

Abstract
Background
Chronic granulomatous disease (CGD) is a rare disease in China, and very little large‐scale studies have been conducted to date. We aimed to investigate the clinical and genetic features of CGD in Chinese pediatric patients.
Methods
Pediatric patients with CGD from Beijing Children's Hospital, Capital Medical University, China, were enrolled from January 2006 to December 2016.
Results
A total of 159 pediatric patients with CGD were enrolled. The median age of clinical onset was 1.4 months, and 73% (116/159) had clinical onset symptoms before the 1 year of age. The most common site of invasion was the lungs. The lymph nodes, liver, and skin were more frequently invaded in X‐linked (XL) CGD patients than in autosomal recessive (AR) CGD patients (P < 0.05). Approximately 64% (92/144) of the pediatric patients suffered from abnormal response to BCG vaccination. The most frequent pathogens were Aspergillus and Mycobacterium tuberculosis. Gene analysis indicated that 132 cases (89%, 132/147) harbored CYBB pathogenic variants, 7 (5%, 7/147) carried CYBA pathogenic variants, 4 (3%, 4/147) had NCF1 pathogenic variants, and 4 (3%, 4/147) had NCF2 pathogenic variants. The overall mortality rate in this study was 43%, particularly the patients were males, with CYBB mutant and did not receive HSCT treatment.
Conclusions
Chronic granulomatous disease is a rare disease affecting Chinese children; however, it is often diagnosed at a later age, and thus, the mortality rate is relatively high. The prevalence and the severity of disease in XL‐CGD are higher than AR‐CGD.
Keywords: children, China, chronic granulomatous disease, clinical, genetics
Key Message.
Chronic granulomatous disease (CGD) is a rare disease in China, and very little large‐scale studies have been conducted to date. We conducted this study to describe the clinical and genetic features of pediatric CGD in China during the past ten years in order to provide scientific and useful data for this disease. A total of 159 pediatric patients with CGD were enrolled. We found in China CGD patients were often diagnosed at a later age and thus the mortality rate is relatively high. The prevalence and the severity of disease in XL‐CGD is higher than AR‐CGD. For male Chinese patients with recurrent infection within one year of age or with BCG disease, CGD should be considered.
Abbreviations
- AR
autosomal recessive
- CGD
chronic granulomatous disease
- GVHD
graft‐versus‐host disease
- NADPH
nicotinamide adenine dinucleotide phosphate oxidase
- XL
X‐linked
1. INTRODUCTION
Chronic granulomatous disease (CGD) is a primary immune deficiency disease that was first described in 1954.1 It is caused by a defect in the nicotinamide adenine dinucleotide phosphate oxidase (NADPH) complex, resulting in the inability of phagocytic cells to kill catalase‐positive and catalase‐negative microorganisms such as Staphylococcus and Aspergillus species.2 NADPH oxidase is composed of five subunits encoded by the CYBB, CYBA, NCF1, NCF2, and NCF4 genes, respectively. Pathogenic variants in the CYBB gene cause X‐linked recessive CGD (XL‐CGD), whereas those involving the other genes lead to autosomal recessive CGD (AR‐CGD).3, 4 However, very little large‐scale study on the genetic features of pediatric cases of CGD in China has been conducted to date. Therefore, we conducted this study with large possible population of Chinese pediatric patients to systematically analyze the clinical and genetic features of CGD to better understand its etiology, epidemiology, and progression.
2. METHODS
We retrospectively reviewed patients <18 years old with CGD from Beijing Children's Hospital, Capital Medical University, Beijing, China. CGD was suspected based on its clinical features and was confirmed by dihydrorhodamine‐1,2,3 (DHR) flow cytometry assays and/or DNA analysis.5 The study design was in accordance with the Helsinki Declaration and was approved by the Beijing Children's Hospital Ethics Committee. Informed consent was obtained from the parents or legal guardians of the pediatric participants.
DHR assays were performed as previously described.6, 7 Stimulation index (SI) was the ratio of geometric mean of DHR from phorbol‐12‐myristate‐14‐acetate (PMA)‐stimulated samples to that from unstimulated samples of the same individual. Genomic DNA was extracted from peripheral blood cells using Mini Blood Kit (Qiagen, Duesseldorf, Germany). Genes relevant to CGD (ie, CYBB, CYBA, NCF1, and NCF2) were amplified using a PCR Kit (Qiagen, Duesseldorf, Germany), followed by sequence analysis. The entire DNA sequences, including bordering intron sequences, of CYBB, CYBA, and NCF2 were analyzed. However, for NCF1, we only sequenced exon 2, to detect patients with c.75_76delGT. The primers of all the genes are available in the Supplementary Material.
All data were analyzed using SPSS ver. 19.0 (International Business Machines Corporation, Chicago, IL, USA). Continuous data were expressed as the mean ± standard deviation or range. Proportions were compared using the chi‐square or Fisher's exact test, and continuous variables within two groups were compared using the independent t test. All tests were two‐tailed, and P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Demographic features
A total of 159 pediatric CGD patients were enrolled in this study, with a male‐to‐female ratio of 7.8:1. The patients were from enrolled 26 provinces of China; the majority were from northern China (62%, 98/159). Approximately 79% (33/159) of the CGD patients were diagnosed after 2010. The average age at diagnosis was 2.4 ± 3 years, ranging from 20 days to 14 years, and the median age was 1.3 years. Nearly 56% (89/159) of the pediatric patients were diagnosed after 1 year of age. The mean age of clinical onset was 7.2 ± 16.8 months (range: 5 days to 11 years), and the median age was 1.4 months. The age of clinical onset of 56 patients (35%, 56/159) was during the neonatal period, whereas that of 116 patients (73%, 116/159) was before 1 year of age. Approximately 21% (33/159) of the patients had family history of CGD. The mean course of hospitalization was 18.0 ± 13.9 days.
Additionally, in our study, 132 pediatric patients had XL‐CGD and 15 with AR‐CGD. Most of the pediatric patients with XL‐CGD were males, whereas the majority of pediatric patients with AR‐GCD were females (P < 0.05; Table 1). The average age at diagnosis for those with XL‐CGD (2.1 ± 2.5 years) was younger than those with AR‐CGD (4.1 ± 4.6 years; t = −2.7, P = 0.007). The mean age of clinical onset in XL‐CGD (6.3 ± 13.3 months) was earlier than that in AR patients (16.9 ± 34.2 months; t = −2.2, P = 0.025).
Table 1.
Clinical features of Chinese pediatric patients with CGD
| Clinical features |
Total %(n/N) |
XL %(n/N) |
AR %(n/N) |
P |
|---|---|---|---|---|
| Male | 89 (141/159) | 99 (131/132) | 40 (6/15) | 0.00 |
| Family history | 21 (33/159) | 20 (27/132) | 20 (3/15) | 0.98 |
| Clinical onset symptoms | ||||
| Fever | 59 (92/155) | 58 (76/131) | 60 (9/15) | 0.85 |
| Cough | 30 (47/155) | 29 (38/131) | 40 (6/15) | 0.33 |
| BCGitis | 10 (16/155) | 11 (14/131) | 13 (2/15) | 0.94 |
| Pustular eruption | 8 (12/155) | 8 (11/131) | 7 (1/15) | 0.81 |
| Perianal abscess | 6 (9/155) | 6 (8/131) | 7 (1/15) | 0.93 |
| Diarrhea | 6 (9/155) | 5 (7/131) | 7 (1/15) | 0.84 |
| Lympholitis | 5 (8/155) | 6 (8/131) | 0 (0/15) | — |
| Cutaneous abscess | 3 (5/155) | 4 (5/131) | 0 (0/15) | — |
| Omphalitis | 1 (1/155) | 1 (1/131) | 0 (0/15) | — |
| Invaded sites | ||||
| Lung | 97 (139/144) | 96 (116/121) | 100 (15/15) | 0.43 |
| Lymph nodes | 57 (82/144) | 63 (76/121) | 33 (5/15) | 0.00 |
| Liver | 48 (69/144) | 53 (64/121) | 27 (4/15) | 0.02 |
| GI | 43 (62/144) | 47 (57/121) | 27 (4/15) | 0.18 |
| Spleen | 37 (53/144) | 40 (49/121) | 27 (4/15) | 0.15 |
| Skin | 37 (53/144) | 42 (51/121) | 13 (2/15) | 0.04 |
| Perianal abscess | 26 (38/144) | 30 (36/121) | 13 (2/15) | 0.22 |
| Diarrhea | 19 (27/144) | 19 (23/121) | 20 (3/15) | 0.84 |
| Oral ulcer | 15 (22/144) | 17 (20/121) | 13 (2/15) | 0.48 |
| Septicemia | 15 (21/144) | 13 (16/121) | 20 (3/15) | 0.38 |
| CNS | 7 (10/144) | 8 (10/121) | 0 (0/15) | — |
| Thrush | 6 (9/144) | 7 (8/121) | 0 (0/15) | — |
| Bone | 6 (9/144) | 7 (8/121) | 7 (1/15) | 0.93 |
| Myocarditis | 6 (8/144) | 6 (7/121) | 7 (1/15) | 0.84 |
| UTI | 3 (5/144) | 3 (4/121) | 7 (1/15) | 0.48 |
| Abnormal response to BCG vaccination | 64 (92/144) | 60 (73/121) | 60 (9/15) | 0.95 |
| Regional abnormalities | 42 (61/144) | 45 (54/121) | 40 (6/15) | 0.54 |
| Axillary lymph node calcification | 26 (34/144) | 24 (29/121) | 20 (3/15) | 0.44 |
3.2. Clinical features
In our study, the most common initial infection site and common invasion site (manifesting with pneumonia) were the lungs (Table 1). Aspergillus infections were relatively common, and the second one was Tuberculosis (TB) infection. Eight pediatric patients were diagnosed with disseminated TB disease. The lymph nodes, liver, and skin were more readily invaded in the CYBB mutant groups than in the non‐CYBB mutant groups (P < 0.05). Perianal abscess was another common clinical manifestation in CGD pediatric patients, accounting for 26.4% (38/144) of the cases. In addition, 52 patients presented with anemia, 22 with malnutrition, three with recurrent suppurative otitis media, and one with inflammatory bowel disease, allergic purpura, or bone marrow invasion, respectively. Of the 159 patients, 144 patients received BCG vaccination at birth or later. Of these, 64% (92/144) of the pediatric patients developed abnormal responses to BCG vaccination, including regional abnormalities (61 cases), axillary lymph node calcification (34 cases), and disseminated BCG (four cases). Compared to the CYBB and non‐CYBB mutant groups, no difference in abnormal responses to BCG vaccination was observed in these two groups (P > 0.05). The other symptoms are shown in Table 1.
3.3. Laboratory findings
A total of 128 pediatric patients underwent blood testing. Among these, 84% (107/128) of the patients had leukocytosis with neutrophils predominant, of which 31% (33/107) of the patients had WBC counts >20 × 109/L. No cases of neutropenia were identified. A total of 116 (91%) pediatric patients had an elevated C‐reactive protein (CRP) levels, and 83% (75/90) showed increased erythrocyte sedimentation rate (ESR). Meanwhile, decreased hemoglobin levels, ranging from 44 to 90 g/L, were observed in 39.8% (51/128) of the patients. There was no difference in the prevalence of leukocytosis, increased CRP and ESR, and decreased hemoglobin between the XL‐CGD and AR‐CGD groups (P > 0.05). Furthermore, five pediatric patients had elevated eosinophil counts, ranging from 8.3% to 19.1%, and two cases had thrombocytopenia (from 68 × 109/L to 88 × 109/L), although these returned to normal once the infection was controlled.
Serum IgA, IgG, IgM, and IgE levels were increased in most pediatric CGD patients in our study, namely 32% (41/128), 60% (78/128), 47% (60/128), and 57% (73/128), respectively. One patient had significantly elevated serum IgE levels (>2000 IU/L). The number of CD4+ T cells, CD8+ T cells, B cells, and NK cells of most of the patients was within the normal range. The neutrophil oxidative function (range: 0.6‐22.8, average of 2.8 ± 4.2; normal range: ≧63.5) in CGD patients was significantly lower than in healthy controls. And, CGD patients with younger age of onset were associated with lower neutrophil oxidative function (r = 0.332, P = 0.010). Additionally, neutrophil oxidative function in the CYBB groups (average of 2.1 ± 3.3) was lower than that in non‐CYBB groups (average of 7.8 ± 6.6; Z = 3.57, P = 0.0004; Figure 1).
Figure 1.
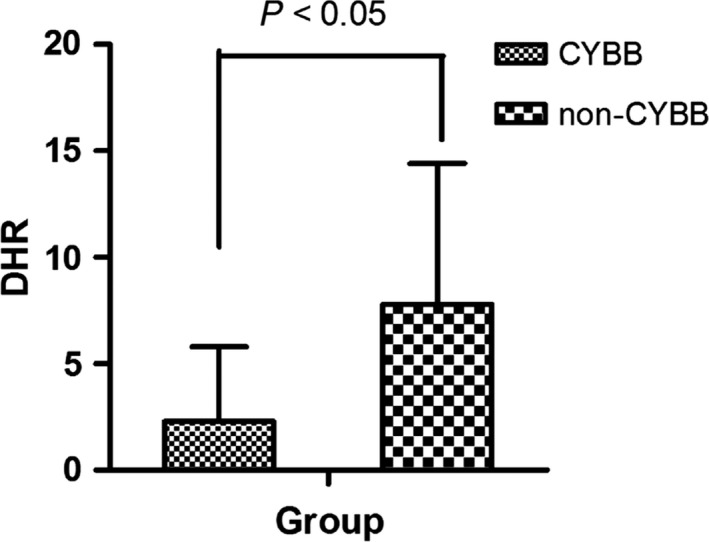
Neutrophil oxidative function results in different groups
Approximately 43% (55/129) of the patients tested positive for pathogens, which consisted of 31 different types. The most common pathogen was Aspergillus spps. (27%, 17/64), and the types were A fumigates (15), A niger (1), and A nidulans (1). The detailed pathogens and infection sites are presented in Table 2.
Table 2.
Distribution of pathogens in Chinese CGD pediatric patients
| Pathogens | Total patients number % (n/N) | Specimens | ||||||
|---|---|---|---|---|---|---|---|---|
| Blood (n) | Sputum (n) | Tissue (n) | BALF (n) | Urine (n) | Stool (n) | Pus (n) | ||
| Aspergillus | 27 (17/64) | 2 | 11 | 2 | 2 | 0 | 0 | 2 |
| Tuberculosis | 20 (13/64) | 0 | 2 | 10 | 2 | 0 | 0 | 0 |
| Klebsiella pneumonia | 11 (7/64) | 0 | 4 | 0 | 1 | 0 | 0 | 2 |
| Candida albicans | 9 (6/64) | 0 | 3 | 0 | 2 | 0 | 0 | 1 |
| Pseudomonas aeruginosa | 8 (5/64) | 1 | 4 | 0 | 0 | 0 | 0 | 0 |
| Staphylococcus aureus | 8 (5/64) | 5 | 0 | 5 | 0 | 0 | 0 | 0 |
| Burkholderia cepacia | 8 (5/64) | 3 | 1 | 0 | 0 | 1 | 0 | 0 |
| EBV | 6 (4/64) | 2 | 0 | 0 | 2 | 0 | 0 | 0 |
| Escherichia coli | 6 (4/64) | 0 | 2 | 0 | 1 | 0 | 0 | 2 |
| CMV | 5 (3/64) | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| Acinetobacter baumanii | 3 (2/64) | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| Aerobacter cloacae | 3 (2/64) | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| Enterococcus | 3 (2/64) | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| Staphylococcus | 3 (2/64) | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Others | 27 (17/64) | 1 with Lophomonas blattarum (BALF), Chrysemonas Luteola (lung tissue), Candida krusei (stool), Candida lusitaniae (sputum), Candida glabrata (sputum), mucor (skin), typhoid (blood and stool), hemolytic staphylococci (blood), Streptococcus pneumonia (sputum), Klebsiella Pneumoniae (sputum), Alternaria Nees (sputum), Brucella (blood), Coagulase‐negative staphylococcus (urine), Branhamellacatarrhalis (sputum), salmonella (blood and stool), human herpesvirus 6 (blood), or nocardiosis (sputum), respectively | ||||||
3.4. Genetic analysis
Genetic testing of pediatric patients with CGD showed that 132 cases (89%, 132/147) harbored pathogenic variants in the CYBB gene, seven patients (5%, 7/147) in the CYBA gene, four patients (3%, 4/147) in the NCF1 gene, and 4 (3%, 4/147) patients in the NCF2 gene. Additionally, in six patients, no gene mutations were detected. In the CYBB variant group, 97 different variants were tested and 24 were novel, and 91% (32/35) of the patients’ mothers were carriers of the CYBB variant (see the attached files). In most cases, the detected CYBB variants were located within exons (78%, 103/132), particularly in exon 7 (27%, 28/105) and exon 9 (19%, 20/105), and rarely seen in exon 8 (2%, 2/105). There were 36 nonsense variants, 36 splice site variants (including 14 suspected splice error), 34 missense variants, 22 deletions (including two large gene deletions [exons 1‐13] and two with McLeod syndrome), and seven insertions. P2 was a male, but we found two different mutations (c.1A>G; c.6delG) in his only X chromosome. A female carrier of XL‐CGD (P71) was found to have a heterozygous mutation in her CYBB gene (c.1212delG), but she had abnormal results of DHR (SI = 1) and CGD clinical symptoms (see the Supplementary Material).
In the CYBA mutant patients, five nonsense variants, two missense variants, two deletions, and one splicing defect variant were identified. The mutation of c.75_76delGT in the NCF1 gene was identified in four patients, wherein two patients had a family history of CGD. The NCF2 variants were identified in four patients: one of them had a homozygous mutation and three had compound heterozygous mutations (Table 3).
Table 3.
Summary of non‐CYBB gene mutations in Chinese CGD pediatric patients
| Patients | Mutant gene | Type | Exon/Intron | Nucleotide change | Mutation type | Predicted codon change | Carrier | SI | References |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CYBA | Compound heterozygous |
Exon 1 Intron 1 |
c.7C>T; c.59 −2A>Ga | Missense/splice site | p.Gln3Ter; skipped exon 2? | Unidentified | — | 30; this study |
| 2 | CYBA | Homozygous | Exon 1 | 7C>T | Missense | p.Gln3Ter | Unidentified | — | 30 |
| 3 | CYBA | Homozygous | Exon 1 | 7C>T | Missense | p.Gln3Ter | Unidentified | — | 30 |
| 4 | CYBA | Homozygous | Exon 5 | c.496_521del26insCa | Deletion | p.Pro156fsTer181 | Unidentified | 4.67 | This study |
| 5 | CYBA | Compound heterozygous | Exons 1, 6 | c.7C>T; c.479delC | Missense/deletion | p.Gln3Ter; p.Pro160ArgfsTer29 | Unidentified | — | 30 |
| 6 | CYBA | Compound heterozygous | Exon 1 | c.1A>G; c.7C>T | Missense | p.Met1Val; p.Gln3Ter | Unidentified | 6 | 30 |
| 7 | CYBA | Compound heterozygous | Exons 1, 3 | del exon 1; c.152T>G | Deletion/missense | skipped exon 1; p.Leu51Arg | Father | — | 6 |
| 8 | NCF1 | Homozygous | Exon 2 | c.75_76delGT | Deletion | p.Tyr26HisfsTer24 | Unidentified | 1 | 4, 6 |
| 9 | NCF1 | Homozygous | Exon 2 | c.75_76delGT | Deletion | p.Tyr26Hisfs Ter24 | Unidentified | 11.65 | 4, 6 |
| 10 | NCF1 | Homozygous | Exon 2 | c.75_76delGT | Deletion | p.Tyr26Hisfs Ter24 | Mother | 1.33 | 4, 6 |
| 11 | NCF1 | Homozygous | Exon 2 | c.75_76delGT | Deletion | p.Tyr26Hisfs Ter24 | Mother, sister | 7.67 | 4, 6 |
| 12 | NCF2 | Compound heterozygous | Exon 1 | c.196 C>T c.304C>T | Missense | p.Arg66Ter Arg102Ter | Unidentified | 1 | 30 |
| 13 | NCF2 | Homozygous | Exon 13 | c.1180T>Ga | Missense | p.Tyr394Asp | Unidentified | 22.1 | This study |
| 14 | NCF2 | Compound heterozygous | Exons 1, 13 | c.304C>T; c.1180T>Ga | Missense | p.Arg102Ter; p.Tyr394Asp | Unidentified | 11.62 | 30 |
| 15 | NCF2 | Compound heterozygous | Exons 1, 13 | c.172‐174delAAG; c.1180T>Ga | Deletion/missense | p.Lys58del; p.Tyr394Asp | Unidentified | 11.44 | 30 |
Novel mutation. —, not examined.
3.5. Treatment and outcomes
In our study, all CGD patients were advised to receive prophylactic treatment with cotrimoxazole (sulfamethoxazole [SMZ] 24 mg/kg/d or trimethoprim [TMP] 4‐6 mg/kg/d, divided twice daily), IFN‐gamma (50 μg/m2/dose, subcutaneous injections three times a week), and itraconazole (patient age <13 years or <50 kg, 100 mg/d; >13 years or >50 kg, 200 mg/d) to prevent bacterial and fungal infections. However, most patients did not persist with prophylactic treatment because of side effects and financial reasons. If TB infection diagnosis occurred or developed BCG disease, the patients would receive anti‐TB treatments. To date, 25 patients with CYBB mutant type have undergone hematopoietic stem cell transplantation (HSCT), with five patients awaiting HSCT. The median age of pediatric patients that received HSCT was 4.1 years (range: 4 months to 12 years).
At the time of manuscript preparation, 43% (52/121) of the CGD patients have died, with a median age of death was 2 years, and the average age of death was 3.20 ± 3.80 years (range: 1 month to 16 years). At the age of 12, nearly 50% of the pediatric CGD patients were dead (Figure 2). Most of them died of infectious disease, including pneumonia, septicemia (four patients were infected with Burkholderia cepacia, two were infected with Aspergillus, but most had unknown pathogens), two patients died from graft‐versus‐host disease (GVHD), one patient died of post‐transplantation infection, and one died of hydrocephalus.
Figure 2.
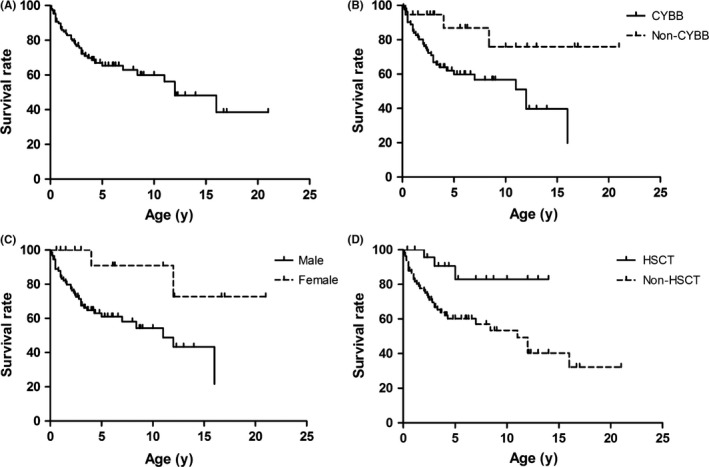
Survival rate of CGD pediatric patients in different groups. A, Survival curves for pediatric patients with CGD; B, Survival curves for male and female group. Significant differences were observed (P = 0.0153); C, Survival curves for CYBB and non‐CYBB groups. Significant differences were observed (P = 0.0184); and D, Survival curves for HSCT and non‐HSCT groups. Significant differences were observed (P = 0.0137)
The mortality rate was higher in non‐HSCT patients (51%, 49/96) than HSCT patients (12% (3/25) (χ2 = 6.07, P < 0.05; Figure 2). Among the HSCT patients, three died of GVHD, infection, and (or) multi‐organ dysfunction. Three of the HSCT patients currently had GVHD, affecting the lungs, skin, and gut. Additionally, a higher mortality rate was observed among males (48%, 49/103) and patients with CYBB gene variants than females (17%, 3/18) and those with non‐CYBB variants (20%, 4/20) (χ2 = 5.45, P < 0.05; χ2 = 5.04, P < 0.05). In addition, neutrophil oxidative function in the deceased patients (average of 1.5 ± 1.6) was lower than in alive patients (average of 3.9 ± 4.9) groups (t = −2.59, P = 0.0131). Finally, 69 patients were alive (including 22 transplanted patients), with a median age of 4.9 years and average age of 6.13 ± 4.54 years (range: 4 months to 21 years).
4. DISCUSSION
Chronic granulomatous disease is a rare and fatal inherited immunodeficiency disease. Among Europeans, the prevalence of CGD is about 1 in 250 000 individuals. However, in China, no data relating to CGD prevalence are available. Most CGD patients might be under‐diagnosed in China due to lack of knowledge. However, with significant developments in medicine, more cases have been diagnosed recently in China. In our study, approximately more than 70% of cases were diagnosed nearly within the past 5 years.
Chronic granulomatous disease patients were predominantly male and usually present symptoms at an early age. In our study, the median age of clinical onset in CGD patients was 1.4 months, but the age of CGD diagnosis in China is often later. However, in the United States and Europe, the age of onset symptoms is older than in China,8, 9 which may be because Chinese children receive BCG vaccination routinely right after birth. Additionally, we found that the age of XL‐CGD clinical onset was younger than that of AR‐CGD, which is similar to the findings of earlier study.10 In addition, previous studies have shown that 30%‐40% of CGD patients have a family history of CGD, whereas in our study, only 21% had a positive family history of CGD. In our study, nearly 64% of the CGD pediatric patients suffered from abnormal responses to BCG vaccination, which was similar to Zhou et al's study.11 They reported a cohort of 169 CGD patients exposed to BCG vaccination and showed a high incidence (59%) of BCG infection in China.11 In Hong Kong and Iran, 47% and 56% of CGD patients develop BCGitis.12, 13 Apart from CGD, BCG infections are reported in other primary immunodeficiency diseases (PID), including severe combined immunodeficiency syndrome (SCID), Mendelian susceptibility to mycobacterial diseases (MSMD), and X‐linked hyper‐IgM syndrome (XHIM).14, 15 For this reason, there is restriction on the BCG vaccination for the PID patients and newborns with a family history of these PID.
In our study, lungs were the most common invasion sites (manifested as pneumonia) during the course of CGD. However, the prevalence of pneumonia among CGD patients in some European countries is lower (eg, Italy, 47%). Aspergillus was the most common pathogen in our study. Previous studies have also reported that Aspergillus was the most common pathogen in CGD patients with pneumonia, with an incidence of 41% in the United States and 18% in Europe.3, 8 TB is also another common pathogen of Chinese CGD patients. In the United States and Israel, deep abscesses, including liver, spleen, and brain, or pulmonary abscesses are the major manifestations, accounting for 46% to 76% of the symptoms, which differs from the results of this study.3, 9 In Europe, the lungs (66% of patients), skin (53%), lymph nodes (50%), gastrointestinal tract (48%), and liver (32%) were the most frequently infected sites.8 Perianal abscesses are a typical symptom of CGD patients; however, this is non‐specific, and in our study, the rate was higher than that of the United Kingdom and Ireland (15%), Italy (8%), and Europe (21%).8, 16 Bortoletto et al3 analyzed CGD patients and reported that XL‐CGD has a significantly higher rate of perirectal abscess, gastric obstruction, and urinary tract obstruction. However, we did not obtain similar results, which might be because of the smaller size of the study population. Further investigations should thus be performed. Additionally, we found one patient with inflammatory bowel disease (IBD) similar to that reported by a previous study and pointed out that the prevalence of CGD‐IBD increases with age, the symptoms are usually mild, and patients show good responses to treatment.17
In this study, we found that most CGD patients have hypergammaglobulinemia. Similar results were also demonstrated by the previous studies.18, 19, 20 The positive detection rate of pathogens was low in our study, which was similar to that observed in a previous study.8 This might be due to previous long‐term antibiotic/antifungal prophylactic therapy. Catalase‐positive bacteria and fungi such as staphylococci, Serratia, Burkholderia, and Aspergillus are the major pathogens in CGD patients.21, 22 In our study, Aspergillus and TB were the two most common pathogens, whereas in the United States, the most common pathogens were Staphylococcus aureus, Serratia marcescens, Burkholderia cepacia, Nocardia, and Aspergillus.3 Burkholderia cepacia was not common in our study, but the patient with this pathogen infection showed poor prognosis.
Previous studies have shown that nearly 70% of the patients with CGD harbored CYBB pathogenic variants (ie, XL‐CGD),3, 8 whereas in our study, 89% of the patients carried CYBB variants. However, studies conducted in Arab/North African countries and Turkey showed that AR‐CGD accounts for 71% and 67% of the cases, respectively, more common than XL‐CGD.8, 12 This difference in prevalence rates may be attributable to the fact that consanguineous marriage is a common practice in these countries. To date, we detected CYBB variants in CGD patients, mostly involving exons 7 and 9, similar to that of previous studies.8, 23 Mutation analysis remains an important tool for genetic counseling and prenatal diagnosis.24 Except for the CYBB variant, a previous study has shown that there are at least three types of AR‐CGD, which include NCF1 (20%), NCF2 (5%), and CYBA (5%).8 However, in our study, we found AR‐CGD is rare in Chinese pediatric patients, and CYBA is more common than the other two types. In NCF1, we only searched for the most common mutation (a GT homozygous deletion), and four patients had this mutation. The mutation in six CGD patients was not determined in our study, but we lost the chance to do further analysis.
The first study of CGD demonstrated a mortality rate of >50% by age 6 and almost 65% mortality by age 7.25 With advances in medicine, decrease in mortality rate has been achieved.3, 26 A recent study reported a median survival of 25 years in 50% of cases after diagnosis.16 In Europe, the median life expectancy of CGD is relatively high (37.8 years for XL‐CGD patients and 49.6 years for AR‐CGD patients).8 However, in our study, the mortality rate of Chinese pediatric CGD patients is 43%, and the median age of death is 2 years. By age 12, the mortality rate is 50%. Additionally, we found that the mortality rate in patients with XL‐CGD, males, or non‐HSCT treatment was higher than those with AR‐CGD, females, or HSCT treatment, respectively. A previous study also demonstrated a higher mortality rate among the XL‐CGD patients than in AR‐CGD patients.8 Antibacterial prophylaxis and immunotherapy with interferon‐γ are helpful in reducing the frequency of infection in CGD patients.9, 27 However, Martire et al demonstrated that IFN‐γ treatment did not reduce the incidence of infections.16 In our study, all patients were recommended for prophylaxis using these methods. Unfortunately, not all of the patients followed this recommendation for different reasons. BCG vaccine‐induced disease is the risk factor for CGD patients. Timely and effective treatment is very important. A previous study showed recombinant human IFN‐gamma treatment together with anti‐TB drugs could provide better control of BCGosis.28 In our study, we treated these patients with IFN‐gamma and anti‐TB drugs (isoniazid, rifampicin, and ethambutol), but because of very few follow‐up patients, we did not get significant statistics. Additionally, we suggested to treat vaccinated CGD patients with anti‐TB drugs in our study, even though there were no symptoms. But no guidelines were demonstrated for the treatment method of these patients until now. It still needs further study. Åhlin and Fasth29 pointed out that bone marrow or stem cell transplantation could reduce mortality of CGD patients. In our study, 25 patients received transplantation, and 22 of them lived a longer life. However, some patients also might die of GVHD and infection after transplantation. Gene therapy may be another curative option. This can correct CGD by gene transfer, and now, some clinical trials have provided some insights into the application of gene therapy for CGD patients.
5. CONCLUSIONS
China continues to face numerous challenges in the diagnosis of this disease. For male Chinese patients with recurrent infection within 1 year of age or with BCG disease, CGD should be considered. Early detection and treatment (including antibacterials and antifungals, and later, if possible, HSCT) may significantly reduce mortality.
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
Li‐Wei Gao, Bao‐Ping Xu, and Jian‐Xin He: Dr Gao, Dr Xu, and Dr He conceptualized and designed the study, drafted the initial manuscript, and approved the final manuscript as submitted. Qing‐Qin Yin, Yue‐Juan Tong, Jin‐Gang Gui, Xiu‐yun Liu, Xue‐li Feng, Ju Yin, Jun Liu, Jun Liu, Yan Guo, Yao Yao, Kun‐Ling Shen, Yu‐Lung Lau, and Zai‐Fang Jiang: Dr Yin, Dr Tong, Dr Gui, Dr Liu, Dr Feng, Dr Yin, Dr Liu, Dr Yan Guo, Dr Yao, Dr Shen, Dr Liu, and Dr Jiang designed the data collection instruments, and coordinated and supervised data collection, critically reviewed the manuscript, and approved the final manuscript as submitted. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Supporting information
ACKNOWLEDGMENTS
We thank the patients and their families, whose cooperation was essential for collection of the data used in this study. We thank all members for helping accomplishing this work.
Gao L‐W, Yin Q‐Q, Tong Y‐J, et al. Clinical and genetic characteristics of Chinese pediatric patients with chronic granulomatous disease. Pediatr Allergy Immunol. 2019;30:378–386. 10.1111/pai.13033
Funding information
This study is supported by Beijing Municipal Science & Technology Commission (NO. Z181100001718061).
Edited by: Marina Atanaskovic‐Markovic
Contributor Information
Bao‐Ping Xu, Email: xubaopingbch@163.com.
Jian‐Xin He, Email: hejianxin1970@126.com.
REFERENCES
- 1. Janeway CA, Craig J, Davidson M, et al. Hypergammaglobulinemia associated with severe, recurrent and chronic non‐specific infection. Am J Dis Child. 1954;88:388‐392. [Google Scholar]
- 2. Roos D, de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol. 2014;175:139‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bortoletto P, Lyman K, Camacho A, Fricchione M, Khanolkar A, Katz BZ. Chronic granulomatous disease: a large, single‐center US experience. Pediatr Infect Dis J. 2015;34:1110‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Köker MY, Camcıoğlu Y, van Leeuwen K, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immunol. 2013;132:1156‐1163.e5. [DOI] [PubMed] [Google Scholar]
- 5. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan‐American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190‐197. [DOI] [PubMed] [Google Scholar]
- 6. Xu H, Tian W, Li SJ, et al. Clinical and molecular features of 38 children with chronic granulomatous disease in mainland china. J Clin Immunol. 2014;34:633‐641. [DOI] [PubMed] [Google Scholar]
- 7. Sun J, Wang Y, Liu D, et al. Prenatal diagnosis of X‐linked chronic granulomatous disease by percutaneous umbilical blood sampling. Scand J Immunol. 2012;76:512‐518. [DOI] [PubMed] [Google Scholar]
- 8. van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS ONE. 2009;4:e5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Oliveira‐Junior EB, Zurro NB, Prando C, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 latin American patients: first report from the LASID registry. Pediatr Blood Cancer. 2015;62:2101‐2107. [DOI] [PubMed] [Google Scholar]
- 10. Wolach B, Gavrieli R, de Boer M, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. 2017;92:28‐36. [DOI] [PubMed] [Google Scholar]
- 11. Zhou Q, Hui X, Ying W, et al. A cohort of 169 chronic granulomatous disease patients exposed to BCG vaccination: a retrospective study from a single center in Shanghai, China (2004–2017). J Clin Immunol. 2018;38:260‐272. [DOI] [PubMed] [Google Scholar]
- 12. Lee PP, Chan KW, Jiang L, et al. Susceptibility to mycobacterial infections in children with X‐linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27:224‐230. [DOI] [PubMed] [Google Scholar]
- 13. Fattahi F, Badalzadeh M, Sedighipour L, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31:792‐801. [DOI] [PubMed] [Google Scholar]
- 14. Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N. Bacillus Calmette‐Guérin (BCG) complications associated with primary immunodeficiency diseases. J Infect. 2012;64:543‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bustamante J, Picard C, Boisson‐Dupuis S, Abel L, Casanova JL. Genetic lessons learned from X‐linked Mendelian susceptibility to mycobacterial diseases. Ann N Y Acad Sci. 2011;1246:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martire B, Rondelli R, Soresina A, et al. Clinical features, long‐term follow‐up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 2008;126:155‐164. [DOI] [PubMed] [Google Scholar]
- 17. Angelino G, De Angelis P, Faraci S, et al. Inflammatory bowel disease in chronic granulomatous disease: An emerging problem over a twenty years' experience. Pediatr Allergy Immunol. 2017;28:801‐809. [DOI] [PubMed] [Google Scholar]
- 18. Hanoglu D, Ozgür TT, Ayvaz D, Köker MY, Sanal O. Chronic granulomatous disease presenting with hypogammaglobulinemia. J Investig Allergol Clin Immunol. 2011;21:310‐312. [PubMed] [Google Scholar]
- 19. Patiroglu T, Gungor HE, Lazaroski S, Unal E. Chronic granulomatous disease with markedly elevated IgE levels mimicking hyperimmunoglobulin E syndrome. Acta Microbiol Immunol Hung. 2013;60:155‐162. [DOI] [PubMed] [Google Scholar]
- 20. Esenboga S, Emiralioglu N, Cagdas D, et al. Diagnosis of interstitial lung disease caused by possible hypersensitivity pneumonitis in a child: think CGD. J Clin Immunol. 2017;37:269‐272. [DOI] [PubMed] [Google Scholar]
- 21. Chiriaco M, Salfa I, Di MG, Rossi P, Finocchi A. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. 2016;27:242‐253. [DOI] [PubMed] [Google Scholar]
- 22. Ben‐Ari J, Wolach O, Gavrieli R, Wolach B. Infections associated with chronic granulomatous disease: linking genetics to phenotypic expression. Expert Rev Anti Infect Ther. 2012;10:881‐894. [DOI] [PubMed] [Google Scholar]
- 23. Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: X‐linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45:246‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kulkarni M, Gupta M, Madkaikar M. Phenotypic prenatal diagnosis of chronic granulomatous disease: a useful tool in the absence of molecular diagnosis. Scand J Immunol. 2017;86:486‐490. [DOI] [PubMed] [Google Scholar]
- 25. Johnston RB, McMurry JS. Chronic familial granulomatosis. Report of five cases and review of the literature. Am J Dis Child. 1967;114:370‐378. [DOI] [PubMed] [Google Scholar]
- 26. Martinez CA, Shah S, Shearer WT, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. 2012;129:176‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med. 1991;324:509‐516. [DOI] [PubMed] [Google Scholar]
- 28. Ying W, Sun J, Liu D, et al. Clinical characteristics and immunogenetics of BCGosis/BCGitis in Chinese children: a 6 year follow‐up study. PLoS ONE. 2014;9:e94485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Åhlin A, Fasth A. Chronic granulomatous disease ‐ conventional treatment vs. hematopoietic stem cell transplantation: an update. Curr Opin Hematol. 2015;22:41‐45. [DOI] [PubMed] [Google Scholar]
- 30. Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. 2010;44:291‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials