registry.15 OPGs are thus not comprehensively covered, but tumors that have been biopsied or treated are more likely to be included. Especially in the older cases, biopsy does not necessarily imply that the tumor has been symptomatic. Because the coding of intracranial tumors is not always fully accurate due to a lack of biopsy or surgery, the medical records of the NF1 patients with tumors of the brain and CNS were manually reviewed in order to identify OPGs. All analyses were carried out with and without OPGs.
Cancer incidence and risk
For the calculation of the standardized incidence ratio (SIR) and cumulative risk, the NF1 patients born during the ascertainment period 1987–2011 were followed up from birth to death, emigration, 20th birthday or the end of follow‐up on December 31st, 2014, whichever occurred first. The follow‐up was started at birth in order to cover the entire spectrum of childhood cancer in NF1. Because the diagnoses of the NF1 children born during the ascertainment period have been comprehensively established,1, 3 restricting the analysis to only those born 1987–2011 should reduce the risk of missing patients. However, it is possible that we have missed some patients whose NF1 has not been diagnosed by the end of the ascertainment period because of mild symptoms. Since children with cancer or other life‐threatening condition are thoroughly examined in hospitals, we believe that undiagnosed NF1 is highly unlikely even among those who die of cancer during their first years of life.
The SIR was calculated as the ratio of observed to expected cases, where the expected cases were obtained by multiplying the person‐years observed in the NF1 cohort with the corresponding population rate of cancer incidence, stratified by age, calendar‐period and gender. The 95% confidence intervals (CI) were computed by assuming that the number of observed cases followed the Poisson distribution. The homogeneity of the SIRs between the genders was tested using the standard likelihood ratio test of the Poisson regression model. The cumulative risk of cancer by age 20 years was calculated with death as a competing risk.
Mortality and survival
In order to avoid immortality bias when assessing the standardized mortality ratio (SMR), the follow‐up of the NF1 patients started at the first NF1‐related hospital visit 1987–2011, i.e., the hospital visit that led to the inclusion of the patient into the cohort. All NF1 patients below the age of 20 years at cohort entry were included in the analysis, irrespective of their year of birth. The follow‐up ended at death, emigration, 20th birthday or the end of follow‐up on December 31st, 2014, whichever occurred first. Only deaths due to cancer were considered as events for the SMR analysis. The SMR for cancer was calculated as the ratio of observed to expected number of deaths using the general population rates matched for age, calendar‐period and gender as a reference. In the SMR analysis related to tumors of the brain and CNS, an event was defined as a cancer‐related death of a patient with a tumor of the brain and CNS. The 95% CI of SMR was obtained using the Poisson distribution, as for SIR.
The survival of patients with NF1 and tumors of the brain and CNS was compared to the survival of matched controls. The controls were patients with tumors of the brain and CNS from the Finnish Cancer Registry matched for gender, age at diagnosis (within 5 years) and diagnosis year (within 5 years). All available controls without NF1 were included. The follow‐up started at the time of tumor diagnosis, when the patient was <20 years of age and ended at death or censoring due to emigration or the end of follow‐up on December 31st, 2014. Cumulative, cancer‐specific survival proportions with 95% CI were calculated using the weighted Kaplan–Meier method in which matched controls of each NF1 patient were weighted using the inverse of the size of the NF1 patient's control group. The matched Cox regression model28 was used to compare the groups. Statistical analyses were conducted with the statistical software R, version 3.4.2 (https://www.r-project.org/) and the popEpi package, version 0.4.4.
Results
Childhood cancer incidence and risk
A total of 524 NF1 patients (238 females, 286 males) were born during the ascertainment period (1987–2011), yielding 8,376 person‐years of follow‐up. The cancer registry linkage revealed 56 tumors diagnosed in 53 patients (31 females, 22 males) aged less than 20 years (Fig. 1). The median age at cancer diagnosis of females was 5.8 years (range 0.67 to 18) and of males 9.3 years (range 2.5 to 19) (p = 0.23). The childhood cancer incidence among NF1 patients was significantly higher than in the general population of similar age and calendar‐period with an SIR of 35.6 (95% CI 27.1 to 45.8) (Table 1). The cancer SIR of females was significantly higher than of males (p = 0.012). The cumulative risk of a patient with NF1 being diagnosed with cancer or an intracranial tumor was 12.1% (95% CI 8.9% to 15.2%) by age 20 years – 9.4% (95% CI 5.4% to 13.1%) among males and 15.3% (95% CI 10.0% to 20.3%) among females. The SIRs stratified by tumor location are shown in Table 2.
Figure 1.
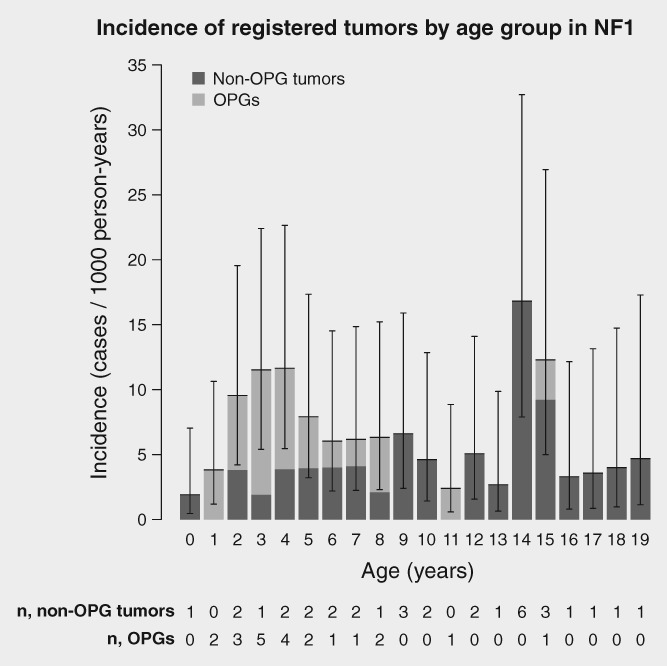
Incidence of tumors registered in the Finnish Cancer Registry by age at diagnosis among patients with neurofibromatosis type 1 (NF1). Total tumor incidence and the proportion of tumors other than optic pathway gliomas (OPGs) are shown. Whiskers show 95% confidence intervals of the overall incidence, computed with the statistical software R package epiR, version 0.9–96. The numbers of tumors diagnosed by patient age are shown under the corresponding bar.
Table 1.
Observed and expected number of cancers and standardized incidence ratios (SIR) among NF1 patients aged <20 years by gender and tumor type
| All tumors | Non‐OPG tumors | |||||||
|---|---|---|---|---|---|---|---|---|
| Population | Expected | Person‐years | Observed | SIR | 95% CI | Observed | SIR | 95% CI |
| Total | 1.57 | 8,376 | 56 | 35.6 | 27.1 to 45.8 | 34 | 21.6 | 15.1 to 29.7 |
| Males | 0.91 | 4,610 | 23 | 25.3 | 16.3 to 37.1 | 15 | 16.5 | 9.5 to 26.3 |
| Females | 0.66 | 3,766 | 33 | 49.7 | 34.6 to 68.6 | 19 | 28.6 | 17.6 to 43.5 |
p Value for comparison between genders for all tumors p = 0.012. OPG: optic pathway glioma; CI: confidence interval
Table 2.
Observed and expected numbers of cancer cases and standardized incidence ratios (SIR) with 95% confidence intervals (CI) among Finnish NF1 patients aged <20 years by cancer location
| Location | ICD‐10 codes | Expected | Observed | SIR | 95% CI |
|---|---|---|---|---|---|
| Brain, CNS | C70‐C72 | 0.39 | 45 | 115.7 | 86.4 to 155.0 |
| non‐OPG tumors in brain, CNS | C70‐C72 | 0.39 | 23 | 59.1 | 39.3 to 89.0 |
| Peripheral nerves and autonomic nervous system | C47 | 0.03 | 8 | 271.7 | 135.9 to 543.2 |
| Soft tissues | C48‐C49 | 0.06 | 2 | 33.8 | 8.5 to 135.3 |
| Bone | C40‐C41 | 0.04 | 1 | 24.7 | 3.5 to 175.6 |
Abbreviations: CNS, central nervous system; ICD‐10, International classification of diseases, 10th edition; OPG, optic pathway glioma.
Out of the 56 tumors, 22 were OPGs. After exclusion of OPGs, the remaining 34 tumors (non‐OPG tumors) yielded an SIR of 21.6 (95% CI 15.1 to 29.7) (Table 1), and the gender difference was no longer significant (p = 0.11). The risk of a patient with NF1 being diagnosed with a non‐OPG tumor was 8.1% (95% CI 5.3% to 10.8%) by age 20 years.
Cancer mortality
Among the 741 NF1 patients (351 females, 390 males) who entered the cohort before age 20 years, 21 deaths occurred under 20 years of age. There were 7,742 person‐years of follow‐up (3,444 among females and 4,298 among males). Seventeen of the 21 deaths were due to cancer, highlighting the significantly higher cancer mortality compared to the general population: the SMR for cancer death was 73.1 (95% CI 42.6 to 117.1). There was no statistically significant difference between the genders (females: 9 deaths, SMR 94.0, 95% CI 45.2 to 169.4; males: 8 deaths, SMR 58.5, 95% CI 26.7 to 108.9; p = 0.33). The risk of death from tumor by age 20 was 4.0% (95% CI 2.5% to 6.6%).
CNS tumors
The most common location of NF1‐associated malignant tumors, according to the Finnish Cancer Registry, was the CNS (ICD‐10 C70‐C72) with 45 tumors out of a total of 56 tumors. The median age of the patients at the time of diagnosis of OPGs was 4.5 years (range 1.3 to 15), and of the 22 patients with OPG, 14 were females and 8 were males. The median age at the time of diagnosis of other tumors of the brain and CNS was 9.3 years (range 2.5 to 18) in 12 females and 11 males. While the overall SIR of tumors of the brain and CNS was 115.7 (95% CI 86.4 to 155.0), the SIR value sank after exclusion of the OPGs to 59.1 (95% CI 39.3 to 89.0) (Table 2).
The detailed locations and grades of the CNS tumors are presented in Table 3. Most of the tumors (11) were pilocytic astrocytomas. One high‐grade (WHO grade III‐IV) glioma was observed during follow‐up. In addition, three high‐grade gliomas in patients aged <20 years were known to have occurred outside the follow‐up time. The locations of the CNS tumors are shown in Figure 2. In addition to gliomas, five neurofibromas, plexiform neurofibromas or neurilemomas, and one MPNST presenting in the spinal cord were located in the CNS. When also the tumors located in the spinal cord were excluded and the analysis was limited to the non‐OPG tumors of the brain only, the SIR remained high (SIR 49.9, 95% CI 29.8 to 77.6).
Table 3.
Localization of central nervous system tumors in patients with NF1 aged <20 years by tumor grade
| Localization | n (total 45) | % | Benign | Grade I | Grade II | High‐grade | Grade not known |
|---|---|---|---|---|---|---|---|
| Optic pathway | 22 | 48.9 | 13 | 3 | 6 | ||
| Brainstem | 4 | 8.9 | 4 | ||||
| Cerebral lobes | 6 | 13.3 | 5 | 1 | |||
| Cerebellum | 2 | 4.4 | 2 | ||||
| Midbrain | 6 | 13.3 | 3 | 3 | |||
| Spinal cord | 5 | 11.1 | 4 | 11 |
MPNST in the spinal cord.
Figure 2.
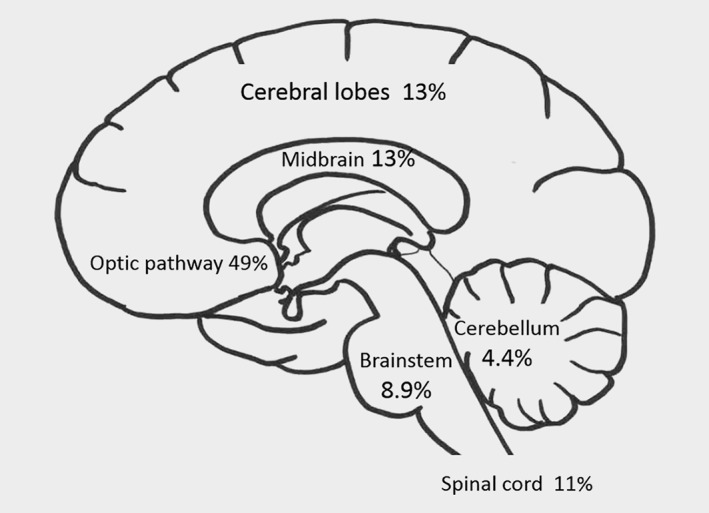
Localization of brain tumors in the study. Numbers of cases in each location are shown in Table 3.
Mortality related to non‐OPG tumors of the CNS at ages below 20 years was significantly higher than in the general population, with 5 deaths and an SMR of 72.1 (95% CI 23.4 to 168.2). Four of those who died were male (SMR 102.1, 95% CI 31.7 to 237.0) and one was female (SMR 33.1, 95% CI 1.9 to 145.8) (p = 0.27). The median age at death was 14 years (range 4.0 to 19). Survival of NF1 patients diagnosed with non‐OPG tumors of the CNS at age <20 years was compared to the survival of patients with matched tumors in the general population. Neither the overall nor the cancer‐specific 5‐year survival of NF1 patients with CNS tumors differed from the general population (hazard ratio 0.64 for overall survival, 95% CI 0.23 to 1.76, p = 0.39).
Malignancies of the peripheral nervous system
MPNSTs (ICD‐O‐3 codes 9540/3 and 9560/3) were detected in 9 patients: 5 females and 4 males, whose median age at diagnosis was 15 years (range 12 to 19). The cases were scattered among the ICD‐10 categories of peripheral nerves and autonomic nervous system (C47), soft tissues (C48‐C49) and CNS (C70‐C72) (Table 2). An MPNST‐specific SIR could not be computed because MPNST is extremely rare in the general population among subjects aged less than 20 years, and thus there was not a sufficient number of controls. The cumulative risk of an MPNST by age 20 years was 2.7% (95% CI 1.4% to 5.3%). Out of the 9 patients who had MPNST, 4 died during the follow‐up.
Other malignancies
One case of adamantinoma of the tibia, one rhabdomyosarcoma and one neuroblastoma were diagnosed. Leukemias, pheochromocytomas and gastrointestinal stromal tumors (GISTs) are often mentioned as being associated with NF1, but none of these were seen in the patients of our study.
Discussion
Malignancies in pediatric patients with NF1 have been reported in case series from specialized referral centers, but cohort studies with data on population‐based incidences of NF1 associated malignancies in childhood and adolescence have been sparse.13, 16, 29 The present study compared the total population‐based Finnish NF1 cohort with general population data collected by the nationwide Finnish Cancer Registry. We believe that the resulting estimates can be generalized to other populations, since the Finnish population is very similar to that of other western countries.
We have previously shown that the cancer risk of patients with NF1 is elevated (SIR 5.03).6 The present study focused on the population aged <20 years and showed that the SIR for tumors registered in the Finnish Cancer Registry is 35.6. Together, our previous study6 and the present study show that the relative risk of cancer among patients with NF1 is the highest in childhood and adolescence, and that children and adolescents with NF1 have a substantial risk of also other tumors than OPG. The present study also suggests a higher cancer SIR among females than males, which is partly due to the seemingly higher incidence of symptomatic OPGs in females. This is in accordance with a previous study showing that females have more symptomatic OPGs, and females with NF1 and OPG are more likely than males to experience visual decline requiring treatment.30, 31 However, gender differences observed in our study and in previous pediatric cohorts may be compromised by sampling bias. Our recent study suggests that girls may be diagnosed with NF1 later than boys.3 Thus, girls with mild NF1 symptoms may more likely than boys be missing from pediatric cohorts. There were fewer females than males also in our study, suggesting that some females with mild symptoms may be missing from the cohort.
The results of our study show that the cancer‐related mortality is high (SMR for cancer death 73.1). Cancers were clearly the most frequent cause of death, since 17 out of the 21 deaths of NF1 patients aged <20 years were due to cancer. In comparison, accidents are the leading cause of death among children and adolescents in general Finnish population, cancer being only the fourth most common cause (http://www.stat.fi/til/ksyyt/index_en.html). There was no significant gender‐related difference in mortality in the NF1 cohort <20 years of age.
The vast majority, 45 of 56 of the registered tumors, were located in the CNS, OPG being the most common type. Even without the OPGs, the incidence of CNS tumors was 59.1‐fold the incidence in the corresponding general population. Because OPGs represent the majority of brain tumors in NF1, it is not surprising that the main focus of previous studies on childhood CNS tumors has been OPG. OPGs have also been included in most of the case series covering all CNS tumors. In our study, OPGs were diagnosed at younger age than non‐OPG tumors of the brain and CNS. The median age at the time of diagnosis of OPGs was 4.5 years, and of non‐OPG tumors 9.3 years (p = 0.001). This agrees with a previous study on MRI findings, which reported that the number of NF1 patients diagnosed with extra‐optic glioma was highest in the age group 10–19 years.32
The results of our study show that 49% of CNS tumors were OPGs and the remaining 51% were evenly distributed to various locations in the CNS. Only 8.9% were brainstem gliomas; previous studies have reported somewhat higher numbers of brainstem gliomas (17–18%).10, 11 This may be due to differences in the inclusion of patients in the present and previous studies, since patients with non‐symptomatic brainstem gliomas may not have been ascertained in our study. Grade I pilocytic astrocytomas represented the most frequent morphology of the tumors (Table 3), and only two high‐grade tumors of the CNS were detected in the cohort. However, outside the ascertainment and follow‐up years of this study, there were three high‐grade (WHO grade III–IV) gliomas in patients aged <20 years. High‐grade gliomas do occur in NF1.22, 33 It is possible that high‐grade tumors are more frequent in adult than pediatric NF1 patients. Glioblastomas are typically associated with constitutional mismatch repair deficiency (CMMRD), which has overlapping clinical findings with NF1. Specifically, multiple café au lait macules have been reported in more than 60% and skinfold freckling in more than 10% of patients with CMMRD.34 In our cohort, the patients with high‐grade gliomas were not suspected of CMMRD because, in addition to café au lait macules, they had neurofibromas and skinfold freckling.
The present study shows that non‐OPG tumors of the CNS are associated with a high SMR (72.1). Previous studies have suggested that gliomas in NF1 patients are associated with better patient survival than gliomas diagnosed in patients without NF1. In these studies, however, patients with OPGs have been included.13, 35, 36 In our study, the survival of patients with non‐OPG tumors of the brain and CNS does not significantly differ from the survival of matched non‐NF1 controls. Inclusion of OPGs, which are associated with a better prognosis of NF1 patients than non‐NF1 patients,13, 37 may have led to an apparently more favorable outcome reported in previous studies.
Our cohort included three patients with OPG and another CNS malignancy: one meningioma, one MPNST eight years after radiation treatment of a symptomatic OPG (outside the follow‐up of our study), and one grade II astrocytoma of the corpus callosum. Unlike OPG patients without NF1, patients with NF1 are also at risk of developing a second CNS tumor in addition to OPG.13
MPNST is an aggressive malignancy specifically associated with NF1. The mortality caused by MPNST in the NF1 population is over 2,000‐fold compared to the general population.6 Case reports and patient series show that MPNSTs may develop already during adolescence, but the risk of MPNST in childhood has not been established.16, 17, 22, 38 The present study identified nine MPNSTs in patients aged less than 20 years. The median age at diagnosis of these patients was 15 years (range 12 to 19). The cumulative risk of having an MPNST by age 20 years was 2.7%, and four MPNSTs were fatal during the follow‐up. Thus, MPNST is not a rarity in young patients with NF1 – it constitutes a marked threat to NF1 patients already during their teenage years. Pheochromocytomas and GISTs are associated with NF1, but based on our study, they are not common in children or teenagers with NF1. However, pheochromocytomas and GISTs may not be comprehensively covered by the Finnish Cancer Registry which hampers our ability to draw conclusions on incidences.
NF1 is claimed to predispose to leukemias.25, 39 Juvenile myelomonocytic leukemia (JMML) is often included in the list of frequent malignancies associated with NF1. Bader and Miller reported originally in 197839 that the ratio of acute lymphocytic leukemia to nonlymphocytic leukemia was 9:20 in children with neurofibromatosis, which is markedly different from the 4:1 ratio in children without neurofibromatosis. This was before the present classification of leukemias in ICD‐O‐2 and ICD‐O‐3. JMML (9946/3 in ICD‐O‐3) is a rare myelodysplastic syndrome of childhood, and 14% of patients with JMML have NF1.40 Furthermore, 9% of all patients with chronic myelomonocytic leukemia (CMML), collected from the network of registers in Great Britain, had NF1.41 Based on these numbers, NF1 patients have been estimated to have a 200‐fold increase in the incidence of CMML compared to the general population.41
Inactivation of both NF1 alleles has been demonstrated in the bone marrow cells of NF1 patients with hematologic malignancies, suggesting a pathogenic role of NF1 in these malignancies.25 However, JMML is a rare leukemia in the general population as well as in NF1. Assuming an overall annual incidence of JMML of 1.2 per million children aged 0–11 years and a 200‐fold increase in the incidence among patients with NF1, 1.4 cases would have been expected to surface in our study, but no leukemias were observed. Thus, we cannot confirm the increased risk for leukemia nor JMML in NF1. There was not a single case of CMML neither in the entire Finnish NF1 cohort6 nor in the large population‐based study from Great Britain7while two fatal cases of JMML were reported in a UK population of 1,186 NF1 patients.16 Thus, although NF1 patients represent an unusually large percentage of patients with JMML, leukemia is not a frequent complication of NF1.
To conclude, brain tumors are the frontline cancers to develop in children with NF1, and MPNSTs are a major concern in adolescence. The risk for myeloid malignancies may not be as marked as previously suggested in the literature.
References
- 1. Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 2015;135:904–6. [DOI] [PubMed] [Google Scholar]
- 2. Huson SM, Compston DA, Clark P, et al. A genetic study of von Recklinghausen neurofibromatosis in south East Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet 1989;26:704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kallionpää RA, Uusitalo E, Leppävirta J, et al. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med 2018;20:1082–1086. [DOI] [PubMed] [Google Scholar]
- 4. Jouhilahti E‐M, Peltonen S, Heape AM, et al. The pathoetiology of neurofibromatosis 1. Am J Pathol 2011;178:1932–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gutmann DH, Ferner RE, Listernick RH, et al. Neurofibromatosis type 1. Nat Rev Dis Prim 2017;3:17004. [DOI] [PubMed] [Google Scholar]
- 6. Uusitalo E, Rantanen M, Kallionpää RA, et al. Distinctive cancer associations in patients with Neurofibromatosis type 1. J Clin Oncol 2016;34:1978–86. [DOI] [PubMed] [Google Scholar]
- 7. Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system and of malignant neoplasms, in people with neurofibromatosis: population‐based record‐linkage study. Br J Cancer 2013;108:193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Listernick R, Charrow J, Greenwald M, et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr 1994;125:63–6. [DOI] [PubMed] [Google Scholar]
- 9. Listernick R, Ferner RE, Liu GT, et al. Optic pathway gliomas in neurofibromatosis‐1: controversies and recommendations. Ann Neurol 2007;61:189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mahdi J, Shah AC, Sato A, et al. A multi‐institutional study of brainstem gliomas in children with neurofibromatosis type 1. Neurology 2017;88:1584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guillamo J‐S, Creange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain 2003;126:152–60. [DOI] [PubMed] [Google Scholar]
- 12. Friedrich RE, Nuding MA. Optic pathway Glioma and cerebral focal abnormal signal intensity in patients with Neurofibromatosis type 1: characteristics, treatment choices and follow‐up in 134 affected individuals and a brief review of the literature. Anticancer Res 2016;36:4095–121. [PubMed] [Google Scholar]
- 13. Singhal S, Birch JM, Kerr B, et al. Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child 2002;87:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Helfferich J, Nijmeijer R, Brouwer OF, et al. Neurofibromatosis type 1 associated low grade gliomas: a comparison with sporadic low grade gliomas. Crit Rev Oncol Hematol 2016;104:30–41. [DOI] [PubMed] [Google Scholar]
- 15. Leinonen MK, Miettinen J, Heikkinen S, et al. Quality measures of the population‐based Finnish cancer registry indicate sound data quality for solid malignant tumours. Eur J Cancer 2017;77:31–9. [DOI] [PubMed] [Google Scholar]
- 16. Evans DGR, O'Hara C, Wilding A, et al. Mortality in neurofibromatosis 1: in north West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet 2011;19:1187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans DGR, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39:311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Raedt T, Brems H, Wolkenstein P, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 2003;72:1288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrari A, Bisogno G, Macaluso A, et al. Soft‐tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 2007;109:1406–12. [DOI] [PubMed] [Google Scholar]
- 20. Friedrich RE, Hartmann M, Mautner VF. Malignant peripheral nerve sheath tumors (MPNST) in NF1‐affected children. Anticancer Res 2007;27:1957–60. [PubMed] [Google Scholar]
- 21. Demir HA, Varan A, Yalçn B, et al. Malignant peripheral nerve sheath tumors in childhood: 13 cases from a single center. J Pediatr Hematol Oncol 2012;34:204–7. [DOI] [PubMed] [Google Scholar]
- 22. Varan A, Şen H, Aydın B, et al. Neurofibromatosis type 1 and malignancy in childhood. Clin Genet 2016;89:341–5. [DOI] [PubMed] [Google Scholar]
- 23. Vasconcelos RAT, Coscarelli PG, Alvarenga RP, et al. Malignant peripheral nerve sheath tumor with and without neurofibromatosis type 1. Arq Neuropsiquiatr 2017;75:366–71. [DOI] [PubMed] [Google Scholar]
- 24. Evans DGR, Salvador H, Chang VY, et al. Cancer and central nervous system tumor surveillance in pediatric Neurofibromatosis 1. Clin Cancer Res 2017;23:e46–53. [DOI] [PubMed] [Google Scholar]
- 25. Side L, Taylor B, Cayouette M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with Neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med 1997;336:1713–20. [DOI] [PubMed] [Google Scholar]
- 26. Miles DK, Freedman MH, Stephens K, et al. Patterns of hematopoietic lineage involvement in children with neurofibromatosis type 1 and malignant myeloid disorders. Blood 1996;88:4314–20. [PubMed] [Google Scholar]
- 27. Giovannoni I, Callea F, Boldrini R, et al. Malignant Pheochromocytoma in a 16‐year‐old patient with Neurofibromatosis type 1. Pediatr Dev Pathol 2014;17:126–9. [DOI] [PubMed] [Google Scholar]
- 28. Cox D. Regression models and life‐tables. JR Stat Soc B 1972;34:187–220. [Google Scholar]
- 29. McGaughran JM, Harris DI, Donnai D, et al. Evans DG. A clinical study of type 1 neurofibromatosis in north West England. J Med Genet 1999;36:197–203. [PMC free article] [PubMed] [Google Scholar]
- 30. Diggs‐Andrews KA, Brown JA, Gianino SM, et al. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 2014;75:309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fisher MJ, Loguidice M, Gutmann DH, et al. Gender as a disease modifier in neurofibromatosis type 1 optic pathway glioma. Ann Neurol 2014;75:799–800. [DOI] [PubMed] [Google Scholar]
- 32. Sellmer L, Farschtschi S, Marangoni M, et al. Non‐optic glioma in adults and children with neurofibromatosis 1. Orphanet J Rare Dis 2017;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huttner AJ, Kieran MW, Yao X, et al. Clinicopathologic study of glioblastoma in children with neurofibromatosis type 1. Pediatr Blood Cancer 2010;54:890–6. [DOI] [PubMed] [Google Scholar]
- 34. Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet 2017;91:507–19. [DOI] [PubMed] [Google Scholar]
- 35. Pollack IF, Mulvihill JJ. Special issues in the management of gliomas in children with neurofibromatosis 1. J Neurooncol 1996;28:257–68. [DOI] [PubMed] [Google Scholar]
- 36. Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol 2008;67:240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deliganis AV, Geyer JR, Berger MS. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen disease) in childhood optic glioma. Neurosurgery 1996;38:1114–8. [DOI] [PubMed] [Google Scholar]
- 38. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res 2002;62:1573–7. [PubMed] [Google Scholar]
- 39. Bader JL, Miller RW. Neurofibromatosis and childhood leukemia. J Pediatr 1978;92:925–9. [DOI] [PubMed] [Google Scholar]
- 40. Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European working group on Myelodysplastic syndromes in childhood (EWOG‐MDS). Blood 1997;89:3534–43. [PubMed] [Google Scholar]
- 41. Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population‐based UKCCSG study. Br J Cancer 1994;70:969–72. [DOI] [PMC free article] [PubMed] [Google Scholar]