

Abstract
Essentials.
Non‐factor VIII (FVIII) therapies for hemophilia A, such as bispecific antibodies (bsAbs), are in development.
Bispecific antibodies are intrinsically different from FVIII and lack many of the same regulatory mechanisms.
These differences complicate assignment and interpretation of FVIII‐equivalent activity.
Inability to assign FVIII equivalence compromises our capacity to assess hemostatic potential of bsAb therapies.
Background
Activated factor VIII (FVIIIa) mimetic bsAbs aim to enable prophylactic treatment of hemophilia A patients with and without inhibitors. With different mechanisms of action, benchmarking their activity against FVIII to determine efficacious yet safe dosage is difficult.
Objective
To compare the activities of sequence identical emicizumab (SI‐Emi) and another bsAb, BS‐027125, to recombinant FVIII (rFVIII) using clinical and nonclinical assays and to evaluate our ability to assign a FVIII‐equivalent value to bsAbs and implications thereof.
Methods
Activities of SI‐Emi, BS‐027125, and rFVIII were measured by one‐stage clotting assay, chromogenic factor Xa generation assay, and thrombin generation assay. We also assessed the activity of anti‐FIXa and anti‐FX bivalent homodimers of each bsAb and probed the effect of different reagents in thrombin generation assay (TGA).
Results
The FVIII‐like activity of SI‐Emi and BS‐027125 ranged greatly across each assay, varying both by parameter measured within an assay and by reagents used. Notably, SI‐Emi anti‐FIXa bivalent homodimer had meaningful activity in several assays, whereas BS‐027125 anti‐FIXa bivalent homodimer only had activity in the chromogenic assay. Surprisingly, SI‐Emi displayed activity in the absence of phospholipids, while BS‐027125 had minimal phospholipid‐independent activity.
Conclusions
Bispecific antibodies demonstrate little consistency between assays tested here owing to intrinsic differences between FVIII and bsAbs. While some trends are shared, the bsAbs also differ in mechanism. These inconsistencies complicate assignment of FVIII‐equivalent values to bsAbs. Ultimately, a deeper mechanistic understanding of bsAbs as well as bsAb‐tailored assays are needed to monitor and predict their hemostatic potential and long‐term efficacy and safety confidently.
Keywords: bispecific antibodies, blood coagulation tests, factor VIII, hemophilia A, hemostasis
1. INTRODUCTION
New hemophilia A (HemA) therapies continue to advance to improve patient outcomes, particularly with the advent of extended half‐life factor VIII (FVIII) replacement concentrates.1 Still, a major challenge in the treatment of HemA remains the development of anti‐FVIII inhibitors, creating the need for alternative therapies that replace or “bypass” the functionality of FVIII.2, 3 Since the 1980s, inhibitor patients have relied on bypass therapies such as activated prothrombin complex concentrates (aPCCs) or recombinant factor VIIa (rFVIIa); however, effectiveness of these treatments in preventing bleeds is often poor.4 More recently, alternative approaches are being explored to inhibit inhibitors of clotting such as anti‐tissue factor pathway inhibitor (TFPI) antibodies5 or RNA interference‐mediated knock‐down of antithrombin III.6 Unlike thrombin generation in the presence of FVIII, with these approaches thrombin generation is driven by the extrinsic FVIIa/tissue factor pathway via either enhanced FXa generation or reduced FXa inhibition, respectively.7, 8 Another approach developed over the last few years is a bsAb, emicizumab,9 which strives to mimic the cofactor function of FVIIIa by binding to activated factor IX (FIXa) and factor X (FX). It represents, currently, the only non‐FVIII treatment approach attempting to restore the functionality of the intrinsic tenase complex.
Critical to the development of FVIII replacement therapies are the preclinical and clinical assays used to evaluate the factor's activity. The one‐stage clotting assay measures time to clot formation in plasma and the chromogenic assay measures the generation of activated factor X (FXa) by the intrinsic tenase complex in a purified system. These two assays are globally accepted as validated methods to assign potency and monitor FVIII levels in the clinic. However, bypassing agents, which have a different mechanism of action compared to FVIII, cannot rely on these assays and either are assigned activity based on clot time reduction in the activated partial thromboplastin time assay (aPTT), as with aPCCs, or are simply dosed based on mass (e.g. rFVIIa). For bsAbs, which aim to mimic rather than bypass FVIII's mechanism, it is natural to try to assign a FVIII‐equivalence value because FVIII activity is a surrogate for effective HemA treatment. However, the utility of standard clinical assays in equating the activity of bsAbs to FVIII needs to be studied given the intrinsic mechanistic differences between these molecules.
Factor VIII has specific characteristics that are not mimicked by bsAbs (reviewed in Reference 10). Upon thrombin activation, FVIII dissociates from von Willebrand factor and binds to phosphatidylserine (PS) on activated platelets. These rate‐limiting steps do not exist for bsAbs since they do not require activation, do not dissociate from vWF, and do not bind to phospholipids. Notably, these rate‐limiting steps vary between clinical assays. While a more global assay such as thrombin generation (TGA) may provide additional metrics by which to compare FVIII to antibodies, it is not a clinical assay and is subject to additional variability owing to a lack of standardization. As such non‐FVIII replacement therapies continue to develop, the importance of measuring and appropriately interpreting FVIII‐like activity, in terms of both efficacy and safety, should not be underestimated. Ultimately, a deeper understanding of the mechanistic differences between bsAbs and FVIII is required to determine safe dosing windows that provide adequate protection from breakthrough bleeds and prevent the accumulation of joint damage over time, as well as safe, efficacious methods for treating breakthrough bleeds, trauma, and use in surgery should the bsAb not provide full restoration of FVIII‐like activity.
To investigate these differences and understand how they manifest in various assays, we focused on comparison of two bsAbs and FVIII. Emicizumab is a bsAb specific for FIX/FIXa and FX/FXa that is currently marketed as Hemlibra.9 While emicizumab can co‐localize FIX/FIXa and FX/FXa, we sought to develop a bsAb that more closely mimics FVIII's specificity for FIXa and FX. We hypothesized that specificity for FIXa and FX prevents the formation of non‐productive trimeric complexes (i.e. FIX‐bsAb‐FXa) and may thereby enable greater efficacy. One of the bsAbs we identified, BS‐027125, is selective for FIXa and specific for FX. Here, we highlight the challenges in assigning a FVIII‐equivalent activity value to non‐FVIII replacement therapies by investigating the activity of two distinct FVIIIa mimetic bsAbs, sequence identical emicizumab and BS‐027125, in three types of assays in comparison to recombinant FVIII (rFVIII) and discuss the potential implications for their therapeutic use.
2. METHODS
2.1. Bispecific antibody production and purification
2.1.1. Sequence identical emicizumab (SI‐Emi)
Emicizumab drug product is reserved for patient use only, so SI‐Emi was produced using the published sequence from the World Health Organization's International Nonproprietary Names database.11 In short, heavy and light chain vectors encoding the anti‐FIX(a) and anti‐FX(a) arms (Addgene.org plasmid ID 11364, 113665, and 113666) were coexpressed in CHO cells and purified by affinity chromatography using a MabSelect SuRe column (GE Healthcare) followed by ion exchange chromatography on a HiTrap SP FF column to separate target bsAb from anti‐FIX(a) or anti‐FX(a) homodimeric byproducts.12 To generate one‐armed antibodies, vectors carrying a truncated form of the anti‐FX(a) or anti‐FIX(a) heavy chain lacking the Fab regions were coexpressed with intact anti‐FIX(a) or anti‐FX(a) heavy chain, respectively, and light chain vector. Target one‐armed antibodies were purified as described previously, followed by size exclusion chromatography. Purified antibodies were dialyzed into phosphate buffered saline and tested at the concentrations indicated for each assay.
2.1.2. BS‐027125
The Adimab yeast‐based platform was utilized to identify antibodies specific for FIXa and FX. Briefly, eight different naive antibody libraries were used in eight parallel selections as previously described.13 Subsequently, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027, respectively, were chosen for further study. Heavy and light chains were coexpressed in CHO cells and purified using a MabSelect SuRe column. Target bsAb and contaminating homodimers were separated on a Mono S 10/100 GL column with a salt gradient. One‐armed antibodies were generated using the same methods as described earlier. Purified antibodies were dialyzed into PBS and tested at the concentrations indicated for each assay.
Purity, monodispersity, sequence identity, and correct chain pairing were confirmed by HPLC and mass spectrometry for both bispecific antibodies and their homodimeric byproducts (data not shown).
2.2. Surface plasmon resonance
Binding kinetics of BS‐027125 for FIX, FIXa, FX, and FXa were determined by multicycle kinetics on a Biacore T200 (GE Healthcare); 250 RU of bsAb was captured on the surface of a Protein A chip. Analytes were applied at concentrations from 3 to 100 nmol/L for FX, 0.2 to 50 nmol/L for FIXa, and 0.6 to 100 nmol/L for FIX in 100 mmol/L HEPES, 150 mmol/L NaCl, 2.5 mmol/L CaCl2, 0.05% Tween20 for 45 or 120 seconds (for FX, or FIX and FIXa, respectively) followed by a 600‐second dissociation in buffer alone. Experiments were performed in triplicate at a flow rate of 100 μL/min at 25°C on the same chip. Raw data were processed by reference flow cell and buffer blank subtraction using the T200 Evaluation Software, and kinetic parameters were derived using a 1:1 interaction model with global fitting, yielding a χ2/Rmax of ≤0.05.
2.3. One‐stage clotting assay
Bispecific antibody and WHO FVIII were analyzed with an aPTT‐based one‐stage assay on a Sysmex CA‐1500. Samples were diluted 1:10 in Tris‐BSA. On the instrument, 50 μL of Siemens FVIII‐depleted plasma was added to 50 μL of diluted sample and incubated for 60 seconds at 37°C, followed by addition of 50 μL activator (ActinFSL, SynthASil, or C.K.Prest) and incubation for 240 seconds at 37°C. Finally, 50 μL of CaCl2 was added and time to fibrin clot formation was measured.
2.4. Chromogenic assay
Factor Xa generation for bsAbs or rFVIII (Advate®) was measured using a modified chromogenic assay. Bovine FIXa and FX from the Chromogenix Coatest SP Factor VIII kit (Diapharma) were replaced with human FIXa and FX (Haematologic Technologies) at final concentrations of 10 and 100 nmol/L, respectively. All other kit components were used per package instructions. Briefly, bsAb or rFVIII dilution series were added to assay wells and incubated with premixed solutions of FIXa, FX, and phospholipids. For FVIII, a 1:4 molar ratio of alpha‐thrombin:FVIII was added to the wells along with the FIXa/FX/phospholipid mixture. After 5 minutes, CaCl2 was added to initiate FXa generation. After 10 minutes, substrate was added and absorbance read at 405 nm with a Synergy2 plate reader (BioTek) to determine initial rates of substrate cleavage. The amount of FXa generated was based on a standard curve of FXa substrate cleavage. In addition, rFVIII and bsAbs were assayed using the commercially available BIOPHEN FVIII:C kit (HYPHEN BioMed) using both the 1:10 and 1:40 dilution protocols per manufacturer instructions.
2.5. Thrombin generation assay (TGA)
Calibrated automated thrombography was performed using congenital HemA plasma (George King Biomedical) and triggered with factor XIa (FXIa, 52 pmol/L final) or tissue factor (TF, Innovin, 1:30 000 final dilution) combined with phospholipid vesicles (phosphatidylcholine (PC): phosphatidylethanolamine (PE): phosphatidylserine (PS), 40:40:20% molar ratio or PC:PS 80:20% molar ratio, 10 μmol/L final). A calibrator reagent (Stago) of known thrombin concentration was used. Data were collected on a Fluoroskan Ascent instrument and TGA parameters were generated by Thrombinoscope software. Factor VIII‐equivalence was calculated for each parameter by nonlinear regression of the linear portion of rFVIII response.
2.6. Statistics
Standard deviations and standard error of the mean were calculated from the means of each group. Comparison of differences in thrombin generation activity on phospholipid vesicles of different compositions was determined by t test at each vesicle concentration with Holm‐Sidak correction to control for multiple comparisons.
3. RESULTS
3.1. Surface plasmon resonance
The affinity of BS‐027125 for FIXa and FIX were determined to be 1.76 ± 0.02 nmol/L and 5.71 ± 0.11 nmol/L, respectively (Figure S1A,B). The BS‐027125 showed a 3.3‐fold higher affinity for FIXa over FIX and was specific for FX with a K D of 49.1 ± 1.3 nmol/L (Figure S1C) and no detectable binding to FXa up to 1 μmol/L. In contrast, the affinities of SI‐Emi for each FIX, FIXa, FX, and FXa were confirmed to a range from 1 to 5 μmol/L (data not shown). These data are consistent with previously published results showing that SI‐Emi cannot distinguish between the active and zymogen forms of FIX or FX and binds with equally low affinity to all antigens.14
3.2. One‐stage assay
To evaluate the activities of SI‐Emi and BS‐027125, we began with the most common clinical assay, the aPTT‐based one‐stage clotting assay.15 A FVIII standard curve was generated from 1.5 to 0.0625 IU/mL and linear regression analysis was applied to a log‐log plot of clotting time versus concentration to determine FVIII‐equivalent activity of the antibodies (Figure S2A). While the two lowest concentrations of SI‐Emi tested (31 and 62 nmol/L) produced 45 ± 7% and 116 ± 17% FVIII‐like activity, respectively, FVIII‐equivalence could not be assigned to higher, clinically relevant concentrations of SI‐Emi as clotting times were out of the range of the FVIII standard curve (Figure 1A and Figure S2A). For each bsAb, the anti‐FIXa and anti‐FX homodimers, byproducts of the purification process, were included as controls. Unexpectedly, both homodimeric byproducts of SI‐Emi displayed shortened clot times. The SI‐Emi anti‐FIXa homodimer exhibited less activity relative to SI‐Emi bsAb, but still produced >100% FVIII‐like activity while the anti‐FX homodimer produced up to 7 ± 1% FVIII‐like activity. BS‐027125, in contrast, produced up to 134 ± 13% FVIII‐like activity at the highest concentrations tested, with the anti‐FIXa and anti‐FX homodimers of BS‐027125 showing no detectable activity (Figure 1B). While the experiments discussed here were performed using ActinFSL, an ellagic acid‐based activator, experiments using silica‐based and kaolin‐based activators (SynthASil and C.K. Prest, respectively) (Figure S3) as well as previous reports from Adamkewicz et al support these conclusions.16
Figure 1.
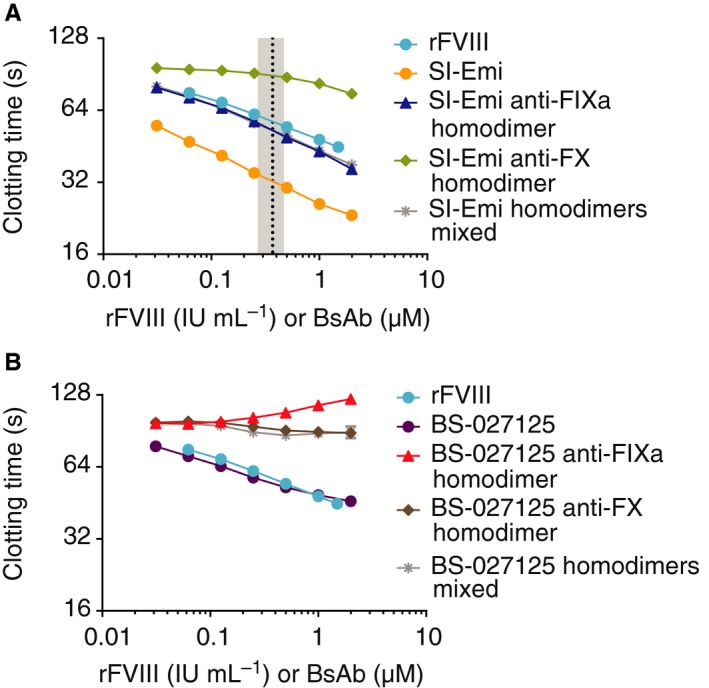
One‐stage clotting times of rFVIII, SI‐Emi, and BS‐027125. A, Dose response of SI‐Emi and SI‐Emi anti‐FIXa and anti‐FX homodimers (mixed and individually) compared to rFVIII. Black dotted line and gray shaded area are mean and range of steady‐state emicizumab concentration of patients receiving emicizumab prophylaxis, respectively.9, 17 B, Dose response of BS‐027125 and BS‐027125 anti‐FIXa and anti‐FX homodimers (mixed and individually) compared to rFVIII. Data are mean ± SD. anti‐FIXa, anti‐factor IXa; anti‐FX, anti‐factor X; BS‐027125, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027; rFVIII, recombinant factor VIII; SI‐Emi, sequence identical emicizumab
3.3. Chromogenic assay
We next tested the activity of bsAbs using a modified form of the second most common clinical assay, the chromogenic FXa generation assay. This modified format enabled the precise variation of individual components including hFIXa, hFX, and phospholipids at defined concentrations. We generated a FVIII standard curve from 0.5 to 0.002 IU/mL and applied linear regression analysis to log‐log plots of activity against concentration (Figure S2B). The resulting linear equation was used to determine FVIII‐equivalent activity of the antibodies. The SI‐Emi achieved up to 46.6 ± 0.2% FVIII‐like activity at 1 μmol/L (Figure 2A). Strikingly, the SI‐Emi anti‐FIXa homodimer, consisting of two FIX(a) binding sites, achieved more activity than 1 IU/mL FVIII, though an equivalence value could not be assigned as this was above the range of the standard curve. No activity was observed for the SI‐Emi anti‐FX homodimer. The BS‐027125 achieved a maximum of 14.3 ± 0.2% FVIII‐like activity while further increases in concentration resulted in reduced activity. The BS‐027125 anti‐FIXa homodimer also showed FVIII‐like activity, but only 35% of the bsAb's activity. The BS‐027125 anti‐FX homodimer showed no activity. Similar data and analyses are shown for FVIIIa, SI‐Emi, and BS‐027125 using the commercially available BIOPHEN FVIII:C kit in Figure S4. Notably, the FVIII‐equivalence determined for each antibody varies not only between assays but also within the same assay depending on the dilution protocol used (Table S1). This behavior can be explained by the differences in relative molar concentrations of FVIII versus antibodies to FIXa and FX, which has been previously discussed by Lenting and colleagues.10 Consistent between assays, however, are the nonparallel dose responses of the bsAbs compared to FVIII.
Figure 2.
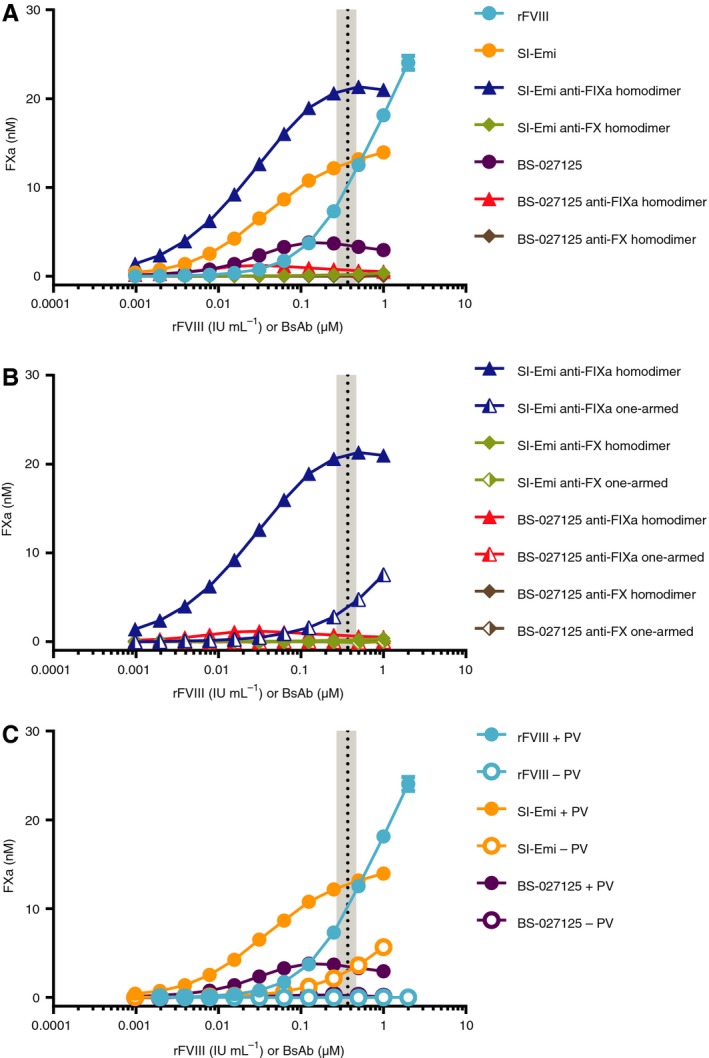
Chromogenic assay for FXa generation by rFVIII, SI‐Emi, and BS‐027125. A, Dose response of rFVIII, SI‐Emi bsAb, SI‐Emi anti‐FIXa and anti‐FX homodimers, BS‐027125 bsAb, and BS‐027125 anti‐FIXa and anti‐FX homodimers. B, Comparison of one‐armed or homodimer versions of SI‐Emi anti‐FIXa and anti‐FX antibodies, and BS‐027125 anti‐FIXa and anti‐FX antibodies. C, FXa generation activity of rFVIII, SI‐Emi, and BS‐027125 in the absence of phospholipid vesicles. Data are mean ± SD. Black dotted line and gray shaded area are mean and range of steady‐state emicizumab concentration of patients receiving emicizumab prophylaxis,9 respectively. anti‐FIXa, anti‐FX, bsAB, bispecific antbody; BS‐027125, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027; FXa, factor Xa; rFVIII, recombinant factor VIII; SI‐Emi, sequence identical emicizumab
Owing to the unexpected activity observed for the homodimers described, we also generated one‐armed antibody controls of the anti‐FIXa and anti‐FX arms. While the BS‐027125 anti‐FIXa one‐armed antibody had no activity, the SI‐Emi anti‐FIXa one‐armed antibody maintained the ability to generate FXa, reaching 26.7 ± 0.2% of FVIII‐like activity at 1 μmol/L (Figure 2B). Neither anti‐FX one‐armed antibody showed activity.
We also assessed whether bsAbs could support FXa generation in the absence of phospholipid vesicles. At concentrations within plasma levels achieved during prophylaxis with emicizumab in humans (0.37 ± 0.1 μmol/L),9, 17 SI‐Emi unexpectedly demonstrated 20 ± 0.2% FVIII‐like activity in the absence of phospholipid vesicles (Figure 2C) whereas BS‐027125 showed 1.3 ± 0.2% FVIII‐like activity.
3.4. Thrombin generation
Finally, we evaluated the activity of bsAbs in TGA. In FXIa‐triggered TGA, rFVIII shortened the lag time and increased the peak relative to baseline HemA plasma (Figure 3A). SI‐Emi and BS‐027125 also generated thrombin, albeit to different extents. To determine the FVIII‐equivalence of these bsAbs, nonlinear regressions of each parameter versus rFVIII concentration were applied, utilizing ranges of rFVIII that represent the linear portion of the standard curves. For some parameters, a high and a low FVIII standard curve could be drawn. Here, we analyzed FVIII‐equivalence against a single standard curve (Figure S2C).
Figure 3.
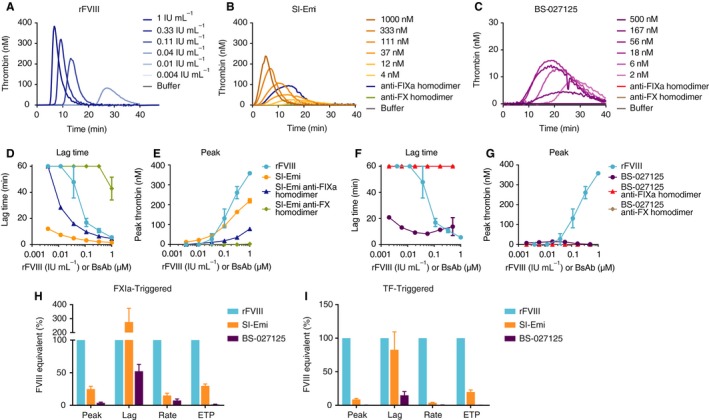
Thrombin generation of rFVIII, and bsAbs SI‐Emi, and BS‐027125. A‐C, Representative thrombograms of FXIa‐ triggered TGA by increasing concentrations of rFVIII, SI‐Emi, and BS‐027125, respectively. Activity of anti‐FIXa and anti‐FX homodimers of SI‐Emi (1 μmol/L, both) and BS‐027125 (20 nmol/L, both) are also shown. D‐E, Dose response by lag time, D, and peak thrombin, E, of SI‐Emi and SI‐Emi anti‐FIXa and anti‐FX homodimers compared to rFVIII. F‐G, Dose response by lag time, F, and peak thrombin, G, of BS‐027125 and BS‐027125 anti‐FIXa and anti‐FX homodimers compared to rFVIII. H‐I, FVIII‐equivalence of SI‐Emi and BS‐027125 across four parameters of TGA in FXIa‐triggered experiments, H, and TF‐triggered experiments, I. Data are mean ± SEM. anti‐FIXa, anti‐factor IXa; anti‐FX, anti‐factor X; bsAbs, bispecific antibodies; BS‐027125, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027; FXIa, factor Xia; rFVIII, recombinant factor VIII; SI‐Emi, sequence identical emicizumab; TF, tissue factor; TGA, thrombin generation assay
The SI‐Emi bsAb shortened the lag time to 275 ± 98% FVIII‐like activity, yet increased peak thrombin to 25 ± 4% of FVIII‐like activity (Figure 3D‐E). The SI‐Emi anti‐FIXa homodimer retained substantial activity by both lag time (up to 120 ± 12% FVIII‐like activity) and peak (up to 6.4 ± 2.8% FVIII‐like activity) while the SI‐Emi anti‐FX homodimer only had a decreased lag time at the highest concentration tested (Figure 3D‐E). The BS‐027125 also shortened the lag time, up to 52 ± 10% of FVIII‐like activity, while it increased peak thrombin up to 3.6 ± 1.1%. No detectable activity was observed with the anti‐FIXa or anti‐FX homodimers of BS‐027125. Other parameters of TGA, rate and endogenous thrombin potential (ETP), also showed variability in FVIII‐equivalence (Figure 3H and Figure S5) for both SI‐Emi and BS‐027125. Notably, in TF‐triggered TGA (Figure 3I and Figures S2D and S6), both SI‐Emi and BS‐027125 showed different FVIII‐equivalence across different parameters compared with FXIa‐triggered TGA.
In TGA, phospholipid vesicles provide the necessary surface for formation of coagulation complexes. The composition of such vesicles has been shown to modulate the activity of these complexes.18, 19, 20 For example, to mimic activated platelet surfaces better, incorporation of PE into PC:PS vesicles has been shown to enhance FVIII binding to PS resulting in greater FXa generation.19 Because bsAbs do not bind phospholipids, we tested how phospholipid vesicle composition and concentration affect the activity of FVIII, SI‐Emi, and BS‐027125 in FXIa‐triggered TGA. Both FVIII and BS‐027125 showed significant yet different sensitivities to phospholipid vesicle composition, while SI‐Emi was not affected (Figure 4A). Because of these effects, determination of FVIII‐equivalence is impacted. For example, when fitting a peak thrombin standard curve to rFVIII with PC:PS or PC:PE:PS vesicles (Figure 4B), the slope is greater on PC:PS than on PC:PE:PS vesicles (270 nmol/L vs. 84 nmol/L thrombin per IU/mL rFVIII, respectively), resulting in eight‐fold higher FVIII‐like activity for SI‐Emi on PC:PS vesicles than on PC:PE:PS vesicles (Figure 4C). The BS‐027125 was similarly affected, producing higher apparent FVIII‐like activity on PC:PS vesicles than on PC:PE:PS vesicles.
Figure 4.
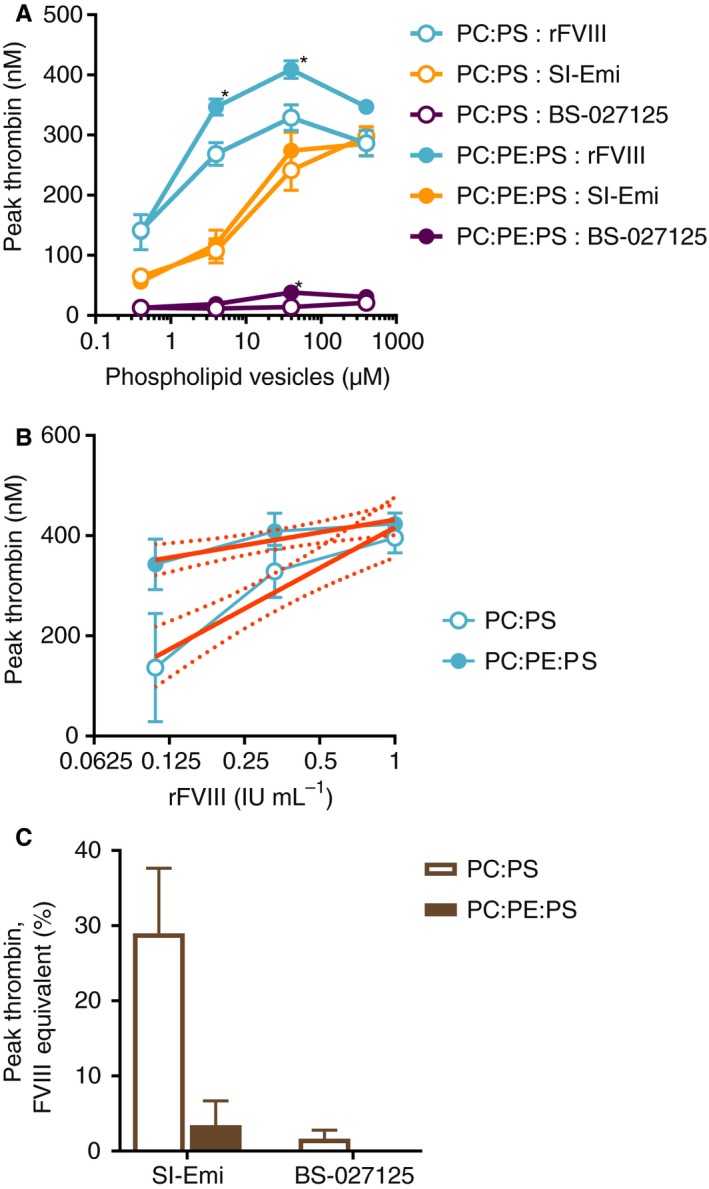
Effect of phospholipid vesicle composition and concentration on FVIII‐equivalence of SI‐Emi and BS‐027125 in FXIa‐triggered TGA. A, Peak thrombin of rFVIII (0.3 IU/mL), SI‐Emi (300 nmol/L), and BS‐027125 (20 nmol/L) with increasing concentrations of PC:PS (80:20%) or PC:PE:PS (40:40:20%) vesicles. *P < 0.04 by t test. B, Interpolation of rFVIII peak thrombin standard curves with 95% confidence intervals on 40 μmol/L PC:PS or PC:PE:PS vesicles. C, Peak thrombin FVIII‐equivalence of SI‐Emi and BS‐027125 on 40 μmol/L PC:PS or PC:PE:PS vesicles. Data are mean ± SEM. BS‐027125, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027; FVIII, factor VIII; FXIa, factor XIa; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; rFVIII, recombinant factor VIII; SI‐Emi, sequence identical emicizumab; TGA, thrombin generation assay
3.5. Summary of FVIII‐equivalence
In Table 1, we summarized the relative FVIII‐equivalence of SI‐Emi, BS‐027125, and their respective anti‐FIXa and anti‐FX homodimers across each assay, including two parameters of TGA for both FXIa‐triggers and TF‐triggers. Depending on the assay and parameter used, the apparent FVIII‐equivalent activity of SI‐Emi (at concentrations achieved at steady state for patients on emicizumab prophylaxis), ranged from 9% to outside the range of the standard curve (>150%), while peak activity of BS‐027125 ranged from <1% to 134%.
Table 1.
Relative FVIII‐equivalence of bsAbs across assays and assay parameters
| Chromogenic | aPTT | TGA (FXIa trigger) | TGA (TF trigger) | |||
|---|---|---|---|---|---|---|
| Lag time | Peak | Lag time | Peak | |||
| rFVIII | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ |
| SI‐Emia | ++++ | ++++++++ | +++++ | ++ | +++ | + |
| Anti‐FIXa homodimer | +++++ | ++++ | ++++ | + | Not determined | Not determined |
| Anti‐FX homodimer | + | + | + | + | Not determined | Not determined |
| BS‐027125b | + | ++++ | +++ | + | ++ | + |
| Anti‐FIXa homodimer | + | No activity | No activity | No activity | Not determined | Not determined |
| Anti‐FX homodimer | No activity | No activity | No activity | No activity | Not determined | Not determined |
Abbreviations: anti‐FIXa, anti‐factor IXa; anti‐FX, anti‐factor X; aPTT, activated partial thromboplastin time;bs‐Abs, bispecific antibodies; BS‐027125, anti‐FIXa and anti‐FX antibodies BS‐125 and BS‐027; rFVIII, recombinant factor VIII; SI‐Emi, sequence identical emicizumab; TF, tissue factor; TGA, thrombin generation assay.
Relative activity at concentrations achieved at steady state in patients on emicizumab prophylaxis.
Relative activity at concentrations giving peak activity.
4. DISCUSSION
While bsAbs are intended to mimic FVIII function, they do not inherently share all features of FVIII. Assay design, reagents used, and variability across different parameters within the same assay further complicate equation of non‐FVIII replacement molecules to FVIII and limit our ability to predict protection from bleeding and safety from thrombosis.
The aPTT‐based one‐stage assay clearly has limitations when comparing bsAbs with FVIII. The time required for FVIII activation and the subsequent activity of FVIIIa are combined into one measurement, clotting time. The activity of bsAbs, which do not require activation, is therefore overestimated in this assay, independent of activator (Figure S3).16 This is particularly apparent in the aPTT results from the HAVEN‐1 Phase 3 trial in inhibitor patients.21 After first dose of emicizumab, clotting times were maximally shortened and showed no dose response. Indeed, this overestimation was noted during both nonhuman primate22 and early human studies23 with emicizumab. Because bsAbs have a kinetic advantage, the one‐stage assay cannot be used to determine whether a patient is protected from bleeding or at risk of thrombosis. Moreover, use of all other aPTT‐based assays, including activated protein C resistance and Bethesda assays, is warned against in emicizumab prescribing information. This limitation has additional practical implications when the need for monitoring is high, for instance, during immune tolerance induction. If a FVIIIa mimetic bsAb is utilized in patients undergoing ITI, neither inhibitory titers nor the restoration of endogenous FVIII activity can be accurately measured with existing aPTT‐based assays. For emicizumab, an alternate, bovine chromogenic‐based Bethesda assay has been developed24 to circumvent this challenge. In addition, Nogami et al recently reported the discovery of anti‐emicizumab antibodies that can be used to neutralize emicizumab activity and restore the utility of the standard Bethesda assay.25 Unfortunately, the bovine chromogenic Bethesda assay is not broadly used and anti‐emicizumab antibodies are not yet commercially available. Furthermore, the use of bypassing agents to treat breakthrough bleeds in patients concurrently on a bsAb therapy and ITI presents an unprecedented safety concern that requires thoughtful evaluation.
In contrast to aPTT‐based assays, the two‐stage chromogenic assay has a FVIII preactivation step that allows a more direct comparison of the FXa‐generating activity of bsAbs to FVIIIa. However, with only FIXa present, specificity for FIXa versus FIX is not considered, nor the potential for bsAbs to interfere with other areas of coagulation where FIX(a) and FX(a) play a role (e.g. TF/FVIIa complex, prothrombinase complex, etc.). Therefore, an assay containing only FIXa and no FIX likely overestimates the activity of bsAbs that lack FIXa specificity. Additionally, the obvious lack of parallelism between FVIII and bsAb dose‐response curves observed in both our modified chromogenic assay and the commercially available BIOPHEN FVIII:C kit (Figures S2 and S4 and Table S1) clearly shows their procoagulant activities do not scale proportionally with one another and limit the use of this assay for assignment of FVIII‐equivalence. However, both bsAbs do show linear responses within certain dose ranges indicating that the chromogenic assay could be used as a surrogate to estimate the amount of bsAb in plasma. Still, this would require use of a self‐standard to eliminate dilution effects (discussed previously) and would not inform on the relationship between bsAb concentration and true procoagulant activity.
As a purified system, the chromogenic assay enables targeted mechanistic studies to better understand the specific interactions and effects of a bsAb. For example, the SI‐Emi anti‐FIX(a) homodimer promoted FXa generation more effectively than the SI‐Emi bsAb. One possible explanation may be cross‐reactivity of the anti‐FIX(a) arm with FX in this avid format, effectively making the anti‐FIX(a) homodimer bispecific. Consistent with this possibility, Kitazawa et al recently reported that emicizumab showed a higher binding response to FX than to FIX in an avid ELISA.14 However, the monovalent SI‐Emi anti‐FIX(a) one‐armed antibody still promoted FXa generation as well, suggesting there may be an additional unknown effect of the antibody on FIXa. Furthermore, it is surprising that SI‐Emi showed phospholipid‐independent activity given its low affinities for its antigens in solution (~1 μmol/L). These data are notable since phospholipid binding is an important regulatory mechanism for clotting. Emicizumab was previously tested for phospholipid‐independent activity and although none was measured, it was only tested up to 300 nmol/L, just below the steady‐state plasma level of emicizumab.14, 26 While BS‐027125 showed only minimal phospholipid‐independent activity, its activity in the presence of phospholipids was also substantially lower. Additional work will be needed to determine whether SI‐Emi, emicizumab, or other bsAbs can generate FXa away from exposed PS in more physiologic plasma‐based or blood‐based assays, and ultimately in vivo.
The most global assay in this study is TGA as it reflects all stages of clotting (initiation, amplification, propagation, and inhibition) and informs on both the kinetics and total amount of thrombin generated. The most standardized trigger reagent for TGA is TF,27 yet FXIa has become popular among groups developing non‐FVIII therapies28, 29 since it appears to be sensitive to FVIII levels. The FXIa‐triggered TGA is likely driven by higher than normal amounts of FIXa,10 which may bias the assay toward the tenase complex. Although this may increase sensitivity to FVIII and bsAbs alike, it may not model in vivo hemostasis, particularly in joints since FXI‐deficient patients generally do not experience joint bleeds. Further, we have shown that varying phospholipid composition can affect FVIII standard curves. Thus, reagent choice impacts FVIII‐equivalence. Finally, it is unclear which TGA parameter best reflects protection from bleeding. Lag time, with a FXIa trigger, gives an advantage to bsAbs since they do not require activation. The ETP may be misleading, because when and where thrombin is generated may be as important as how much is generated. This complexity highlights the limits of our ability to assign and monitor hemostatic potential by TGA.
Potency assignment of FVIII molecules to the WHO FVIII international standard relies on parallel dose responses.30 However, mechanistic differences between FVIII and bsAbs lead to nonparallelism that precludes determination of FVIII equivalence. For emicizumab, target dosing was based on ex vivo TGA correlations between porcine FVIII and emicizumab in an acquired HemA model in nonhuman primates.17, 22 Once in humans, the primary endpoint for efficacy became annualized bleeding rate for treated bleeds, but the connection between TGA parameters and human clinical efficacy was not explored further.9 In short, no single assay or assay parameter has been shown to correlate with therapeutic efficacy or safety, making it difficult to determine optimal dosing strategies for prophylactic treatment with bsAbs as well as safe and efficacious methods for treating breakthrough bleeds, trauma, or surgery.31 Our inability to assign FVIII‐equivalence to bsAbs and the higher incidence of thrombotic complications observed when combining bypassing agents with emicizumab9 compared to rFVIIa32, 33 or aPCCs34, 35 alone warrant improved monitoring and further study to understand the precise mechanisms of FVIIIa mimetic bsAbs.36, 37 Control of bleeds during surgery or trauma is an area of great concern. To ensure the patient neither bleeds nor suffers a thrombotic event it is crucial to be able to assess and monitor the procoagulant activity associated with bsAbs, alone and in combination with FVIII or bypassing agents. Discovery of more clinically relevant measures, such as clot quality, as well as the development of bsAb‐tailored assays and new animal models, are needed to fill these gaps.
While FVIIIa mimetic bsAbs have the potential to change the treatment paradigm for HemA patients, the field needs to expand our capabilities to assess and predict the efficacy and safety of these molecules more accurately. We must also continue to discuss and educate on the challenges and limitations of using existing metrics to characterize non‐FVIII therapies. This is particularly true if additional bsAbs are developed in the future since all will have unique properties (affinity, specificity, etc.) that will affect their behaviors differently in existing assays.
In short, bsAbs differ from FVIII in several important features making direct comparison to FVIII in existing assays challenging. The apparent procoagulant activities of bsAbs vary substantially between assays routinely used to assess the activity of FVIII, and a single FVIII‐equivalent activity value cannot be assigned to a non‐FVIII molecule with the currently available tools.
CONFLICTS OF INTEREST
The authors are employees of Sanofi, in Waltham, MA.
AUTHOR CONTRIBUTIONS
N. C. Leksa and M.M. Aleman designed and performed experiments and wrote the manuscript. A. G. Goodman and D. Rabinovich designed and performed experiments and edited the manuscript. R. Peters and J. Salas oversaw the study and edited the manuscript. All authors approved the final version.
Supporting information
ACKNOWLEDGMENTS
Funding for this study was provided by Bioverativ, a Sanofi company.
Leksa NC, Aleman MM, Goodman A, Rabinovich D, Peters R, Salas J. Intrinsic differences between FVIIIa mimetic bispecific antibodies and FVIII prevent assignment of FVIII‐equivalence. J Thromb Haemost. 2019;17:1044–1052. 10.1111/jth.14430
Manuscript handled by: Robert Gosselin and Pieter Reitsma
Final decision: Robert Gosselin and Pieter Reitsma, 12 March 2019
REFERENCES
- 1. Peters R, Harris T. Advances and innovations in haemophilia treatment. Nat Rev Drug Discov. 2018;17:493–508. [DOI] [PubMed] [Google Scholar]
- 2. Shima M, Lillicrap D, Kruse‐Jarres R. Alternative therapies for the management of inhibitors. Haemophilia. 2016;22(suppl 5):36–41. [DOI] [PubMed] [Google Scholar]
- 3. Peyvandi F, Garagiola I, Biguzzi E. Advances in the treatment of bleeding disorders. J Thromb Haemost. 2016;14:2095–106. [DOI] [PubMed] [Google Scholar]
- 4. Leissinger CA. Advances in the clinical management of inhibitors in hemophilia A and B. Semin Hematol. 2016;53:20–7. [DOI] [PubMed] [Google Scholar]
- 5. Chowdary P, Lethagen S, Friedrich U, Brand B, Hay C, Abdul Karim F, et al. Safety and pharmacokinetics of anti‐TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. 2015;13:743–54. [DOI] [PubMed] [Google Scholar]
- 6. Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377:819–28. [DOI] [PubMed] [Google Scholar]
- 7. Sehgal A, Barros S, Ivanciu L, Cooley B, Qin J, Racie T, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21:492–7. [DOI] [PubMed] [Google Scholar]
- 8. Waters EK, Sigh J, Friedrich U, Hilden I, Sorensen BB. Concizumab, an anti‐tissue factor pathway inhibitor antibody, induces increased thrombin generation in plasma from haemophilia patients and healthy subjects measured by the thrombin generation assay. Haemophilia. 2017;23:769–76. [DOI] [PubMed] [Google Scholar]
- 9. Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809–18. [DOI] [PubMed] [Google Scholar]
- 10. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130:2463–8. [DOI] [PubMed] [Google Scholar]
- 11. Recommended International Nonproprietary Names : List 75. WHO Drug Information: World Health Organization, 2016;30:111‐3. [Google Scholar]
- 12. Sampei Z, Igawa T, Soeda T, Okuyama‐Nishida Y, Moriyama C, Wakabayashi T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE. 2013;8:e57479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu TY, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS‐based, high‐throughput selection and analytical tool. Protein Eng Des Sel. 2013;26:663–70. [DOI] [PubMed] [Google Scholar]
- 14. Kitazawa T, Esaki K, Tachibana T, Ishii S, Soeda T, Muto A, et al. Factor VIIIa‐mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117:1348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kitchen S, Signer‐Romero K, Key NS. Current laboratory practices in the diagnosis and management of haemophilia: a global assessment. Haemophilia. 2015;21:550–7. [DOI] [PubMed] [Google Scholar]
- 16. Adamkewicz JI, Soeda T, Kotani N, Calatzis A, Levy G. Effect of emicizumab (ACE910) – a humanized bispecific antibody mimicking FVIIIa cofactor function – on coagulation assays commonly in use for monitoring of hemophilia A patients (abstract). Haemophilia. 2017;23:4.27990784 [Google Scholar]
- 17. Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII‐mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374:2044–53. [DOI] [PubMed] [Google Scholar]
- 18. Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–1052. [PubMed] [Google Scholar]
- 19. Gilbert GE, Arena AA. Phosphatidylethanolamine induces high affinity binding sites for factor VIII on membranes containing phosphatidyl‐L‐serine. J Biol Chem. 1995;270:18500–5. [DOI] [PubMed] [Google Scholar]
- 20. Tavoosi N, Davis‐Harrison RL, Pogorelov TV, Ohkubo YZ, Arcario MJ, Clay MC, et al. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286:23247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adamkewicz JI, Schmitt C, Calatzis A, Young G, Negrier C, Callaghan M, et al. Pharmacodynamic data and coagulation biomarkers in persons with hemophilia A (PwHA) with inhibitors: results from the HAVEN 1 Emicizumab (ACE910) phase 3 study. Res Pract Thromb Haemost. 2017;1:162.30046686 [Google Scholar]
- 22. Muto A, Yoshihashi K, Takeda M, Kitazawa T, Soeda T, Igawa T, et al. Anti‐factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12:206–13. [PubMed] [Google Scholar]
- 23. Uchida N, Sambe T, Yoneyama K, Fukazawa N, Kawanishi T, Kobayashi S, et al. A first‐in‐human phase 1 study of ACE910, a novel factor VIII‐mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamkewicz JKB, Steinbuesch D, Calatzis A. Measurement of FVIII Inhibitor Titer Using a Chromogenic Bethesda Assay (CBA) in the Presence of Emicizumab (ACE910), a Humanized Bispecific Antibody Mimicking FVIIIa Cofactor Function. 2017 Scientific Symposium of the Hemostasis and Thrombosis Research Society Scottsdale, AZ: Haemophilia, 2017, 23 (Suppl. S3), 1044–49. [Google Scholar]
- 25. Nogami K, Soeda T, Matsumoto T, Kawabe Y, Kitazawa T, Shima M. Routine measurements of factor VIII activity and inhibitor titer in the presence of emicizumab utilizing anti‐idiotype monoclonal antibodies. J Thromb Haemost. 2018;16:1383–90. [DOI] [PubMed] [Google Scholar]
- 26. Kitazawa T, Igawa T, Sampei Z, Muto A, Kojima T, Soeda T, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–4. [DOI] [PubMed] [Google Scholar]
- 27. Dargaud Y, Wolberg AS, Gray E, Negrier C, Hemker HC; Subcommittee on Factor VIII, Factor IX, and Rare Coagulation Disorders . Proposal for standardized preanalytical and analytical conditions for measuring thrombin generation in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2017;15:1704–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muto A, Yoshihashi K, Takeda M, Kitazawa T, Soeda T, Igawa T, et al. Anti‐factor IXa/X bispecific antibody ACE910 prevents joint bleeds in a long‐term primate model of acquired hemophilia A. Blood. 2014;124:3165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waters EK, Hilden I, Sorensen BB, Ezban M, Holm PK. Thrombin generation assay using factor XIa to measure factors VIII and IX and their glycoPEGylated derivatives is robust and sensitive. J Thromb Haemost. 2015;13:2041–52. [DOI] [PubMed] [Google Scholar]
- 30. Hubbard AR, Dodt J, Lee T, Mertens K, Seitz R, Srivastava A, et al.; Factor VIII and Factor IX Subcommittee of The Scientific and Standardisation Committee of The International Society on Thrombosis and Haemostasis . Recommendations on the potency labelling of factor VIII and factor IX concentrates. J Thromb Haemost. 2013;11:988–1052. [DOI] [PubMed] [Google Scholar]
- 31. Tripodi A, Chantarangkul V, Novembrino C, Peyvandi F. Advances in the treatment of hemophilia: implications for laboratory testing. Clin Chem. 2019;65:254–62. [DOI] [PubMed] [Google Scholar]
- 32. Abshire T, Kenet G. Safety update on the use of recombinant factor VIIa and the treatment of congenital and acquired deficiency of factor VIII or IX with inhibitors. Haemophilia. 2008;14:898–902. [DOI] [PubMed] [Google Scholar]
- 33. O'Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295:293–8. [DOI] [PubMed] [Google Scholar]
- 34. Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA): 10‐year compilation of thrombotic adverse events. Haemophilia. 2002;8:83–90. [DOI] [PubMed] [Google Scholar]
- 35. Gomperts ED. FEIBA safety and tolerability profile. Haemophilia. 2006;12:14–1052. [Google Scholar]
- 36. Shapiro AD, Mitchell IS, Nasr S. The future of bypassing agents for hemophilia with inhibitors in the era of novel agents. J Thromb Haemost. 2018;16:2362–74. [DOI] [PubMed] [Google Scholar]
- 37. Al‐Samkari H, Croteau SE. Shifting landscape of hemophilia therapy: implications for current clinical laboratory coagulation assays. Am J Hematol. 2018;93:1082–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials