

Summary
The succinate dehydrogenase (SDH) enzyme complex functions as a key enzyme coupling the oxidation of succinate to fumarate in the citric acid cycle. Inactivation of this enzyme complex results in the cellular accumulation of the oncometabolite succinate, which is postulated to be a key driver in tumorigenesis. Succinate accumulation inhibits 2‐oxoglutarate‐dependent dioxygenases, including DNA and histone demethylase enzymes and hypoxic gene response regulators. Biallelic inactivation (typically resulting from one inherited and one somatic event) at one of the four genes encoding the SDH complex (SDHA/B/C/D) is the most common cause for SDH deficient (dSDH) tumours. Germline mutations in the SDHx genes predispose to a spectrum of tumours including phaeochromocytoma and paraganglioma (PPGL), wild type gastrointestinal stromal tumours (wtGIST) and, less commonly, renal cell carcinoma and pituitary tumours. Furthermore, mutations in the SDHx genes, particularly SDHB, predispose to a higher risk of malignant PPGL, which is associated with a 5‐year mortality of 50%. There is general agreement that biochemical and imaging surveillance should be offered to asymptomatic carriers of SDHx gene mutations in the expectation that this will reduce the morbidity and mortality associated with dSDH tumours. However, there is no consensus on when and how surveillance should be performed in children and young adults. Here, we address the question: “What age should clinical, biochemical and radiological surveillance for PPGL be initiated in paediatric SDHx mutation carriers?”.
1. INTRODUCTION
Prior to the turn of this century, it was commonly considered that 10% of phaeochromocytomas and paragangliomas (PPGLs) were familial. However the recent molecular revolution has brought with it an understanding that PPGL have a rich hereditary background, as 30% of PPGL are now known to be familial.1 Germline mutations in the SDHx genes account for 30‐40% of hereditary PPGL cases and mutations in the SDHB gene in particular predict a higher risk of malignant potential.2 Although there is is general agreement that biochemical and imaging surveillance should be offered to asymptomatic carriers of SDHx gene mutations in the expectation that this will reduce the morbidity and mortality associated with SDH deficient tumours,3 there is at present no consensus on when and how surveillance should be performed in children and young adults. Here, we address the question: "What age should clinical, biochemical and radiological surveillance for PPGL be initiated in paediatric SDHx mutation carriers?".
2. METHODS
In order to address this important clinical question, a thorough review of the literature was performed. MEDLINE was searched via PubMed using the following search terms; (a) SDH or succinate dehydrogenase and (b) child or children or paediatric or adolescent and (c) tumour or cancer or paraganglioma or phaeochromocytoma or PPGL or GIST. This search yielded 413 results, of which 43 papers were relevant. An additional five papers were identified through the initial 43 manuscripts. Papers were included in this review if they contained paediatric index cases, detailed phenotypes including age of presentation, and in the case of having both index and nonindex or asymptomatic carriers, and a clear discrimination between the two was required for inclusion in this literature review. A second search was performed to identify the literature addressing the issue of surveillance in paediatric SDHx carriers and the term “surveillance” was added to the above search terms. This search yielded 275 results, of which 11 were relevant.
2.1. What is the prevalence of related disease in childhood SDHx carriers?
Whilst there are several reports of tumour development in paediatric SDHx mutation carriers, the prevalence of disease among paediatric nonprobands remains low. A review of the literature identified 105 paediatric (<18 years of age) index cases for which detailed data on the phenotype were available. A review of the genotype information demonstrated that 77 (73.3%) patients had a germline pathogenic SDHB variant, 25 (23.8%) cases a pathogenic SDHD variant and three cases (2.9%) a pathogenic SDHC variant (Figure 1). The variant type was available for 93 patients and included 20 (21.6%) nonsense variants, 28 (30.1%) missense variants, 13 (13.9%) splice site variants, 26 (27.9%) frameshift variants. A copy number variant was identified in 6 (6.4%) cases (one gene duplication, five exonic deletions and one whole gene deletion). Notably, the frequency of reported truncating variants (nonsense/frameshift and splice site variants) identified in this paediatric population was 72% compared to a reported frequency of 52% in an adult population presenting with PPGL.1
Figure 1.
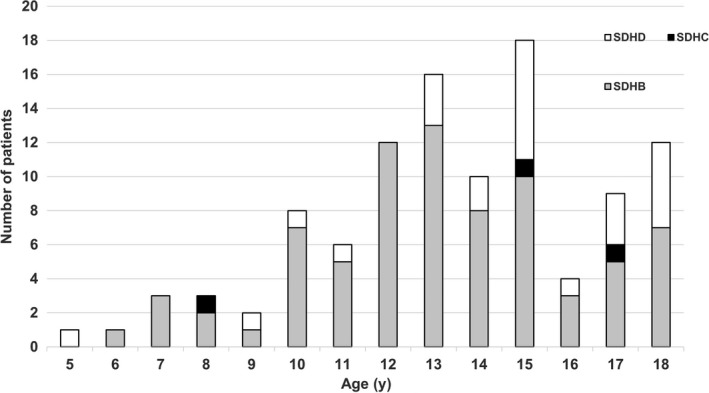
Illustrates the individual succinate dehydrogenase gene subunit variant carried by the index cases and the age of presentation with a phaeochromocytoma and paraganglioma (PPGL)
The mean age of this cohort was 13.5 years (range 5‐18 years). There was no significant difference in the mean age at presentation with PPGL in those patients with germline SDHB variants (13.1 years) compared to those with SDHC (13.3 years) or SDHD variants (14.6 years) (P = 0.78), and there was no significant difference in the mean age at presentation of those patients presenting with malignant tumours vs those presenting with localized tumours (12.8 vs 13.4 years, P = 0.9). Over a third of identified cases (34.3%) had multiple synchronous or metachronous PPGL (Table 1 and Table S1 in supplementary data). An extra‐adrenal paraganglioma was reported in 65 (61.9%), 31 (29.5%) patients presented with a phaeochromocytoma, 8 (7.6%) patients presented with both tumour types, and for one case the exact tumour type was not stated. The secretory status of the tumour was reported for 73 patients: 63 (86.3%) patients with biochemically functioning tumours and 10 (13.7%) cases had a nonsecretory tumour. Four patients in this cohort also developed a non‐PPGL tumour including one renal cell carcinoma, one renal oncocytoma, one nephrogenic adenoma and one pituitary tumour (Table 1 and Table S1). Malignant tumours were reported in 26 patients (24.8%), and the majority of malignant cases were identified in patients with a pathogenic SDHB germline variant (22 patients, 84.6%). Of note, this is a lower figure than what King et al4 found in their study of metastatic PPGL related to primary tumour development in childhood and adolescence, where 85.2% (n = 23) of their paediatric and adolescent patients with SDHB mutations developed metastatic disease. This review of the literature highlights not only the importance of identifying a germline mutation in children presenting with PPGL, but also the possibility of SDHx mutation penetrance at a young age. However, an appropriate surveillance protocol must balance the occurrence of paediatric tumour development against the increasing evidence that the penetrance of SDHB is lower than originally reported and the potential adverse effects of screening.
Table 1.
Summary of paediatric index cases with a succinate dehydrogenase gene subunit (SDHx) mutation reported in literature (data based on references5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52)
| SDHx subunit gene | |
| SDHB | 77 (73.3%) |
| SDHC | 3 (2.9%) |
| SDHD | 25 (23.8%) |
| Variant type | |
| NA | 12 |
| Nonsense | 20 (21.6%) |
| Missense | 28 (30.1%) |
| Splice site | 13 (13.9%) |
| Frameshift | 26 (27.9%) |
| Copy number variation | 6 (6.4%) |
| Tumour type | |
| PGL | 65 (61.9%) |
| PCC | 29 (27.6%) |
| Both | 10 (9.5%) |
| Not known | 1 (1.0%) |
| Non‐PPGL | 4 |
| Multifocal disease | |
| Present | 36 (34.3%) |
| Absent | 69 (65.7%) |
| Functional status | |
| NA | 32 |
| Functioning | 63 (86.3%) |
| Nonfunctioning | 10 (13.7%) |
| Malignant disease | |
| Present | 26 (24.8%) |
| Absent | 79 (75.2%) |
NA, not available; PCC, phaeochromocytoma; PGL, paraganglioma; PPGL, phaeochromocytoma and paraganglioma.
2.2. What are the current recommendations for the age at which to commence surveillance for SDHx mutation carriers?
The Endocrine Society recommends that surveillance should comprise annual biochemistry (urinary or plasma metanephrines) and sporadic cross‐sectional imaging of the skull base, neck, thorax, abdomen and pelvis (MRI is the preferred radiation sparing imaging modality).3 There are no recommendations as to the lower age limit of genetic testing of children in SDHx mutation families. In the UK, genetic testing for inherited neoplasia syndromes is usually conducted around the time when clinical, biochemical and radiological surveillance would begin.53 It is also important to note that as SDHD variants have a preferential paternal transmission pattern of inheritance, clinical surveillance is only recommended for those carriers that inherit an SDHD variant from their father.
To our knowledge, no guidance exists to inform the most appropriate starting age for clinical, biochemical and radiological surveillance in paediatric SDHx mutation carriers. A review of the literature identified 13 manuscripts that attempted to address this issue. Eight studies argued for surveillance to start before or at 10 years including one expert opinion,54 three case series with less than five patients,6, 55, 56 three retrospective studies with 32, 92 and 116 patients, respectively5, 57, 58), and one case‐control study with 241 patients.59 Three reports recommended starting from the second decade of life (one systematic review of 95 papers suggesting a start age between 11‐20 years, one retrospective study with 91 patients suggesting an age of 27.1 years based on HNPGL penetrance calculations and one case series of three families suggested a starting age of 18 years for HNPGL screening).15, 60, 61 However, two further studies suggested that the starting age should either be between 5 and 10 years or alternatively a minimum of one decade before the earliest age of disease onset in that kindred (two expert opinions).62, 63
When considering at what age a screening programme should be commenced it is relevant to consider (a) what ages have SDHx‐related tumours occurred? (b) what is the estimated risk at different ages? and (c) what is the risk threshold at which screening is deemed appropriate? Ideally, this decision would be based on a robust cost‐benefit analysis (including the health costs of false positive screening diagnoses) but for rare disorders such SDHx‐related tumours evidence is necessarily limited and therefore decisions will involve a major element of expert judgement.
Recent studies, focused on more accurately predicting the clinical penetrance of SDHx genes, have adopted methods to control for ascertainment bias and suggest an estimated clinical penetrance of around 20% by age 50 years for SDHB mutation carriers.64
In the largest study of SDHB and SDHD mutation carriers yet reported, Andrews et al65 estimated the risks of PPGL and head and neck paraganglioma (HNPGL) by ages at 5, 10, 16 and 18 years in SDHB mutation carriers (n = 598 with clinical information available) and in SDHD carriers (n = 137 with clinical information available) (Table 2). There are some limitations to this analysis. Firstly, the figures include probands who are affected with the disease and therefore will increase penetrance estimates, and therefore, the analysis was also performed with probands excluded (Table 2). The second limitation of this study is that as these are clinical penetrance risks, they will not include asymptomatic tumours that might have been detected by screening. Using a different methodology, Benn et al recently estimated the lifetime penetrance of pathogenic SDHA, SDHB and SDHC variants by adopting an algorithm which compared allelic frequencies in 1815 cases (including cases from ref. [65]) vs controls from ExAC (http://exac.broadinstitute.org). The estimated lifetime penetrance of pathogenic SDHA and SDHC variants was low (1.7% and 8.3%, respectively), whilst the penetrance for SDHB was similar to that previously reported at 20.2%.66 The authors also speculated that SDHx gene penetrance may be directly proportional to the risk of multifocal tumours as SDHB gene mutations are more commonly associated with multifocal tumours compared to SDHA gene mutations.
Table 2.
Estimated risk of PPGL and HNPGL for SDHB and SDHD carriers by age 5, 10, 16 and 18 y, respectively, in data from Andrews et al65
| SDHx subunit gene | Penetrance at age 5 y | Penetrance at age 10 y | Penetrance at age 16 y | Penetrance at age 18 y | ||||
|---|---|---|---|---|---|---|---|---|
| PPGL | HNPGL | PPGL | HNPGL | PPGL | HNPGL | PPGL | HNPGL | |
|
SDHB n = 598 |
0.17% [95% CI 0.0‐0.51] |
0% |
1.7% [95% CI 0.67‐2.8] |
0% |
7.6% [95% CI 5.4‐9.8] |
0.38% [95% CI 0.0‐0.90] |
10.2% [95% CI 7.6‐12.7 |
0.58% [95% CI 0.0‐1.2] |
|
SDHD n = 137 |
0% | 0% |
0.28% [95% CI 0.0‐0.82] |
0% |
3.1% [95% CI 0.062‐6.0] |
1.6% [95% CI 0.0‐3.7] |
7.0% [95% CI 2.5‐11.3] |
3.2% [95% CI 0.063‐6.1] |
|
SDHB a n = 371 |
0% | 0% |
0.28% [95% CI 0.0‐0.82] |
0% |
1.2% [95% CI 0.023‐2.4] |
0% |
2.2% [95% CI 0.56‐3.7] |
0.32% [95% CI 0.0‐0.94] |
|
SDHD a n = 67 |
0% | 0% | 0% | 0% | 0% | 0% |
6.4% [95% CI 0.13‐12.3] |
1.7% [95% CI 0.0‐4.9] |
NB. No confidence intervals are given before the first noncensored event (if no children in the cohort have experienced tumours by the age specified).
HNPGL, head and neck paraganglioma; PPGL, phaeochromocytoma and paraganglioma.
Nonproband gene carriers only (probands excluded from analysis).
2.3. What are the controversies and/or risks of early surveillance?
Several factors complicate the decision when surveillance should be commenced in paediatric SDHx mutation carriers. Important considerations include the growing awareness that the lifetime penetrance of SDHB mutations is significantly lower (see above) than that estimated when the gene was first identified (originally estimated at 70%‐80%). In addition, surveillance programmes can be disruptive to patients’ lives, requiring them (and their parents) to take time off school or work and incur travel costs (and screening costs if not covered by a national health system or health insurance). The potential for the identification of incidental lesions on imaging raises several ethical and medicolegal questions also. Onwubiko and Mooney67 found that 35.7% (n = 86) of paediatric patients attending their level 1 paediatric trauma centre had at least one incidental finding on computed tomography studies of the thorax, abdomen and pelvis. However, only 30 (63.8%) of the clinically significant findings were reported, with follow‐up recommended by the radiologist in 14 cases (29.8%), and only four patients (4.6%) required further investigations. Incidental findings may be associated with a psychological burden and additional health costs.68 Finally, there is extremely limited information on the specificity and sensitivity of surveillance modalities for child and adolescent SDHx mutation carriers. Though the use of MRI instead of CT imaging reduces exposure to ionizing radiation, there is a lack of conclusive data on the potential long‐term consequences of exposure to the strong magnetic fields employed by MRI.69 Some groups have adapted their surveillance programmes at younger ages. Rijken et al64 started surveillance in a 7‐year‐old SDHB mutation carrier by urinary catecholamines measurements, allowing for surveillance to commence without the stress of imaging or phlebotomy. However, 24‐hour urinary collections can be difficult to obtain in young children. Tufton et al57 replaced MRI imaging with ultrasound in children under 10 years of age if they were unable to tolerate MRI. In doing so, the patients could experience the potential benefits of earlier surveillance whilst minimizing the anxiety involved during surveillance. However it must also be considered that ultrasound has a lower sensitivity and specificity compared with MRI in PPGL detection.3
The Endocrine Society clearly state that the morbidity associated with SDHB gene mutations requires particular attention,3 and therefore, more stringent screening efforts may be necessary in order to reduce the morbidity and mortality associated with SDHB‐mutated PPGL. It is notable that in the paediatric population studied in this review (similar to adult populations), SDHB is the most common SDHx subunit gene to be implicated in index cases presenting with PPGL (77/105, 73.3%) and malignant PPGL (22/26, 84.6%).
Finally, parental anxiety may also influence the decision as to when genetic testing should be performed and when clinical surveillance should be commenced and this can be a complex issue to manage in clinical practice. It is clear that when possible, a balance should be struck between being flexible and open to the needs and concerns of a family, whilst still keeping the interests and the long‐term welfare of the child at the forefront.53
3. CONCLUSION
Despite the occurrence of SDHx driven tumours in children, the majority of these tumours present in adulthood. The absolute risks of a “clinical PPGL” in the paediatric age group are estimated to exceed 1% and 5% at ages 10 years and 16 years in SDHB mutation carriers and ages 16 and 18 years in SDHD carriers, respectively.65 Reviewing those paediatric index cases with SDHx variants reported in the literature, 9.5% (10/105) of cases presented before the age of 10 years and 70% of those cases were SDHB gene mutation carriers. Furthermore, in cases for which detailed phenotypic data on paediatric index cases were available (Table 1 and Supporting Information Table S1), 86.3% (63/73) of tumours reported were secretory and therefore could be diagnosed on biochemical screening.
Importantly, this data would suggest that the approach to clinical surveillance should be tailored to the SDHx subunit gene affected, as recent studies would suggest a higher risk of PPGL (and possibly of synchronous and multifocal tumours) with SDHB gene mutations. Based on the available data reviewed herein both for asymptomatic SDHx carriers and index cases, we propose that for SDHB mutation carriers annual physical examination (with blood pressure) and biochemical screening should generally commence at the age of 5 years and at age 10 years for SDHA, SDHC and SDHD carriers. Radiological surveillance using MRI (or ultrasound for those who find MRI intolerable) and MRI of head and neck should be commenced from the age of 10 years for SDHB and 15 years for SDHA, SDHC and SDHD carriers and repeated every 2‐3 years if annual biochemical testing is normal. A MRI scan would be performed earlier if biochemical investigations are abnormal (the majority of early onset PPGL are biochemically active). Genetic testing can be offered to families from the ages at which surveillance would be initiated (5 or 10 years). Clinical and genetic investigations should be performed earlier if clinical symptoms develop, and an earlier age for starting MRI surveillance could be adopted in those rare families in which a PPGL has occurred in a child (or young adult) or based on the judgement of an experienced endocrinologist or paediatric endocrinologist. Finally, this clinical review has highlighted the need for a large prospective multicentre study to better inform existing surveillance and management strategies for paediatric and adolescent SDHx mutation carriers.
CONFLICT OF INTEREST
The authors have no conflict of interests and nothing to declare.
Supporting information
4. ACKNOWLEDGEMENTS
Eamonn R. Maher acknowledges support from the European Research Council (Advanced Researcher Award), the NIHR (Senior Investigator Award and Cambridge NIHR Biomedical Research Centre), the Cancer Research UK Cambridge Cancer Centre, and the Medical Research Council (Infrastructure Award). The University of Cambridge has received salary support for Eamonn R. Maher from the National Health Service (NHS) in the East of England through the Clinical Academic Reserve. The views expressed herein are those of the authors and not necessarily those of the NHS or the Department of Health.
Wong MY, Andrews KA, Challis BG, et al. Clinical Practice Guidance: Surveillance for phaeochromocytoma and paraganglioma in paediatric succinate dehydrogenase gene mutation carriers. Clin Endocrinol (Oxf). 2019;90:499–505. 10.1111/cen.13926
Funding information
Dr Casey receives funding from GIST Support UK.
REFERENCES
- 1. Evenepoel L, Papathomas TG, Krol N, et al. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ‐line mutations. Genet Med. 2015;17(8):610‐620. [DOI] [PubMed] [Google Scholar]
- 2. Gimenez‐Roqueplo A‐P, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra‐adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63(17):5615‐5621. [PubMed] [Google Scholar]
- 3. Lenders J, Duh Q‐Y, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99(6):1915‐1942. [DOI] [PubMed] [Google Scholar]
- 4. King KS, Prodanov T, Kantorovich V, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29(31):4137‐4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Srirangalingam U, Walker L, Khoo B, et al. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH‐B) gene mutation carriers. Clin Endocrinol (Oxf). 2008;69(4):587‐596. [DOI] [PubMed] [Google Scholar]
- 6. Prodanov T, Havekes B, Nathanson KL, Adams KT, Pacak K. Malignant paraganglioma associated with succinate dehydrogenase subunit B in an 8‐year‐old child: the age of first screening? Pediatr Nephrol. 2009;24(6):1239‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Imamura H, Muroya K, Tanaka E, et al. Sporadic paraganglioma caused by de novo SDHB mutations in a 6‐year‐old girl. Eur J Pediatr. 2016;175(1):137‐141. [DOI] [PubMed] [Google Scholar]
- 8. Benn DE, Croxson MS, Tucker K, et al. Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene. 2003;22(9):1358‐1364. [DOI] [PubMed] [Google Scholar]
- 9. Neumann H, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943‐951. [DOI] [PubMed] [Google Scholar]
- 10. Bockenhauer D, Rees L, Neumann H, Foo Y. A sporadic case of paraganglioma undetected by urine metabolite screening. Pediatr Nephrol. 2008;23(10):1889‐1891. [DOI] [PubMed] [Google Scholar]
- 11. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69(1):49‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Astuti D, Hart‐Holden N, Latif F, et al. Genetic analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and phaeochromocytoma susceptibility. Clin Endocrinol (Oxf). 2003;59(6):728‐733. [DOI] [PubMed] [Google Scholar]
- 13. Graham D, Gooch M, Ye Z, et al. Pheochromocytoma in a twelve‐year‐old girl with sdhb‐related hereditary paraganglioma‐pheochromocytoma syndrome. Case Rep Genet. 2014;2014:273423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santiago AH, Campbell BT, Estrada DE. Early presentation of familial paraganglioma with SDHB mutation in a 13 year old child and its mother. J Pediatr Endocrinol Metab. 2010;23(4):419‐422. [DOI] [PubMed] [Google Scholar]
- 15. Fish JH, Klein‐Weigel P, Biebl M, Janecke A, Tauscher T, Fraedrich G. Systematic screening and treatment evaluation of hereditary neck paragangliomas. Head Neck. 2007;29(9):864‐873. [DOI] [PubMed] [Google Scholar]
- 16. Luiz HV, da Silva TN, Pereira BD, et al. Malignant paraganglioma presenting with hemorrhagic stroke in a child. Pediatrics. 2013;132(6):e1709. [DOI] [PubMed] [Google Scholar]
- 17. Timmers H, Kozupa A, Eisenhofer G, et al. Clinical presentations, biochemical phenotypes, and genotype‐phenotype correlations in patients with succinate dehydrogenase subunit b‐associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92(3):779‐786. [DOI] [PubMed] [Google Scholar]
- 18. McDonnell CM, Benn DE, Marsh DJ, Robinson BG, Zacharin MR. K40E: a novel succinate dehydrogenase (SDH)B mutation causing familial phaeochromocytoma and paraganglioma. Clin Endocrinol. 2004;61(4):510‐514. [DOI] [PubMed] [Google Scholar]
- 19. Bayley JP, Grimbergen AE, van Bunderen PA, van der Wielen M, Kunst HP, Lenders JW. The first Dutch SDHB founder deletion in paraganglioma – pheochromocytoma patients. BMC Med Genet. 2009;10(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park S‐A, Lee DH, Kim HS. Aggressive imaging features in a malignant pheochromocytoma with a novel mutation of the SDHB gene. Clin Nucl Med. 2017;42(9):687‐689. [DOI] [PubMed] [Google Scholar]
- 21. Pandit R, Khadilkar K, Sarathi V, et al. Germline mutations and genotype–phenotype correlation in Asian Indian patients with pheochromocytoma and paraganglioma. Eur J Endocrinol. 2016;175(4):311‐323. [DOI] [PubMed] [Google Scholar]
- 22. Martucci VL, Lorenzo ZG, Weintraub M, et al. Association of urinary bladder paragangliomas with germline mutations in the SDHB and VHL genes. Urologic Oncology. 2015;33(4):167.e13‐167.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choat H, Derrevere K, Knight L, Brown W, Mack EH. SDHB‐Associated paraganglioma in a pediatric patient and literature review on hereditary pheochromocytoma‐paraganglioma syndromes. Case Rep Endocrinol. 2014;2014:502734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cascón A, Inglada‐Pérez L, Comino‐Méndez I, et al. Genetics of pheochromocytoma and paraganglioma in spanish pediatric patients. Endocr Relat Cancer. 2013;20(3):L1‐6. [DOI] [PubMed] [Google Scholar]
- 25. Lefebvre S, Borson‐Chazot F, Boutry‐Kryza N, et al. Screening of mutations in genes that predispose to hereditary paragangliomas and pheochromocytomas. Horm Metab Res. 2012;44(05):334‐338. [DOI] [PubMed] [Google Scholar]
- 26. Domingues R, Montalvão P, Magalhães M, Santos R, Duarte L, Bugalho MJ. Identification of three new variants of SDHx Genes in a cohort of Portuguese patients with extra‐adrenal paragangliomas. J Endocrinol Invest. 2012;35(11):975‐980. [DOI] [PubMed] [Google Scholar]
- 27. Muth A, Abel F, Jansson S, Nilsson O, Ahlman H, Wängberg Bo. Prevalence of Germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: a population‐based study in Western Sweden. World J Surg. 2012;36(6):1389‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Musil Z, Puchmajerová A, Křepelová A, et al. Paraganglioma in a 13‐year‐old girl: a novel SDHB gene mutation in the family? Cancer Genet Cytogenet. 2010;197(2):189‐192. [DOI] [PubMed] [Google Scholar]
- 29. Neumayer C, Moritz A, Asari R, et al. Novel SDHD germ‐line mutations in pheochromocytoma patients. Eur J Clin Invest. 2007;37(7):544‐551. [DOI] [PubMed] [Google Scholar]
- 30. Daniel E, Jones R, Bull M, Newell‐Price J. Rapid‐sequence MRI for long‐term surveillance for paraganglioma and phaeochromocytoma in patients with succinate dehydrogenase mutations. Eur J Endocrinol. 2016;175(6):561‐570. [DOI] [PubMed] [Google Scholar]
- 31. Mora J, Cascón A, Robledo M, Catala A. Pediatric paraganglioma: an early manifestation of an adult disease secondary to germline mutations. Pediatr Blood Cancer. 2006;47(6):785‐789. [DOI] [PubMed] [Google Scholar]
- 32. Novosel A, Heger A, Lohse P, Schmidt H. Multiple pheochromocytomas and paragangliomas in a young patient carrying a SDHD gene mutation. Eur J Pediatr. 2004;163(12):701‐703. [DOI] [PubMed] [Google Scholar]
- 33. Cascón A, Landa Í, López‐Jiménez E, et al. Molecular Characterisation of a common SDHB deletion in paraganglioma patients. J Med Genet. 2008;45(4):233‐238. [DOI] [PubMed] [Google Scholar]
- 34. Ghayee HK, Havekes B, Corssmit E, et al. Mediastinal paragangliomas: association with mutations in the succinate dehydrogenase genes and aggressive behavior. Endocr Relat Cancer. 2009;16(1):291‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Majumdar S, Friedrich CA, Koch CA, Megason GC, Fratkin JD, Moll GW. Compound heterozygous mutation with a novel splice donor region DNA sequence variant in the succinate dehydrogenase subunit b gene in malignant paraganglioma. Pediatr Blood Cancer. 2010;54(3):473‐475. [DOI] [PubMed] [Google Scholar]
- 36. Armstrong R, Greenhalgh KL, Rattenberry E, et al. Succinate Dehydrogenase subunit B (SDHB) gene deletion associated with a composite paraganglioma/neuroblastoma. J Med Genet. 2009;46(3):215‐216. [DOI] [PubMed] [Google Scholar]
- 37. Hammond PJ, Murphy D, Robert Carachi D, Davidson F, McIntosh D, “Childhood, . Phaeochromocytoma and paraganglioma: 100% incidence of genetic mutations and 100% survival. J Pediatr Surg. 2010;45(2):383‐386. [DOI] [PubMed] [Google Scholar]
- 38. Dimachkieh AL, Dobbie A, Olson DR, Lovell MA, Prager JD. Tracheal paraganglioma presenting as stridor in a pediatric patient, case report and literature review. Int J Pediatr Otorhinolaryngol. 2018;107:145‐149. [DOI] [PubMed] [Google Scholar]
- 39. Hermsen MA, Sevilla MA, Llorente JL, et al. “Relevance of germline mutation screening in both familial and sporadic head and neck paraganglioma for early diagnosis and clinical management. Cell Oncol. 2010;32(4):275‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neychev V, Straughan D, Pacak K, Kebebew E. Multidisciplinary management of locally advanced and widely metastatic paraganglioma in a patient with life‐threatening compressive symptoms. Head Neck. 2015;37(12):E205‐E208. [DOI] [PubMed] [Google Scholar]
- 41. Kim MS, Muratore C, Snelling L, et al. Ischemic stroke and rhabdomyolysis in a 15 year‐old girl with paraganglioma due to an SDHB Exon 6 (Q214X) mutation. J Pediatr Endocrinol Metab. 2011;22(6):565‐572. [DOI] [PubMed] [Google Scholar]
- 42. Prasad P, Kant JA, Wills M, O’Leary M, Lovvorn H, Yang E. Loss of heterozygosity of succinate dehydrogenase B mutation by direct sequencing in synchronous paragangliomas. Cancer Genet Cytogenet. 2009;192(2):82‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sait S, Pandit‐Taskar N, Modak S. Failure of MIBG scan to detect metastases in SDHB‐mutated pediatric metastatic pheochromocytoma. Pediatr Blood Cancer. 2017;64(11):e26549. [DOI] [PubMed] [Google Scholar]
- 44. Cascón A, Montero‐Conde C, Ruiz‐Llorente S, et al. Gross SDHB deletions in patients with paraganglioma detected by multiplex PCR: a possible hot spot? Genes Chromosom Cancer. 2006;45(3):213‐219. [DOI] [PubMed] [Google Scholar]
- 45. Else T, Marvin ML, Everett JN, et al. The clinical phenotype of SDHC‐associated hereditary paraganglioma syndrome (PGL3). J Clin Endocrinol Metab. 2014;99(8):E1482‐E1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schiavi F, Savvoukidis T, Trabalzini F, et al. Paraganglioma syndrome. Ann N Y Acad Sci. 2006;1073(1):190‐197. [DOI] [PubMed] [Google Scholar]
- 47. Marvin ML, Bradford CR, Sisson JC, Gruber SB. Diagnosis and management of hereditary paraganglioma syndrome due to the F933>X67 SDHD mutation. Head Neck. 2009;31(5):689‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Srirangalingam U, Khoo B, Matson M, et al. SDHD‐related chromaffin tumours: disease localisation to genetic dysfunction. Horm Res Paediatr. 2010;73(2):135‐139. [DOI] [PubMed] [Google Scholar]
- 49. Timmers HJ, Pacak K, Bertherat J, et al. Mutations associated with succinate dehydrogenase D‐related malignant paragangliomas. Clin Endocrinol. 2008;68(4):561‐566. [DOI] [PubMed] [Google Scholar]
- 50. Badenhop RF, Jansen JC, Fagan PA, et al. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J Med Genet. 2004;41(7):e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fakhry N, Niccoli‐Sire P, Barlier‐Seti A, Giorgi R, Giovanni A, Zanaret M. Cervical paragangliomas: is SDH genetic analysis systematically required? Eur Arch Otorhinolaryngol. 2008;265(5):557‐563. [DOI] [PubMed] [Google Scholar]
- 52. Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ‐line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Can Res. 2000;60(24):6822‐6825. [PubMed] [Google Scholar]
- 53. Clarke A. What is at stake in the predictive genetic testing of children? Fam Cancer. 2010;9(1):19‐22. [DOI] [PubMed] [Google Scholar]
- 54. Timmers HJ, Gimenez‐Roqueplo A‐P, Mannelli M, Pacak K. Clinical aspects of SDHx‐related pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2009;16(2):391‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lefebvre M, Foulkes W. Pheochromocytoma and paraganglioma syndromes: genetics and management update. Curr Oncol. 2014;21(1):e8‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Renard L, Godfraind C, Boon LM, Vikkula M. A novel mutation in the SDHD gene in a family with inherited paragangliomas? Implications of genetic diagnosis for follow up and treatment. Head Neck. 2003;25(2):146‐151. [DOI] [PubMed] [Google Scholar]
- 57. Tufton N, Shapiro L, Srirangalingam U, et al. Outcomes of annual surveillance imaging in an adult and paediatric cohort of succinate dehydrogenase B mutation carriers. Clin Endocrinol (Oxf). 2017;86(2):286‐296. [DOI] [PubMed] [Google Scholar]
- 58. Benn DE, Gimenez‐Roqueplo A‐P, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827‐836. [DOI] [PubMed] [Google Scholar]
- 59. Jochmanova I, Wolf KI, King KS, et al. SDHB‐related pheochromocytoma and paraganglioma penetrance and genotype–phenotype correlations. J Cancer Res Clin Oncol. 2017;143(8):1421‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma‐paraganglioma syndromes. J Intern Med. 2009;266(1):19‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eijkelenkamp K, Osinga TE, de Jong MM, et al. Calculating the optimal surveillance for head and neck paraganglioma in SDHB‐mutation carriers. Fam Cancer. 2017;16(1):123‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. National Cancer Institute . (2018). Genetics of endocrine and neuroendocrine neoplasias. [online] https://www.cancer.gov/types/thyroid/hp/medullary-thyroid-genetics-pdq (Accessed 11 March 2018). [PubMed]
- 63. Else T, Greenberg S, Fishbein L. Hereditary paraganglioma‐pheochromocytoma syndromes 2008 May 21 [Updated 2018 Oct 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, seattle; 1993. –2019. [Google Scholar]
- 64. Rijken JA, Niemeijer ND, Corssmit E, et al. Low penetrance of paraganglioma and pheochromocytoma in an extended kindred with a germline SDHB exon 3 deletion. Clin Genet. 2016;89(1):128‐132. [DOI] [PubMed] [Google Scholar]
- 65. Andrews KA, Ascher DB, Pires D, et al. Tumour risks and genotype–phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD . J Med Genet. 2018;55:384‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Benn DE, Zhu Y, Andrews KA, et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J Med Genet. 2018;55:729‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Onwubiko C, Mooney DP. The prevalence of incidental findings on computed tomography of the abdomen/pelvis in pediatric trauma patients. Eur J Trauma Emerg Surg. 2018;44(1):15‐18. [DOI] [PubMed] [Google Scholar]
- 68. Schmidt CO, Hegenscheid K, Erdmann P, et al. Psychosocial consequences and severity of disclosed incidental findings from whole‐body MRI in a general population study. Eur Radiol. 2013;23(5):1343‐1351. [DOI] [PubMed] [Google Scholar]
- 69. Hartwig V, Giovannetti G, Vanello N, Lombardi M, Landini L, Simi S. Biological effects and safety in magnetic resonance imaging: a review. Int J Environ Res Public Health. 2009;6(6):1778‐1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials