

Summary
Background
Data on the prevalence and clinical features of Austrian patients with hereditary angioedema (HAE) with C1‐inhibitor (C1‐INH) deficiency (HAE‐1) or dysfunction (HAE‐2) are lacking.
Methods
Current baseline data were collected in a national survey. The records of HAE patients at the Medical University of Graz were analyzed with regard to clinical characteristics.
Results
A total of 137 patients were identified, yielding a prevalence of 1 : 64,396. The median age at the onset of symptoms was 6.5 years, and the median age at the time of correct diagnosis 21.0 years. The median delay in diagnosis was 15.0 years for newly diagnosed patients without a family history of HAE. Patients with a family history of HAE received an immediate diagnosis. HAE patients without a family history of HAE and born before 1960 had to wait a median of 16.0 years until they were diagnosed correctly. Patients born after 1980 still experienced a median diagnostic delay of 6.5 years.
Conclusion
Patients with this condition still face an excessive diagnostic delay in some parts of Austria, or their disorder may even remain unrecognized by specialists. This underlines the need for better awareness of the disease.
Introduction
Hereditary angioedema (HAE) is a rare autosomal dominant genetic condition with an estimated prevalence of 1 : 50,000 1. Several mutations in the SERPING1 gene have been reported to cause HAE‐1, which is characterized by low functional and antigen levels of the C1 inhibitor (C1‐INH), or HAE‐2, which is marked by low functional levels but normal or elevated antigen levels 2. HAE‐1 accounts for about 85 % of cases of inherited bradykinin‐induced angioedema, whereas 15 % of mutations in the SERPING1 gene result in HAE‐2. Approximately 25 % of C1‐INH‐HAE patients have typical symptoms of angioedema with no family history of HAE, and may therefore be classified as de novo mutants 3. Low levels of functional C1‐INH result in excessive generation of bradykinin (the key mediator of HAE) due to uncontrolled activation of the plasma contact system. This leads to a transient increase in local vascular permeability and blood vessel dilatation in the deeper layers of the skin and mucous membranes, causing episodes of edema 4. Recurrent angioedema attacks typically have a non‐pruritic, non‐pitting nature and may occur at virtually any site of the body. Frequent locations are the extremities as well as the genitourinary and gastrointestinal tracts, where untreated angioedema attacks may not only be disfiguring but especially painful as well. Angioedema attacks in the upper airways may lead to asphyxiation and death if not treated appropriately with a timely response 5. Hereditary angioedema is a rare condition and is frequently misdiagnosed or overlooked, thus delaying the patients' access to appropriate treatment. The swelling that is characteristic of HAE is frequently misdiagnosed as an allergy and treated inappropriately 6.
The available treatment options for acute treatment as well as short‐term and long‐term prophylaxis include a variety of options that have emerged over the past decade. Less than ten years ago, attenuated androgens were the standard choice for long‐term prophylaxis in HAE patients. However, this treatment was viewed skeptically because of its potential side effects. Since the risk of adverse events with the use of androgens increases in a dose‐ and time‐dependent manner, patients had to be monitored closely and physicians needed to be mindful of adverse effects. Attenuated androgens are no longer available in Germany or Austria. The fact that these agents have to be imported from abroad makes it difficult for the physician to prescribe them 7, 8. In the absence of a curative treatment for C1‐INH‐HAE, Austrian patients are currently given C1‐INH concentrates in an acute setting as short‐term treatment and long‐term prophylaxis. Attacks may be treated with icatibant, which antagonizes the bradykinin B2 receptor and has proved to be effective in adolescents as well as children 9, 10. All of the above‐mentioned therapy options are available in Austria. To the best of our knowledge, fresh‐frozen plasma and tranexamic acid are no longer used for the treatment of HAE in Austria. Attenuated androgens are still used by some patients who took them in the past for long‐term prophylaxis and still undergo regular check‐ups. Research on new oral kallikrein inhibitors for the prevention of HAE attacks is currently in progress and has yielded positive results 11.
National surveys of the prevalence of C1‐INH‐HAE are scarce. Studies in Sweden, Italy and Norway, based on 146, 983, and 67 patients, respectively, reported minimum prevalences of 1 : 64,028, 1 : 66,284 and 1 : 66,597 respectively 12, 13, 14. Publications from Denmark, Spain, and Greece reported national prevalences of 1 : 72,671, 1 : 91,162 and 1 : 93,235, respectively 15, 16, 17. Two recent reports from Slovenia and the Western Cape Province, South Africa, reported lower prevalence rates of 1 per 105,000 and 1 per 140,000 inhabitants 18, 19. The prevalence of C1‐INH‐HAE in Austria has not yet been surveyed. In order to determine the clinical characteristics of patients in Austria, we retrospectively analyzed medical records at the Medical University of Graz. The aim was to obtain reliable disease‐specific information about HAE‐1/2 patients in Austria.
With regard to clinical characteristics, a retrospective study encompassing a large number of C1‐INH‐HAE patients revealed that the vast majority of them experienced their first symptoms in childhood or adolescence; these symptoms then persisted throughout their lives 20. More recent observations based on an analysis of the Icatibant Outcome Survey (IOS) registry data, including 171 patients from eight centers in Germany, Spain, Italy, France, the UK, Denmark, Israel and Sweden, showed that the median age of patients at the onset of symptoms was 12.0 years (mean 14.5 years), with a median age at diagnosis of 24.3 years (mean 27.9 years), and a median diagnostic delay of 8.5 years (mean 12.9 years) 21.
Methods
As of August 2017, we had sparse data on the prevalence of HAE in Austria. A survey was initiated at Austrian departments of dermatology, among local HAE experts and among physicians from other disciplines who were known to be aware of HAE and were listed on the homepage of the Austrian patient organization. Dermatologists are frequently the first doctors to diagnose HAE in Austria, because patients with a swelling commonly consult a dermatologist 22. A total of 11 facilities and doctors throughout Austria were contacted via email and requested to participate in a short survey concerning HAE patients identified by them. Austrian patients with HAE tend to consult different doctors for their condition, and we wished to avoid multiple counting of patients. We asked for the following data: gender, initials, year of birth, postal code, HAE type 1 or 2, and whether the type of HAE had been identified. Only patients with confirmed C1‐INH‐HAE according to the consensus report of the Hereditary Angioedema International Working Group were included in the analysis 1. Patients with other types of bradykinin‐mediated angioedema such as AAE‐C1‐INH or HAE nC1‐INH (HAE‐FXII, HAE‐ANGPTI, HAE‐PLG, HAE‐UNK) were not included. Existing patient records at the Medical University of Graz were analyzed with regard to the onset of symptoms, age at diagnosis and diagnostic delay.
Different numbers of patients were analyzed for the individual parameters as only complete and confirmed data were processed; only patients who were alive at the time of data analysis were included. Descriptive analysis of epidemiological data was performed; the results are presented as means and standard deviations (SD), median, Q1 and Q3 as well as minimum and maximum values. We used the Mann‐Whitney U‐test to compare the different categories of the onset of symptoms, age at diagnosis, and delay from the onset of symptoms to the correct diagnosis. The level of significance was set to an alpha level of 0.05. Given the small quantity of existing patient data for analysis, medians and means could differ because outliers are liable to influence means and the low patient numbers may influence median values. It therefore seems meaningful to view both mean and median data in the present report. For the calculation of prevalence, we collected demographic data in September 2016 and in August 2018 from Statistik Austria, which is the official statistics register in Austria (http://www.statistik.at).
Ethics approval was not required for the retrospective analysis of clinical records or for the survey. Since Austrian law ensures the anonymity of patients, no ethics approval was needed for data collection or analysis.
Results
The survey identified 137 C1‐INH‐HAE patients (43.8 % male) living in Austria, amounting to a prevalence of 1 : 64,396 or 1.55 per 100,000 inhabitants. As shown in Table 1 and in Figure 1 with a corresponding color code, the prevalence varies throughout the federal provinces of Austria: from highest prevalence with 1 per 29,529 inhabitants in Styria to provinces with few or no diagnosed HAE patients. A detailed analysis of Styrian patients revealed 42 patients in 17 unrelated families, with the extended family ranging from 1 to 8 affected persons. Prevalence data were determined twice (2016 and 2018), and showed an increase in the number of patients. The type of C1‐INH‐HAE is known for 96 of the 137 patients. Of these 96 patients, 80.2 % (n = 77) had HAE‐1 and 19.8 % (n = 19) had HAE‐2. A total of 72 patients were identified in 27 unrelated families. The term ‘patients with a family history’ was defined as more than one affected family member (horizontal/vertical relatives, dead or alive). The term “patient without a family history” was defined as the patient being the sole individual in the family with HAE. Fourteen patients belonged to the latter category. Genealogical information was not available for 51 patients. The patients’ median age at the onset of symptoms was 6.5 years (range 3.0–67.0 years, mean 12.6, SD 13.0). No significant difference was noted between male and female patients with regard to the onset of symptoms: the median age of males at onset was 6.5 years (range 3.0–67.0 years, mean 12.3 years, SD 14.6), and the median age of females was 9.0 years (range 3.0–51.0 years, mean 12.9, SD 11.7). Figure 2 shows that HAE manifested early in life in the majority of patients. Our patient records at the Medical University of Graz revealed that no patient developed symptoms before the age of 3.0 years. Further analysis of age at the onset of symptoms revealed that patients born before 1960 reported the onset of symptoms significantly later in life (mean 20.4 years, SD 18.4) than patients born after 1960 (mean 8.8 years, SD 7.4, p = 0.004) and those born after 1980 (mean 7.6 years, SD 5.2, p = 0.007). HAE‐1 patients reported the onset of symptoms at a median age of 6.0 years (range 3.0–51.0 years, mean 10.7, SD 10.7), and HAE‐2 patients reported symptom onset at a median age of 13.0 years (range 4.0–67.0 years, mean 18.4, SD 20.2). The difference was not significant (p = 0.131). The overall median age of patients at the time of correct diagnosis for those with and without a family history of HAE was 21.0 years (range 0.0–69.0 years, mean 22.9, SD 16.4).
Table 1.
HAE prevalence in Austria as of August 2018. Provinces are ranked from lowest to highest prevalence
| Austria | Population (2016) | Patients (09/2016) | Prevalence | Population (2018) | Patients (08/2018) | Prevalence |
|---|---|---|---|---|---|---|
| Vorarlberg | 381,000 | 0 | – | 391,741 | 0 | – |
| Tyrol | 732,671 | 1 | 1: 732,671 | 751,140 | 1 | 1: 751,140 |
| Carinthia | 558,612 | 2 | 1: 279,306 | 560,898 | 2 | 1: 280,449 |
| Burgenland | 289,262 | 3 | 1: 96,421 | 292,675 | 3 | 1: 97,558 |
| Lower Austria | 1,643,001 | 11 | 1: 149,364 | 1,670,668 | 22 | 1: 75,939 |
| Vienna | 1,814,225 | 12 | 1: 151,185 | 1,888,776 | 30 | 1: 62,959 |
| Upper Austria | 1,444,122 | 18 | 1: 80,229 | 1,473,576 | 24 | 1: 61,399 |
| Salzburg | 541,439 | 6 | 1: 90,240 | 552,579 | 13 | 1: 42,506 |
| Styria | 1,225,187 | 37 | 1: 33,113 | 1,240,214 | 42 | 1: 29,529 |
| Total | 8,629,519 | 90 | 1: 95,884 | 8,822,267 | 137 | 1: 64,396 |
Figure 1.
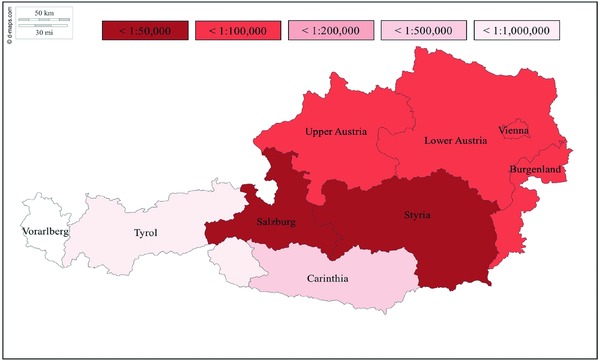
Color‐coded prevalence map of HAE in Austria as of August 2018. (Original map source: https://d-maps.com/m/europa/austria/autriche/autriche68.pdf).
Figure 2.
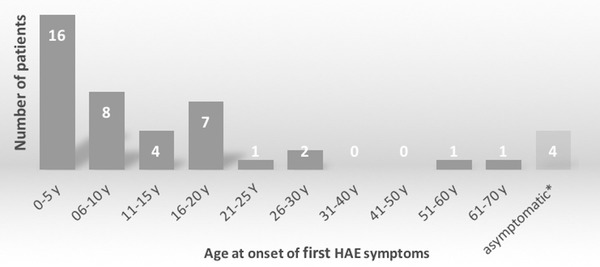
Age at the onset of symptoms in 44 Austrian HAE patients diagnosed with type I or type II HAE. *Four patients (aged 2, 3, 10, and 38 years) are still asymptomatic (as of January 2018).
The determination of diagnostic delay is important, because patients who are symptomatic but have not yet been correctly diagnosed with the condition will be given inappropriate treatment or no treatment at all. Analyzing patients without a family history of HAE and those with a family history of HAE who were the first to be diagnosed with the disease in their family, we registered a median diagnostic delay of 15.0 years (range 1.0–33.0 years, mean 15.0, SD 9.9). Patients born before 1960 were diagnosed with a median delay of 16.0 years (range 1.0–33.0 years, mean 17.6 years, SD 11.8). This long period was reduced to a median of 12.0 years (range 1.0–27.0 years, mean 12.5, SD 7.6) for patients born after 1960. A nearly significant difference in diagnostic delay was noted for patients born before 1960 and those born after 1980 (median 16.0 years [range 1.0–33.0] versus 6.5 years [range 1.0–15.0]; p = 0.057). If HAE had already been diagnosed in a family member, the median delay in diagnosis for other affected family members was 0.0 years (range –6.0 to 16.0, mean 0.5, SD 4.7) (Table 2). The negative values in delay result from pre‐symptomatic testing in some family members.
Table 2.
Epidemiological data of Austrian HAE patients as of August 2018
| Characteristics of HAE patients in Austria | N | [%] | ||||||
|---|---|---|---|---|---|---|---|---|
| Total number (as of August 2018) | 137 | 100 | ||||||
| Male | 60 | 43.8 | ||||||
| Female | 77 | 56.2 | ||||||
| Diagnosis | N a | [%] b | ||||||
| HAE‐1 (confirmed) | 77 | 80.2 | ||||||
| HAE‐2 (confirmed) | 19 | 19.8 | ||||||
| Patients without HAE‐1/2 type identification | 41 | |||||||
| Inheritance | N a | [%] | ||||||
| Unrelated HAE families | 27 | |||||||
| Patients with a family history of HAEc | 72 | 83.7 | ||||||
| Patients without a family history of HAEd | 14 | 16.3 | ||||||
| Patients with no information on inheritance | 51 | |||||||
| Age at onset of HAE symptoms [y] | Mean | ±SD | N a | Median | Q1 | Q3 | Min | Max |
| Total | 12.6 | (±13.0) | 40 | 6.5 | 4.75 | 17.25 | 3.0 | 67.0 |
| Male | 12.3 | (±14.6) | 20 | 6.5 | 5.0 | 12.75 | 3.0 | 67.0 |
| Female | 12.9 | (±11.7) | 20 | 9.0 | 4.75 | 17.25 | 3.0 | 51.0 |
| HAE patients born before 1960 | 20.4 | (±18.4) | 13 | 16.0 | 9.0 | 19.0 | 5.0 | 67.0 |
| HAE patients born after 1960 | 8.8 | (±7.4) | 27 | 5.0 | 4.5 | 11.5 | 3.0 | 28.0 |
| HAE patients born after 1980 | 7.6 | (±5.2) | 11 | 6.0 | 5.0 | 13.5 | 3.0 | 51.0 |
| HAE‐1 | 10.7 | (±10.7) | 27 | 6.0 | 5.0 | 13.5 | 3.0 | 51.0 |
| HAE‐2 | 18.4 | (±20.2) | 8 | 13.0 | 9.0 | 16.0 | 4.0 | 67.0 |
| Age at correct diagnosis [y] | Mean | ±SD | N a | Median | Q1 | Q3 | Min | Max |
| HAE patients (total) | 22.9 | (±16.4) | 55 | 21.0 | 8.5 | 31.0 | 0.0 | 69.0 |
| Diagnostic delay [y] | Mean | ±SD | N a | Median | Q1 | Q3 | Min | Max |
| Totale | 15.0 | (±9.9) | 23 | 15.0 | 8.0 | 19.5 | 1.0 | 33.0 |
| HAE patients born before 1960e | 17.6 | (±11.8) | 11 | 16.0 | 9.5 | 29.0 | 1.0 | 33.0 |
| HAE patients born after 1960e | 12.5 | (±7.6) | 12 | 12.0 | 3.0 | 15.75 | 1.0 | 27.0 |
| HAE patients born after 1980e | 7.2 | (±5.9) | 6 | 6.5 | 2.25 | 11.5 | 1.0 | 15.0 |
| Patients with a family history of HAE | 0.5 | (±4.7) | 17 | 0.0 | –1.0 | 0.0 | –6.0 | 16.0 |
Number of analyzed patients varies, since only complete and confirmed data have been processed.
Percentage refers to patients with confirmed HAE‐1/2 diagnosis (n = 96).
Positive family history = more than one affected person in the family (horizontal/vertical, dead or alive).
Patients with no affected family member reported in the previous, same or next generation.
Calculated on the basis of data from patients without a family history of HAE and data from patients with a family history of HAE who were the first in their families to be diagnosed with HAE.
Discussion
Our nationwide survey revealed an overall prevalence of 1 in 64,396 inhabitants, with a prevalence of at least 1 : 29,529 or 3.39 per 100,000 inhabitants in Styria. Styria is one of nine federal provinces in Austria, with 42 HAE patients in a population of more than 1.2 million. The high prevalence in Styria may be explained by the fact that, in Austria, HAE is primarily diagnosed by dermatologists. Styria has just one hospital department of dermatology (at the Medical University of Graz) which performs biochemical C1‐INH antigen and function testing. The department is well known throughout the federal province for its expertise in the field of angioedema. Thus, patients anywhere in Styria are likely to be referred to this department for clarification of any ambiguous angioedema‐like symptoms. The 42 Styrian patients belong to 17 unrelated families. New mutations causing C1‐INH‐HAE at a rate of 1 : 72,953 have been detected in the Styrian population. This de novo mutation rate is based on a manageable number of unrelated families. We estimate a de novo mutation rate of about 1 : 50,000 to 1 : 100,000 in the SERPING1 gene region, amounting to a high overall prevalence of about 1 : 30,000 to 1 : 50,000 for Austria (based on our data showing an average of 2.5 patients per HAE family). This is indicative of a large number of unreported, unknown, or undiagnosed patients. Since our survey yielded 137 patients, we assume that 22–53 % are not yet diagnosed. Assuming a C1‐INH‐HAE prevalence range of 1 : 30,000 to 1 : 50,000, we may estimate a total of 176 to 294 C1‐INH‐HAE patients in Austria. This would be consistent with previously published data on percentages of diagnosed versus non‐diagnosed patients 23.
Prevalence data from other federal provinces of Austria revealed lower prevalence rates than those in Styria. This has several reasons. First, although we contacted HAE specialists at hospital departments as well as physicians who were known to be aware of HAE, listed on the homepage of the Austrian HAE patients’ organization and therefore with expertise in the subject, other physicians may also treat HAE patients. Secondly, our nationwide survey yielded a response rate of only close to 45 % (5/11), with responses from Vienna, Upper Austria and Salzburg. Furthermore, we are aware of patients from certain regions of Austria who visit specialists in a neighboring federal province with an established HAE care center, indicating cross‐regional patient care. The survey responses revealed 15 patients from outside Styria who had attended our department. Other reasons for the low prevalence rates may be misdiagnosis of patients with similar symptoms, undiagnosed patients, or the fact that C1‐INH‐HAE is a very rare disease in these regions. All these factors may influence – to a greater or lesser extent – the difference in prevalence rates among the federal provinces of Austria.
Switzerland, a neighboring country, has nearly the same number of inhabitants as Austria and similar numbers of C1‐INH‐HAE patients; an investigation yielded 111 affected patients, who are believed to constitute 70 % of all HAE patients in Switzerland 24. A handful of national studies have been performed on the prevalence of HAE. The highest prevalence was noted in Sweden (1.56 patients per 100,000 inhabitants), followed by Italy and Norway with confirmed prevalence rates of 1.51 per 100,000 and 1.50 per 100,000 respectively 12, 13, 14. A recent comparison of national prevalence rates for C1‐INH‐HAE revealed that the epidemiology of this condition is stable; its prevalence was about 1.50 per 100.000 across the six European countries that were compared 25. Accurate estimates of the prevalence of C1‐INH‐HAE in a given population are hindered by the rare occurrence of the disease, poor awareness of the condition, the absence of established registries, the small number of studies on the prevalence and epidemiology of HAE, and diverse methods of data collection (registries, data from patient organizations or surveys).
Laboratory data in our Austrian patient population revealed HAE‐1 in 80.2 % of patients and HAE‐2 in 19.8 %, which is similar to published reports (85 % and 15 %, respectively) 26. Information on C1‐INH‐HAE type identification was not available in 41 out of 137 patients, meaning that C1‐INH antigenic levels have not been determined in those patients to distinguish between C1‐INH‐HAE type 1 and type 2. Nevertheless, diagnosis of C1‐INH‐HAE has been made according to the guidelines 1. In our survey we did not ask whether or not mutation analysis on the patients’ SERPING1 gene had been performed in order to confirm C1‐INH‐HAE 1/2 diagnosis, as biochemical testing is sufficient to diagnose the condition. The patients’ median age at the onset of symptoms was 6.5 years, which is lower than that of patients enrolled in the IOS study (12.0 years) 21. Detailed analysis revealed a significant difference in age at onset between HAE patients born before or after 1960 and those born after 1980 (p = 0.004; p = 0.007). This may be explained by increasing awareness of the symptoms, especially in patients with a family history of HAE, whereas patients born before 1960 may not associate the symptoms experienced in childhood with manifestations of HAE. Our comparison of mean values showed a slight difference: we registered a mean age of 12.6 years (SD 13.0) compared to the published figure of 14.5 years (SD 12.3) 21. The high standard deviation was due to a few extreme outliers, such as an onset of symptoms at the age of 67 years. The overall median age at the time of correct diagnosis for patients with and without a family history of HAE was 21.0 years (range 0.0–69.0 years, mean 22.9, SD 16.4). Thus, the difference between the median age at the time of correct diagnosis and the median age at the onset of symptoms does not concur with the median diagnostic delay in years. The delay in correct diagnosis was derived from the data of patients without a family history of HAE and those with a family history of HAE who were the first to be diagnosed with the disease in their family. In patients with a positive family history we registered a median diagnostic delay of 0.0 years (range –6 to 16 years), which would bias the delay in favor of a misleading low median value. As a result, we obtained an overall median diagnostic delay of 15.0 years, which is nearly double the time period reported in the literature (8.5 years) 21.
Comparing the diagnostic delay in subgroups of patients born before 1960 and after 1980, we noted a nearly significant drop in the delay between the former and the latter (median 16.0 years vs 6.5 years; p = 0.057). It seems likely that HAE is diagnosed correctly at an earlier point in time after manifestation of the initial symptoms in patients born after 1980 without a family history of HAE than in those born before 1960.
Conclusion
This first‐ever nationwide survey of the prevalence of C1‐INH‐HAE in Austria yielded 1 patient per 64,396 inhabitants with remarkable local differences. Patients with C1‐INH‐HAE appear to be quite unequally distributed among the nine federal provinces of Austria with very low to very high prevalence rates. Comparing the diagnostic delay in subgroups of patients over three generations, we noted a reduction in the diagnostic delay, but younger patients without a family history of the disease still face an unacceptable delay of 6.5 years from experiencing their first attack to correct diagnosis of HAE. The delay has clearly been shortened, but further awareness of the disease is needed in order to distinguish HAE from other common causes of angioedema. This would ensure that patients receive targeted therapy for their condition on a timely basis. Our data also demonstrate that higher awareness and better information and communication among doctors is essential in order to improve the management and quality of life of those affected by this rare disease.
Conflict of interest
None.
References
- 1. Cicardi M, Aberer W, Banerji A et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 2014; 69: 602–16. [DOI] [PubMed] [Google Scholar]
- 2. Germenis AE, Speletas M. Genetics of Hereditary Angioedema Revisited. Clin Rev Allergy Immunol 2016; 51: 170–82. [DOI] [PubMed] [Google Scholar]
- 3. Pappalardo E, Cicardi M, Duponchel C et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106: 1147–54. [DOI] [PubMed] [Google Scholar]
- 4. Nussberger J, Cugno M, Amstutz C et al. Plasma bradykinin in angio‐oedema. Lancet 1998; 351: 1693–7. [DOI] [PubMed] [Google Scholar]
- 5. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1‐INH deficiency. J Allergy Clin Immunol 2012; 130: 692–7. [DOI] [PubMed] [Google Scholar]
- 6. Zanichelli A, Longhurst HJ, Maurer M et al. Misdiagnosis trends in patients with hereditary angioedema from the real‐world clinical setting. Ann Allergy Asthma Immunol 2016; 117: 394–8. [DOI] [PubMed] [Google Scholar]
- 7. Maurer M, Magerl M. Long‐term prophylaxis of hereditary angioedema with androgen derivates: a critical appraisal and potential alternatives. J Dtsch Dermatol Ges 2011; 9: 99–107. [DOI] [PubMed] [Google Scholar]
- 8. Greve J, Strassen U, Gorczyza M et al. Prophylaxis in hereditary angioedema (HAE) with C1 inhibitor deficiency. J Dtsch Dermatol Ges 2016; 14: 266–75. [DOI] [PubMed] [Google Scholar]
- 9. Krause K, Metz M, Zuberbier T et al. Successful treatment of hereditary angioedema with bradykinin B2‐receptor antagonist icatibant. J Dtsch Dermatol Ges 2010; 8: 272–4. [DOI] [PubMed] [Google Scholar]
- 10. Farkas H, Reshef A, Aberer W et al. Treatment Effect and Safety of Icatibant in Pediatric Patients with Hereditary Angioedema. J Allergy Clin Immunol Pract 2017; 5: 1671–8.e2. [DOI] [PubMed] [Google Scholar]
- 11. Aygoren‐Pursun E, Bygum A, Grivcheva‐Panovska V et al. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N Engl J Med 2018; 379: 352–62. [DOI] [PubMed] [Google Scholar]
- 12. Nordenfelt P, Dawson S, Wahlgren CF et al. Quantifying the burden of disease and perceived health state in patients with hereditary angioedema in Sweden. Allergy Asthma Proc 2014; 35: 185–90. [DOI] [PubMed] [Google Scholar]
- 13. Zanichelli A, Arcoleo F, Barca MP et al. A nationwide survey of hereditary angioedema due to C1 inhibitor deficiency in Italy. Orphanet J Rare Dis 2015; 10: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stray‐Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol 2000; 20: 477–85. [DOI] [PubMed] [Google Scholar]
- 15. Bygum A. Hereditary angio‐oedema in Denmark: a nationwide survey. Br J Dermatol 2009; 161: 1153–8. [DOI] [PubMed] [Google Scholar]
- 16. Roche O, Blanch A, Caballero T et al. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol 2005; 94: 498–503. [DOI] [PubMed] [Google Scholar]
- 17. Psarros F, Koutsostathis N, Farmaki E et al. Hereditary angioedema in Greece: the first results of the greek hereditary angioedema registry. Int Arch Allergy Immunol 2014; 164: 326–32. [DOI] [PubMed] [Google Scholar]
- 18. Rijavec M, Korosec P, Silar M et al. Hereditary angioedema nationwide study in Slovenia reveals four novel mutations in SERPING1 gene. PLoS One 2013; 8: e56712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coovadia KM, Chothia MY, Baker SG et al. Hereditary angio‐oedema in the Western Cape Province, South Africa. S Afr Med J 2018; 108: 283–90. [DOI] [PubMed] [Google Scholar]
- 20. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119: 267–74. [DOI] [PubMed] [Google Scholar]
- 21. Zanichelli A, Magerl M, Longhurst H et al. Hereditary angioedema with C1 inhibitor deficiency: delay in diagnosis in Europe. Allergy Asthma Clin Immunol 2013; 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aberer W. Angioedema is not just ‘deep urticaria’ but an entity of its own. Allergy 2014; 69: 549–52. [DOI] [PubMed] [Google Scholar]
- 23. Lumry WR, Castaldo AJ, Vernon MK et al. The humanistic burden of hereditary angioedema: Impact on health‐related quality of life, productivity, and depression. Allergy Asthma Proc 2010; 31: 407–14. [DOI] [PubMed] [Google Scholar]
- 24. Steiner UC, Keller M, Schmid P et al. Mutational spectrum of the SERPING1 gene in Swiss patients with hereditary angioedema. Clin Exp Immunol 2017; 188: 430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aygoren‐Pursun E, Magerl M, Maetzel A, Maurer M. Epidemiology of bradykinin‐mediated angioedema: a systematic investigation of epidemiological studies. Orphanet J Rare Dis 2018; 13: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359: 1027–36. [DOI] [PubMed] [Google Scholar]