

Abstract
Background
Atopic dermatitis (AD) is one of the most common skin diseases with a multifactorial etiology. Mutations leading to loss of skin barrier function are associated with the development of AD with group 2 innate lymphoid cells (ILC2) promoting acute skin inflammation. Filaggrin‐mutant (Flgft/ft) mice develop spontaneous skin inflammation accompanied by an increase in skin ILC2 numbers, IL‐1β production, and other cytokines recapitulating human AD. Here, we investigated the role of ILC2, effector cytokines, inflammasome activation, and mast cell function on the development of chronic AD‐like inflammation in mice.
Methods
Mice with a frameshift mutation in the filaggrin gene develop spontaneous dermatitis. Flgft/ft mice were crossed to cell‐ or cytokine‐deficient mouse strains, or bred under germ‐free conditions. Skin inflammation was scored, and microbiome composition was analyzed. Skin protein expression was measured by multiplex immunoassay. Infiltrating cells were analyzed by flow cytometry.
Results
Wild‐type and Flgft/ft mice significantly differ in their microbiome composition. Furthermore, mutant mice do not develop skin inflammation under germ‐free conditions. ILC2 deficiency did not ameliorate chronic dermatitis in Flgft/ft mice, which was also independent of IL‐4, IL‐5, IL‐9, IL‐13, IL‐17A, and IL‐22. Inflammation was independent of NLRP3 inflammasome activation but required IL‐1β and IL‐1R1‐signaling. Mechanistically, IL‐1β promoted hyperactivation of IL‐1R1‐expressing mast cells. Treatment with anti‐IL‐1β‐antibody alleviated dermatitis exacerbation, while antibiotic intervention ameliorated dermatitis in neonatal mice but not in adults with established inflammation.
Conclusions
In summary, we identified a critical role for the microbiome and IL‐1β mediating chronic inflammation in mice with an impaired skin barrier.
Keywords: atopic dermatitis, filaggrin, IL‐1β, innate lymphoid cells, microbiome
Filaggrin deficiency leads to skin dysbiosis early after birth altering adult immune responses, while mice raised under germ‐free conditions remain disease‐free. NLR Family Pyrin Domain Containing 3‐independent processing of IL‐1 in the skin promotes atopic dermatitis (AD)‐like ILC2‐independent inflammation. IL‐1 deficiency or targeting IL‐1 by monoclonal antibodies ameliorates dermatitis. IL‐1R1‐expressing dermal mast cells are key responders to IL‐1, acquire a hyperactive phenotype, and promote AD‐like inflammation.

1. INTRODUCTION
Atopic dermatitis (AD) is a common eczematous pruritic disease with an onset at an early age, affecting up to 30% of children in the Western world. Etiology of AD is multifactorial including genetic predisposition, environmental factors, and immune status, leading to a high complexity in clinical presentation.1, 2, 3 Predisposing genetic factors for the development of AD include mutations in genes affecting the integrity of the skin barrier, such as mattrin (TMEM79) and filaggrin (filament aggregation protein, FLG).4, 5, 6 Filaggrin mutations were found in 30% of AD patients in Poland,7 China (26.0%),8 and Korea (15.7%),9 while healthy individuals had none. Importantly, filaggrin expression is downregulated in AD patients independent of their FLG genotype as a consequence of increased type 2 cytokines contributing to the aggravation of disease.10
We have previously separated and described the two mutated genes—Tmem79/mattrin and filaggrin—leading to the allergic skin phenotype of flaky tail mice.5, 11 Single mutant mice both have a defective skin barrier, and both spontaneously develop AD‐like inflammation. Pathogenesis in Tmem79ft/ft mice is dependent on adaptive immunity, while Flgft/ft mice develop dermatitis through innate immune cells.11 However, the mechanisms underlying inflammation are unclear.
As an atopic disorder, AD is classically considered a type 2‐driven immunopathology involving type 2 T helper (Th2) cells, interleukin (IL)‐4, IL‐5, IL‐9, and IL‐13, as well as IgE, mast cells, basophils, and eosinophils—with more recent data expanding this view to include Th17 and IL‐22 cellular responses in the genesis of AD.12 While T cells are promoting inflammation in certain instances,13, 14, 15 they are largely dispensable in the Flgft/ft model. Mast cells have long been associated with AD, and increased numbers are found in the skin of atopic patients.16 Upon activation by cytokines, FcεRI‐bound IgE, or pathogen‐ and danger‐associated molecular patterns, mast cells can release large amounts of pro‐inflammatory mediators, such as tumor necrosis factor (TNF).17, 18 Furthermore, increased numbers of group 2 innate lymphoid cells (ILC2) in the skin of Flgft/ft mice and patients with mutations in FLG 11 suggest a central role for ILC2 in genesis of skin inflammation in this model. ILC2 are potent innate regulators of type 2 immune responses19, 20, 21, 22, 23, 24 and have been shown to promote inflammation in a model of acute dermatitis induced by topical application of MC903 (calcipotriol, vitamin D3 analogue).25, 26, 27, 28 However, their role in spontaneous dermatitis in Flgft/ft mice is unknown.
Another hallmark of AD is skin dysbiosis, with a shift toward a pathogenic microbiome, in which beneficial commensals such as Propionibacteria or Staphylococcus epidermidis are displaced by other species such as S. aureus, and the patients’ overall skin microbiota diversity decreases.29, 30, 31, 32 Results from flaky tail mice (Flgft/ftTmem79ft/ft) suggested that the microbiota promotes upregulation of IL‐17A and the infiltration of neutrophils and eosinophils into the skin.33
Polymorphisms in members of the IL‐1 family of cytokines and their receptors are associated with skin disorders, such as cutaneous lupus erythematosus, psoriasis, and atopic dermatitis.34, 35 We have previously reported increased IL‐1α, IL‐1β, and IL‐1R1 expression in skin of Flgft/ft mice and AD patients with mutations in FLG.36
In the present study, we set out to investigate the mechanisms underlying AD‐like inflammation in Flgft/ft mice. We discovered that ILC2—while required for acute MC903‐induced dermatitis—were dispensable for spontaneous AD‐like inflammation in Flgft/ft mice with an impaired skin barrier. Instead, the development of skin inflammation was dependent on an interplay between microbiota, IL‐1β, and mast cells.
2. MATERIAL AND METHODS
2.1. Mice
The following mice were backcrossed onto the Flgft/ft (initially isolated from flaky tail mice, JR#9078, Jackson Laboratories, Bar Harbor, ME11) BALB/c background for >8 generations: Rag1−/− (JAX: 002216),37 Rag2−/−γc−/− 38 (JAX; 014593), Il7rCre,39 Roraflox,23 sg/sg mice,40 Il4KN2,41 Il5cer/cer,11 ll13−/−,42 Il9cit/cit,43 Il17a−/−,44 Il22−/−,45 Nlrp3−/−,46 Il1a−/−,47 Il1b−/− 47, Il1r1−/− (JAX:003245),48 Il18−/− (JAX:004130),49 Asc−/−,50 Aim2−/−,51 KitW‐sh/W‐sh (JAX:005051),52 and Il36r−/−.53 Mice were housed in a specific pathogen‐free facility, with irradiated diet and water ad libitum. Experiments under germ‐free conditions were conducted at the Instituto Gulbenkian De Ciência in Portugal. Animal experiments were approved by Trinity College Dublin BioResources and Instituto Gulbenkian de Ciência ethical review board and performed in compliance with EU Directive 2010/63/EU, Irish Medicine's Board and The Health Products Regulatory Authority.
2.2. Scoring of skin inflammation
Severity of skin inflammation was clinically scored (total range: 0‐12) by macroscopic diagnostic criteria as previously described.5 The total score is the sum of individual scores ranging from 0 to 3 (0, none; 1, mild; 2, moderate; 3, severe) that were applied to edema, erythema, scaling, and erosion.
2.3. Preparation of bone marrow–derived mast cells (BMMC) and stimulation
Bone marrow was isolated from the femur and tibia of donor mice and cultured in media (RPMI + 10% FBS + L‐glutamine + penicillin/streptomycin + HEPES + non‐essential amino acids) containing 10 ng/mL SCF and 10 ng/mL IL‐3 (R&D systems) for a total of 4 weeks with media changes twice a week.54, 55 3 × 105 mast cells/well were left untreated or were stimulated with plate‐bound anti‐FcεRIα (10 µg/mL, clone MAR‐1, Thermo Scientific) in the presence or absence of IL‐1β (10 ng/mL, R&D systems) for 24 hours. For adoptive transfers, BMMC were resuspended in sterile PBS. Mice received 1 × 106 mast cells via intradermal injection into the ear.
2.4. Microbiome analysis
Skin microbiome samples were acquired by exposing sterile swabs to the ear skin of Flgft/ft and wild‐type mice, as previously published.56 Mice were kept in the same or adjacent cages, looked after by the same person using the same products. Same surface area was sampled for all age‐ and sex‐matched mice. To avoid cross‐contamination, sterile gloves were changed between each sample collected. Samples were instantly frozen in liquid nitrogen, and 16S rRNA gene sequencing and microbiome analysis was performed by Second Genome (San Francisco, CA), as previously described.57
2.5. Axenic mouse model generation
Male and female Flgft/ft mice were shipped from Trinity College Dublin to the Instituto Gulbenkian De Ciência in Portugal and re‐derived by embryo transfer from a quarantine facility into SPF housing. Subsequent litters were generated by timed‐pregnancies. Fetuses were transferred to GF isolators and fostered by GF C3H mothers, as described in the relevant EMMA protocol (http://strains.emmane-t.org/protocols/GermFree_0902.pdf). Germ‐free and age‐matched SPF control litters were raised and maintained under strictly identical conditions (food, water, humidity, temperature), except the microbiological status.
2.6. Statistics
GraphPad Prism (version 7) was used to generate graphs and for statistical analysis. Area under curve (AUC), Student's t test, and ANOVA were used to determine statistical significance. P‐values < 0.05 were considered statistically significant.
Please refer to the Supporting Information for additional materials and methods.
3. RESULTS
3.1. Flgft/ft mice develop spontaneous atopic dermatitis–like skin inflammation
We sought to investigate the innate mechanisms that elicit inflammation in Flgft/ft mice. Flgft/ft mice develop spontaneous skin inflammation as neonates, with a second phase of overt inflammation—most prominent around the eyes and ears—progressing from 8 weeks of age evidenced by a constant increase in clinical severity (Figure 1A,B). The impaired skin barrier in Flgft/ft mice is represented by significantly increased transepidermal water loss (TEWL), and development of skin inflammation is accompanied by increased circulating IgE (Figure 1C,D). Skin histology of Flgft/ft mice shows dermal and epidermal thickening, scaling, and cellular infiltration into the skin (Figure 1E). Among skin infiltrating immune cells, we find significantly increased numbers of ILC2, eosinophils, basophils, Th2 cells, and also mast cells (Figure 1F)—similar to the localized immune cell repertoire in AD patients.11, 27, 58, 59, 60
Figure 1.
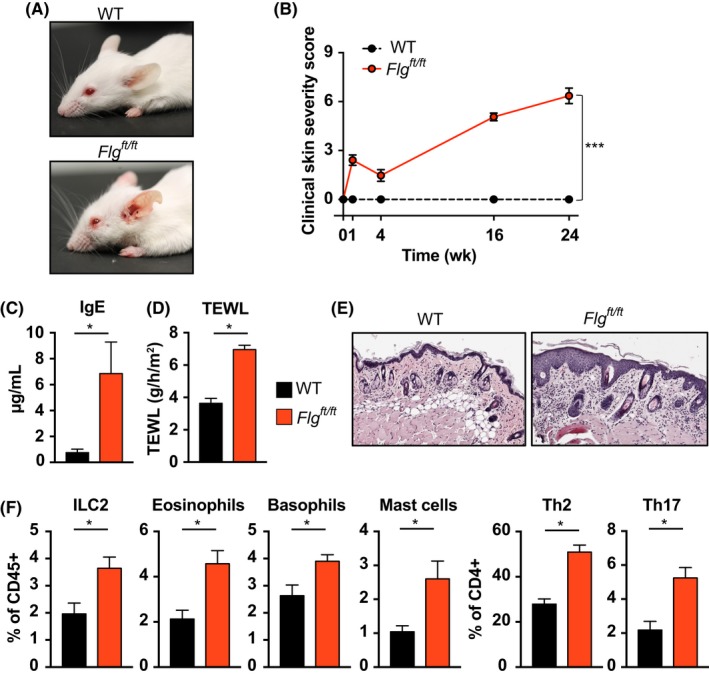
Flgft/ft mice develop spontaneous atopic dermatitis associated with increased cellular infiltration. A, Representative photographs of wild‐type Balb/c (WT, top) and filaggrin‐mutant Flgft/ft mice (bottom) at ten weeks of age. B, Macroscopic clinical scoring of Balb/c (black) and Flgft/ft (red). Graph shows the mean ± SEM from 20 mice per group. ***, P < 0.001, t test of AUC. C, Total serum IgE concentrations from adult wild‐type (black) and 12‐wk‐old Flgft/ft mice (red). Bars show the mean ± SEM of seven mice. *, P < 0.05. D, Transepidermal water loss (TEWL) at 12 wk in wild‐type (black) and Flgft/ft mice (red). Bars show the mean ± SEM of nine mice. *, P < 0.05. E, Representative photomicrograph of H&E‐stained skin from Flgft/ft (right) and wild‐type mice (left). 20x original magnification. (F) Frequencies of indicated cell types isolated from ear skin of wild‐type (black bars) and Flgft/ft mice (red bars) and analyzed by flow cytometry. Bars show the mean + SEM of 6‐8 mice per group of two independent experiments. *, P < 0.05
3.2. Pathogenic cutaneous microbiome promotes chronic inflammation in Flgft/ft mice
In order to investigate whether Flgft/ft mice faithfully reflect the dysbiosis apparent in the skin microbiome of AD patients, we analyzed their skin microbiota. 16S rRNA‐sequencing revealed a significantly altered microbiome in both adult and neonatal Flgft/ft mice compared to wild‐type animals kept in the same facility (Figure 2A). The defective skin barrier integrity in Flgft/ft mice led to decreased microbial diversity with pronounced overrepresentation of firmicutes (Figure 2B), including Staphylococcus species (Figure 2C). Strikingly, Flgft/ft mice raised in germ‐free (GF) conditions developed marked skin inflammation as neonates (Figure 2D,E) that resolved in adults (Figure 2F,G). Consistent with the lack of evident pathology in adult GF mice, serum IgE was reduced to WT level (Figure 2H). Expression of genes associated with AD (Il33, Il1a, Il1b) was significantly altered in the absence of microbiota (Figure 2I). Importantly, while Il33 and Il1a were downregulated independent of filaggrin deficiency, Il1b was significantly increased in the skin of 12‐week‐old adult Flgft/ft mice. Il1b expression was restored to WT levels when mice were kept under germ‐free conditions (Figure 2I). Similarly, treatment of pregnant Flgft/ft females and their litters with broad‐spectrum antibiotics (ABX) from day E14 to P21 significantly decreased clinical severity of skin inflammation in adult offspring at 12 weeks of age (Figure S1A). In contrast, when adult 8‐9‐week‐old Flgft/ft mice that had already developed skin inflammation were treated with ABX for 4 weeks, there was no amelioration of dermatitis (Figure S1B). These data indicate that the development of AD‐like skin inflammation in Flgft/ft mice is microbe‐mediated in neonatal stages, while in adult animals—once skin inflammation is established—antibiotic intervention cannot alter skin disease. However, the cellular events leading up to AD development in this mouse model remain unclear.
Figure 2.
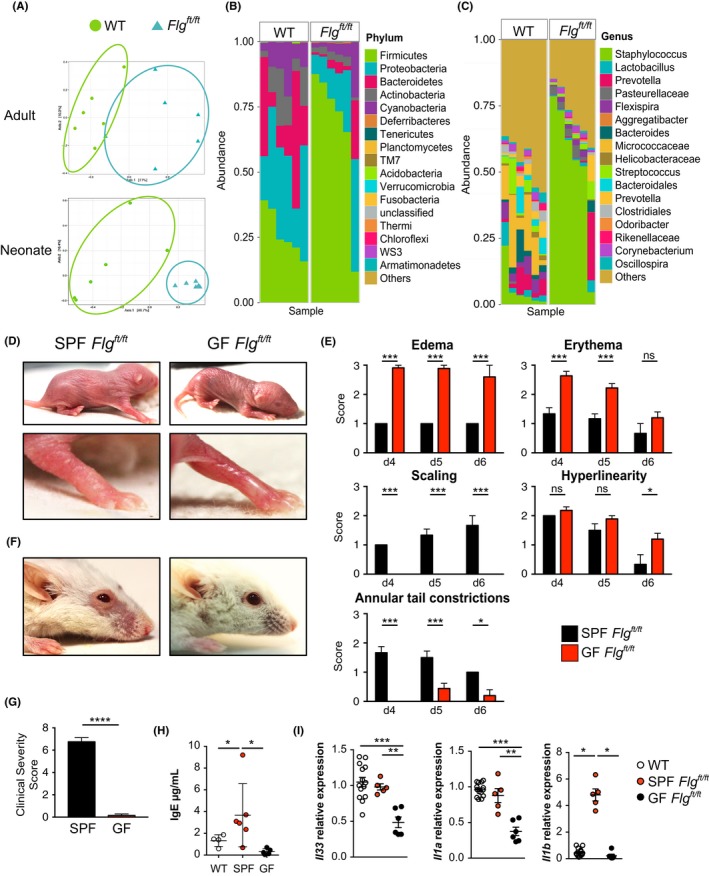
Pathogenic cutaneous microbiome promotes chronic inflammation in Flgft/ft mice. A, Principal component analysis of microbiome samples isolated from skin swaps of wild‐type (blue triangles) and Flgft/ft mice (green circles). Non‐lesional ear skin was swabbed at 12 wk (upper panel) and 3‐4 d (lower panel) of age. B,C, Mean relative abundance of bacterial phyla (B) and genera (C) of bacteria colonizing the skin of adult wild‐type and Flgft/ft mice. D, Representative photographs of neonatal Flgft/ft mice in a specific pathogen‐free (SPF, left panels) or germ‐free (GF, right panels) environment. E, Macroscopic scores for edema, erythema, scaling, hyperlinearity, and annular tail constrictions in neonatal SPF (black) and GF (red) Flgft/ft mice. Bar graphs show the mean + SEM of 9 mice per group. ns, not significant; *P < 0.05, ***P < 0.001. F, Photograph of adult Flgft/ft raised in an SPF (left) or GF (right) environment. Representative of 9 mice per group. G, Macroscopic scoring of SPF (black) and GF (red) Flgft/ft adult mice. ***P < 0.001. H, Serum IgE concentrations in SPF WT (open circles), SPF Flgft/ft (red), and GF Flgft/ft (black) adult mice. I, Relative quantification of Il33, Il1a, Il1b in the skin of adult WT (open circles) and adult Flgft/ft mice raised under SPF (red) or GF (black) conditions. Bars show the mean + SEM of 6 mice per group
3.3. Atopic dermatitis is independent of group 2 innate lymphoid cells
The appearance and severity of dermatitis in Flgft/ft mice is dependent on cells of the innate immune system (Figure 3A)11 with a significant increase in dermal ILC2 numbers in Flgft/ft mice (Figure 1F). Therefore, we tested whether ILC‐deficient mice on Flgft/ft background developed AD. Treatment of Rag1−/−Flgft/ft mice with anti‐CD90 mAb to deplete ILC2 did not alter development of skin inflammation (data not shown). Rag2−/−Il2rg−/−Flgft/ft mice developed skin inflammation comparable to Flgft/ft mice and Rag2−/−Flgft/ft mice (Figure 3B). As Rag2−/−Il2rg−/− are not only deficient in ILC but also NK, T, and B cells, we wanted to confirm our results by using a more specific model of ILC deficiency. Therefore, ILC2‐deficient Rorafl/sgIl7raCre/+ mice23 were crossed onto the Flgft/ft background. ILC2‐deficient Flgft/ft mice developed severe skin inflammation (Figure 3C). These data from three separate and distinct models of ILC2 deficiency led us to conclude that ILC2 were dispensable for skin inflammation in this spontaneous and chronic model of AD‐like inflammation in mice with a defective skin barrier.
Figure 3.
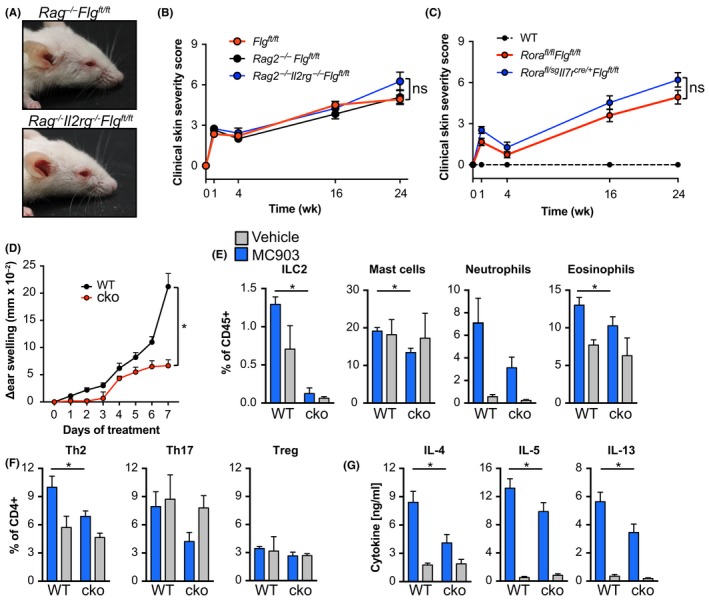
Dermatitis in Flgft/ft mice is independent of ILC2, while acute skin inflammation is ILC2‐dependent. A, Representative photographs of Rag−/−Flgft/ft (upper panel) and Rag2−/−Il2rg−/−Flgft/ft (lower panel) mice. B, Macroscopic clinical scoring of Flgft/ft (red), Rag2−/−Flgft/ft (black), and Rag2−/−Il2rg−/−Flgft/ft (blue). Graph shows the mean ± SEM from 6 to 7 mice per group. C, Macroscopic clinical scoring of wild‐type (dashed black), Rorafl/flFlgft/ft (red), and Rorafl/sgIl7rCre/+Flgft/ft (blue) mice. Graph shows the mean + SEM from 9 mice per group. D, Skin inflammation was induced in Rorafl/sgIl7rCre/+ (cko; red line) and control (WT; black line) mice by daily topical application of 4 nmol MC903 in 100% ethanol onto the right ear. The left ear was treated with ethanol and served as internal control. Ear thickness was measured daily. Mean ± SEM from at least 6 mice per group from two independent experiments is depicted. *P < 0.05, t test of AUC. E, Frequency of ILC2, mast cells, neutrophils, and eosinophils isolated from the ears of MC903‐ (blue) and ethanol‐treated (Vehicle, gray) Rorafl/sgIl7rCre/+ (cko) and control (WT) mice. F, Frequency of T helper cell subsets isolated from the ears of MC903‐ (blue) and ethanol‐treated (gray) Rorafl/sgIl7rCre/+ (cko) and control (WT) mice. Bar graphs show the mean ± SEM from six mice of two independent experiments. *P < 0.05. G, Draining (MC903, blue) and non‐draining (Vehicle, gray) lymph node cells were restimulated with anti‐CD3/anti‐CD28 for 72 h and indicated cytokines in the supernatant were measured by ELISA. Bar graphs show the mean + SEM from 6 to 8 mice per group from two independent experiments. ns, not significant, t test of AUC
3.4. Group 2 innate lymphoid cells promote acute skin inflammation
Daily application of MC903 elicits acute AD‐like skin inflammation28 (Figure 3D). We, and others, have previously shown using antibody‐mediated depletion models as well as bone marrow chimeric mice that development of MC903‐elicited skin inflammation is dependent on ILC2.25, 27 Indeed, using ILC2‐deficient Rorafl/sgIl7raCre/+ mice, we could confirm that ear swelling was ameliorated in acute dermatitis (Figure 3D). As expected, ILC2 were not present in the inflamed skin of Rorafl/sgIl7raCre/+ mice (Figure 3E). Moreover, cellular infiltration was blunted in ILC2‐deficient mice including the recruitment of granulocytes, Th2 and Th17 cells (Figure 3E,F). Furthermore, type 2‐associated cytokine production in draining LN was impaired (Figure 3G). Because of the divergent roles ILC2 play in the initiation processes of acute chronic dermatitis, we sought to determine which other factors promote AD in the clinically more relevant Flgft/ft model.
3.5. Impaired skin barrier–induced dermatitis operates independently of type 2 and type 17 cytokines
Atopic dermatitis in humans is generally accompanied by an increase in type 2‐ and type 17‐associated cytokines,61, 62, 63 with IL‐13 and IL‐5 reported as important systemic biomarkers for infant AD.64 Indeed, analysis of cytokines in the skin of Flgft/ft mice revealed a significant increase of IL‐4, IL‐5, IL‐9, IL‐13, IL‐17A, and IL‐22 (Figure 4A,B). To investigate whether the development of AD‐like inflammation in Flgft/ft mice was dependent on the cardinal type 2 cytokines, we generated IL‐4‐, IL‐5‐, IL‐9‐, and IL‐13‐knockout mice on the Flgft/ft background. However, Th2 cytokine‐deficient Flgft/ft mice developed inflammation comparable to that observed in cytokine‐sufficient Flgft/ft mice (Figure 4C). Similarly, knockout of IL‐17A or IL‐22 on the Flgft/ft background did not ameliorate skin inflammation (Figure 4D). Because neither knockout of individual type 2 nor type 17 cytokines protected from skin inflammation, we investigated cytokines upstream in the inflammatory cascade.
Figure 4.
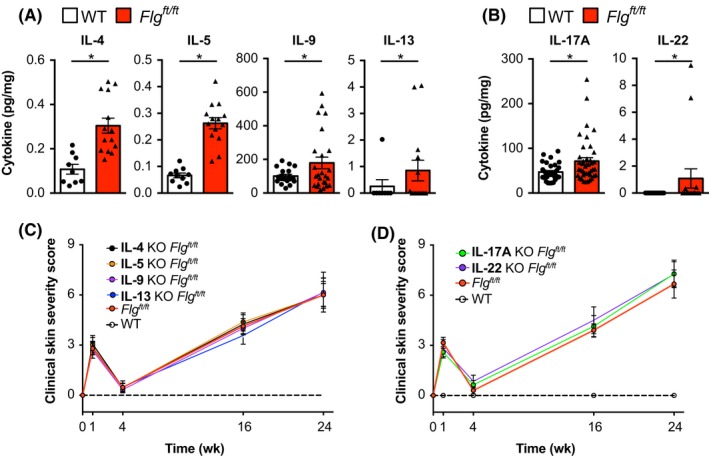
Dermatitis in Flgft/ft mice is independent of type 2‐ and type 17‐associated effector cytokines. A,B, Indicated cytokines isolated from ear skin of age‐matched wild type (open bars) and Flgft/ft (red bars) depicted as pg cytokine per mg of total isolated protein. C, Macroscopic clinical scoring of wild‐type (dashed black), Flgft/ft (red), IL‐4−/−Flgft/ft (black), IL‐5−/−Flgft/ft (orange), IL‐9−/−Flgft/ft (violet), and IL‐13−/−Flgft/ft (blue) mice. Graph shows the mean + SEM from at least 8 mice per group. D, Macroscopic clinical scoring of wild‐type (dashed black), Flgft/ft (red), IL‐17A−/−Flgft/ft (green), and IL‐22−/−Flgft/ft (violet) mice. Graph shows the mean + SEM from at least 6 mice per group from two to three independent experiments
3.6. Inflammasome‐independent IL‐1β‐mediated IL‐1R1 signaling is required for inflammation
IL‐1β is significantly elevated in skin blisters of patients with AD that have FLG mutations (Figure 5A) and in the skin of Flgft/ft mice (Figure 5B). We crossed Il1a−/−, Il1b−/−, and Il1r1−/− mice onto the Flgft/ft background to investigate their contribution to spontaneous skin inflammation (Figure 5C,D). While IL‐1α deficiency did not decrease the inflammatory score of Flgft/ft mice (P = 0.942), IL‐1β‐deficient Flgft/ft mice were protected from the development of skin inflammation (Figure 5C,D; P = 0.013). Importantly, IL‐1R1 was required for IL‐1β‐mediated inflammation as Flgft/ft mice deficient in IL‐1R1 were comparably protected (Figure 5C,D; P = 0.009). Furthermore, inhibition of IL‐1β by treatment with anti‐IL‐1β‐antibodies ameliorated MC903 elicited acute exacerbation of skin disease in Flgft/ft mice (Figure 5E,F). We addressed contribution of other IL‐1 cytokine family members to spontaneous skin inflammation in Flgft/ft mice but development of dermatitis in Flgft/ft mice was independent of IL‐33, IL‐18, and IL‐36 (Figure S2A). These results reveal an important role for IL‐1β and IL‐1R1 in the development of spontaneous skin inflammation in this model.
Figure 5.
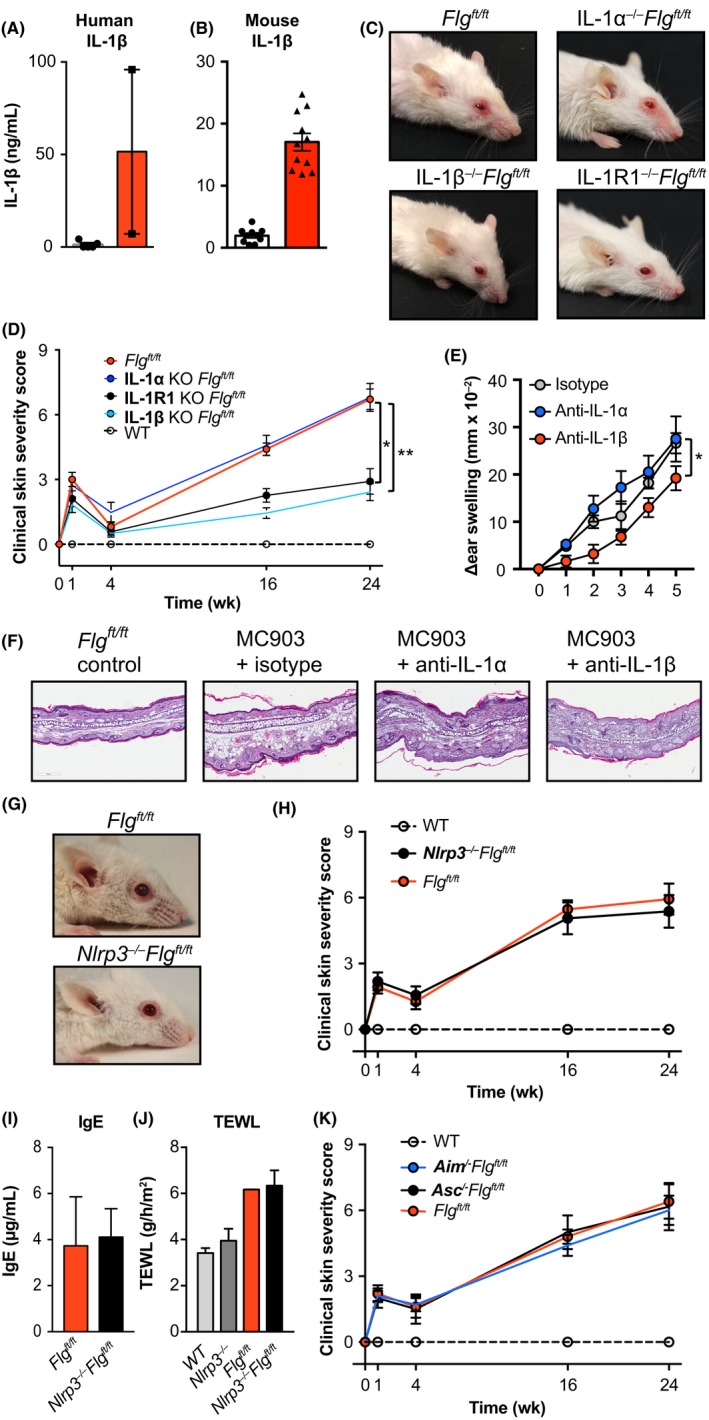
Inflammation in Flgft/ft mice is dependent on IL‐1β and IL‐1R1 signaling but occurs inflammasome‐independent. A, IL‐1β was measured in skin biopsies of patients with or without FLG mutation. B, IL‐1β protein concentration in ear skin of age‐matched wild type (open bars) and Flgft/ft (red bars) depicted as pg cytokine per mg of total isolated protein. C, Representative photographs of Flgft/ft (upper left), IL‐1α−/−Flgft/ft (upper right), IL‐1β−/−Flgft/ft (lower left), and IL‐1R1−/−Flgft/ft (lower right). D, Macroscopic clinical scoring of wild‐type (open circles), Flgft/ft (red), IL‐1R1−/−Flgft/ft (black), IL‐1β−/−Flgft/ft (light blue), and IL‐1α−/−Flgft/ft (blue) mice. Graph shows the mean + SEM from 8 mice per group. *P < 0.05; **P < 0.01; t test of AUC against Flgft/ft. E, Flgft/ft mice were treated daily with isotype control mAb (gray circles), anti‐IL‐1α‐(blue) or anti‐IL‐1β‐mAb (red) and MC903 was topically applied to induce dermatitis exacerbation. Ear thickness was measured daily. Graph shows the mean + SD of 4‐5 mice per group. *P < 0.05, AUC. F, Representative H&E‐stained sections of untreated (left) and MC903‐treated ears from (E). G, Representative photographs of Flgft/ft (upper panel) and Nlrp3−/−Flgft/ft (lower panel) mice. H, Macroscopic clinical scoring of wild‐type (open circles), Flgft/ft (red), and Nlrp3−/−Flgft/ft (black) mice. Graph shows the mean + SEM from 7 mice per group. I, Serum IgE levels in Flgft/ft (red), and Nlrp3−/−Flgft/ft (black) mice. J, Transepidermal water loss in wild‐type (gray), Nlrp3−/− (dark gray), Flgft/ft (red), and Nlrp3−/−Flgft/ft (black) mice. Bar graphs show the mean + SEM from 4 to 6 mice per group. K, Macroscopic clinical scoring of wild‐type (open circles), Flgft/ft (red), Aim−/−Flgft/ft (blue), and Asc−/−Flgft/ft (black) mice. Graph shows the mean + SEM from 6 to 10 mice per group from three independent experiments
IL‐1β requires processing by the NLRP3 inflammasome to be cleaved from pro‐IL‐1β to become bioactive IL‐1β. We generated Nlrp3−/−Flgft/ft mice to investigate the contribution of the NLRP3 inflammasome to inflammation in our model (Figure 5G,H). However, inflammasome‐mediated maturation was not required as these mice developed skin inflammation comparable to that seen in Flgft/ft mice (Figure 5G,H). NLRP3 deficiency did not alter the defective skin barrier or generation of IgE in mutant mice (Figure 5I,J). Indeed, there was also no role for ASC and AIM2 (Figure 5K). We further addressed whether inflammation in Flgft/ft mice can be targeted therapeutically with the potent inflammasome inhibitor MCC95065 during a period of MC903‐induced exacerbation of skin inflammation. Chemical inhibition of the inflammasome did not ameliorate the acute development of skin inflammation in Flgft/ft mice (Figure S2B), which was consistent with our results on spontaneous and chronic inflammation in Nlrp3−/−Flgft/ft mice. These data demonstrate the development of skin inflammation in mutant mice is independent of the NLRP3 inflammasome.
3.7. Dermal mast cells promote inflammation in mice with impaired skin barrier
A study in mice with NLRP3‐dependent skin inflammation analyzed the interplay between microbiota, TNFα, and IL‐1β and demonstrated a critical role for mast cells in skin inflammation.66 Histological analysis of the skin of adult Flgft/ft mice showed elevated numbers of connective tissue mast cells (Figure 6A,B). As we observed significant amelioration in response to IL‐1β deficiency, we were interested whether IL‐1β was directly promoting mast cell activation and inflammation in Flgft/ft mice. Therefore, we generated bone marrow–derived mast cells from wild‐type and Il1r1−/− donors and stimulated them through FcεRI in the presence and absence of IL‐1β costimulation (Figure 6C). While FcεRI‐crosslinking induced the release of pro‐inflammatory IL‐6 and TNFα, as well as CCL2 and IL‐13, costimulation with IL‐1β further increased cytokine release in an IL‐1R1‐dependent manner (Figure 6C). This result suggests a hyperresponsive phenotype of mast cells and a pro‐inflammatory role in dermatitis exacerbation in response to IL‐1β.
Figure 6.
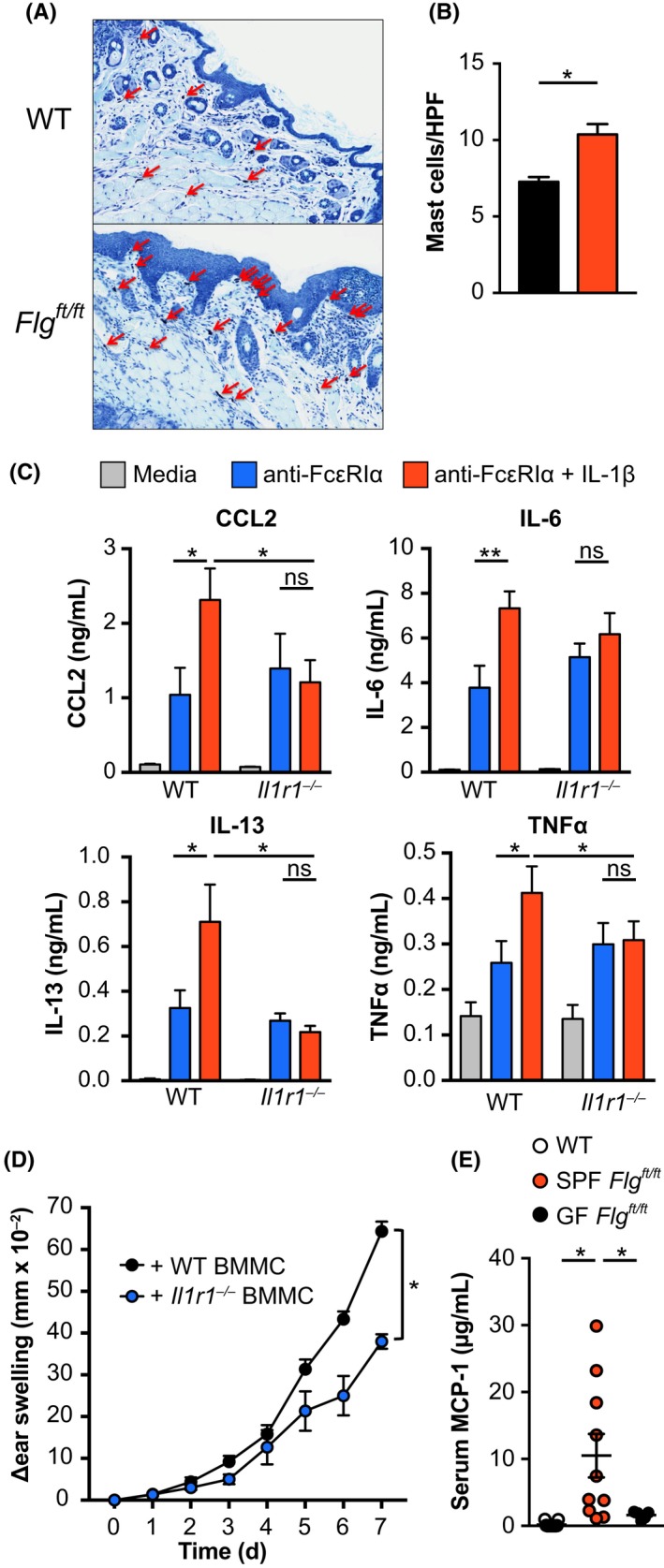
IL‐1β drives hyperactivation of mast cells in AD‐like inflammation. A, Toluidine blue staining of wild‐type (upper panel) and Flgft/ft (lower panel) skin. Red arrows indicate connective tissue mast cells. B, Quantification of mast cells per HPF from toluidine blue stained skin samples. Bar graphs show the mean + SEM from 8 mice per group. *P < 0.05, ttest. D, Bone marrow–derived mast cells from wild‐type (left) and Il1r1−/− (right) mice were stimulated with media (light gray) or plate‐bound anti‐FcεRIα (10 µg/mL) in the presence (red) or absence (blue) of recombinant IL‐1β (10 ng/mL). CCL2, IL‐6, IL‐13, and TNFα were measured by ELISA. Bars show the mean + SEM of nine replicates from three independent experiments. *P < 0.05. E, Wild‐type (black) and IL‐R1−/− (blue) mast cells were transferred intradermally into the ears of KitW‐shFlgft/ft mice. Six weeks later, MC903 or 100 EtOH was applied onto the ears and ear thickness was measured daily. Graph shows the mean + SEM of 5 mice per group from two experiments. *P < 0.05, t test of AUC. F, Serum mast cell protease 1 levels from adult (10 wk) wild‐type (open circles), Flgft/ft (red), and germ‐free Flgft/ft (black) mice. *P < 0.05
In order to confirm the pathogenic role of IL‐1β‐responsive mast cells in skin inflammation, we transferred wild type‐ or Il1r1−/−‐derived BMMC into the ears of mast cell–deficient KitW‐sh mice67 on a Flgft/ft background (Figure 6D). Indeed, when we induced inflammation in these mice using MC903, IL‐1β‐responsive mast cells were sufficient to aggravate inflammation (Figure 6D). Interestingly, germ‐free Flgft/ft mice had significantly decreased serum levels of IgE compared to their SPF counterparts (Figure 2H) and—lacking the IgE‐mediated activation of mast cells—also showed decreased levels of MCP‐1 (Figure 6F). Importantly, GF mice showed reduced expression of IL‐1β further reducing activation of mast cells (Figure 2I). These data indicate that IL‐1β‐mediated hyperactivation of mast cells contributes significantly to dermatitis in Flgft/ft mice.
4. DISCUSSION
In the present study, we have shown that the development of skin inflammation in Flgft/ft mice is independent of group 2 innate lymphoid cells but requires an interplay between the microbiome, IL‐1β, and mast cells. ILC2 are implicated in AD, with AD skin lesions showing higher expression of ILC2 genes RORA, IL1R1, IL17RB, TSLPR, and AREG, and increased numbers of ILC2.25, 27 We have reported elevated ILC2 in skin blisters of AD patients with mutation in FLG.11 Here, mice deficient in ILC2 developed significantly ameliorated acute skin inflammation but despite the higher numbers of ILC2 present in the skin of Flgft/ft mice,11 ILC2 play a redundant role in the pathogenesis of chronic dermatitis. While MC903‐induced ILC2 function could be targeted by anti‐IL‐18,68 we show that chronic inflammation is independent of IL‐18, further corroborating the observed redundancy of ILC2.69, 70 Although ILC2 may contribute to genesis of acute skin inflammation, their increase in numbers under chronic inflammatory conditions is probably a consequence of an overall increase in infiltrating immune cell populations.
A similar general increase of type 2‐ and 17‐associated cytokine production is observed in the inflamed skin of Flgft/ft mice; however, no functional roles for individual cytokines were observed in mutant mice. This is perhaps surprising as IL‐4 and IL‐13 are known to be involved upstream of filaggrin in the development of AD by promoting skin barrier disintegration71, 72 and dermal fibrosis73—possibly via TSLP.74 IL‐5 and IL‐9 are effector cytokines promoting single aspects of AD, such as eosinophilia,56 neuronal stimulation,75 and survival of T cells and ILC2.76 IL‐17A can promote skin inflammation77 and was shown to decrease expression of filaggrin and tight junction proteins.78 Similarly, IL‐22 is able to regulate keratinocyte function.63, 79 Despite these cytokines being upregulated in the skin, the single knockout of each cytokine did not alleviate disease. This highlights that single target therapy may not be useful to therapeutically deplete cytokines during chronic disease. In this context, dupilumab, targeting the IL‐4Rα‐chain and thereby the actions of IL‐4 and IL‐13, has recently been approved by the FDA for the treatment of moderate‐to‐severe AD patients (reviewed in80) and is proving to be efficacious.81, 82 Interestingly, in the context of this study, in patients undergoing dupilumab treatment IL‐1β was shown to be considerably downregulated,83 while markers of skin integrity, including FLG, are restored.84
We show that chronic dermatitis in Flgft/ft mice developed independently of IL‐1α but was instead dependent on IL‐1β. Importantly, treatment with anti‐IL‐1β ‐antibody could alleviate MC903‐induced skin flares supporting findings that canakinumab can prevent TSLP release from keratinocytes.85 In a recent study, it was shown that 6‐8‐week‐old Flgft/ft mice without overt dermatitis constitutively expressed increased amounts of IL‐1α in the epidermis but 21 days after acute mechanical skin injury released less IL‐1α and more IL‐1β.86 These results indicate important, but divergent, roles for IL‐1 family cytokines in acute and chronic dermatitis. One of our next steps will therefore include the analysis what impact IL‐1‐family members have on expression of key skin barrier proteins.
In Flgft/ft mice, we observed that IL‐1β enhances FcεRI‐mediated signaling in mast cells confirming previous reports.87 Recently, it was shown that mast cells with a mutated NLRP3 inflammasome and higher caspase‐1 activity produced IL‐1β in response to TNFα or LPS stimulation.66 Using kitw‐shFlgft/ft mice as mast cell–recipient mice, we show that IL‐1β‐responsive mast cells promote inflammation via IL‐1R1‐signaling. Because kitw‐sh mice certainly have phenotypes independent of mast cell deficiency,88 further studies using more sophisticated models of mast cell deficiency are required to analyze their in vivo function in mice with skin barrier defects.
Treatment with the NLRP3‐inflammasome inhibitor MCC950 did not ameliorate inflammation in our multifactorial model of AD when we triggered an acute inflammatory response with MC903. Furthermore, inflammation was unaltered in NLRP3‐deficient, ASC‐deficient, and AIM2‐deficient Flgft/ft mice. Therefore, IL‐1β processing occurs independent of the NLRP1, NLRP3, NLRP6, and AIM2 inflammasome. To date, we cannot rule out a role for the NLRC4 inflammasome (or NAIP‐NLRC4) that can directly recruit caspase‐1. In a recent study, NLRC4 was found in the scale extracts of psoriasis patients; however, no inflammasome components (including NLRC4) were found in atopic dermatitis patients.89 Inflammasome‐independent processing of IL‐1β has been observed in a variety of settings (reviewed in90). In particular, neutrophil‐ and mast cell–derived proteases play prominent roles in the extracellular processing of IL‐1‐family cytokines,91, 92 with human mast cell protease 1 shown to process pro‐IL‐1.93 Increased mast cell chymase activity may therefore contribute to IL‐1β maturation in the Flgft/ft model; however, further investigations are required to fully understand the role of mast cell proteases in AD.
Skin microbiome sequencing revealed a shift toward pathogenic staphylococci, which has been reported in AD patients.30 While the microbiota in flaky tail mice induced IL‐17A‐mediated neutrophilia, we could show that in Flgft/ft mice IL‐17A was redundant.33 Indeed, IL‐17A‐mediated inflammation is observed in Tmem79ft/ft (Saunders et al, unpublished) but not in Flgft/ft mice. Our results indicate that the pathogenic microbiome present in neonates imprinted a hyperresponsive phenotype in mast cells. This phenotype is maintained when adult mice were treated with antibiotics. When we treated neonate Flgft/ft mice with antibiotics before inflammation is established, or when mice are raised in GF conditions, the subsequent adult dermatitis phenotype is significantly ameliorated. A recent study in a cohort of newborn children suggests that early colonization with commensal staphylococci genera protects from AD and that microbiotic changes only occur after the onset of AD.94 Therefore, the Flgft/ft mouse model provides a basis for future investigations into the extrinsic factors and intrinsic mechanisms of neonatal inflammation before AD‐like inflammation develops. While the broad application of state‐of‐the‐art techniques gives a comprehensive analysis of the AD‐like inflammation in mutant mice on the Flgft/ft background, future approaches need to target the separate pathways leading to disease and untangle the relative contribution to pathogenesis.
In summary, we revealed a critical role of IL‐1β in the initiation and maintenance of skin inflammation in a mutant mouse model of defective skin barrier. Further investigations will be required to assess the contribution of IL‐1β‐responsive mast cells, as well as roles of the integrity of the skin barrier and interplay of the microbiome, in the generation of inflammation in AD patients. These endeavors will lead to the development of novel management options for children suffering from AD.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
CS is a Long‐Term EMBO Fellow (ALTF 587‐2016). PGF was supported by Science Foundation Ireland (10/IN.1/B3004), the Wellcome Trust (092530/Z/10/Z), and National Children's Research Centre. Mice axenization was supported by the EC FP7‐Infrastructure project INFRAFRONTIER‐I3 (312325) at the Gulbenkian Institute, EMMA Portugal. We thank Hans‐Reimer Rodewald and Stephen Galli for reagents and helpful advice. We are grateful to Emily Hams, Ruairi Kavanagh and colleagues in the group for assistance.
Schwartz C, Moran T, Saunders SP, et al. Spontaneous atopic dermatitis in mice with a defective skin barrier is independent of ILC2 and mediated by IL‐1β. Allergy. 2019;74:1920–1933. 10.1111/all.13801
REFERENCES
- 1. Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109‐1122. [DOI] [PubMed] [Google Scholar]
- 2. Otsuka A, Nomura T, Rerknimitr P, Seidel JA, Honda T, Kabashima K. The interplay between genetic and environmental factors in the pathogenesis of atopic dermatitis. Immunol Rev. 2017;278(1):246‐262. [DOI] [PubMed] [Google Scholar]
- 3. Cabanillas B, Brehler AC, Novak N. Atopic dermatitis phenotypes and the need for personalized medicine. Curr Opin Allergy Clin Immunol. 2017;17(4):309‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Palmer C, Irvine AD, Terron‐Kwiatkowski A, et al. Common loss‐of‐function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441‐446. [DOI] [PubMed] [Google Scholar]
- 5. Saunders SP, Goh C, Brown SJ, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol. 2013;132(5):1121‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315‐1327. [DOI] [PubMed] [Google Scholar]
- 7. Wozniak M, Kaczmarek‐Skamira E, Romanska‐Gocka K, Czajkowski R, Kaluzna L, Zegarska B. The prevalence of mutations in the gene encoding filaggrin in the population of Polish patients with atopic dermatitis. Postepy Dermatol Alergol. 2016;33(2):128‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li M, Liu Q, Liu J, et al. Mutations analysis in filaggrin gene in northern China patients with atopic dermatitis. J Eur Acad Dermatol Venereol. 2013;27(2):169‐174. [DOI] [PubMed] [Google Scholar]
- 9. On HR, Lee SE, Kim S‐E, et al. Filaggrin mutation in Korean patients with atopic dermatitis. Yonsei Med J. 2017;58(2):395‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124(3 suppl 2):R7‐R12. [DOI] [PubMed] [Google Scholar]
- 11. Saunders SP, Moran T, Floudas A, et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J Allergy Clin Immunol. 2016;137(2):482‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brunner PM, Guttman‐Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad‐spectrum and targeted therapies. J Allergy Clin Immunol. 2017;139(4S):S65‐S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008;128(11):2625‐2630. [DOI] [PubMed] [Google Scholar]
- 14. Woodward AL, Spergel JM, Alenius H, et al. An obligate role for T‐cell receptor alphabeta+ T cells but not T‐cell receptor gammadelta+ T cells, B cells, or CD40/CD40L interactions in a mouse model of atopic dermatitis. J Allergy Clin Immunol. 2001;107(2):359‐366. [DOI] [PubMed] [Google Scholar]
- 15. Akdis M, Simon HU, Weigl L, Kreyden O, Blaser K, Akdis CA. Skin homing (cutaneous lymphocyte‐associated antigen‐positive) CD8+ T cells respond to superantigen and contribute to eosinophilia and IgE production in atopic dermatitis. J Immunol. 1999;163(1):466‐475. [PubMed] [Google Scholar]
- 16. Irani AM, Sampson HA, Schwartz LB. Mast cells in atopic dermatitis. Allergy. 1989;44(suppl 9):31‐34. [PubMed] [Google Scholar]
- 17. Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF‐alpha/cachectin. Nature. 1990;346(6281):274‐276. [DOI] [PubMed] [Google Scholar]
- 18. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. 2014;14(7):478‐494. [DOI] [PubMed] [Google Scholar]
- 19. Roediger B, Kyle R, Yip KH, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. 2013;14(6):564‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Molofsky AB, Nussbaum JC, Liang H‐E, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210(3):535‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brestoff JR, Kim BS, Saenz SA, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519(7542):242‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type‐2 immunity. Nature. 2010;464(7293):1367‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oliphant CJ, Hwang YY, Walker JA, et al. MHCII‐mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwartz C, Khan AR, Floudas A, et al. ILC2s regulate adaptive Th2 cell functions via PD‐L1 checkpoint control. J Exp Med. 2017;214(9):2507‐2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim BS, Siracusa MC, Saenz SA, et al. TSLP elicits IL‐33‐independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwartz C, Eberle JU, Hoyler T, Diefenbach A, Lechmann M, Voehringer D. Opposing functions of thymic stromal lymphopoietin‐responsive basophils and dendritic cells in a mouse model of atopic dermatitis. J Allergy Clin Immunol. 2016;138(5):1443–1446. [DOI] [PubMed] [Google Scholar]
- 27. Salimi M, Barlow JL, Saunders SP, et al. A role for IL‐25 and IL‐33‐driven type‐2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low‐calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci USA. 2006;103(31):11736–11741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bjerre RD, Bandier J, Skov L, Engstrand L, Johansen JD. The role of the skin microbiome in atopic dermatitis: a systematic review. Br J Dermatol. 2017;177(5):1272–1278. [DOI] [PubMed] [Google Scholar]
- 30. Kong HH, Oh J, Deming C, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16(3):143–155. [DOI] [PubMed] [Google Scholar]
- 32. Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoff S, Oyoshi MK, Macpherson A, Geha RS. The microbiota is important for IL‐17A expression and neutrophil infiltration in lesional skin of Flg(ft/ft) mice. Clin Immunol. 2015;156(2):128–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oyoshi MK, He R, Kumar L, Yoon J, Geha RS. Cellular and molecular mechanisms in atopic dermatitis. Adv Immunol. 2009;102:135–226. [DOI] [PubMed] [Google Scholar]
- 35. Behniafard N, Gharagozlou M, Sotoudeh S, et al. Association of single nucleotide polymorphisms of interleukin‐1 family with atopic dermatitis. Allergol Immunopathol (Madr). 2014;42(3):212–215. [DOI] [PubMed] [Google Scholar]
- 36. Kezic S, O'Regan GM, Lutter R, et al. Filaggrin loss‐of‐function mutations are associated with enhanced expression of IL‐1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129(4):1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG‐1‐deficient mice have no mature B and T lymphocytes. Cell 1992;68(5):869–877. [DOI] [PubMed] [Google Scholar]
- 38. Song J, Willinger T, Rongvaux A, et al. A mouse model for the human pathogen Salmonella typhi. Cell Host Microbe. 2010;8(4):369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schlenner SM, Madan V, Busch K, et al. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity. 2010;32(3):426–436. [DOI] [PubMed] [Google Scholar]
- 40. Mamontova A, Seguret‐Mace S, Esposito B, et al. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circulation. 1998;98(24):2738–2743. [DOI] [PubMed] [Google Scholar]
- 41. Mohrs K, Wakil AE, Killeen N, Locksley RM, Mohrs M. A two‐step process for cytokine production revealed by IL‐4 dual‐reporter mice. Immunity 2005;23(4):419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKenzie GJ, Bancroft A, Grencis RK, McKenzie AN. A distinct role for interleukin‐13 in Th2‐cell‐mediated immune responses. Curr Biol. 1998;8(6):339–342. [DOI] [PubMed] [Google Scholar]
- 43. Gerlach K, Hwang YY, Nikolaev A, et al. TH9 cells that express the transcription factor PU.1 drive T cell‐mediated colitis via IL‐9 receptor signaling in intestinal epithelial cells. Nat Immunol. 2014;15(7):676–686. [DOI] [PubMed] [Google Scholar]
- 44. Nakae S, Komiyama Y, Nambu A, et al. Antigen‐specific T cell sensitization is impaired in IL‐17‐deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17(3):375–387. [DOI] [PubMed] [Google Scholar]
- 45. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin‐22 but not interleukin‐17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27(4):647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. [DOI] [PubMed] [Google Scholar]
- 47. Horai R, Asano M, Sudo K, et al. Production of mice deficient in genes for interleukin (IL)‐1alpha, IL‐1beta, IL‐1alpha/beta, and IL‐1 receptor antagonist shows that IL‐1beta is crucial in turpentine‐induced fever development and glucocorticoid secretion. J Exp Med. 1998;187(9):1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Glaccum MB, Stocking KL, Charrier K, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL‐1. J Immunol. 1997;159(7):3364–3371. [PubMed] [Google Scholar]
- 49. Takeda K, Tsutsui H, Yoshimoto T, et al. Defective NK cell activity and Th1 response in IL‐18‐deficient mice. Immunity. 1998;8(3):383–390. [DOI] [PubMed] [Google Scholar]
- 50. Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature. 2004;430(6996):213–218. [DOI] [PubMed] [Google Scholar]
- 51. Rathinam V, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell‐deficient W‐sash c‐kit mutant Kit W‐sh/W‐sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167(3):835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Russell SE, Horan RM, Stefanska AM, et al. IL‐36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016;9(5):1193–1204. [DOI] [PubMed] [Google Scholar]
- 54. Galli SJ, Tsai M, Gordon JR, Geissler EN, Wershil BK. Analyzing mast cell development and function using mice carrying mutations at W/c‐kit or Sl/MGF (SCF) loci. Ann N Y Acad Sci. 1992;664:69–88. [DOI] [PubMed] [Google Scholar]
- 55. Gaudenzio N, Sibilano R, Starkl P, Tsai M, Galli SJ, Reber LL. Analyzing the functions of mast cells in vivo using 'Mast Cell Knock‐in' mice. J Vis Exp. 2015;99:52753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Floudas A, Saunders SP, Moran T, et al. IL‐17 receptor A maintains and protects the skin barrier to prevent allergic skin inflammation. J Immunol. 2017;199(2):707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miezeiewski M, Schnaufer T, Muravsky M, et al. An in vitro culture model to study the dynamics of colonic microbiota in Syrian golden hamsters and their susceptibility to infection with Clostridium difficile. ISME J. 2015;9(2):321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim BS, Wang K, Siracusa MC, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193(7):3717–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kiehl P, Falkenberg K, Vogelbruch M, Kapp A. Tissue eosinophilia in acute and chronic atopic dermatitis: a morphometric approach using quantitative image analysis of immunostaining. Br J Dermatol. 2001;145(5):720–729. [DOI] [PubMed] [Google Scholar]
- 60. Bateman EA, Ardern‐Jones MR, Ogg GS. Persistent central memory phenotype of circulating Fel d 1 peptide/DRB1*0101 tetramer‐binding CD4+ T cells. J Allergy Clin Immunol. 2006;118(6):1350–1356. [DOI] [PubMed] [Google Scholar]
- 61. Gittler JK, Shemer A, Suárez‐Fariñas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130(6):1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Esaki H, Brunner PM, Renert‐Yuval Y, et al. Early‐onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol. 2016;138(6):1639–1651. [DOI] [PubMed] [Google Scholar]
- 63. Lou H, Lu J, Choi EB, et al. Expression of IL‐22 in the skin causes Th2‐biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial Th2 cytokines and the GRP pathway. J Immunol. 2017;198(7):2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McAleer MA, Jakasa I, Hurault G, et al. Systemic and stratum corneum biomarkers of severity in infant AD include markers of innate and Th‐related immunity and angiogenesis. Br J Dermatol. 2018180(3):586‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Coll RC, Robertson A, Chae JJ, et al. A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Nunez G. Critical role for mast cells in interleukin‐1beta‐driven skin inflammation associated with an activating mutation in the nlrp3 protein. Immunity. 2012;37(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Becker M, Reuter S, Friedrich P, et al. Genetic variation determines mast cell functions in experimental asthma. J Immunol. 2011;186(12):7225–7231. [DOI] [PubMed] [Google Scholar]
- 68. Ricardo‐Gonzalez RR, Van Dyken SJ, Schneider C, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol. 2018;19(10):1093‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Vély F, Barlogis V, Vallentin B, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17(11):1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brasseit J, Kwong Chung C, Noti M, et al. Divergent roles of interferon‐gamma and innate lymphoid cells in innate and adaptive immune cell‐mediated intestinal inflammation. Front Immunol. 2018;9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kamsteeg M, Bergers M, de Boer R, et al. Type 2 helper T‐cell cytokines induce morphologic and molecular characteristics of atopic dermatitis in human skin equivalent. Am J Pathol. 2011;178(5):2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morizane S, Yamasaki K, Kajita A, et al. TH2 cytokines increase kallikrein 7 expression and function in patients with atopic dermatitis. J Allergy Clin Immunol. 2012;130(1):259–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zheng T, Oh MH, Oh SY, Schroeder JT, Glick AB, Zhu Z. Transgenic expression of interleukin‐13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol. 2009;129(3):742–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Oh M‐H, Oh SY, Yu J, et al. IL‐13 induces skin fibrosis in atopic dermatitis by thymic stromal lymphopoietin. J Immunol. 2011;186(12):7232–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Foster EL, Simpson EL, Fredrikson LJ, et al. Eosinophils increase neuron branching in human and murine skin and in vitro. PLoS One. 2011;6(7):e22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Turner J‐E, Morrison PJ, Wilhelm C, et al. IL‐9‐mediated survival of type 2 innate lymphoid cells promotes damage control in helminth‐induced lung inflammation. J Exp Med. 2013;210(13):2951–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nakajima S, Kitoh A, Egawa G, et al. IL‐17A as an inducer for Th2 immune responses in murine atopic dermatitis models. J Invest Dermatol. 2014;134(8):2122–2130. [DOI] [PubMed] [Google Scholar]
- 78. Yuki T, Tobiishi M, Kusaka‐Kikushima A, Ota Y, Tokura Y. Impaired tight junctions in atopic dermatitis skin and in a skin‐equivalent model treated with interleukin‐17. PLoS One. 2016;11(9):e0161759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nograles KE, Zaba LC, Shemer A, et al. IL‐22‐producing "T22" T cells account for upregulated IL‐22 in atopic dermatitis despite reduced IL‐17‐producing TH17 T cells. J Allergy Clin Immunol. 2009;123(6):1244–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vangipuram R, Tyring SK. Dupilumab for moderate‐to‐severe atopic dermatitis. Skin Therapy Lett. 2017;22(6):1–4. [PubMed] [Google Scholar]
- 81. Gooderham MJ, Hong HC, Eshtiaghi P, Papp KA. Dupilumab: a review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol. 2018;78(3S1):S28–S36. [DOI] [PubMed] [Google Scholar]
- 82. Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate‐to‐severe atopic dermatitis. N Engl J Med. 2014;371(2):130–139. [DOI] [PubMed] [Google Scholar]
- 83. Hamilton JD, Suárez‐Fariñas M, Dhingra N, et al. Dupilumab improves the molecular signature in skin of patients with moderate‐to‐severe atopic dermatitis. J Allergy Clin Immunol. 2014;134(6):1293–1300. [DOI] [PubMed] [Google Scholar]
- 84. Guttman‐Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in atopic dermatitis patients. J Allergy Clin Immunol. 2019;143(1):155‐172. [DOI] [PubMed] [Google Scholar]
- 85. Bernard M, Carrasco C, Laoubi L, et al. IL‐1beta induces thymic stromal lymphopoietin and an atopic dermatitis‐like phenotype in reconstructed healthy human epidermis. J Pathol. 2017;242(2):234–245. [DOI] [PubMed] [Google Scholar]
- 86. Archer NK, Jo JH, Lee SK, et al. Injury, dysbiosis and filaggrin deficiency drive skin inflammation via keratinocyte IL‐1alpha release. J Allergy Clin Immunol. 2019;143(4):1426‐1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hultner L, Kolsch S, Stassen M, et al. In activated mast cells, IL‐1 up‐regulates the production of several Th2‐related cytokines including IL‐9. J Immunol. 2000;164(11):5556–5563. [DOI] [PubMed] [Google Scholar]
- 88. Feyerabend TB, Gutierrez DA, Rodewald HR. Of mouse models of mast cell deficiency and metabolic syndrome. Cell Metab. 2016;24(1):1–2. [DOI] [PubMed] [Google Scholar]
- 89. Hiruma J, Harada K, Motoyama A, et al. Key component of inflammasome, NLRC4, was identified in the lesional epidermis of psoriatic patients. J Dermatol. 2018;45(8):971–977. [DOI] [PubMed] [Google Scholar]
- 90. Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome‐independent regulation of IL‐1‐family cytokines. Annu Rev Immunol. 2015;33:49–77. [DOI] [PubMed] [Google Scholar]
- 91. Joosten LA, Netea MG, Fantuzzi G, et al. Inflammatory arthritis in caspase 1 gene‐deficient mice: contribution of proteinase 3 to caspase 1‐independent production of bioactive interleukin‐1beta. Arthritis Rheum. 2009;60(12):3651–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Clancy DM, Sullivan GP, Moran H, et al. Extracellular neutrophil proteases are efficient regulators of IL‐1, IL‐33, and IL‐36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018;22(11):2937–2950. [DOI] [PubMed] [Google Scholar]
- 93. Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL‐1 beta) to an active IL‐1 species by human mast cell chymase. J Exp Med. 1991;174(4):821–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kennedy EA, Connolly J, Hourihane JO, et al. Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol. 2017;139(1):166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials