

Abstract
Mutations in KRAS are often associated with resistance to EGFR‐targeting antibody therapy. Using comprehensive systems analyses, GNB5 has been identified as a potential target to overcome therapy resistance targeting the EGFR signaling pathways, whereby the AKT signaling pathway (PI3K) rather than the ERK signaling pathway (RAS) might be dominantly affected. Personalized mathematical modeling and simulations of this signaling pathway/network and respective perturbations are of great utility to customize therapy for patients.
Abbreviations
- CMS
consensus molecular subtypes
- CRC
colorectal cancer
- EGFR
epidermal growth factor receptor
- GPCRs
G protein‐coupled receptors
- GSEA
gene set enrichment analysis
- mCRC
metastatic colorectal cancer
- NSCLC
nonsmall cell lung cancer
- ODE
ordinary differential equations
- RECIST
response evaluation criteria in solid tumors
- TCGA
The Cancer Genome Atlas
- TKI
tyrosine kinase inhibitors
Cetuximab and panitumumab are FDA‐approved monoclonal antibodies against epidermal growth factor receptor (EGFR), which are commonly used for patients with RAS wild‐type metastatic colorectal cancer (mCRC) 1. For different indications such as nonsmall cell lung cancer (NSCLC) there are also third‐generation EGFR tyrosine kinase inhibitors targeting specific resistance mutations in the tyrosine kinase domain (T790M) approved 2. KRAS is a downstream signaling molecule of EGFR and is mutated in approximately 40% of CRC patients. However, only a small fraction of patients respond to this therapy and drug resistance remains a major issue 1. EGFR inhibitors such as cetuximab bind to the extracellular domain of EGFR, which is a transmembrane receptor tyrosine kinase. RAS‐RAF‐MAPK, PI3K‐PTEN‐AKT, and JAK/STAT are the three major downstream signaling pathways activated by EGFR. In the last decade much effort has been invested into the analyses of these signaling components and it has been shown that alterations in these downstream molecules might be involved in anti‐EGFR therapy resistance mechanisms 1, 3, 4 (Fig. 1A). So far, KRAS mutations have been the common predictors of resistance to cetuximab and panitumumab. However, recent studies, including the work from Park et al. 5 indicate that the KRAS status might not be sufficient to predict anti‐EGFR therapy response. Therefore, other molecules, independent of the downstream signaling components, must also be taken into account. In this issue of The FEBS Journal, Park et al. 5 were able to identify DUSP4, ETV5, GNB5, NT5E, and PHLDA1 as potential drug targets to overcome cetuximab resistance in KRAS wild‐type cells, using an comprehensive systems approach including mathematical modeling and simulation of treatment responses (Fig. 1B). Furthermore, they provide evidence that the knockdown of GNB5 increases cetuximab sensitivity even in KRAS mutant cells.
Figure 1.
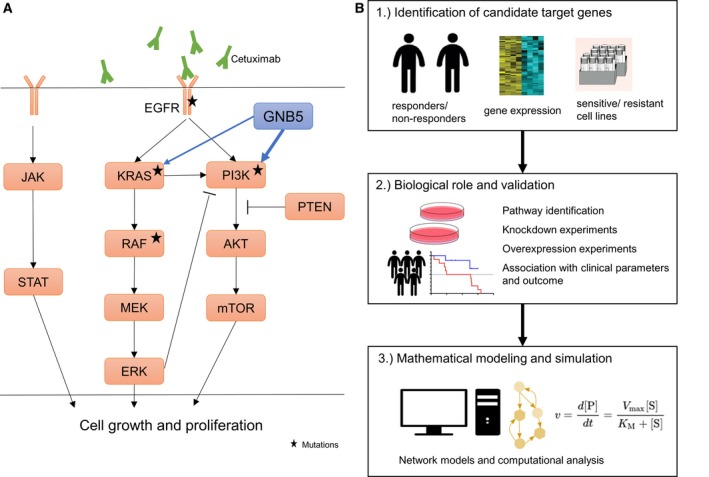
Epidermal growth factor receptor signaling pathways and common mutations associated with therapy resistance (A) and systemic approach for the identification and validation of therapy resistance targets as well as mathematical modeling and simulation of therapy response (B).
As a first step, responders and nonresponders are typically identified by progression‐free survival or according to the response evaluation criteria in solid tumors (RECIST). Significantly differentially expressed genes between these two groups may be determined using RNA sequencing or microarray analyses. Classification approaches such as random forest analyses or regularized logistic regression may be useful to narrow down the number of gene candidates with highest impact into the prediction model 6. Park et al. were able to verify five of the differently expressed genes, which were also significantly upregulated in cetuximab‐resistant cell lines. RNAi‐based knockdown of these candidates showed that only one candidate, GNB5, a downstream molecule of G protein‐coupled receptors (GPCRs), increased sensitivity to both the EGFR inhibitor cetuximab and the EGFR tyrosine kinase inhibitor erlotinib in KRAS mutant cell lines. Analysis in large patient cohorts, for example, data from The Cancer Genome Atlas (TCGA), is useful to associate the expression of a target with clinical parameters including CRC sidedness, TNM‐staging, microsatellite instability, consensus molecular subtypes (CMS1‐4), and with clinical outcome. Interestingly, patients with high GNB5 expression were associated with worse overall survival. Gene set enrichment analysis (GSEA) may be applied to RNA sequencing data to address if gene ontology terms/pathways, hallmarks or oncogenic signatures are enriched in tumors with high expression of the target versus tumors with low expression of the target. In the present study 5, the signaling effect was investigated in a GNB5 overexpression cell model and by using results from phosphorylation measurements, colony‐forming assays, and viability analyses. Prompted by these results, the authors suggest that GNB5 could contribute to cetuximab resistance and proliferation by dominantly affecting the Akt signaling rather than the ERK signaling.
The work by Park et al. 5 demonstrates that mathematical modeling [based on Michaelis–Menten kinetics and ordinary differential equations (ODE)] not only makes it possible to analyze and simulate specific signaling networks but also to investigate the effects of different perturbations 7. It is important to analyze how sensitive (or robust) the model is against variation in the estimated parameters. A model is always a simplification of reality and typically the essential factors are extracted and translated into a network, whereby the connections are derived from consensus knowledge as evident in literature and databases. In this context, the question arises whether the result (cell survival) is robust/stable against a change in the network connections, considering the variations in different cell models or cancer types. In particular, for the EGFR signaling network structural variations were considered to include or to exclude activating connection between EGFR and PI3K 8, or inhibiting connection between ERK and PI3K 9 and no changes in cell survival were observed. Sensitivity/resistance to cetuximab was shown to depend only on KRAS mutation, but could be reversed by GNB5 overexpression or GNB5 knockdown, respectively 5. Logical (Boolean) modeling 8, 10 could be an alternative in testing changes of individual factors or rules within a signaling network topology, especially patient/cell type‐specific adaptations are of interest.
However, a number of open questions still remain to be clarified, for example, if targets can be identified, which are contributing to the resistance mechanism of other tyrosine kinase inhibitors (TKI). This may include also factors from the tumor microenvironment able to rescue cancer cells from kinase inhibitors, as shown by (phospho)proteome profiling 11. Especially in the investigation of signaling networks, this technology in combination with phenotypic responses based on perturbation experiments provides a useful extension for network inference and modeling 7. Since recent observations show that targeting the EGFR signaling pathway can also trigger immunogenic cell death 12 or influence the tumor immune environment via the JAK/STAT3 axis, possible effects for immunotherapy or combination therapy are also of great interest.
Ultimately, systemic analysis and, in particular, personalized mathematical models—for example, taking into account the KRAS mutation status or the activation of EGFR variants of a patient—and corresponding simulations (validated, at least in some cases, by the treatment of patient‐related tumor organoid models), could help to customize therapy for each patient, and that is exactly what we expect from precision medicine.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgement
Christina Plattner is a recipient of a DOC fellowship of the Austrian Academy of Sciences.
References
- 1. Zhao B, Wang L, Qiu H, Zhang M, Sun L, Peng P, Yu Q & Yuan X (2017) Mechanisms of resistance to anti‐EGFR therapy in colorectal cancer. Oncotarget 8, 3980–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murtuza A, Bulbul A, Shen JP, Keshavarzian P, Woodward BD, Lopez‐Diaz FJ, Lippman SM & Husain H (2019) Novel third‐generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res 79, 689–698. [DOI] [PubMed] [Google Scholar]
- 3. Okamoto I (2010) Epidermal growth factor receptor in relation to tumor development: EGFR‐targeted anticancer therapy. FEBS J 277, 309–315. [DOI] [PubMed] [Google Scholar]
- 4. Leto SM & Trusolino L (2014) Primary and acquired resistance to EGFR‐targeted therapies in colorectal cancer: impact on future treatment strategies. J Mol Med 92, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park SM, Hwang CY, Cho SH, Lee D, Gong JR, Lee S, Nam S & Cho KH (2019) Systems analysis identifies potential target genes to overcome cetuximab resistance in colorectal cancer cells. FEBS J. 10.1111/febs.14773. [DOI] [PubMed] [Google Scholar]
- 6. Hastie T, Tibshirani R & Friedman JH (2009) The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd edn. Springer, New York. [Google Scholar]
- 7. Korkut A, Wang W, Demir E, Aksoy BA, Jing X, Molinelli EJ, Babur O, Bemis DL, Onur Sumer S, Solit DB et al (2015) Perturbation biology nominates upstream‐downstream drug combinations in RAF inhibitor resistant melanoma cells. Elife 4, e04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eduati F, Doldan‐Martelli V, Klinger B, Cokelaer T, Sieber A, Kogera F, Dorel M, Garnett MJ, Bluthgen N & Saez‐Rodriguez J (2017) Drug resistance mechanisms in colorectal cancer dissected with cell type‐specific dynamic logic models. Cancer Res 77, 3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Won JK, Yang HW, Shin SY, Lee JH, Heo WD & Cho KH (2012) The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J Mol Cell Biol 4, 153–163. [DOI] [PubMed] [Google Scholar]
- 10. Klamt S, Saez‐Rodriguez J & Gilles ED (2007) Structural and functional analysis of cellular networks with Cell NetAnalyzer. BMC Syst Biol 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koch H, Wilhelm M, Ruprecht B, Beck S, Frejno M, Klaeger S & Kuster B (2016) Phosphoproteome profiling reveals molecular mechanisms of growth‐factor‐mediated kinase inhibitor resistance in EGFR‐overexpressing cancer cells. J Proteome Res 15, 4490–4504. [DOI] [PubMed] [Google Scholar]
- 12. Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, Sigismund S, Ravenda PS, Bonaldi T, Zampino MG et al (2016) The EGFR‐specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med 22, 624–631. [DOI] [PubMed] [Google Scholar]