

Abstract
Premise of the Study
A key question in evolutionary biology is why some clades are more successful by being widespread geographically, biome diverse, or species‐rich. To extend understanding of how shifts in area, biomes, and pollinators impact diversification in plants, we examined the relationships of these shifts to diversification across the mega‐genus Salvia.
Methods
A chronogram was developed from a supermatrix of anchored hybrid enrichment genomic data and targeted sequence data for over 500 of the nearly 1000 Salvia species. Ancestral areas and biomes were reconstructed using BioGeoBEARS. Pollinator guilds were scored, ancestral pollinators determined, shifts in pollinator guilds identified, and rates of pollinator switches compared.
Key Results
A well‐resolved phylogenetic backbone of Salvia and updated subgeneric designations are presented. Salvia originated in Southwest Asia in the Oligocene and subsequently dispersed worldwide. Biome shifts are frequent from a likely ancestral lineage utilizing broadleaf and/or coniferous forests and/or arid shrublands. None of the four species diversification shifts are correlated to shifts in biomes. Shifts in pollination system are not correlated to species diversification shifts, except for one hummingbird shift that precedes a major shift in diversification near the crown of New World subgen. Calosphace. Multiple reversals back to bee pollination occurred within this hummingbird clade.
Conclusions
Salvia diversified extensively in different continents, biomes, and with both bee and bird pollinators. The lack of tight correlation of area, biome, and most pollinator shifts to the four documented species diversification shifts points to other important drivers of speciation in Salvia.
Keywords: adaptive radiation, BioGeoBEARS, evolution, historical biogeography, hummingbird, Lamiaceae, long‐distance dispersal, niche, phylogenomics, species diversification
A long‐standing and central objective within evolutionary biology has been to elucidate and describe mechanisms governing speciation (Darwin, 1859; Gottlieb, 1984; Schluter, 2001; Rieseberg and Willis, 2007; Kay and Sargent, 2009; Givnish, 2010; Sobel et al., 2010; Edwards and Donoghue, 2013; Ricklefs, 2014). In this context, one of the key questions raised is why some organismal lineages are more successful (widespread geographically, biome diverse, or species‐rich) than others. An often‐espoused hypothesis has been adaptive radiation—the rise of a diversity of ecological roles and role‐specific adaptations within a lineage (Givnish, 2015). Others have emphasized the role of dispersals to new areas and subsequent diversification into new ecological zones (“dispersification” of Moore and Donoghue, 2007). However, definitively evaluating lineage success and adaptive radiations has been elusive due to the myriad factors responsible for speciation and evolutionary success, suboptimal phylogenetic contexts, and lack of analytical and statistical frameworks.
Recent and continuing intellectual and computational advancements have facilitated the integration of phylogenetics, biogeography, ecology, morphometrics, diversification, and character transitions, thus enabling evolutionary biologists to explore and perhaps more rigorously test hypotheses in ways not possible a decade ago. In the process, these advances have transformed the study of plant diversification from a field of oft‐speculative narrative to a complex discipline that uses sophisticated computational methods to meld, correlate, and interpret results from these disparate fields (e.g., Crisp et al., 2009; Drummond et al., 2012; van der Niet and Johnson, 2012; Beaulieu et al., 2013; Cornwell et al., 2014; Givnish et al., 2014, 2015; Linder et al., 2014; Zanne et al., 2014; Uribe‐Convers and Tank, 2015; Lagomarsino et al., 2016; Roalson and Roberts, 2016; Rose et al., 2016; Spalink et al., 2016a, b; Cardillo et al., 2017; Vamosi et al., 2018). Additionally, the underlying assumptions and thus interpretation of such correlated findings are now the subject of healthy debate (Maddison and FitzJohn, 2014; Rabosky and Goldberg, 2015; Uyeda et al., 2018).
Salvia L. (sages, Lamiaceae), with a nearly worldwide distribution (Fig. 1), multiple diversity hotspots, extraordinary evolutionary success in terms of both numbers of species and ecological amplitude, diverse vegetative and floral forms, and a unique staminal architecture to promote outcrossing provides an excellent model to address these questions (Claßen‐Bockhoff et al., 2003, 2004; Walker et al., 2004, 2015; Walker and Sytsma, 2007; Wester and Claßen‐Bockhoff, 2007; Takano and Okada, 2011; Jenks et al., 2012; Li et al., 2013; Wang et al., 2013; Celep et al., 2014; Drew et al., 2017a; Will and Claßen‐Bockhoff, 2017). Salvia (sensu Drew et al., 2017a), defined in part by only two stamens with at least some elongation in the anther connective, is sister to a relatively depauperate four‐stamen lineage comprising Lepechinia Willd. and Melissa L.; these three genera together form the subtribe Salviinae within the species‐rich tribe Mentheae (Walker and Sytsma, 2007; Drew and Sytsma, 2012). The five small, previously recognized genera, Dorystaechas Boiss. & Heldr., Meriandra Benth., Perovskia Kar., Rosmarinus L., and Zhumeria Rech.f. & Wendelbo are embedded within Salvia, share a suite of synapomorphic, morphological traits with Salvia (including having only two stamens with some connective elongation), and have thus been recently included in a recircumscribed Salvia (Drew et al., 2017a). Breaking up Salvia s.l. into upward of 11 genera, although not all are confirmed monophyletic, has been recently advocated (Will et al., 2015; Will and Claßen‐Bockhoff, 2017). We disagree with this approach following the arguments of Drew et al. (2017a) based on phylogenetic, taxonomic, and practical considerations and instead recognize subgenera within a broad Salvia (Drew et al., 2017a; Hu et al., 2018; Table 1).
Figure 1.
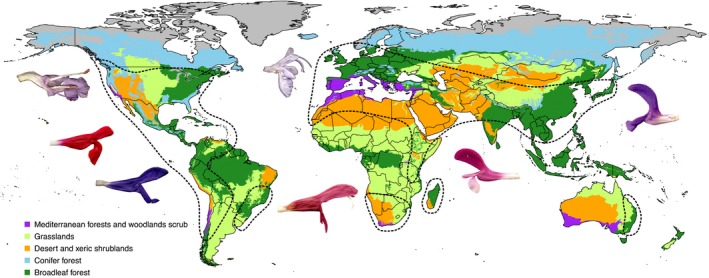
Biogeographic, ecological, and floral diversity of Salvia. Dashed lines represent general distribution of Salvia (see Fig. 6 for specific biogeographical regions). The five broad biome regions of the world occupied by Salvia and used in this study are depicted. Images illustrate some of the range of floral diversity seen in Salvia across different biogeographical regions.
Table 1.
Subgeneric classification of Salvia s.l. used in this study, following in part Drew et al. (2017a) and as updated by Hu et al. (2018). For an alternative classification system of Salvia s.l. in which up to 11 genera (some not monophyletic by the present study) are recognized, see Will and Claßen‐Bockhoff (2017). The “Salvia aegyptiaca clade” of Drew et al. (2017a) is placed informally for now with subgen. Zhumeria. The informal name “Heterosphace” is provided for now to New World sect. Salviastrum, the Old World “Salvia verticillata group” of Will and Claßen‐Bockhoff (2017), and their close relatives
| Subgenus | Estimated number of species | Biogeographic distribution | Notes | Clade number of Will and Claßen‐Bockhoff (2014) | Staminal type based on Walker and Sytsma (2007) |
|---|---|---|---|---|---|
| Audibertia (Benth.) J.B.Walker, B.T.Drew, & K.J.Sytsma | 19 | Western North America to northern Mexico | See Walker et al. (2015); originally considered sect. Audibertia Epling or as the genus Audibertia Benth. (incorrectly) or the genus Ramona Greene; now comprises two sections (Echinosphace Benth. and Audibertia) | Clade II | H, I |
| Calosphace (Benth.) Epling | 550 | North America, Central America, Caribbean, South America | Will and Claßen‐Bockhoff (2017) refer to this as the genus Lasemia Raf. | Clade II | E, F, G |
| Dorystaechas (Boiss. & Heldr. ex Benth.) J.B.Walker, B.T.Drew, & J.G.González | 1 | Endemic to Antalya region of Turkey | See Drew et al. (2017a); originally named as the genus Dorystaechas Boiss. & Heldr. ex Benth. | K | |
| Glutinaria (Raf.) G.X.Hu, C.L.Xiang, & B.T.Drew | 100 | East Asia, with outliers in SE Asia, eastern Australia, and central Eurasia | See Hu et al. (2018); previously named as the genus Glutinaria Raf.; now comprises eight named sections. Six distinct stamen types now recognized. | Clade IV | N |
| “Heterosphace” | 43 | E, S, SW North America, southern Africa and Madagascar, NE Africa, SW Asia, Mediterranean region, Europe | Includes the genus Salviastrum Scheele, Salvia sect. Hemisphace Benth., sect. Heterosphace Benth.; also includes (in part) Hymenosphace Benth. and Eusphace Benth. (see Will and Claßen‐Bockhoff 2017) | Clade I | A |
| Meriandra (Benth.) J.B.Walker, B.T.Drew, & J.G.González | 2 | NE Africa and Indian Himalayas | See Drew et al. (2017a); originally named as the genus Meriandra Benth. | J | |
| Perovskia (Kar.) J.B.Walker, B.T.Drew, & J.G.González | 8 | Central and SW Asia | See Drew et al. (2017a); originally named as the genus Perovskia Kar. | D | |
| Rosmarinus (L.) J.B.Walker, B.T.Drew, & J.G.González | 3 | Mediterranean region | see Drew et al. (2017a); originally named as the genus Rosmarinus L. | C | |
| Salvia L. | 70 | SW Asia, Europe, Mediterranean region | contains the type species for Salvia L., S. officinalis L.; includes sect. Hymenosphace Benth. (in part) and most of sect. Eusphace Benth. (see Will and Claßen‐Bockhoff 2017) | Clade I | A |
| Sclarea (Moench) Benth. | 120 | SW Asia, Europe, Mediterranean region, with outliers in southern Africa | Comprises Salvia sect. Aethiopis Benth. and sect. Plethiosphace Benth.; (see Will and Claßen‐Bockhoff 2017) | Clade I | B |
| Zhumeria (Rech. f. & Wendelbo) J.B.Walker, B.T.Drew, & J.G.González | 31 | Central and SW Asia, Mediterranean region | See Drew et al. (2017a); originally named as the monotypic genus Zhumeria Rech. f. & Wendelbo; we include informally for now other Salvia which were previously placed in genera Pleudia Raf. and Polakia Staf; this study demonstrates that Polakia is not monophyletic. | Clade III (in part) | L, M |
Salvia as circumscribed in the broader sense, with nearly 1000 species, is one of the largest genera of flowering plants and displays a remarkable range of morphological, ecological, and geographical variation. The distribution of Salvia is noteworthy not only because the lineage is young and widespread (ca. early Oligocene for stem clade, Drew and Sytsma, 2012), but also because it has diversity hotspots on four continents. Salvia is hyperdiverse from northern Mexico through central South America (subgen. Calosphace), but also has centers of diversity in East Asia, as well as Southwest Asia and the Mediterranean region (extending to southern Africa). Perhaps more surprisingly, Salvia has apparently evolved variants of a staminal lever mechanism via an elongated connective independently in each of these three geographic regions (Claßen‐Bockhoff et al., 2003, 2004; Walker et al., 2004, 2015; Walker and Sytsma, 2007; Drew et al., 2017a; Hu et al., 2018). In this background of geographic and floral diversification, Salvia has radiated into different biomes and adapted to different pollinators (Wester and Claßen‐Bockhoff, 2006, 2007, 2011; Claßen‐Bockhoff, 2007; Celep et al., 2014; Walker et al., 2015; Fragoso‐Martínez et al., 2018).
We explore patterns of geographical and species diversification in Salvia by leveraging anchored hybrid enrichment genomic data (Buddenhagen et al., 2016 [preprint]; Cardillo et al., 2017; Fragoso‐Martínez et al., 2017; Mitchell et al., 2017), along with an emerging phylogenetic data set and framework for the genus based on targeted gene sequencing (e.g., Drew et al., 2017a), fossil evidence for calibrating this phylogeny, online distributional data for most of the species, and a rich pollinator data set. We evaluate correlations between biogeographic change (i.e., dispersals into new areas), transitions into biomes or pollination systems, and shifts in rates of species diversification across this time‐calibrated tree. Specifically, we examine the hypothesis that dispersals to new areas are an important first and over‐arching step in subsequent habitat, morphological, pollination, and species diversification. Secondarily, we examine the role that shifts in biomes and/or pollination system might have had in subsequent shifts in the ability of Salvia subclades to explore and diversify in different ecological niches. For example, we ask: Are shifts to bird pollination frequent and correlated to shifts in diversification? Are shifts to new biomes repeated across continents in a parallel fashion and correlated to subsequent shifts in pollinator and/or to shifts in species diversification? The analytical design used to evaluate correlates of diversification in Salvia involves four main steps. The first step entails estimating the phylogenetic tree and divergence times for Salvia and its subclades. The second step locates significant shifts in species diversification rate (speciation and extinction). The third step identifies episodes of biogeographic dispersal across areas and continents and correlation of such events to species diversification. The fourth step evaluates the role of biome and/or pollinator shifts in species diversification and whether such shifts are correlated to exploration of new geographical areas. We implement a novel, model‐based approach to estimate biome transitions through time, taking into account historical evidence for the availability of a specific biome through the Neogene and Quaternary.
MATERIALS AND METHODS
Species sampling in Salvia and outgroups
Our aim was to construct an overall nuclear genome phylogeny using both anchored hybrid enrichment (AHE) and nuclear ribosomal DNA (nrDNA) of Salvia where tip relationships are less affected by possible reticulation events involving chloroplast DNA (cpDNA). Although low‐copy nuclear genes, nrDNA and cpDNA generally provide congruent relationships among the major lineages in the genus (e.g., Walker and Sytsma, 2007; Drew and Sytsma, 2012; Drew et al., 2017a; Hu et al., 2018), hybridization and/or chloroplast capture can occur within these well‐supported Salvia clades (e.g., Sudarmono, 2007; Walker et al., 2015). We largely followed the taxon sampling in the largest phylogenetic analysis of Salvia to date (Drew et al., 2017a) and enlarged the sampling with additional species from the Old World (Will and Claßen‐Bockhoff, 2017; Hu et al., 2018) and subgenus Calosphace (Fragoso‐Martínez et al., 2018). Our final nrDNA data set thus had 528 species including 519 (~52%) species of Salvia as well as seven species of Lepechinia and two species of Melissa, which were used as a monophyletic outgroup. The complete nrDNA region (ITS/ETS) for 38 species from Salvia, Lepechinia, and Melissa was obtained via the raw reads from the anchored hybrid enrichment method (see below).
We subsampled 38 species from this 528 set to obtain sequences for 498 single‐copy genes via anchored hybrid enrichment. Those selected included 35 representatives from all major clades of Salvia identified by Drew et al. (2017a); this sampling was done to obtain a well‐resolved backbone for the Salvia‐wide tree. Three close outgroups (two Lepechinia and one Melissa) were included to complete sampling of the subtribe Salviinae. To verify the relationships of the two outgroup genera Melissa and Lepechinia to Salvia (Drew and Sytsma, 2011, 2012, 2013), we sampled and used two genera (Prunella and Agastache) from the tribe Mentheae as a monophyletic, ultimate outgroup (based on Drew and Sytsma, 2012). Species and subgeneric placements (see Table 1) are provided in [Link], [Link].
Anchored hybrid enrichment: library preparation, enrichment, and sequencing
The anchored hybrid enrichment (AHE) method (Lemmon and Lemmon, 2012; Buddenhagen et al., 2016 [preprint]) was used to target 498 moderately conserved, low‐copy nuclear loci. DNA was extracted from silica gel‐dried or fresh leaf tissue using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). DNA concentrations were verified using the Qubit 2.0 Fluorometer (Life Technologies, Eugene, OR, USA). Library preparation and enrichment were performed at the Florida State University Center for Anchored Phylogenomics (http://www.anchoredphylogeny.com) following the methods of Lemmon et al. (2012) and Prum et al. (2015) as described by Buddenhagen et al. (2016 [preprint]). In summary, DNA was sonicated with a Covaris E220 Focused‐ultrasonicator (Woburn, MA, USA) with Covaris microTUBES to obtain ca. 200–800 bp fragments. Libraries were prepared and indexed (8 bp) with a liquid‐handling robot (Beckman‐Coulter Biomek FXp, Brea, CA, USA), samples were pooled in equal quantities, and each pool was enriched using AHE Angiosperm v.1 kit. After enrichment, the libraries were sequenced on one lane (PE150bp, 48Gb output) of an Illumina HiSeq2500 at the Translational Science Laboratory in the College of Medicine at Florida State University.
After quality filtering using the high‐chastity filter in Illumina's Casava software, overlapping read pairs were merged and sequencing adapters were trimmed following Rokyta et al. (2012). Reads were then assembled using the quasi‐denovo approach described in Hamilton et al. (2016), but with Arabidopsis thaliana (L.) Heynh., Billbergia nutans H.Wendl. ex Regel, and Carex lurida Wahlenb. serving as references for the assembly. Assembled contigs derived from fewer than 20 reads were removed to avoid the effects of possible contamination and/or misindexing. The orthology of homologous contigs passing this filter was then assessed using pairwise distances as done by Hamilton et al. (2016). Orthologous sequences at each locus were then aligned using MAFFT (v. 7.023b, Katoh and Standley, 2013) and trimmed and masked using the methods of Hamilton et al. (2016), with MINGOODSITES=15, MINPROPSAME=0.3, and MISSINGALLOWED=38. Alignments were then manually inspected in Geneious R9 (Biomatters Ltd., Aukland, New Zealand; Kearse et al., 2012) and misaligned regions were masked. The final alignments consisted of both target regions and variable flanks for 498 target loci.
Finally, nrDNA (ITS and ETS) was obtained from the raw reads of each of the 38 species (eliminating the two ultimate outgroups) sequenced via AHE. The complete ITS sequence of Salvia hydrangea DC. ex Benth. (KP294355) and the ETS sequence of Salvia namaensis Schinz (KJ584290) were used as references to match and assemble reads in Geneious v7.1.7 (http://www.geneious.com, Kearse et al., 2012). Reads were assembled using an iterative mapping approach in the default Geneious mapper under medium sensitivity. Mapping was conducted for a maximum of 5 iterations. For calculating consensus sequences, minimum coverage was set to 2. If coverage was 0, no base was called. Residues in consensus sequences were called based on a minimum of 65% identical residues to account for paralogy; otherwise, ambiguity codes were used. Subsequently, these sequences were aligned using MAFFT with other sequences of ITS and ETS. We concatenated this alignment with an alignment of ITS and ETS from previously sequenced or obtained from GenBank for a total of 538 taxa. GenBank accession information for both AHE and nrDNA are provided in Appendix 2; the aligned data sets for both AHE and nrDNA sequencing are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8m40rb0.
Construction of the Salvia phylogenetic and temporal framework
To generate a well‐resolved backbone topology for Salvia based on AHE sequences, to which all ITS/ETS sequences of 528 species would ultimately be added, three different maximum likelihood (ML) or Bayesian analyses were conducted. First, both coalescent and concatenated ML approaches were performed on the 40 species AHE data. Second, only ITS/ETS data for all 528 species were analyzed under ML. And third, Bayesian estimates of topology and branching times were done with the 528 species ITS/ETS data set, but including topological prior constraints based on well‐supported nodes obtained with the AHE data.
AHE phylogenetic analyses
The concatenated AHE data set was used to estimate a phylogeny under the GTRGAMMA model implemented in RAxML v8.1.21 (Stamatakis, 2014; with default parameters), with the GTR model and branch lengths being allowed to vary across loci. One hundred bootstrap replicates were collected to estimate phylogenetic support. In addition, the species tree was estimated under the coalescent model as implemented by ASTRAL‐II (v.4.9.7, Mirarab and Warnow, 2015), using bootstrapped gene trees estimated under the GTRGAMMA model in RAxML v8.1.21 (Stamatakis, 2014).
nrDNA phylogenetic analyses
The nrDNA (ITS/ETS) data set was analyzed with RAxML under the GTRGAMMA model. To explore the robustness of relationships among major clades and placement of species within in each clade based solely on nrDNA, we performed an additional RAxML analysis by implementing the “‐g” topology constraint parameter in RAxML. The AHE ASTRAL‐II tree (only nodes with 100% support) was used as a constraint tree, and all other 480 species were unconstrained.
Bayesian time‐calibrated phylogenetic analysis
BEAST 2.4.3 (Bouckaert et al., 2014) in the XSEDE interface of CIPRES (Miller et al., 2010) was used to generate a time‐calibrated tree by incorporating an uncorrelated log‐normal clock and both yule and separately birth–death speciation processes. Following previous recommendations (Baele et al., 2013; Condamine et al., 2015), we compared the yule and birth–death (BD) priors by estimating both AICM using the method‐of‐moments estimator (Baele et al., 2013) and the differences in log marginal likelihoods (and thus log Bayes Factors) using the smoothed harmonic mean estimator (Newton and Raftery, 1994; Suchard et al., 2001) in Tracer v.1.6 (Rambaut et al., 2014) using 1000 bootstrap replicates. BEAUti files were generated with the 528 taxa ITS/ETS nrDNA data set only, as the immense size of the supermatrix data set precluded efficient exploration under the Bayesian framework in BEAST. Because ML analyses of both the AHE and AHE plus nrDNA data sets provided high support for all major clades, we enforced monophyly of clades as tree priors for major backbone (not internal) nodes with 100% bootstrap values in both the AHE coalescent and concatenated trees. The GTR + Γ + I model of sequence evolution was used. Based on the larger time‐calibrated phylogeny of subfamily Nepetoideae (Drew and Sytsma, 2012), both the root of the tree (crown of subtribe Salviinae or stem of Salvia) and the crown of Salvia were set to normal distributions: offsets of 34 Ma (95% quantile = 32.4–35.6 Ma) and 30 Ma (95% quantile = 28.6–31.4 Ma), respectively. In addition, we employed two published fossil records to calibrate internal nodes with log‐normal distributions. Following Lancaster and Kay (2013) who employed the fossil recorded by Mai and Walter (1988), we constrained the MRCA of Salvia officinalis, S. aucheri, and S. fruticosa with an offset of 2.6 Ma, mean of 2.0, and SD of 1.0. Second, we placed late Miocene Salvia pollen from Alaska at the stem of S. greatae, S. funerea, and S. californica following Emboden (1965) with an offset of 5.3 Ma, mean of 1.0, and SD of 1.5. The shape and size of the fossil pollen grains are very similar to these three species of Salvia subgen. Audibertia sect. Echinospace but not to S. carduacea, the fourth and earlier‐diverging species within the section (Walker et al., 2015). The location of these fossil pollen well outside the present distribution of sect. Echinospace supports placement of the fossil prior on the stem of the three species.
For each separate analysis using the yule or BD models, we ran four independent analyses of 100,000,000 generations each in BEAST, saving 10,000 trees from each run. This number was necessary to get effective sampling of all parameters as visualized in output log files in Tracer v.1.6 (Rambaut et al., 2014). After removing 20% of samples as burn‐in, independent runs for the best‐fitting model (yule or BD) were combined and a maximum clade‐credibility (MCC) tree was constructed using TreeAnnotator v.1.8.4 (Bouckaert et al., 2014). Node heights were based on common ancestor height values rather than the default median height values to avoid generating zero or negative length branches among recently diverged tip species. To examine additional BEAST trees for several downstream analyses (and to test whether results were conservative by being consistent across the most different topologies), we randomly sampled 1000 trees from the posterior probability distribution (PPD). Treespace (Kendall and Colijn, 2016) in R was used to identify 10 clusters comprising distinct groves of trees in this subset of PPD trees. The median tree of each of the 10 clusters was identified and saved.
Diversification shifts in Salvia
We used BAMM v2.5 (Bayesian Analysis of Macroevolutionary Mixtures; Rabosky, 2014, 2018; Rabosky et al., 2014, 2017) to estimate rates of speciation (λ), extinction (μ), and net diversification (r) within Salvia. Our specific focus was to identify clades exhibiting significant rate shifts in speciation and to conduct rate‐through‐time analysis of these rates. Although we strived for even coverage across Salvia in our taxa sampling, the incomplete sampling using 519 of a total 989 Salvia species (our latest count) required the use of a sampling probability file in which 12 clades (terminals) were identified to which all unsampled species could reliably be placed. The three largest clades, representing radiations in Asia, in the Mediterranean/Southwest Asian/African region, and in Central to South America, included 51%, 27%, and 36% of the species, respectively. Likewise, two additional terminals were constructed for Melissa and Lepechinia in which all unsampled outgroup species were placed in each. BAMM models the placements of unsampled species within their designated clade using the existing topology and branch lengths. Terminals, proportion of extant species sampled for each, and justification for clade numbers are provided in Appendix S3.
Priors for BAMM were generated using the package BAMMtools v.2.1 (Rabosky et al., 2014) for the R statistical environment (R Core Team, 2018). BAMM analyses were run for 500 million generations sampling every 100,000 generations. Metropolis coupled MCMC was implemented with four Markov chains and the numbers of generations to propose a chain swap set to 1000. The expected number of shifts was set to 50 given the size of the phylogeny. Separate runs with 1, 4, or 10 expected shifts were conducted to test the sensitivity of the resulting shifts to this prior. After removing 20% of the samples from the posterior distribution sampled by BAMM, we analyzed the output using BAMMtools and computed the 95% credible rate shift configurations. We also estimated the rate shift configuration with the maximum a posteriori probability as the single best shift configuration. We explored clade specific evolutionary rates by obtaining rates‐through‐time plots (λ, μ, and r) for Salvia subclades of interest. All these diversification analyses were done on the BEAST MCC tree as well as the as the 10 grove trees selected from the PPD tree distribution.
Ancestral range estimation in Salvia
Ancestral range estimation (ARE) for Salvia was done for the MCC tree and each of the 10 grove trees using the DEC (Ree and Smith, 2008) and DECj models in BioGeoBEARS (Matzke, 2013, 2014) in R v3.3.1. The “j” or jump parameter allows for a daughter lineage to immediately occupy via long‐distance dispersal a new area that is different from the parental lineage. The jump parameter has been recently questioned, as well as the statistical (nested) relationship between DEC and DECj (Ree and Sanmartín, 2018). We explored both models to evaluate any major differences in the inferred biogeographical history of Salvia. We identified seven broad geographic areas (see Figs. 1 and 7) important in the context of the distributions of Salvia: (1) North America, including northern and central Mexico; (2) South America, including southern Mexico, Central America, and the West Indies; (3) western Eurasia, including Europe, Mediterranean Africa; (4) Southwest Asia, from Turkey, Syria, Israel east through Iran, Pakistan, and to Bhutan; (5) northeastern Africa and southern Arabia; (6) southern Africa including Madagascar; and (7) east Asia, including the area from India, Southeast Asia, into eastern Australia for one species, S. plebeia R.Br.
Figure 7.
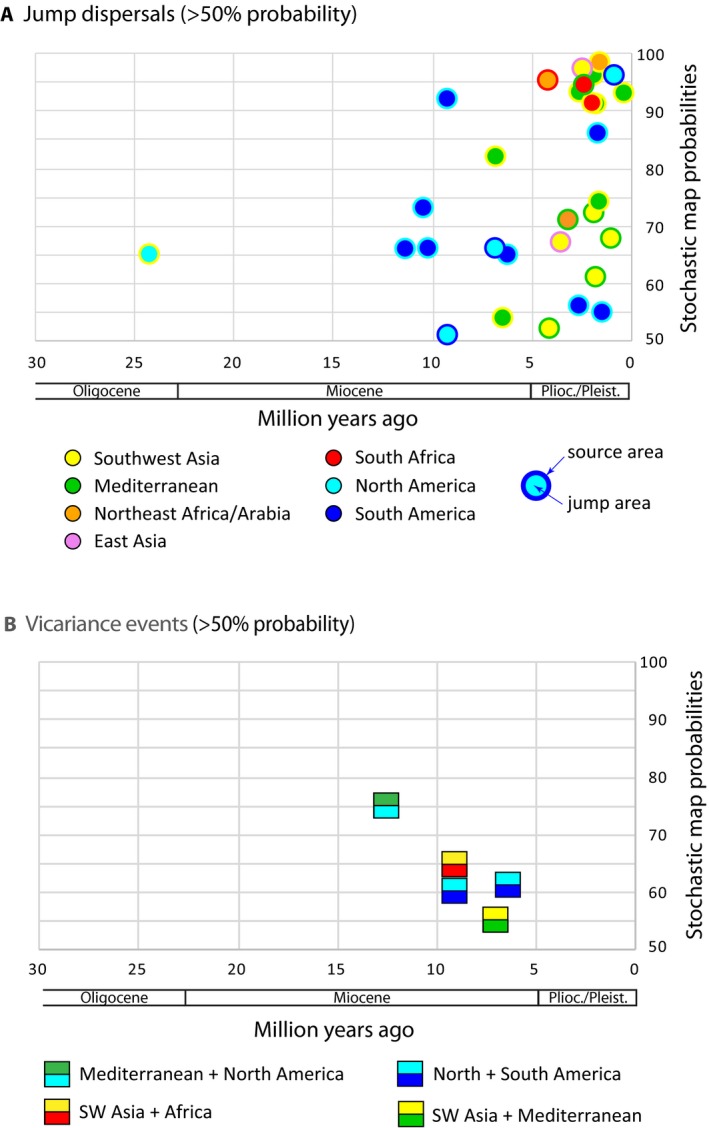
Summary of 100 stochastic mappings using the DECj model in BioGeoBEARS for Salvia ancestral area estimation. Stochastic mapping probabilities >50% are shown for (A) jump dispersals and (B) vicariance events relative to stem node ages. Jump dispersals are color coded by source and jump areas. The four different types of vicariance events are color coded by original combined areas and resulting separated areas.
The floristic patterns involving the horn of Africa, Arabia, and Southwest Asia (including the Irano‐Turanian region) are complicated (Hedge, 1957, 1986; de Winter, 1971; Thulin, 1991; Jurgens, 1997; Salvo et al., 2011; Djamali et al., 2012), and thus we followed the arguments for dividing this region as in Manafzadeh et al. (2014, 2017). The separation between Mediterranean Europe and northern Africa from Southwest Asia is particularly contentious, especially in the context of Asia Minor (Anatolia) and the Palestine region. Thus, we ran parallel analyses with different definitions of western Eurasia and Southwest Asia; species occurring in the area comprising Turkey south to Israel were alternatively placed in one or the other of these two broad areas. Similarly, separation of the North, Central, and South American regions for Salvia is problematic. Salvia is especially diverse in Mexico and is represented there by Madrean‐pine oak, subtropical, and tropical lineages. Broad floristic (and faunistic) areas meet in southern Mexico, and thus we followed the emerging biogeographic consensus in placing most of Mexico north of the Isthmus of Tehuantepec with North America (Axelrod, 1958, Axelrod, 1975; Graham, 1999; Morrone, 2010; Escalante and Morrone, 2013; Berger et al., 2016). Areas south of the Isthmus of Tehuantepec in Mexico, Central America, and the Caribbean were placed with South America.
Species distributions were obtained from primary literature, previous analyses of groups within Salvia, and from the Global Biodiversity Information Facility (GBIF; http://www.gbif.org) as necessary following established protocols to ensure accurate species distributions from this data depository (Maldonado et al., 2015; Spalink et al., 2016a, b). Species scoring for area is provided in Appendix S2. We allowed the inferred ancestors to occupy up to four areas based on the fact that no extant species occupies more than three, and max=4 then would be very conservative. Dispersal probabilities between allowed pairs of areas were specified for three separate time slices (37–15 Ma, 15–5 Ma, 5–0 Ma) to factor in the timing of possible important geological events affecting Salvia dispersal (e.g., closure of the Mediterranean Sea, northern hemisphere land bridges, formation of the Panamanian Isthmus, separation of eastern and western Asia). Many of these events have been modeled specifically in DEC and DECj analyses elsewhere (e.g., Buerki et al., 2011; Drew and Sytsma, 2012; Sulman et al., 2013; Berger et al., 2016; Cardinal‐McTeague et al., 2016; Spalink et al., 2016a, b). We conducted 100 biogeographical stochastic mapping (BSM) replicates in BioGeoBEARS (Matzke, 2016; Dupin et al., 2017) on the MCC tree and on each of the 10 grove trees. We summarized the probability of each class of cladogenetic event (vicariance, sympatry, subset‐sympatry, and jump dispersals) given the DEC or DECj model and distribution data across the 11 trees. Additional details regarding model development, temporal stratification, and dispersal probabilities among the geographical regions through time are provided in Appendix S4.
Biome shifts in Salvia
In addition to shifts in biogeographic areas, we were also interested in assessing the level of shifts between biome types as Salvia diversified in time and space. Specifically, are biome shifts more frequent following dispersal between areas (or continents), are biome shifts correlated with increased species diversification, and are shifts in pollinators linked to shifts in biomes? To accomplish this, we used the 14‐biome framework of Olson et al. (2001) that are delimited within eight biogeographical realms (http://www.worldwildlife.org/wildworld). Species were scored for one or more biomes by extracting the biome type corresponding to the latitude/longitude coordinates of specimens obtained in GBIF or, when absent in GBIF, with georeferenced localities based on type specimens and the literature. The extraction of biomes included representation in all but the tundra biome, and both the boreal forest (including taiga) as well as the mangrove biomes were represented by only six and two species, respectively. To reduce the number of polymorphic taxa for biome type, we removed these three poorly represented biomes. We also combined the categories of tropical and temperate coniferous forest to a broader coniferous forest biome, tropical and temperate broadleaf forest to broadleaf forest, the four grassland types into one grassland biome, and left the desert/xeric shrublands and Mediterranean forests/ woodlands scrub biomes as demarcated. The final data set thus included five combined biome types (Fig. 1): arid/desert, Mediterranean, grassland, broadleaf forest, and coniferous forest (biome coding for each species is provided in Appendix S2).
We evaluated biome evolution in a phylogenetic context by using BioGeoBEARS to implement a Markov‐k (Mk) model for a standard unordered character (i.e., biome) model with equal rates character evolution (but this model extended as described below). We used BAYAREALIKE with a (range‐switching) implemented, but with d (anagenetic dispersal) and e (anagenetic extinction) = 0 (as well as both vicariance and jump dispersals parameters = 0, as default in BAYAREALIKE). We extended the biome shift model in BAYAREALIKE with two novel approaches. First, we used a “manual biome switching probability” file to allow greater probability of shifts between specific biome types (as done between adjacent areas with ARE). We used the logic that biome types shared by the same species should have higher transition probabilities. We constructed a Jaccard's similarity coefficient (Jaccard, 1912) matrix for all biomes based on shared presence among the 528 species sampled and scaled these values so that the highest value was given a probability of 1.0 in the “manual biome switching probability” file. Thus, we mimic the “all rates different” (but maintaining symmetry) model as implemented in other packages (e.g., ace).
Second, we incorporated time slices and a “stratified” occurrence file to permit the rise of biomes only in specific time slices—Mediterranean biome from 4 Ma and grassland biome from 14 Ma to present. The Mediterranean climate and biomes of Europe and of California are known to have arisen only recently in the Pliocene or Pleistocene (Suc, 1984; Ackerly, 2009), and this approach ensures that shifts to biomes do not occur until more recent time intervals when there is geological evidence for their presence. The Mediterranean fynbos climate of South Africa likely initiated at the end of the Miocene, although fynbos plant clades have been dated as recent (Pliocene) to older (early to mid‐Miocene) radiations (e.g., Linder, 2005; Verboom et al., 2009; Linder and Verboom, 2015). Because we demonstrate that southern African Salvia fynbos lineages are Pliocene in age, using a 4 Ma stratification for all Mediterranean biomes is appropriate. Likewise, northern hemisphere grasslands were constrained to the late Tertiary as that biome only became dominant following the mid‐Miocene climatic optimum (Anderson, 2006; Edwards et al., 2010; Strömberg, 2011). Southern African grasslands also have been suggested to be older than northern hemisphere counterparts with early origins from openings in broadleaf forests during the Oligocene and again in the late Miocene (Linder and Verboom, 2015). Thus, our mid‐Miocene stratification date (14 Ma) may be too conservative generally, but should be appropriate for Salvia as all southern African lineages are later radiating. To verify the use of these two biome constraints in the context of southern Africa, we compared the results to an analysis without the biome stratifications enforced.
All biome shift analyses were done with the MCC tree as well as with the 10 grove trees obtained from the PP set. As BSM under the BAYAREALIKE (pure a+, d=e=0) models is still not fully implemented (Dupin et al., 2017), variation in biome shifts was assessed by comparing biome shifts across all 11 trees. Summary statistics of biome shifts through time were obtained across 11 trees by estimating the biome with the highest probability at each node of the phylogeny and counting the number of times switches from one biome to another in adjacent nodes. Additional details regarding model development, temporal stratification, and shift probabilities among the biomes through time are provided in Appendix S5.
Pollinator shifts in Salvia
We reconstructed the pollinators of Salvia onto the 11 trees used in the biogeographic analyses with maximum‐likelihood by calculating the marginal ancestral states for all nodes of each phylogeny with the rayDISC function of the R package corHMM (Beaulieu et al., 2013, 2017). This function allows for taxa to be coded polymorphic, which was ideal with our data set that includes several undetermined species. Pollinators were coded from the literature as being bee‐ or bird‐pollinated, or polymorphic for the two (Wester and Claßen‐Bockhoff, 2007, 2011); pollinator coding is provided in Appendix S2. Before the reconstructions, a series of evolutionary models for discrete traits were tested using the fitMK function of the R library phytools (Revell, 2012) and compared with the Akaike information criterion (AIC). These were the equal rates (ER), symmetric (SYM), all rates different (ARD), and two ordered models. The method of Yang (2006) was used to fix the root state probabilities. In this approach, the estimated transition rates are used to set the weights at the root (Yang, 2006; Beaulieu et al., 2013, 2017). To better estimate the number of transitions in pollinator type at different time intervals, we increased the random sampling of trees to 100 from the PP, and then discretized the node states using a 50% threshold.
RESULTS
Anchored hybrid enrichment of nuclear loci provides strong support among major clades of Salvia
The AHE sequencing generated an average of 453 gene loci per accession (of 498 screened loci) at an average length of 623 bp. Across all 40 species, 438 loci were shared, and these were used for all subsequent analyses. The coalescent species tree based on ASTRAL‐II (Fig. 2) is 100% supported at most nodes (branches with asterisks in Fig. 2 are used as prior constraints in subsequent BEAST analyses). Only four nodes within Salvia are not fully supported. Additionally, the sister relationship of Melissa and Lepechinia is weakly supported. The RAxML bootstrap tree of concatenated AHE data is identical to the coalescent tree except in the weakly supported placement of Melissa sister to Lepechinia + Salvia (see Appendix S6). Thus, these analyses indicate strong relationships among the major clades within Salvia.
Figure 2.
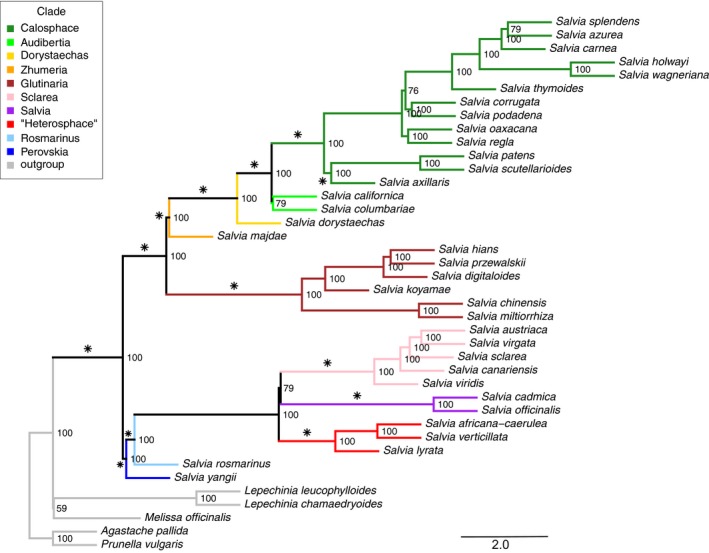
Coalescent ASTRAL II tree of anchored hybrid enrichment genomic data based on 316 nuclear genes across 35 species of Salvia and five outgroup genera in the tribe Mentheae. Support values are provided next to each node. Branch lengths are expressed in coalescent units. Asterisks indicate clades that are designated as monophyletic in the BEAST analyses of 528 species based on nrDNA. Edges are colored by subgenus (see Table 1). (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives). The bootstrap RAxML tree based on concatenation of the 316 nuclear genes is provided in Appendix S6.
Salvia comprises three well‐supported major clades. These three clades include 11 named subgenera (see Discussion), which are provided in Table 1 and Fig. 2 (and subsequent figures and tables) to facilitate discussion of results. The first clade, including subgen. Salvia, Sclarea, and “Heterosphace” (informal designation for this group), comprises largely southwestern Asian and Mediterranean species, but also species from south and eastern Africa, Madagascar, and the eastern to southwestern United States and adjacent Mexico. Subgenera Perovskia and Rosmarinus (both formerly recognized at the generic level) represent early diverging lineages of this first clade. The second clade (subgen. Glutinaria) is almost exclusively eastern Asian with several species more broadly distributed into central and western Eurasia. The third clade, sister to the Asian subgen. Glutinaria, comprises the two Old World subgen. Zhumeria (and close relatives not currently placed in a subgenus) and Dorystaechas (Old World subgen. Meriandra not sampled here) and the exclusively New World subgen. Audibertia (western North America) and Calosphace (primarily Mexico to South America).
nrDNA data with AHE‐supported monophyletic clade priors in BEAST generate the most complete phylogenetic framework to date for Salvia
The RAxML likelihood tree based only on nrDNA (ITS and ETS) for 528 species is similar, but not entirely congruent with the broad relationships provided by AHE nuclear loci (Appendix S7). The three main clades in Salvia are maintained although relationships within each differ to some extent, although none of these differences are well supported. Notably in the first clade, subgen. Rosmarinus is sister to the remainder and subgen. Salvia is sister to subgen. “Heterosphace” + Sclarea. The RAxML likelihood trees based on nrDNA data and invoking the “‐g” AHE topology constraint (Appendix S7) and on the supermatrix of nrDNA and AHE data (tree not shown), however, are similar to each other and recover all the major clades seen with the AHE data alone (Fig. 2). Importantly, in each of the three analyses, all 528 species are placed consistently within one of the 11 subgeneric clades. Additionally, subgen. Zhumeria clearly comprises the former and recently recognized genera Polakia and Pleudia in addition to Zhumeria, this clade distributed from Southwest Asia, northeast Africa and Arabia, to the Mediterranean.
Based on these results, the nrDNA data set for 528 species was used to generate BEAST trees with topological constraints supported by AHE analyses added as priors. The yule model had a larger marginal likelihood and lower AICM value compared to the birth–death model (Table 2), and thus had a better fit with the data with both estimates. All subsequent downstream analyses are based on the yule BEAST trees. The combined log files from four independent BEAST runs had high effective sampling sizes (ESS > 200). The BEAST MCC tree is depicted in Fig. 3 (the BEAST tree with taxon labels and 95% confidence nodal time intervals is available in Appendix S8). The PCoA 1 and 2 distribution of 1000 PP BEAST trees and the 10 identified grove tree sets (and each median tree) from Treespace analysis is shown in Appendix S9.
Table 2.
Birth death and yule model comparison for BEAST analyses of Salvia nrDNA. Marginal likelihood test uses the smoothed harmonic mean estimator. Provided are the marginal likelihoods, standard error (SE) estimated from bootstrap replicates, and differences between log marginal likelihoods (log Bayes factors). Positive differences (* yule model) indicate better fit of the row's model compared to the column's model. AICM test uses the methods‐of‐motion estimator. Provided are AICM values, SE estimated from bootstrap replicates, and differences between AICM values. Lower values indicate better model fit. Positive differences (* yule model) indicate better relative fit of the row's model compared to the column's model
| Model | Marginal likelihoods | SE | Birth death | yule |
|---|---|---|---|---|
| Birth death | −104596.418 | ±0.189 | — | ±977.098 |
| * yule | ±103619.319 | ±0.1 | 977.098 | — |
| Model | AICM | SE | Birth death | yule |
|---|---|---|---|---|
| Birth death | 1707250.552 | ±58760.263 | — | ±231290.49 |
| * yule | 1475960.062 | ±79188.808 | 231290.49 | — |
Figure 3.
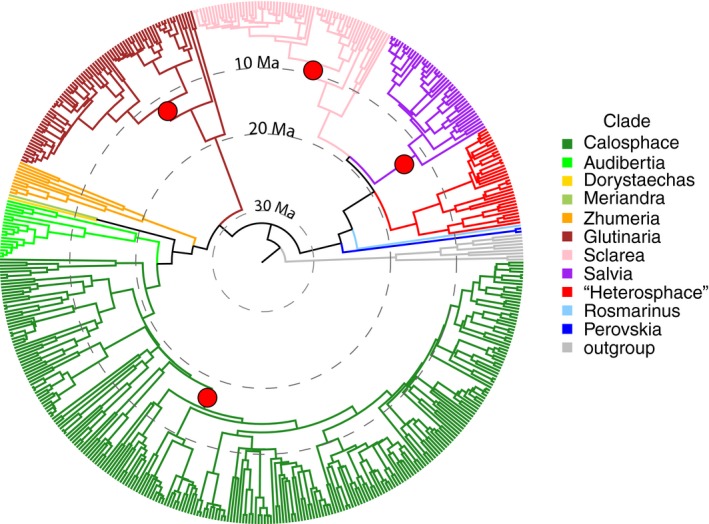
BEAST maximum clade credibility tree (yule model) using Salvia nrDNA and rooted with a monophyletic outgroup of Lepechinia and Melissa. Major clades that were well supported based on AHE genomic data (see Fig. 2) were used as topological constraints in BEAST. Edges are colored by subgenus. (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives; the “Salvia aegyptiaca clade” is informally placed here with previously recognized subgen. Zhumeria). The edges with the four BAMM species diversification shifts (see Fig. 4) are indicated. This MCC tree is used for all other subsequent analyses. An enlarged BEAST tree with species terminals and 95% nodal age intervals is provided in Appendix S8.
Four diversification shifts evident in time‐calibrated Salvia tree
Divergence time estimates for major subclades within Salvia are largely overlapping, with respect to 95% confidence intervals, among the MCC and 10 median grove BEAST trees, and thus, all subsequent results and discussion will focus on mean times from the MCC BEAST tree. BAMM analyses achieved sufficient effective samples sizes after 100,000,000 generations. The best shift configuration models across the MCC BEAST and 10 grove trees sampled from the PP favored four shifts in speciation rate to account for the pattern and levels of diversity across the subtribe Salviinae (Fig. 4, Table 3). Shift configurations across eight of the grove trees exhibit all four of the shifts. Two grove trees only showed one shift (subgen. Sclarea) in the best shift configuration, but both exhibited alternative credible shift configurations with the four shifts. The MCC BEAST tree showed three shifts but lacked the shift in subgen. Salvia, although an alternative but less likely credible shift configuration displayed all four. The overall rate of speciation in Salvia increased slowly but steadily from 32 to 15 Ma (λ going from 0.15 to 0.20), but beginning with the large shift in subgen. Calosphace around 15 Ma, the rate of speciation accelerated almost 3‐fold during the last 15 Myr (λ going from 0.20 to 0.33 (Fig. 5)).
Figure 4.
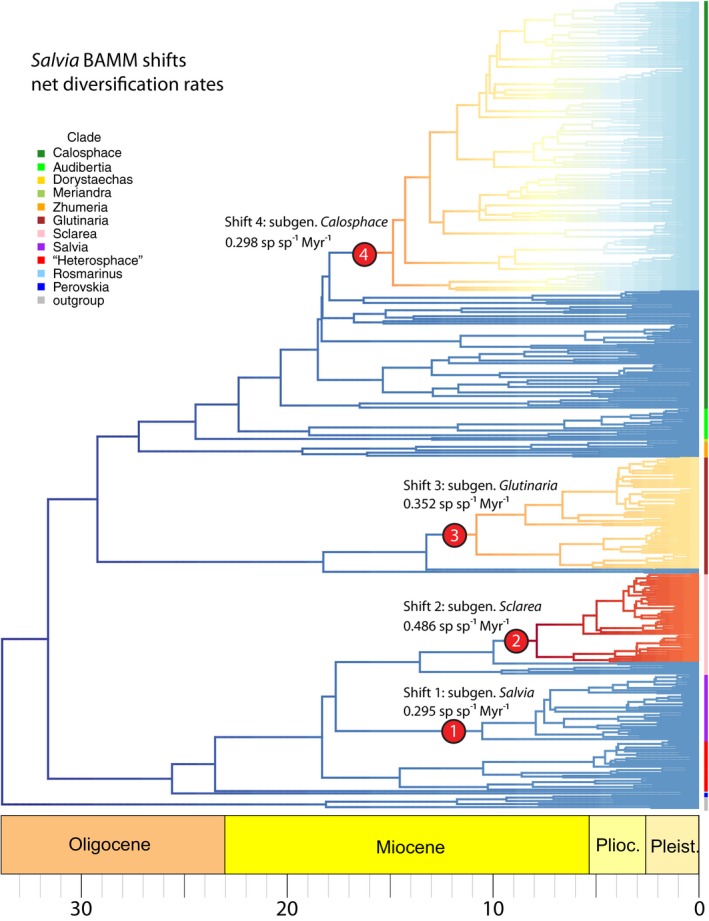
Rates of diversification across Salvia using BAMM based on the MCC BEAST tree. Edges colored in red and blue exhibit high and low rates of net diversification, respectively. The four edges with shifts seen across the MCC BEAST trees and 10 grove trees from the PP are designated by number and include net diversification (r) values—the MCC tree does not show Shift 1 but 8 grove trees do (see Table 3). Terminals are color coded by subgenus. (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives; the “Salvia aegyptiaca clade” is informally placed here with previously recognized subgen. Zhumeria). An enlarged BEAST tree with species terminals is provided in Appendix S8.
Table 3.
BAMM diversification shift sets across the MCC and 10 median grove trees from the BEAST PP distribution (numbered as in Fig. 4). Clade names are based on the subgeneric designations (see Table 1, Fig. 5). Asterisks designate slightly different shift positions in clade. Estimated rates (mean and 95% quantile) of speciation (λ), extinction (μ), and net diversification (r) are provided for the four shifts
| Tree/Clade | 1. Salvia | 2. Sclarea | 3. Glutinaria | 4. Calosphace |
|---|---|---|---|---|
| MCC | — | √ | √ | √ |
| Grove 1 | √ | √ | √ | √ |
| Grove 2 | — | √ | — | — |
| Grove 3 | √ | √* | √ | √ |
| Grove 4 | √* | √ | √ | √ |
| Grove 5 | √* | √ | √ | √ |
| Grove 6 | √* | √ | √ | √* |
| Grove 7 | √ | √ | √ | √ |
| Grove 8 | √ | √ | √ | √ |
| Grove 9 | √* | √ | √ | √ |
| Grove 10 | — | √ | — | — |
| Ages (Ma) | 17.5–7.4 | 9.9–7.8 | 13.1–10.7 | 17.8–14.7 |
| λ |
0.350 (0.219–0.520) |
0.535 (0.395–0.657) |
0.400 (0.268–0.515) |
0.328 (0.275–0.379) |
| μ |
0.055 (0.003–0.186) |
0.049 (0.004–0.137) |
0.048 (0.004–0.141) |
0.030 (0.003–0.081) |
| r | 0.295 | 0.486 | 0.352 | 0.298 |
Figure 5.
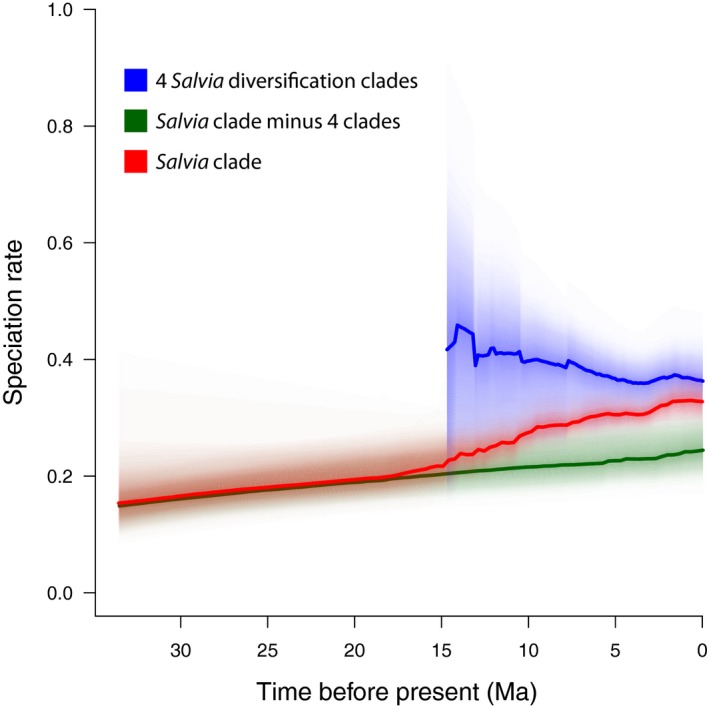
Overall rate of speciation in Salvia (red graph) slowly increasing from 32 to 15 Ma, but accelerating 3‐fold beginning with the first large shift in subgen. Calosphace around 15 Ma. Speciation rate of just the four clades exhibiting significant BAMM shifts on their edges (see Fig. 4) is shown as the blue graph. The background rate of speciation in Salvia minus these four clades is shown as the green graph.
Rate shift 1 (crown of subgen. Salvia) occurred before 10.4 Ma in Southwest Asia, increased mean net diversification rates (r) to 0.295 (mean speciation rate or λ = 0.350, mean extinction rate or μ = 0.055), and gave rise to a large number of species endemic to Turkey. Shift 2 (two nodes within subgen. Sclarea) occurred between 9.9–7.8 Ma in Southwest Asia and/or the Mediterranean region (the latter has less than 50% probability in ARE) and gave rise to a large number of species (including many Turkish and Iranian endemics) in both the broad Mediterranean and Southwest Asia regions, and to a small lineage in eastern and southern Africa. This rate shift, the second most recent, largest of the four in Salvia, and found across all grove trees, increased r to 0.486 (λ = 0.535, μ = 0.049). Shift 3 (two nodes within subgen. Glutinaria) occurred in East Asia during the mid‐Miocene (13.1–10.7 Ma), increased r to 0.352 (λ = 0.400, μ = 0.048), and gave rise to almost all eastern Asian Salvia. Shift 4 (five nodes internal to the crown of subgen. Calosphace) occurred in Mexico at 17.8–14.1 Ma, the oldest of any rate shift in Salvia, and increased r to 0.298 (λ = 0.328, μ = 0.030). The placement of this shift is constant in the core subgen. Calosphace but the timing of this shift by BAMM within the radiation of subgen. Calosphace in Mexico is less certain, relative to the preceding three, considering the proportional lack of species sampling (266 of ~600), extremely short branch lengths, and different topologies among grove trees evident in the core subgen. Calosphace.
Temporal and spatial differentiation within Salvia
The DECj model (Ln L = −498.01), incorporating the jump dispersal parameter along with those for dispersal, extinction, and cladogenesis, relative to the DEC model (Ln L = −526.06) was significantly better (p = 6.9e‐14) assuming they are nested models. DECj was also supported for all 10 median grove BEAST trees. The inferred ancestral region estimation (ARE) for almost all nodes are identical with both DEC and DECj. The MCC BEAST tree showing ARE under the DECj model is depicted in Fig. 6. The alternative scoring of Asia Minor with Europe and Mediterranean Africa rather than with Southwest Asia gave nearly similar ARE for the important nodes provided here and discussed later, and thus will no longer be considered. Similarly, ARE for the 10 median grove BEAST trees gave similar results relative to ARE using the MCC BEAST tree. We found no instances where the most likely area estimated for nodes of interest in the MCC tree differed from the most commonly estimated area among these trees from the BEAST posterior. We base all subsequent discussions on the MCC tree constructed on the DECj model (Fig. 6).
Figure 6.
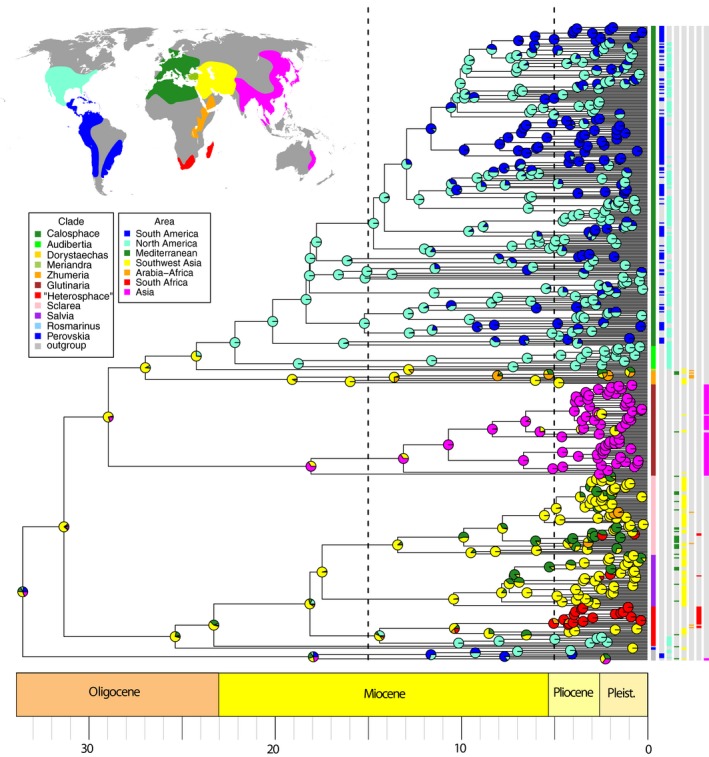
BioGeoBEARS DECj tree showing ancestral region estimation for Salvia using seven defined biogeographical regions and based on the BEAST MCC tree (see Fig. 4). Worldwide distribution of Salvia in the map of biogeographical regions is based in part on Hedge (1974, 1986), Claßen‐Bockhoff et al. (2003), Walker et al. (2004), Froissart (2007), Wester and Claßen‐Bockhoff (2011), Wei et al. (2015), Will et al. (2015), Will and Claßen‐Bockhoff (2014, 2017). Dashed lines separate the three time intervals used in BioGeoBEARS. Circles in the tree indicate frequencies of each area estimated for the node and are color coded by area. Terminals are color coded by subgenus. (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives; the “Salvia aegyptiaca clade” is informally placed here with previously recognized subgen. Zhumeria.) To the right of the terminals and their subgeneric designations are seven columns indicating area scoring(s) for each terminal. Species names for each terminal are provided in Appendix S8.
The 100 biogeographical stochastic mappings (BSM) in BioGeoBEARS provided a probability distribution across the branches of the MCC BEAST tree for each of the different cladogenetic events. Given the parameters of this model, 91% of cladogenetic events involve sympatry, 3% involve vicariance, and 6% involve jump‐dispersals. All 30 jump dispersals mapped between areas are depicted by nodal age and frequency of occurrence (<50% frequency were removed) from the 100 BSM (Fig. 7A). The oldest jump dispersal (24 Ma), at the Oligocene–Miocene transition, occurred between Southwest Asia and eastern North America giving rise to the large American radiation of subgen. Audibertia and Calosphace, the first of two movements of Salvia into the New World. Seven of the nine Miocene jump dispersals are within subgen. Calosphace and involve crossing the Isthmus of Panama (see Discussion on the disputed age of this geological event). During the Pliocene and Pleistocene the majority of the remaining jump dispersals occurred between Southwest Asia and Mediterranean (10 dispersals), with dispersals between southern Africa and either Southwest Asia, Mediterranean, or Northeast Africa/Arabia accounting for three jump dispersal events, and an additional four and two dispersals occurred, respectively, between North and South America and between Southwest Asia and eastern Asia. The six vicariance events in the 100 BSM (>50% frequency) are all mid‐late Miocene in age (Fig. 7B). The oldest (14 Ma) vicariant event involved an ancestral area of Mediterranean plus North America and gave rise to the second North American restricted lineage, within subgen. “Heterosphace”. A widespread flora from Southwest Asia to southern Africa fragmented at 9.5 Ma, resulting in a separate southern African area. Two vicariant events involving North and South America are inferred in the late Miocene. Many other vicariance events are alternative, lower probability (<<50%) stochastic mappings at nodes with inferred jump dispersals.
The subtribe Salviinae diversified in the early Oligocene (34 Ma), and the crown radiation of Salvia s.l. occurred soon after (31 Ma). The diversification events for both the stem and crown of Salvia are unambiguously estimated as occurring in Southwest Asia. The radiation of the first large clade (subgen. Perovskia, Rosmarinus, Salvia, Sclarea, and “Heterosphace”) from the Old World (excluding the East Asian clade) is largely reconstructed as Southwest Asia (Fig. 6). Within this first clade, multiple shifts to Europe and Mediterranean Africa occurred in the mid‐Miocene to Pliocene, and one range expansion (combined area) comprised Mediterranean to eastern North America (within subgen. “Heterosphace”). Three independent movements in this clade to Northeast and/or southern Africa are evident. The first, inferred as a vicariant event (at 10 Ma, see above), occurred within subgen. “Heterosphace” with the subsequent African radiation commencing at the Miocene–Pliocene boundary (5 Ma). The first split within this clade separated Northeast Africa/Arabia (e.g., S. nilotica) and southern Africa/Madagascar. Our sampling of the six Malagasy species is limited, but the two sampled species represent a very recent Pleistocene divergence from at most a Pliocene dispersal event from mainland Africa. The origin of the other two southern African clades involve Pliocene jump dispersals from Southwest Asia (subgen. Salvia) and from Mediterranean (subgen. Sclarea).
Radiation of the second clade, in East Asia (subgen. Glutinaria), involved a dispersal event from Southwest Asia between the late Oligocene and mid‐Miocene. The crown radiation of the East Asian clade occurred at 18 Ma in the mid‐Miocene, with much of the species diversification during the Pliocene (Fig. 6). Barriers to subsequent dispersals between Southwest Asia and East Asia are implied by the fact that only four (of ca. 90) species (S. glutinosa, S. hians, S. nubicola, S. plebeia) potentially overcame this geographic barrier. Multiple dispersals from mainland East Asia to Japan occurred in the Pliocene (including but not limited to S. nipponica + S. glabrescens, S. lutescens + S. ranzaniana, and S. japonica).
The third large Salvia clade, including the Old World subgen. Zhumeria, Dorystaechas, and Meriandra and the New World subgen. Audibertia and Calosphace, diversified first in Southwest Asia in the late Oligocene (27 Ma) (Fig. 6). Subgenus Zhumeria, sister to the remainder, exhibited a subsequent Miocene (18 Ma) crown radiation in Southwest Asia and later Miocene shifts into Northeast Africa, southern Arabia, and more recently into the Mediterranean region (Fig. 6). The exclusively New World subclade, comprising subgen. Audibertia (western North America) and subgen. Calosphace (largely Mexico to South America), is sister to a species‐poor subclade comprising subgen. Dorystaechas and Meriandra that are sporadically distributed in Southwest Asia (Dorystaechas; Antalya region of Turkey) to India and Northeast Africa. Our ARE places the jump dispersal from the Old World to the New World in a relatively narrow time interval in the late Oligocene (24–22 Ma), thus anywhere from 8 to 10 Myr earlier than the movement to eastern North America in the first Salvia clade (subgen. “Heterosphace”). At the end of this time interval, Salvia had already diversified into stem lineages of the western North American subgen. Audibertia and the Mexican to South American subgen. Calosphace. Although both species sampling and resolution within subgen. Calosphace are presently limited with this data, ARE strongly indicates a Mexican (southern North America) origin for the subclade. Many independent jump dispersal (and some back dispersal) events to Central America and into South America are inferred (Fig. 6). These dispersal events started no earlier than 12 Ma in the mid‐Miocene but continued throughout the late Miocene and into the Pliocene.
Biome diversification within Salvia: convergent shifts evident in different clades
Extracting biome type for each species by using georeferenced accessions in the framework of five combined biome types (arid/desert, Mediterranean, grassland, broadleaf forest, coniferous forest) was a conservative approach; i.e., scoring erred on the side of polymorphism. Species coded as polymorphic were often in the mainly European and Southwest Asian clades (e.g., subgen. Salvia, Sclarea, “Heterosphace” in part). Species in the New World subgen. Audibertia and Calosphace and especially in the East Asian subgen. Glutinaria were less polymorphic. Biome shifts across Salvia modeled using BAYAREALIKE (with a+, d=e=0) and summarized across the MCC and 10 grove trees are depicted on the MCC tree in Fig. 8A. Many of the earliest nodes (20–33 Ma) are polymorphic for broadleaf forest + coniferous forest + desert/xeric shrubland biomes, although broadleaf forest was the dominant biome in these early nodes across all 11 trees. The diversification of most subgenera, however, exhibits a focusing of biome specializations, but these often in a convergent fashion. Specialization to desert and xeric shrublands in the New World is pronounced in subgen. Audibertia in western North America during the late Miocene, less so in its sister clade Calosphace, and common in the unrelated subgen. “Heterosphace” in eastern to southwestern North America. Shifts predominantly to these xeric biomes in the Old World are seen in subgen. Zhumeria (especially in the Arabian/northeast African region), Sclarea (Southwest Asia), and less so in “Heterosphace” (southern Africa). Shifts to Mediterranean biome occur independently, all within the last 4 Myr, in subgen. Audibertia (western North America), Sclarea and Salvia (Mediterranean, Middle East, especially Turkey), and “Heterosphace” (southern Africa). Shifts to grasslands are seen in many subgenera during the Miocene, but are most dominant in Calosphace (mostly South America), Sclarea and Salvia (Southwest Asia), and “Heterosphace” (southern Africa).
Figure 8.
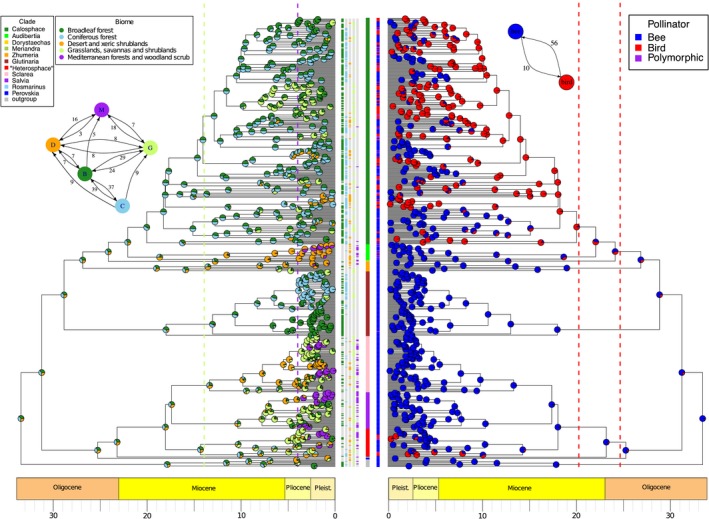
Reconstruction of (A) biome and (B) pollinator shifts in Salvia using mirror BEAST MCC trees (see Fig. 4). Terminals are color coded by subgenus in the center column. (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives; the “Salvia aegyptiaca clade” is informally placed here with previously recognized subgen. Zhumeria). Species names for each terminal are provided in Appendix S8. (A) Biome shifts were obtained with a modified BAYAREALIKE model in BioGeoBEARS. Frequency of biomes reconstructed at each node are summarized across the MCC and 10 grove trees. Terminals are scored for each of the five biomes by color coding in the five columns to their right. Dashed lines represent the time constraints placed on the origin of grassland (14 Ma) and Mediterranean (4 Ma) biomes. Inset A depicts the number of transitions between biomes. (B) Pollinator shifts were obtained using the function rayDISC of the R package corHMM, estimated transition rates were used to set the weights at the root, and the MCC and grove trees were sampled. Terminals are scored for bee, bird, or polymorphic by color coding in the column to their left. Dashed lines indicate the 95% confidence interval of the crown radiation of hummingbirds (McGuire et al., 2014). Inset B depicts the number of pollinator transitions between bird and bee pollination systems.
Broadleaf and coniferous forests largely dominate the East Asian subgen. Glutinaria, and each of these biomes are restricted to specific subclades (Fig. 8A). The two forest biomes dominate the diversification of both Mexican and South American species of Calosphace (with more recent shifts to arid and grassland biomes as noted above), but there is no specialization of the two biomes within Calosphace lineages as seen in East Asian Glutinaria. Broadleaf and coniferous forest biomes are minor in the other clades of Salvia, although most clades exhibit some signal of forest biomes earlier in their diversifications (e.g., mid‐Miocene). Transitions between broadleaf and coniferous forest biomes, as averaged over the MCC and 10 grove trees, account for the majority of biome shifts in Salvia (Fig. 8A, inset). Given the nearly equal representation (1/3 each) of these two forest biomes and the desert/arid shrubland biome in the early biome diversification of Salvia, the asymmetry in shifts giving rise to grassland and Mediterranean biomes is striking (Fig. 8A, inset). Grassland biome lineages arose from broadleaf forest biome lineages >4× more often than from either coniferous forest or desert/arid biome lineages, and of all biomes, the broadleaf forest is the only one that shows a transition back from the grassland biome. The Mediterranean (and the most recent) biome only shows transitions from the grassland and desert/arid shrubland biomes, never directly from either broadleaf or coniferous forest biomes. In addition, the Mediterranean biome only exhibits a few back transitions to grasslands, indicating that radiation into the more recent Mediterranean biome has so far been mostly unidirectional in Salvia.
Relative to the four inferred BAMM diversification shifts (see Figs. 3 and 4), biome shifts do not appear to be directly related to increased speciation in Salvia. Two diversification shifts (Shift 4 in subgen. Calosphace, Shift 3 in subgen. Glutinaria) occur in mixed broadleaf and coniferous forest biomes, but these nodes are imbedded in clades largely scored for these forest biomes with no biome shifts nearby. Likewise, Shift 2 in subgen. Sclarea occurs within a larger clade scored for desert/arid shrubland, although a later (~5 Myr) shift to the grassland biome did occur. Shift 1 in subgen. Salvia is placed on a branch polymorphic for forests, desert, and grassland biomes, but with subsequent narrowing to grassland or Mediterranean biomes.
Pollinator shifts within Salvia
The preferred model of discrete character evolution chosen across trees using AIC was the all rates different model (ARD). Using this model, the root state was consistently estimated as being bee pollinated (Fig. 8B). Across the 11 trees (grove trees and MCC tree), there was an average of 10 subsequent changes (range 9–11) from bee to bird pollination, and an average of 56 changes (range 53–58) from bird back to bee pollination (Fig. 8B, inset). Across trees, there were consistent changes in the New World from bee to bird pollination (hummingbirds) in subgen. Calosphace and “Heterosphace” (and to a lesser extent in Audibertia with several species polymorphic for pollination), and multiple independent shifts (all southern African) to bird pollination (hummingbirds and passerines) in subgen. “Heterosphace”. All shifts back to bee pollination occurred in New World subgen. Calosphace with a few species in these bee‐pollinated clades re‐evolving hummingbird pollination. Shifts to bird pollination are not linked to shifts in biomes. Hummingbird pollination likely arose in forested biomes of Mexico within subgen. Calosphace (Fig. 8B), as were most of the subsequent shifts back to bee pollination. The more recent shifts to hummingbird pollination (often polymorphic) in subgen. Audibertia occur in either desert/arid shrubland or Mediterranean biomes. In eastern North American subgen. “Heterosphace”, shifts to hummingbird pollination occurred in mixtures of forest, grassland, and desert biomes. Similarly, the rise of passerine bird pollination in southern African subgen. “Heterosphace” occurred in a mixture of forest, grassland, and Mediterranean biomes.
Most shifts in pollinator also do not appear directly related to shifts in diversification. BAMM Shifts 1–3 (Figs. 3 and 4) occur in bee‐pollinated branches (subgen. Salvia, Sclarea, Glutinaria), whereas Shift 4 occurs in hummingbird‐pollinated branches (subgen. Calosphace). None of the four diversification shifts in Salvia are correlated directly with branches with pollinator shifts. However, the shift to hummingbird pollination at the base of subgen. Calosphace is followed by a shift in diversification (Shift 4) several edges later.
DISCUSSION
We present here the largest phylogenetic analysis of Salvia s.l. to date (519 of what we now estimate to be 989 species) for which we more broadly sampled all the major lineages identified in previous studies (e.g., Drew et al., 2017a; Will and Claßen‐Bockhoff, 2017). Using the anchored hybrid enrichment (AHE) method of generating sequence data of hundreds of nuclear loci for a subset of 40 taxa, we obtained a highly resolved topology of the major clades of Salvia. We leveraged this AHE tree to add in a total of 528 (519 Salvia plus outgroups) species based on nrDNA to provide for the first time a comprehensive, Salvia‐wide phylogenetic framework with 100% support for most nodes. This framework provides the basis for a coherent, subgeneric classification of most of the genus. However, two clades within Salvia will require further attention regarding formal subgeneric designation: the circumscription of Zhumeria with respect to species related to this former genus, and the circumscription of the Old World and New World members of the informal “Heterosphace”. We infer for the first time a statistically based biogeographical ancestral area estimation of this remarkable radiation across different continents and biomes over the last 34 Myr. We then coupled this historical biogeographical scenario with the evaluation of two putative features driving this radiation—shifts in biomes and pollinator type—and their possible correlation with four identified BAMM shifts in diversification across Salvia.
A robust phylogenetic framework across Salvia expands a model system for addressing evolutionary phenomena
Our AHE NGS approach expands (40 species) on the more limited (12 species of subgen. Calosphace and one Lepechinia) AHE study of Fragoso et al. (2017) and documents the ability of AHE to provide high‐resolution signal across a diversifying lineage (Fig. 2). The only nodes not well supported in subtribe Salviinae are (1) Melissa and Lepechinia relative to Salvia and (2) subgen. Sclarea + Salvia. Increased taxa sampling with the AHE approach (our unpublished data; Drew et al., 2018) indicates continued lack of resolution at the base of subtribe Salviinae but increased support for subgen. Sclarea + Salvia. We are now also expanding this AHE approach to obtain both nuclear and entire plastome sequences (Drew et al., 2018) to better examine the role of incomplete lineage sorting, hybridization, and introgression that is known to occur in Salvia but localized to within some well‐defined clades (e.g., Walker et al., 2015).
Phylogenetic relationships among major lineages within the broadly defined Salvia (sensu Drew et al., 2017a), using sequence data gathered via the combination of NGS and targeted locus approaches, reflect but greatly expand on the results of Drew et al. (2017a) and Will and Claßen‐Bockhoff (2017). Five small subgenera, recently considered separate genera (Dorystaechas, Meriandra, Perovskia, Rosmarinus, and Zhumeria), are placed within Salvia as proposed by Drew et al. (2017a), although with more precision. Subgenus Zhumeria is closely related to a small group of species from Southwest Asia and adjacent Arabia/Africa previously referred to as the “Salvia aegyptiaca clade” (Drew et al., 2017a), although we do not now formally place them within subgen. Zhumeria. For the first time, subgen. “Zhumeria” (with the Salvia aegyptiaca clade included) is strongly placed as sister to two small Old World subgenera (Dorystaechas and Meriandra) plus the large New World radiation of subgen. Audibertia and Calosphace. Sister to this more inclusive clade is the East Asian subgen. Glutinaria. As also demonstrated by Hu et al. (2018), subgen. Glutinaria is monophyletic despite its constituents containing a diversity of stamen types and previously being placed in three separate subgenera (Salvia, Sclarea, and the former Allagospadonopsis).
The remaining third clade in Salvia is now better resolved, with the two small subgenera Perovskia and Rosmarinus diverging consecutively relative to three larger subclades, two of which are subgen. Salvia and Sclarea. We provide for the first time strong support, based on both AHE and nrDNA data, for the monophyly of the third subclade, which comprises three distinct lineages in Salvia. The first lineage includes all eastern and southwestern North American species previously placed in sects. Salviastrum Scheele and Heterosphace Benth. (clade I‐B of Will and Claßen‐Bockhoff, 2017; see Walker and Elisens, 2001); the second includes species derived from the major diversification in South (and East) Africa and Madagascar previously placed in sects. Eusphace Benth., Heterosphace, and Hymenosphace Benth. (clade I‐A in part of Will and Claßen‐Bockhoff, 2017); and the third is sect. Hemisphace Benth. from Europe, North Africa, and Southwest Asia (“S. verticillata group” of Will and Claßen‐Bockhoff, 2017). Interestingly, this clade was supported at 100% in the cpDNA tree presented in Will and Claßen‐Bockhoff (2017) without further discussion.
The diversification of Salvia since mid‐Oligocene was driven in part by intercontinental dispersal events and parallel biome specializations out of Southwest Asia
Our biogeographical analysis expands on the results of Will and Claßen‐Bockhoff (2017) and, for the first time, addresses the biogeographical diversification of the entire genus Salvia with statistical analyses (DEC and DECj) on a well‐resolved phylogenetic framework for the major clades (Fig. 7). The ancestral area for the crown radiation of Salvia is inferred to be Southwest Asia at 32 Ma in the early Oligocene rather than the Mediterranean region as previously suggested (Claßen‐Bockhoff et al., 2004; Will and Claßen‐Bockhoff, 2017). This ancestral region for Salvia largely encompasses the western portion of the Irano‐Turanian (IT) floristic region, a vast and diverse area considered an Old World hotspot for evolutionary and floristic diversity (Manafzadeh et al., 2014, 2017). The complex and heterogeneous geological history of the IT region (Rogl, 1999; Mouthereau et al., 2012; Manafzadeh et al., 2017) is key to understanding the early patterns of biome switches and dispersal events in Salvia. During the late Eocene, the Tethys Ocean between Africa and Europe was in the process of closing, and the humid‐mesic broadleaf deciduous and coniferous forests of the Paleogene broadly distributed across the northern hemisphere (Wolfe, 1975) were probably already fragmenting in the IT region with intervening arid regions (see Manafzadeh et al., 2014). The abrupt cooling and aridification event at the Eocene–Oligocene (~34 Ma) transition (Fig. 8c; Zachos et al., 2001), coupled with the subsequent Arabia–Eurasia plate collisions (~18 and 11 Ma) and concurrent uplift of the Iranian and Anatolian plateaus (Mouthereau et al., 2012), accelerated the rise of the arid IT flora.
Biome reconstructions for the early stages of Salvia diversification in the Oligocene and Miocene (Fig. 8A) indicate ancestral mixed broadleaf deciduous and coniferous forest along with desert/arid shrublands in Southwest Asia, this reconstruction fitting with the geological, climatic, and paleovegetation evidence for the early Neogene in the IT region (Akhmetiev and Zaporozhets, 2014). Southwest Asia continued the primary biogeographical home for much of the radiations of subgen. Salvia and Sclarea. Two of the four diversification shifts within Salvia occurred in these clades in Southwest Asia almost simultaneously (9.8–6.8 Ma) following major geological events of plate contact, plateau uplifts, and subsequent biome changes (see below). These two radiations appear to be tied to transitions to either arid biomes (subgen. Sclarea) or grassland biomes (subgen. Salvia).
Salvia subsequently radiated out from Southwest Asia toward the close of the Oligocene and more rapidly during the early to mid‐Miocene by dispersing to different areas at different times (Mediterranean region, Arabia/Northeast Africa, southern Africa, East Asia, and North America) and by specializing to a diversity of biome types. This early radiation roughly mirrors the source area and time for Brassicaceae (Franzke et al., 2011), Apiaceae (Banasiak et al., 2013), and Halophyllum in Rutaceae (Manafzadeh et al., 2014). The invasion of the Saharo‐Arabian region occurred sporadically in the history of Salvia, but most pronounced in one lineage of the subgen. Zhumeria clade between 19–14 Ma. This time interval spans the first (18–16 Ma) and second (13–11 Ma) African/Arabian plate collisions with Eurasia (Manafzadeh et al., 2017), events that would have facilitated these colonizations. The dispersals to the Mediterranean region (subgen. Sclarea, subgen. Salvia in part) would have been aided via land corridors available during the early to late Miocene as previously proposed based on floristics (Zohary, 1973) and more recently on ancestral region estimation (Manafzadeh et al., 2014). All of the dispersals to the Anatolian Plateau, giving rise to the many Turkish Salvia endemics in subgen. Salvia and Sclarea, are timed from 6 Ma and into the Pliocene. This time is consistent with the initial volcanism and beginning of the uplift ca. 11 Ma to form the 2‐km high eastern Anatolian plateau (Barazangi et al., 2006) following the last of the African/Arabian plate collisions with Eurasia. For both the Mediterranean Sea basin and the Anatolian Plateau, diversification in the more recent (Pliocene) Mediterranean biome followed shifts from arid, forest, or grassland biomes.
Southwest Asia was also the inferred area for long‐distance dispersal events of Salvia to at least East Asia (one time), to North America (two times), and possibly southern Africa in two of its three separate colonizations. The dispersal to east Asia occurred somewhere between the Late Oligocene Warming and the Mid Miocene Climatic Optimum. The later crown radiation of the East Asian subgen. Glutinaria at 18 Ma would have followed the uplift of the Kunlun, Tianshan, and Altay mountain ranges on the west edge of the Qinghai‐Tibetan Plateau (Favre et al., 2015; Caves et al., 2017). These uplifts apparently were effective in blocking further dispersals in Salvia between East Asia and Southwest Asia as only four (of ca. 90) species (S. glutinosa, S. hians, S. nubicola, S. plebeia) potentially overcame this geographic barrier. However, the first three species currently have a distribution that only extends to the western Himalayas, and are thus not part of the Sino‐Japanese flora, and the origin of S. plebeia is unclear. It is therefore possible that there are no extant examples of species of Salvia dispersing between Southwest Asia and East Asia since orogenic barriers arose in the mid‐Miocene. The subsequent BAMM diversification shift within subgen. Glutinaria (ca. 13–11 Ma) immediately precedes the radiation of the two large subclades with each adapted primarily to either broadleaf or coniferous forest, suggesting a possible biome specialization as driving this diversification. The diversification of subgen. Glutinaria may also be driven by staminal evolution (Hu et al., 2018).
Dispersal to southern Africa, and then to Madagascar, has occurred three times in Salvia, and our results largely support previous suggestions of origins and timing for these lineages (Will and Claßen‐Bockhoff, 2014, 2017). The oldest (between 10–5 Ma) and largest radiation occurred within subgen. “Heterosphace”. Although ambiguous in the BioGeoBEARS reconstructions (Fig. 8), the ARE of this subgenus is clearly Southwest Asia based on stochastic mapping and that the southern African clade is sister to species mainly from Southwest Asia (few either Northeast Africa/Arabia or Mediterranean North Africa). As some species (e.g., S. nilotica) do show the disjunct eastern RAND (Pokorny et al., 2015) distribution between Northeast Africa/Arabia and southern Africa, or apparent dispersals back to Northeast Africa/Arabia (e.g., S. somalensis), more complete sampling is warranted to examine the nature and timing of this recurring East African pattern. The other two independent dispersal events to southern Africa, one each in subgen. Salvia and Sclarea, are late Pliocene in origin and derive from Southwest Asia and the Mediterranean, respectively. All three independent southern African long‐distance dispersals, however, derive from a grassland type biome (not desert or Mediterranean biomes) based on stochastic mapping. However, once reaching southern Africa and Madagascar, they have diversified into forest, grassland, and Mediterranean biomes.
Salvia dispersed twice to the New World. The most recent involved the dispersal to eastern North America (sect. Salviastrum of subgen. “Heterosphace”) from within an Old World lineage during the mid‐Miocene. Stochastic mapping suggests that this involved expansion first to the Mediterranean from Southwest Asia and then to eastern North America, presumably as long‐distance dispersal via the North Atlantic Land Bridge (see also Will and Claßen‐Bockhoff, 2017). This bridge was especially suitable geologically and climatically for temperate migrations into the Miocene (Manchester, 1999; Tiffney and Manchester, 2001; Milne, 2006; Tiffney, 2008), and probably late Miocene (Denk et al., 2010; Drew et al., 2017b). This dispersal event was followed by a limited diversification of six Salvia species into eastern and southern North American coniferous forests, grasslands, and arid biomes.
The older dispersal event from the Old World to the New World was a jump dispersal from Southwest Asia in a relatively small time interval in the late Oligocene (24–22 Ma). This event gave rise to subgen. Audibertia (western North America) and subgen. Calosphace (largely Mexico to South America). Although biome reconstruction is ambiguous at the crown of this neotropical dispersal event (Fig. 9A), stochastic mapping favors either coniferous or broadleaf forests. During the late Oligocene to early Miocene, the Bering Land Bridge was suitable for exchanges of temperate deciduous plants and remained available for floristic exchanges until the Pliocene (Tiffney, 1985; Manchester, 1999; Tiffney and Manchester, 2001; Wen et al., 2016). Our data supports the use of this land bridge by Salvia, as previously suggested (Will and Claßen‐Bockhoff, 2017). Based on the presence of fossils clearly attributed to subgen. Audibertia as far north as Alaska, it is likely that the early members from this dispersal event were adapted to various forest biomes of both the more temperate Arcto‐Tertiary flora (Axelrod et al., 1991; Graham, 1999; Manchester, 1999; Milne, 2006) and the Madro‐Tertiary flora of southwestern North America and northern Mexico (Axelrod, 1958, 1975; Graham, 1999; Wen and Ickert‐Bond, 2009). Subgenus Audibertia then later diversified extensively into desert and eventually Mediterranean biomes of western and southwestern North America, primarily in the California Floristic Province (Walker et al., 2015). Early lineages of subgen. Calosphace were centered in Mexico and then radiated south into Central America and South America where they mainly diversified in broadleaf and coniferous forest but also grassland biomes. The BAMM shift within subgen. Calosphace does not appear to be correlated with changes in area or biome as the shift occurs on a branch nested with other Mexican species from broadleaf and/or coniferous forest biomes.
Figure 9.
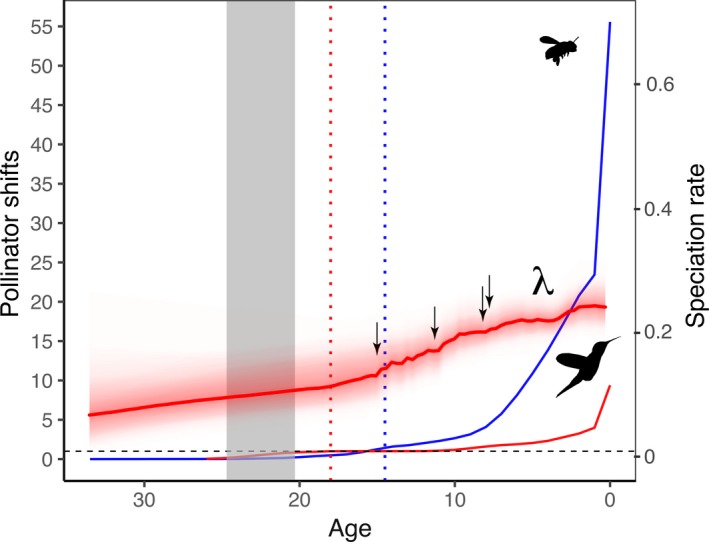
Cumulative number of shifts to bird (red graph) or bee (blue graph) pollination through time in Salvia averaged across 100 PP trees (left axis). Ancestral state for the subtribe Salviinae is bee pollination. Dashed gray horizontal line indicates the threshold when on average one transition to either bee or bird pollination occurs across the 100 PP trees. The red and blue vertical dashed lines indicate when the first transition to either bird or bee pollination, respectively, occurs on average across the 100 PP tree. Overall speciation (λ) rate (right axis) for Salvia from BAMM analysis (thick red graph; see Fig. 5) and ages of the four BAMM shifts (arrows; see Fig. 4) in diversification are provided for context. The vertical gray rectangle depicts the 95% confidence interval of the crown radiation of hummingbirds (McGuire et al., 2014).
The timing of an established overland migration route through the Panamanian Isthmus is of particular interest for Salvia due to the ongoing debate of the timing of this closure based on both geological evidence and time‐calibrated phylogenetic trees. Current and prevailing geological evidence argues for an early Pleistocene closure (O'Dea et al., 2016), although minimally “stepping‐stone” islands may have existed between North America and South America earlier in the Miocene (Wegner et al., 2011; Montes et al., 2012, 2015). On the other hand, phylogenetic chronograms from a growing list of plant clades point to earlier—late Miocene or Pliocene—movements across the Isthmus of Panama (Bacon et al., 2013, 2015; see Drew et al., 2017b, for discussion in context of the Menthineae). Salvia now can be added to this list, as our results indicate many independent jump dispersal (and some back dispersal) events across the Isthmus region starting as early as 12 Ma in the mid‐Miocene and continuing through the Pliocene (Fig. 6).
The diversification of Salvia in the New World was driven in part by repeated shifts between hummingbird and bee pollination
Plant groups that exhibit shifts between different pollinator guilds offer evolutionary insight into the role these shifts play in species diversification and influence convergent or parallel changes in floral color, shape, and structure (Schmidt‐Lebuhn et al., 2007; Tripp and Manos, 2008; Smith, 2010; Lagomarsino et al., 2017; Joly et al., 2018). Salvia is a unique model system to address these questions due to its large size, four identified shifts in species diversification (Figs. 3, 4, 5), frequent shifts between bee and bird pollination (Figs. 8, 9), and extensive field and laboratory data on pollinator vectors, floral architecture, and pollination mechanisms (Claßen‐Bockhoff et al., 2003, 2004; Wester and Claßen‐Bockhoff, 2006, 2007, 2011). Bee pollination is clearly ancestral in Salvia (Fig. 8B; see also Fragoso‐Martínez et al., 2018), but shifts to bird pollination are seen in three subgenera: twice in “Heterosphace” (southern Africa and eastern North America), Audibertia (western North America), and most pronounced in Calosphace (Mexico to South America). In addition, fly pollination has been reported in S. virgata (subgen. Sclarea) and S. verticillata subsp. amasiaca (sect. Hemisphace,“Heterosphace”) in Old World species from central Anatolia in Southwest Asia (Celep et al., 2014).
The diversification of Salvia throughout Mexico and then subsequently further south into Central and South America is a history of specialization to pollinators, both bee and hummingbird. This diversification mirrors that seen in the South American radiation of subtribe Menthinae, one of the sister lineages to subtribe Salviinae, in which hummingbird pollination, especially in arid biomes, was important (Drew and Sytsma, 2012; Drew et al., 2017b). However, this diversification in pollinator modes in Salvia has four unusual attributes. First, it is apparent that the edge exhibiting the main shift in species diversification within the crown of subgen. Calosphace (Figs. 3, 4, 5) comes shortly after, not at the same time, of a clear shift to hummingbird pollination. Although it is clear that shifts between bees and hummingbirds are important in the diversification of Salvia, this shift does not appear to be directly causal for the identified point of increased speciation within subgen. Calosphace. However, the earlier shift to hummingbird pollination precipitated other floral changes (Wester and Claßen‐Bockhoff, 2007) that may be directly linked to the later edge showing the speciation rate shift (see below). If so, these results in Salvia would not support the conclusions of Abrahamczyk and Renner (2015, p. 1) in which they argue “diversification within hummingbird‐pollinated clades in the temperate regions of the Americas appears mainly due to habitat specialization and allopatric speciation, not bird pollination per se.”
Second, we reconstructed the history of pollinator shifts on the phylogenetic trees and identified hummingbird pollination as the ancestral state at both the stem and crown of subgen. Calosphace (ca. 22 and 20 Ma, respectively). In a less‐sampled survey focusing largely on subgen. Calosphace and thus without the larger Salvia perspective we provide, Fragoso‐Martínez et al. (2018) indicated an ancestral state of bee pollination for the clade. This study may have inadvertently forced this reconstruction by not including outgroups and by assigning a prior probability of mainly bee pollination to the root of the tree. Their topology at the base of subgen. Calosphace, critical for any subsequent character reconstruction (pollinator, area, biome), is also clearly at odds with the overwhelming support based on nuclear AHE next‐generation and nrDNA sequences presented here. In their study, the sole member of sect. Axillares is sister to the rest of subgen. Calosphace, whereas the AHE next‐generation and nrDNA data place this section as sister to a clade of mainly hummingbird‐pollinated species in sects. Blakea, Hastatae, and Standleyana, with this clade in turn sister to the rest of subgen. Calosphace. More importantly, the discordant positions of these sections in Fragoso‐Martínez et al. (2018) are based on tree differences between cpDNA and nrDNA (not discussed in that paper) and their use of concatenation of both data sets.
Third, the BEAST chronogram provides strong evidence that the shift to hummingbird pollination in the New World clade (excluding sect. Salviastrum of “Heterosphace”) occurred early in the diversification of the lineage of hummingbirds. The stem of the hummingbird clade dates to at least the Oligocene (34–28 Ma) based on fossils in Europe (Mayr, 2004, 2007), or even older (54–42 Ma) based on genomic evidence (Jarvis et al., 2014; McGuire et al., 2014; Prum et al., 2015). However, the crown of the extant hummingbirds (in South America with later dispersal north) dates only to 22 Ma in the Miocene (McGuire et al., 2014). The dates of extant hummingbirds are consistent with the stem and crown node of subgen. Calosphace (ca. 22 and 20 Ma) both being scored as hummingbird pollinated in our reconstruction, and with the stochastic mapping results summarized across all 100 PP trees indicating an average of ca. 18 Ma for the first hummingbird shift (Fig. 9). Thus, along with New World Gesnerioideae, which shows a switch to hummingbird pollination at ca. 18.5 Ma (Serrano‐Serrano et al., 2017), evidence is mounting that plant lineages quickly adapted to the newly arrived hummingbirds in the early Miocene in South America with the attendant floral changes required for effective bird pollination. The emerging biogeographical and temporal scenario (McGuire et al., 2014) is that hummingbirds (1) used the Beringian dispersal pathway between 28–22 Ma to enter North America, (2) invaded South America over open water before 22 Ma and diversified, (3) subsequently went extinct in North America and Europe, and (4) later recolonized North America from the south ca. 12 Ma by more xeric‐adapted descendants of the extant hummingbirds commonly referred to as Bees and Mountain Gems. As Salvia was also likely entering the New World from Eurasia across Beringia ca. 24 Ma (giving rise to both subgen. Audibertia and Calosphace), it is likely that both Salvia and early hummingbirds were co‐dispersing into and down North America and possibly already interacting at that time. It is noteworthy that hummingbird pollination is far more restricted and more recent in the western North American subgen. Audibertia. This restriction would suggest that any early Miocene Salvia–hummingbird interaction in North America was not effectively maintained and that the reintroduction of hummingbirds into North America 10 Myr later was required for the minor shift to bird pollination in the California Floristic Province Salvia (Walker et al., 2015).
And fourth, we demonstrate that there were actually few shifts to bird pollination in Salvia. Mapping across the 11 trees (or the 100 PP trees) shows an average of 10 shifts from bees, whereas there are ~5× that for shifts from bird to bee (Figs. 8B, 9). Switches from the ancestral hummingbird state in subgen. Calosphace to bee pollination have occurred repeatedly, and the rate of switches from bird to bees increases dramatically from the end of the Miocene through the Pliocene (Fig. 9). The shift back to insect pollination in neotropical hummingbird‐pollinated clades has been demonstrated as well in Fuchsia (Berry et al., 2004), Costus (Salzman et al., 2015), and Gesnerioideae (Serrano‐Serrano et al., 2017). Considering these shifts back to bee pollination in otherwise ancestrally hummingbird‐pollinated clades, it would be fruitful to examine more critically the nature of changes in floral architecture and stamen/style morphology. Many of the Salvia subgenera exhibit a variety of staminal types (Walker and Sytsma, 2007), including the unique lever mechanism (Claßen‐Bockhoff et al., 2003, 2004). The shift to hummingbird pollination in subgen. Calosphace was accompanied not only by distinct changes in staminal morphology but also by the associated loss of the staminal lever mechanism (Wester and Claßen‐Bockhoff, 2007). Thus, a careful re‐analysis of floral evolution in this clade would be crucial, using the backdrop of this new phylogenetic and evolutionary framework presented here, to check for a “footprint” of ancestral hummingbird floral morphology in clades of subgen. Calosphace that have apparently reverted, multiple times independently, to bee pollination.
CONCLUSIONS
This integrated examination within Salvia of changes in biogeographic area, biome specializations, and pollination system switches against the backdrop of a broad phylogenetic perspective has provided deep insight into the diversification of Salvia over a ca. 30 Myr timeframe. However, tight correlations of changes in these three categorical features are not apparent, as hypothesized in the introduction, to each other or to the four BAMM shifts in species diversification identified in Salvia (subgen. Salvia, Sclarea, Glutinaria, and Calosphace). Shifts to bird pollination are not linked to shifts in biomes. Shifts in pollinator also do not appear directly related to shifts in species diversification, excepting perhaps the shift to hummingbird pollination at the base of subgen. Calosphace, which is several nodes distant but temporally close to the large diversification shift in subgen. Calosphace.
We have not explicitly used recent statistical methods for modeling the relationships among character states, speciation rates, and extinction rates on phylogenetic trees (e.g., with BiSSE, MuSSE, GeoSSE, and related phylogenetic comparative models) for an explicit reason. The problem of how to discover plausible histories of singular or near singular events in the context of histories of other characters is a recurring problem in the field of phylogenetic comparative methods that links several seemingly unrelated controversies (Maddison and FitzJohn, 2014; Uyeda et al., 2018). Most importantly, the issue of “phylogenetic pseudoreplication” might be inherent with the categorical features examined here in Salvia. Pollination systems in Salvia, for example, exhibit few shifts to birds (and one of the four does not involve hummingbirds); likewise, dispersal from the Old World to the New World has occurred only twice. These few shifts raise the issue of pseudoreplication for evaluating rigorously bird pollination and area shifts to North America—for both, there are not enough shifts to make meaningful comparisons with other features or with edges exhibiting species diversification. It does appear that the main shift to hummingbirds in North America at the base of subgen. Calosphace set the stage, perhaps with subsequent shifts that followed in area and biomes and in the concomitant changes in corolla and staminal architecture, for this remarkable radiation. However, available tests today would not pick up this one‐off event—an example of “Darwin's scenario” which, along with the “unreplicated burst” scenario, do not provide good evidence for an adaptive/functional relationship among features compared (see fig. 1 of Maddison and FitzJohn, 2014). With comparative data alone, we cannot rule out the existence of a third variable that might be co‐distributed with the node of interest or another feature, and which might be the ultimate driver of the processes that we are trying to examine.
These results in Salvia probably point toward other “hidden” variables (sensu Beaulieu et al., 2013) driving key components of its extraordinary diversification through time, among continents and biomes, and with different pollinators. Specifically, we suggest that corolla shape, staminal lever types, anther connective elongation, and style/stigma shape may be better correlated to these other features and to shifts in species diversification. For example, staminal levers from elongate connectives such as those seen in subgen. Sclarea may allow for pollen deposition in a more isolated dorsal area of the pollinator's body, freeing species in this clade from competition for pollinators (Claßen‐Bockhoff et al., 2004). Moreover, these floral characters can be treated as continuous characters, are not susceptible to most of the problems discussed relative to categorical features, and can be analyzed with an innovative suite of emerging tools for phylogenetic comparative approaches (e.g., Chartier et al., 2014; Khabbazian et al., 2016; Rose et al., 2016; Kriebel et al., 2017; Joly et al., 2018).
AUTHOR CONTRIBUTIONS
R.K, B.T.D., and K.J.S. conceived and undertook the project; R.K., B.T.D., F.C., C.P.D., J.P.R., M.M.M., E.M.L., and A.R.L. assisted with data collection; F.C., B.T.D., J.G.G., C.‐L.X., and J.B.W. provided systematic data; R.K., B.T.D., and K.J.S. analyzed the data; K.J.S. led the writing with contributions from all authors.
Supporting information
Appendix S1. Species names and GenBank information for nrDNA data (ETS and ITS). Species with *** were sampled for AHE and the data are deposited in data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8m40rb0.
Appendix S2. Species names, subgeneric or clade designations, vouchers, GenBank accessions, biogeographical area scoring (see Appendix S4 for area names), biome scoring (see Appendix S5 for biome names), and pollinator scoring. The “Salvia aegyptiaca clade” is informally listed here with subgen. Zhumeria. Species in the informal “Heterosphace” are listed here under two subgroups, New World “Salviastrum” or Old World “Verticillata”.
Appendix S3. Terminals, proportion of extant species sampled for each clade for BAMM analyses of Salvia. The “Salvia aegyptiaca clade” is informally listed here with subgen. Zhumeria. The informal “Heterosphace” are listed here under New World “heterosphaceA” or Old World “heterosphaceB”.
Appendix S4. BioGeoBEARS area: details regarding model development, temporal stratification, and dispersal probabilities among the geographical regions through time for BioGeoBEARS analyses of Salvia. See Appendix S2 for species coding.
Appendix S5. BioGeoBEARS biome: details regarding model development, temporal stratification, and dispersal probabilities among biomes through time for BioGeoBEARS analyses of Salvia. See Appendix S2 for species coding.
Appendix S6. RAxML tree of anchored hybrid enrichment genomic data based on 316 concatenated nuclear genes across 35 species of Salvia and five outgroup genera in the tribe Mentheae. Bootstrap support values are provided next to each node. Asterisks indicate clades that are designated as monophyletic in the BEAST analyses of 528 species based on nrDNA. Edges are colored by subgenus (see Table 1). (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives.) See Fig. 2 for ASTRAL II coalescent bootstrap tree based on anchored hybrid enrichment genomic data.
Appendix S7. RAxML bootstrap tree of 528 taxa of Salvia and two outgroup genera based on analysis of nrDNA ITS/ETS only (provided no topological constraints). Phylogram showing all species tips, their major subgeneric clades designations, and RAxML bootstrap values for each branch. Relationships among major clades are not completely congruent with those based on anchored hybrid enrichment genomic data (see Fig. 2, Appendix S6).
Appendix S8. BEAST maximum clade credibility tree (yule model) using Salvia nrDNA and rooted with a monophyletic outgroup of Lepechinia and Melissa. Major clades that were well supported based on anchored hybrid enrichment genomic data (see Fig. 2) were used as topological constraints in BEAST. This tree is same as shown in Fig. 4 but with species terminals and 95% nodal age intervals provided.
Appendix S9. The PCoA 1 and 2 distribution of 1000 PP BEAST trees from Salvia nrDNA and the 10 identified grove tree sets (and the MCC tree from Fig. 3 and Appendix S8) from the Treespace analysis (Kendall, M., and C. Colijn. 2016. Mapping phylogenetic trees to reveal distinct patterns of evolution. Molecular Biology and Evolution 33: 2735–2743).
ACKNOWLEDGEMENTS
We thank Sarah Friedrich for assistance with figures, Sean Holland and Michelle Kortyna at the FSU Center for Anchored Phylogenomics for assistance in data collection and analysis, Elena Conti for information on evolution in the Mediterranean region, the Systematics and Evolution group in the Botany Department, University of Wisconsin for constructive comments, and two anonymous reviewers for their insightful help. We also acknowledge Aaron Jenks for providing access to published sequences not on GenBank. This work was funded in part by a University of Wisconsin Graduate School grant, the University of Wisconsin Botany Department Hofmeister Endowment, an NSF‐DEB grant to K.J.S (DEB‐1046355), and an NSF‐DEB grant to K.J.S. and B.T.D. (DEB‐1655606).
Kriebel, R. , Drew B. T., Drummond C. P., González‐Gallegos J. G., Celep F., Mahdjoub M. M., Rose J. P., et al. 2019. Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: leveraging anchored hybrid enrichment and targeted sequence data. American Journal of Botany 106(4): 573–597.
DATA ACCESSIBILITY
Species, vouchers, and GenBank accessions for targeted sequences are provided in Appendix S1. AHE raw sequences and final alignments are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8m40rb0 (Kriebel et al., 2019). Species data for area, biome, and pollinator are provided in Appendix S2. The R scripts, aligned data, trees, and data necessary to reproduce the analyses in this paper are deposited in Dryad or in [Link], [Link], [Link].
LITERATURE CITED
- Abrahamczyk, S. , and Renner S. S.. 2015. The temporal build‐up of hummingbird/ plant mutualisms in North America and temperate South America. BMC Evolutionary Biology 15: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerly, D. D. 2009. Evolution, origin and age of lineages in the Californian and Mediterranean floras. Journal of Biogeography 36: 1221–1233. [Google Scholar]
- Akhmetiev, M. A. , and Zaporozhets N. I.. 2014. Paleogene events in Central Eurasia: their role in the flora and vegetation cover evolution, migration of phytochore boundaries, and climate changes. Stratigraphy and Geological Correlation 22: 312–335. [Google Scholar]
- Anderson, R. C. 2006. Evolution and origin of the Central Grassland of North America: climate, fire, and mammalian grazers 1. Journal of the Torrey Botanical Society 133: 626–647. [Google Scholar]
- Axelrod, D. I. 1958. Evolution of the Madro‐Tertiary Geoflora. Botanical Review 24: 433–509. [Google Scholar]
- Axelrod, D. I. 1975. Evolution and biogeography of Madrean‐Tethyan sclerophyll vegetation. Annals of the Missouri Botanical Garden 62: 280–334. [Google Scholar]
- Axelrod, D. I. , Arroyo M. T. K., and Raven P. H.. 1991. Historical development of temperate vegetation in the Americas. Revista Chilena de Historia Natural 64: 413–446. [Google Scholar]
- Bacon, C. D. , Mora A., Wagner W. L., and Jaramillo C. A.. 2013. Testing geological models of evolution of the Isthmus of Panama in a phylogenetic framework. Botanical Journal of the Linnean Society 171: 287–300. [Google Scholar]
- Bacon, C. D. , Silvestro D., Jaramillo C., Smith B. T., Chakrabarty P., and Antonelli A.. 2015. Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proceedings of the National Academy of Sciences, USA 112: 6110–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele, G. , Li W. L. S., Drummond A. J., Suchard M. A., and Lemey P.. 2013. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Molecular Biology and Evolution 30: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasiak, Ł. , Piwczynski M., Ulinski T., Downie S. R., Watson M. F., Shakya B., and Spalik K.. 2013. Dispersal patterns in space and time: a case study of Apiaceae subfamily Apioideae. Journal of Biogeography 40: 1324–1335. [Google Scholar]
- Barazangi, M. , Sandvol E., and Seber D.. 2006. Structure and tectonic evolution of the Anatolian plateau in eastern Turkey. Geological Society of America Special Paper 409: 463–473. [Google Scholar]
- Beaulieu, J. M. , O'Meara B. C., and Donoghue M. J.. 2013. Identifying hidden rate changes in the evolution of a binary morphological character: the evolution of plant habit in campanulid angiosperms. Systematic Biology 62: 725–737. [DOI] [PubMed] [Google Scholar]
- Beaulieu, J. M. , Oliver J. C., and O'Meara B. C.. 2017. corHMM: Analysis of Binary Character Evolution. R package version 1.22. https://CRAN.R-project.org/package=corHMM.
- Berger, B. A. , Kriebel R., Spalink D., and Sytsma K. J.. 2016. Divergence times, historical biogeography, and shifts in speciation rates of Myrtales. Molecular Phylogenetics and Evolution 95: 116–136. [DOI] [PubMed] [Google Scholar]
- Berry, P. E. , Hahn W. J., Sytsma K. J., Hall J. C., and Mast A.. 2004. Phylogenetic relationships and biogeography of Fuchsia (Onagraceae) based on noncoding nuclear and chloroplast DNA data. American Journal of Botany 91: 601–614. [DOI] [PubMed] [Google Scholar]
- Bouckaert, R. , Heled J., Kühnert D., Vaughan T., Wu C. H., Xie D., Suchard M. A., et al. 2014. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10: e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddenhagen, C. , Lemmon A. R., Lemmon E. M., Bruhl J., Cappa J., Clement W. L., Donoghue M., et al. 2016. Anchored phylogenomics of angiosperms I: assessing the robustness of phylogenetic estimates. bioRxiv 086298 [Preprint]. 10.1101/086298. [DOI] [Google Scholar]
- Buerki, S. , Forest F., Alvarez N., Nylander J. A. A., Arrigo N., and Sanmartín I.. 2011. An evaluation of new parsimony‐based versus parametric inference methods in biogeography: a case study using the globally distributed plant family Sapindaceae. Journal of Biogeography 38: 531–550. [Google Scholar]
- Cardillo, M. , Weston P. H., Reynolds Z. K. M., Olde P. M., Mast A. R., Lemmon E. M., Lemmon A. R., et al. 2017. The phylogeny and biogeography of Hakea (Proteaceae) reveals the role of biome shifts in a continental plant radiation. Evolution 71: 1928–1943. [DOI] [PubMed] [Google Scholar]
- Cardinal‐McTeague, W. M. , Sytsma K. J., and Hall J. C.. 2016. Biogeography and diversification of Brassicales: a 103 million year tale. Molecular Phylogenetics and Evolution 99: 204–224. [DOI] [PubMed] [Google Scholar]
- Caves, J. K. , Bayshashov B. U., Zhamangara A., Ritch A. J., Ibarra D. E., Sjostrom D. J., Mix H. T., et al. 2017. Late Miocene uplift of the Tian Shan and Altai and reorganization of Central Asia climate. GSA Today 27: 4–10. [Google Scholar]
- Celep, F. , Atalay Z., Dikmen F., Doğan M., and Claßen‐Bockhoff R.. 2014. Flies as pollinators of melittophilous Salvia species (Lamiaceae). American Journal of Botany 101: 2148–2159. [DOI] [PubMed] [Google Scholar]
- Chartier, M. , Jabbour F., Gerber S., Mitteroecker P., Sauquet H., von Balthazar M., Staedler Y., et al. 2014. The floral morphospace—a modern comparative approach to study angiosperm evolution. New Phytologist 204: 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claßen‐Bockhoff, R. 2007. Floral construction and pollination biology in the Lamiaceae. Annals of Botany 100: 359–360. [Google Scholar]
- Claßen‐Bockhoff, R. , Wester P., and Tweraser E.. 2003. The staminal lever mechanism in Salvia L. (Lamiaceae)—a review. Plant Biology 5: 33–41. [Google Scholar]
- Claßen‐Bockhoff, R. , Speck T., Tweraser E., Wester P., Thimm S., and Reith M.. 2004. The staminal lever mechanism in Salvia L. (Lamiaceae): a key innovation for adaptive radiation? Organisms Diversity & Evolution 4: 189–205. [Google Scholar]
- Condamine, F. L. , Nagalingum N. S., Marshall C. R., and Morlon H.. 2015. Origin and diversification of living cycads: a cautionary tale on the impact of the branching process prior in Bayesian molecular dating. BMC Evolutionary Biology 15: 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell, W. K. , Westoby M., Falster D. S., FitzJohn R. G., O'Meara B. C., Pennell M. W., McGlinn D. J., et al. 2014. Functional distinctiveness of major plant lineages. Journal of Ecology 102: 345–356. [Google Scholar]
- Crisp, M. D. , Arroyo M. T. K., Cook L. G., Gandolfo M. A., Jordan G. J., Mcglone M. S., Weston P. H., et al. 2009. Phylogenetic biome conservatism on a global scale. Nature 458: 754–756. [DOI] [PubMed] [Google Scholar]
- Darwin, C. 1859. The origin of species by means of natural selection. Random House, London, UK. [Google Scholar]
- Denk, T. , Grimsson F., and Zetter R.. 2010. Episodic migration of oaks to Iceland: evidence for a North Atlantic “land bridge” in the latest Miocene. American Journal of Botany 97: 276–287. [DOI] [PubMed] [Google Scholar]
- Djamali, M. , Brewer S., Breckle S. W., and Jackson S. T.. 2012. Climatic determinism in phytogeographic regionalization: a test from the Irano‐Turanian region, SW and Central Asia. Flora 207: 237–249. [Google Scholar]
- Drew, B. T. , and Sytsma K. J.. 2011. Testing the monophyly and placement of Lepechinia in the tribe Mentheae (Lamiaceae). Systematic Botany 36: 1038–1049. [Google Scholar]
- Drew, B. T. , and Sytsma K. J.. 2012. Phylogenetics, biogeography, and staminal evolution in the tribe Mentheae (Lamiaceae). American Journal of Botany 99: 933–953. [DOI] [PubMed] [Google Scholar]
- Drew, B. T. , and Sytsma K. J.. 2013. The South American radiation of Lepechinia (Lamiaceae): phylogenetics, divergence times and evolution of dioecy. Botanical Journal of the Linnean Society 171: 171–190. [Google Scholar]
- Drew, B. T. , González‐Gallegos J. G., Xiang C. L., Kriebel R., Drummond C. P., Walker J. B., and Sytsma K. J.. 2017a. Salvia united: the greatest good for the greatest number. Taxon 66: 133–145. [Google Scholar]
- Drew, B. T. , Liu S., Bonifacino J. M., and Sytsma K. J.. 2017b. Amphitropical disjunctions in New World Menthinae: three Pliocene dispersals to South America following late Miocene dispersal to North America from the Old World. American Journal of Botany 104: 1695–1707. [DOI] [PubMed] [Google Scholar]
- Drew, B. T. , Kriebel R., Drummond C. P., Lemmon A. R., Lemmon E. M., and Sytsma K. J.. 2018. Anchored phylogenomics resolve deep relationships within Salvia (Lamiaceae). Botany 2018: Annual Meeting of the Botanical Society of America, Rochester, MN, USA [online abstract, http://www.botanyconference.org/engine/search/index.php?func=detailandaid=779].
- Drummond, C. S. , Eastwood R. J., Miotto S. T. S., and Hughes C. E.. 2012. Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): testing for key innovation with incomplete taxon sampling. Systematic Biology 61: 443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin, J. , Matzke N. J., Särkinen T., Knapp S., Olmstead R. G., Bohs L., and Smith S. D.. 2017. Bayesian estimation of the global biogeographical history of the Solanaceae. Journal of Biogeography 44: 887–899. [Google Scholar]
- Edwards, E. J. , and Donoghue M. J.. 2013. Is it easy to move and easy to evolve? Evolutionary accessibility and adaptation. Journal of Experimental Botany 64: 4047–4052. [DOI] [PubMed] [Google Scholar]
- Edwards, E. J. , Osborne C. P., Strömberg C. A. E., Smith S. A., and C4 Grasses Consortium . 2010. The origins of C4 grasslands: integrating evolutionary and ecosystem science. Science 328: 587–591. [DOI] [PubMed] [Google Scholar]
- Emboden, W. A. 1965. Pollen morphology of the genus Salvia, section Audibertia . Pollen et Spores 6: 527–536. [Google Scholar]
- Escalante, T. , and Morrone J. J.. 2013. Biogeographic regions of North American mammals based on endemism. Biological Journal of the Linnean Society 110: 485–499. [Google Scholar]
- Favre, A. , Päckert M., Pauls S. U., Jähnig S. C., Uhl D., Michalak I., and Muellner‐Riehl A. N.. 2015. The role of the uplift of the Qinghai‐Tibetan Plateau for the evolution of Tibetan biotas. Biological Reviews 90: 236–253. [DOI] [PubMed] [Google Scholar]
- Fragoso‐Martínez, I. , Salazar G. A., Martínez‐Gordillo M., Magallón S., Sánchez‐Reyes L., Lemmon E. M., Lemmon A. R., et al. 2017. A pilot study applying the plant Anchored Hybrid Enrichment method to New World sages (Salvia subgenus Calosphace; Lamiaceae). Molecular Phylogenetics and Evolution 117: 124–134. [DOI] [PubMed] [Google Scholar]
- Fragoso‐Martínez, I. , Martínez‐Gordillo M., Salazar G. A., Sazatornil F., Jenks A. A., García Peña M. del R., Barrera‐Aveleida G., et al. 2018. Phylogeny of the Neotropical sages (Salvia subg. Calosphace; Lamiaceae) and insights into pollinator and area shifts. Plant Systematics and Evolution 304: 43–55. [Google Scholar]
- Franzke, A. , Lysak M. A., Al‐Shehbaz I. A., Koch M. A., and Mummenhoff K.. 2011. Cabbage family affairs: The evolutionary history of Brassicaceae. Trends in Plant Science 16: 108–116. [DOI] [PubMed] [Google Scholar]
- Froissart, C. 2007. Salvia plebeia, les grands voyages d'une petite plante. Académie d'Orléans Agriculture, Sciences, Belles‐Lettres et Arts 6(17): 55–65. [Google Scholar]
- Givnish, T. J. 2010. Ecology of plant speciation. Taxon 59: 1326–1366. [Google Scholar]
- Givnish, T. J. 2015. Adaptive radiation versus ‘radiation’ and ‘explosive diversification’: why conceptual distinctions are fundamental to understanding evolution. New Phytologist 207: 297–303. [DOI] [PubMed] [Google Scholar]
- Givnish, T. J. , Barfuss M. H. J., Van Ee B., Riina R., Schulte K., Horres R., Gonsiska P. A., et al. 2014. Adaptive radiation, correlated and contingent evolution, and net species diversification in Bromeliaceae. Molecular Phylogenetics and Evolution 71: 55–78. [DOI] [PubMed] [Google Scholar]
- Givnish, T. J. , Spalink D., Ames M., Lyon S. P., Hunter S. J., Zuluaga A., Iles W. J. D., et al. 2015. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proceedings of the Royal Society, B, Biological Sciences 282: 20151553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb, L. D. 1984. Genetics and morphological evolution in plants. American Naturalist 123: 681–709. [Google Scholar]
- Graham, A. 1999. Late Cretaceous and Cenozoic history of North American vegetation, north of Mexico. Oxford University Press, Oxford, UK. [Google Scholar]
- Hamilton, C. A. , Lemmon A. R., Lemmon E. M., and Bond J. E.. 2016. Expanding anchored hybrid enrichment to resolve both deep and shallow relationships within the spider tree of life. BMC Evolutionary Biology 16: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedge, I. C. 1957. Studies in east Mediterranean species of Salvia . Notes from Royal Botanic Garden, Edinburgh 22: 173–188. [Google Scholar]
- Hedge, I. C. 1974. A revision of Salvia in Africa including Madagascar and the Canary Islands. Notes from Royal Botanic Garden, Edinburgh 33: 573–121. [Google Scholar]
- Hedge, I. C. 1986. Labiatae of South‐West Asia: diversity, distribution and endemism. Proceedings of the Royal Society of Edinburgh 89B: 23–35. [Google Scholar]
- Hu, G. X. , Takano A., Drew B. T., Liu E. D., Soltis D. E., Soltis P. S., Peng H., et al. 2018. Phylogeny and staminal evolution of Salvia (Lamiaceae, Nepetoideae) in East Asia. Annals of Botany 122: 649–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaccard, P. 1912. The distribution of the flora in the alpine zone. New Phytologist 11: 37–50. [Google Scholar]
- Jarvis, E. D. , Mirarab S., Aberer A. J., Li B., Houde P., Li C., Ho S. Y., et al. 2014. Whole‐genome analyses resolve early branches in the tree of life of modern birds. Science 346: 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks, A. A. , Walker J. B., and Kim S. C.. 2012. Phylogeny of New World Salvia subgenus Calosphace (Lamiaceae) based on cpDNA (psbA‐trnH) and nrDNA (ITS) sequence data. Journal of Plant Research 126: 483–496. [DOI] [PubMed] [Google Scholar]
- Joly, S. , Lambert F., Alexandre H., Clavel J., Leveille‐Bourret E., and Clark J. L.. 2018. Greater pollination generalization is not associated with reduced constraints on corolla shape in Antillean plants. Evolution 72: 244–260. [DOI] [PubMed] [Google Scholar]
- Jurgens, N. 1997. Floristic biodiversity and history of African arid regions. Biodiversity and Conservation 6: 495–514. [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, K. M. , and Sargent R. D.. 2009. The role of animal pollination in plant speciation: integrating ecology, geography, and genetics. Annual Review of Ecology, Evolution, and Systematics 40: 637–656. [Google Scholar]
- Kearse, M. , Moir R., Wilson A., Stones‐Havas S., Cheung M., Sturrock S., Buxton S., et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall, M. , and Colijn C.. 2016. Mapping phylogenetic trees to reveal distinct patterns of evolution. Molecular Biology and Evolution 33: 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabbazian, M. , Kriebel R., Rohe K., and Ané C.. 2016. Fast and accurate detection of evolutionary shifts in Ornstein‐Uhlenbeck models. Methods in Ecology and Evolution 7: 811–824. [Google Scholar]
- Kriebel, R. , Khabbazian M., and Sytsma K. J.. 2017. Shifts in pollen shape and size in the order Myrtales using Ornstein‐Uhlenbeck models. PLoS ONE 12: e0187228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriebel, R. , Drew B. T., Drummond C. P., González‐Gallegos J. G., Celep F., Mahdjoub M. M., Rose J. P., et al. 2019. Data from: Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: leveraging anchored hybrid enrichment and targeted sequence data. Dryad Digital Repository. 10.5061/dryad.8m40rb0. [DOI] [PMC free article] [PubMed]
- Lagomarsino, L. P. , Condamine F. L., Antonelli A., Mulch A., and Davis C. C.. 2016. The abiotic and biotic drivers of rapid diversification in Andean bellflowers (Campanulaceae). New Phytologist 210: 1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagomarsino, L. P. , Forrestel E. J., Muchhala N., and Davis C. C.. 2017. Repeated evolution of vertebrate pollination syndromes in a recently diverged Andean plant clade. Evolution 71: 1970–1985. [DOI] [PubMed] [Google Scholar]
- Lancaster, L. T. , and Kay K. M.. 2013. Origin and diversification of the California flora: re‐examining classic hypotheses with molecular phylogenies. Evolution 67: 1041–1054. [DOI] [PubMed] [Google Scholar]
- Lemmon, A. R. , and Lemmon E. M.. 2012. High‐throughput identification of informative nuclear loci for shallow‐scale phylogenetics and phylogeography. Systematic Biology 61: 745–761. [DOI] [PubMed] [Google Scholar]
- Lemmon, A. R. , Emme S. A., and Lemmon E. M.. 2012. Anchored hybrid enrichment for massively high‐throughput phylogenomics. Systematic Biology 61: 727–744. [DOI] [PubMed] [Google Scholar]
- Li, Q. Q. , Li M. H., Yuan Q. J., Cui Z. H., Huang L.‐Q., and Xiao P. G.. 2013. Phylogenetic relationships of Salvia (Lamiaceae) in China: evidence from DNA sequence datasets. Journal of Systematics and Evolution 51: 184–195. [Google Scholar]
- Linder, H. P. 2005. Evolution of diversity: the Cape flora. Trends in Plant Science 10: 536–541. [DOI] [PubMed] [Google Scholar]
- Linder, H. P. , and Verboom G. A.. 2015. The evolution of regional species richness: the history of the southern African flora. Annual Review of Ecology, Evolution, and Systematics 46: 393–412. [Google Scholar]
- Linder, H. P. , Rabosky D. L., Antonelli A., Wüest R. O., and Ohlemüller R.. 2014. Disentangling the influence of climatic and geological changes on species radiations. Journal of Biogeography 41: 1313–1325. [Google Scholar]
- Maddison, W. P. , and FitzJohn R. G.. 2014. The unsolved challenge to phylogenetic correlation tests for categorical characters. Systematic Biology 64: 127–136. [DOI] [PubMed] [Google Scholar]
- Mai, D. H. , and Walther H.. 1988. Die pliozaenen floren von Thüringen, Deutsche Demokratische Republik. Quartaerpalaeontologie 7: 55–297. [Google Scholar]
- Maldonado, C. , Molina C. I., Zizka A., Persson C., Taylor C. M., Albán J., Chilquillo E., et al. 2015. Estimating species diversity and distribution in the era of Big Data: To what extent can we trust public databases? Global Ecology and Biogeography 24: 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manafzadeh, S. , Salvo G., and Conti E.. 2014. A tale of migrations from east to west: the Irano‐Turanian floristic region as a source of Mediterranean xerophytes. Journal of Biogeography 41: 366–379. [Google Scholar]
- Manafzadeh, S. , Staedler Y. M., and Conti E.. 2017. Visions of the past and dreams of the future in the Orient: the Irano‐Turanian region from classical botany to evolutionary studies. Biological Reviews 92: 1365–1388. [DOI] [PubMed] [Google Scholar]
- Manchester, S. R. 1999. Biogeographical relationships of North American Tertiary floras. Annals of the Missouri Botanical Garden 86: 472–522. [Google Scholar]
- Matzke, N. J. 2013. BioGeoBEARS: BioGeography with Bayesian (and likelihood) evolutionary analysis in R Scripts, CRAN: The Comprehensive R Archive Network, Vienna, Austria. http://cran.r-project.org/package=BioGeoBEARS.
- Matzke, N. J. 2014. Model selection in historical biogeography reveals that founder‐event speciation is a crucial process in island clades. Systematic Biology 63: 951–970. [DOI] [PubMed] [Google Scholar]
- Matzke, N. J. 2016. Stochastic mapping under biogeographical models. PhyloWiki BioGeoBEARS website http://phylo.wikidot.com/biogeobears#stochastic_mapping.
- Mayr, G. 2004. Old World fossil record of modern‐type hummingbirds. Science 304: 861–864. [DOI] [PubMed] [Google Scholar]
- Mayr, G. 2007. New specimens of the early Oligocene Old World hummingbird Eurotrochilus inexpectatus . Journal of Ornithology 148: 105–111. [Google Scholar]
- McGuire, J. A. , Witt C. C., Remsen J. V. Jr, Corl A., Rabosky D. L., Altshuler D. L., and Dudley R.. 2014. Molecular phylogenetics and the diversification of hummingbirds. Current Biology 24: 910–916. [DOI] [PubMed] [Google Scholar]
- Miller, M. A. , Pfeiffer W., and Schwartz T.. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 573–8.
- Milne, R. I. 2006. Northern Hemisphere plant disjunctions: a window on Tertiary land bridges and climate change? Annals of Botany 98: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab, S. , and Warnow T.. 2015. ASTRAL‐II: coalescent‐based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31: i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, N. , Lewis P. O., Lemmon E. M., Lemmon A. R., and Holsinger K. E.. 2017. Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L. (Proteaceae). American Journal of Botany 104: 102–115. [DOI] [PubMed] [Google Scholar]
- Montes, C. , Cardona A., McFadden R., Moron S. E., Silva C. A., Restrepo‐Moreno S., Ramírez D. A., et al. 2012. Evidence for middle Eocene and younger land emergence in central Panama: implications for Isthmus closure. Geological Society of America Bulletin 124: 780–799. [Google Scholar]
- Montes, C. , Cardona A., Jaramillo C., Pardo A., Silva J. C., Valencia V., Ayala C., et al. 2015. Middle Miocene closure of the Central American Seaway. Science 348: 226–229. [DOI] [PubMed] [Google Scholar]
- Moore, B. , and Donoghue M.. 2007. Correlates of diversification in the plant clade Dipsacales: geographic movement and evolutionary innovations. American Naturalist 170: 28–55. [DOI] [PubMed] [Google Scholar]
- Morrone, J. J. 2010. Fundamental biogeographic patterns across the Mexican Transition Zone: an evolutionary approach. Ecography 33: 355–361. [Google Scholar]
- Mouthereau, F. , Lacombe O., and Vergés J.. 2012. Building the Zagros collisional orogen: timing, strain distribution and the dynamics of Arabia/Eurasia plate convergence. Tectonophysics 532–535: 27–60. [Google Scholar]
- Newton, M. , and Raftery A. E.. 1994. Approximate Bayesian inference by the weighted likelihood bootstrap. Journal of the Royal Statistical Society, B, Methodological 56: 3–48. [Google Scholar]
- O'Dea, A. , Lessios H. A., Coates A. G., Eytan R. I., Restrepo‐Moreno S. A., Cione A. L., Collins L. S., et al. 2016. Formation of the Isthmus of Panama. Science Advances 2: e1600883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson, D. M. , Dinerstein E., Wikramanayake E. D., Burgess N. D., Powell G., Underwood E. C., D'Amico J. A., et al. 2001. Terrestrial ecoregions of the worlds: a new map of life on Earth. BioScience 51: 933–938. [Google Scholar]
- Pokorny, L. , Riina R., Mairal M., Meseguer A. S., Culshaw V., Cendoya J., Serrano M., et al. 2015. Living on the edge: timing of Rand Flora disjunctions congruent with ongoing aridification in Africa. Frontiers in Genetics 6: 315–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prum, R. O. , Berv J. S., Dornburg A., Field D. J., Townsend J. P., Lemmon E. M., and Lemmon A. R.. 2015. A comprehensive phylogeny of birds (Aves) using targeted next‐generation DNA sequencing. Nature 526: 569–573. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2018. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Website https://www.R-project.org/. [Google Scholar]
- Rabosky, D. L. 2014. Automatic detection of key innovations, rate shifts, and diversity‐dependence on phylogenetic trees. PLoS ONE 9: e89543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky, D. L. 2018. BAMM at the court of false equivalency: a response to Meyer and Wiens. Evolution 72: 2246–2256. [DOI] [PubMed] [Google Scholar]
- Rabosky, D. L. , and Goldberg E. E.. 2015. Model inadequacy and mistaken inferences of trait‐dependent speciation. Systematic Biology 64: 340–355. [DOI] [PubMed] [Google Scholar]
- Rabosky, D. L. , Grundler M., Anderson C., Title P., Shi J. J., Brown J. W., Huang H., and Larson J. G.. 2014. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods in Ecology and Evolution 5: 701–707. [Google Scholar]
- Rabosky, D. L. , Mitchell J. S., and Chang J.. 2017. Is BAMM flawed? Theoretical and practical concerns in the analysis of multi‐rate diversification models. Systematic Biology 66: 477–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Suchard M. A., Xie D., and Drummond A. J.. 2014. Tracer v1.6, Available from http://beast.bio.ed.ac.uk/Tracer.
- Ree, R. H. , and Sanmartín I.. 2018. Conceptual and statistical problems with the DEC+J model of founder‐event speciation and its comparison with DEC via model selection. Journal of Biogeography 65: 583–589. [Google Scholar]
- Ree, R. H. , and Smith S. A.. 2008. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Systematic Biology 57: 4–14. [DOI] [PubMed] [Google Scholar]
- Revell, L. J. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution 3: 217–223. [Google Scholar]
- Ricklefs, R. E. 2014. Reconciling diversification: random pulse models of speciation and extinction. American Naturalist 184: 268–276. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L. H. , and Willis J. H.. 2007. Plant speciation. Science 317: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roalson, E. H. , and Roberts W. R.. 2016. Distinct processes drive diversification in different clades of Gesneriaceae. Systematic Biology 65: 662–684. [DOI] [PubMed] [Google Scholar]
- Rogl, F. 1999. Mediterranean and Paratethys. Facts and hypotheses of an Oligocene to Miocene paleogeography (short overview). Geologica Carpathica 50: 339–349. [Google Scholar]
- Rokyta, D. R. , Lemmon A. R., Margres M. J., and Arnow K.. 2012. The venom‐gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genomics 13: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, J. P. , Kriebel R., and Sytsma K. J.. 2016. Shape analysis of moss (Bryophyta) sporophytes: Insights into land plant evolution. American Journal of Botany 103: 652–662. [DOI] [PubMed] [Google Scholar]
- Salvo, G. , Manafzadeh S., Ghahremaninejad F., Tojibaev K., Zeltner L., and Conti E.. 2011. Phylogeny, morphology, and biogeography of Haplophyllum (Rutaceae), a species‐rich genus of the Irano‐Turanian floristic region. Taxon 60: 513–527. [Google Scholar]
- Salzman, S. , Driscoll H. E., Renner T., André T., Shen S., and Specht C. D.. 2015. Spiraling into history: a molecular phylogeny and investigation of biogeographic origins and floral evolution for the genus Costus . Systematic Botany 40: 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter, D. 2001. Ecology and the origin of species. Trends in Ecology & Evolution 16: 372–380. [DOI] [PubMed] [Google Scholar]
- Schmidt‐Lebuhn, A. N. , Kessler M., and Hensen I.. 2007. Hummingbirds as drivers of plant speciation? Trends in Plant Science 12: 329–331. [DOI] [PubMed] [Google Scholar]
- Serrano‐Serrano, M. L. , Rolland J., Clark J. L., Salamin N., and Perret M.. 2017. Hummingbird pollination and the diversification of angiosperms: an old and successful association in Gesneriaceae. Proceedings of the Royal Society, B, Biological Sciences 284: 20162816–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. D. 2010. Using phylogenetics to detect pollinator‐mediated floral evolution. New Phytologist 188: 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel, J. M. , Chen G. F., Watt L. R., and Schemske D. W.. 2010. The biology of speciation. Evolution 64: 295–315. [DOI] [PubMed] [Google Scholar]
- Spalink, D. , Drew B. T., Pace M. C., Zaborsky J. G., Li P., Cameron K. M., Givnish T. J., et al. 2016a. Evolution of geographical place and niche space: patterns of diversification in the North American sedge (Cyperaceae) flora. Molecular Phylogenetics and Evolution 95: 183–195. [DOI] [PubMed] [Google Scholar]
- Spalink, D. , Drew B. T., Pace M. C., Zaborsky J. G., Starr J. R., Cameron K. M., Givnish T. J., et al. 2016b. Biogeography of the cosmopolitan sedges (Cyperaceae) and the area‐richness correlation in plants. Journal of Biogeography 43: 1893–1904. [Google Scholar]
- Stamatakis, A. 2014. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strömberg, C. A. E. 2011. Evolution of grasses and grassland ecosystems. Annual Review of Earth and Planetary Sciences 39: 517–544. [Google Scholar]
- Suc, J. P. 1984. Origin and evolution of the Mediterranean vegetation and climate in Europe. Nature 307: 429–432. [Google Scholar]
- Suchard, M. A. , Weiss R. E., and Sinsheimer J. S.. 2001. Bayesian selection of continuous‐time Markov chain evolutionary models. Molecular Biology and Evolution 18: 1001–1013. [DOI] [PubMed] [Google Scholar]
- Sudarmono, O. H. 2007. Speciation process of Salvia isensis (Lamiaceae), a species endemic to serpentine areas in the Ise‐Tokai district, Japan, from the viewpoint of the contradictory phylogenetic trees generated from chloroplast and nuclear DNA. Journal of Plant Research 120: 483–490. [DOI] [PubMed] [Google Scholar]
- Sulman, J. D. , Drew B. T., Drummond C., Hayasaka E., and Sytsma K. J.. 2013. Systematics, biogeography, and character evolution of Sparganium (Typhaceae): diversification of a widespread, aquatic lineage. American Journal of Botany 100: 2023–2039. [DOI] [PubMed] [Google Scholar]
- Takano, A. , and Okada H.. 2011. Phylogenetic relationships among subgenera, species, and varieties of Japanese Salvia L. (Lamiaceae). Journal of Plant Research 124: 245–252. [DOI] [PubMed] [Google Scholar]
- Thulin, M. 1991. Aspects of disjunct distributions and endemism in the arid parts of the Horn of Africa, particularly Somalia In Seyani J. H. and Chikuni A. C. [eds.], Proceedings of the 13th plenary meeting of the Association for the Taxonomic Study of the Flora of Tropical Africa [AETFAT], vol. 2, 1105–1120, Zomba, Malawi. [Google Scholar]
- Tiffney, B. H. 1985. The Eocene North Atlantic land bridge: its importance in Tertiary and modern phytogeography of the Northern Hemisphere. Journal of the Arnold Arboretum 66: 243–273. [Google Scholar]
- Tiffney, B. H. 2008. Phylogeography, fossils, and northern hemisphere biogeography: the role of physiological uniformitarianism. Annals of the Missouri Botanical Garden 95: 135–143. [Google Scholar]
- Tiffney, B. H. , and Manchester S. R.. 2001. The use of geological and paleontological evidence in evaluating plant phylogeographic hypotheses in the Northern Hemisphere Tertiary. International Journal of Plant Sciences 162 (6 supplement): 3–17. [Google Scholar]
- Trip, E. A. , and Manos P. S.. 2008. Is floral specialization and evolutionary dead‐end? Pollination system transitions in Ruellia (Acanthaceae). Evolution 62: 1712–1737. [DOI] [PubMed] [Google Scholar]
- Uribe‐Convers, S. , and Tank D. C.. 2015. Shifts in diversification rates linked to biogeographic movement into new areas: an example of a recent radiation in the Andes. American Journal of Botany 102: 1854–1869. [DOI] [PubMed] [Google Scholar]
- Uyeda, J. C. , Zenil‐Ferguson R., and Pennell M. W.. 2018. Rethinking phylogenetic comparative methods. Systematic Biology 67: 1092–1109. [DOI] [PubMed] [Google Scholar]
- Vamosi, J. C. , Magallón S., Mayrose I., Otto S. P., and Sauquet H.. 2018. Macroevolutionary patterns of flowering plant speciation and extinction. Annual Review of Plant Biology 69: 685–706. [DOI] [PubMed] [Google Scholar]
- van der Niet, T. , and Johnson S. D.. 2012. Phylogenetic evidence for pollinator‐driven diversification of angiosperms. Trends in Ecology and Evolution 27: 353–361. [DOI] [PubMed] [Google Scholar]
- Verboom, G. A. , Archibald J. K., Bakker F. T., Bellstedt D. U., Conrad F., Dreyer L. L., Forest F., et al. 2009. Origin and diversification of the Greater Cape flora: ancient species repository, hot‐bed of recent radiation, or both? Molecular Phylogenetics and Evolution 51: 44–53. [DOI] [PubMed] [Google Scholar]
- Walker, J. B. , and Elisens W. J.. 2001. A revision of Salvia sect. Heterosphace (Lamiaceae) in western North America. Sida 19: 571–589. [Google Scholar]
- Walker, J. B. , and Sytsma K. J.. 2007. Staminal evolution in the genus Salvia (Lamiaceae): molecular phylogenetic evidence for multiple origins of the staminal lever. Annals of Botany 100: 375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J. , Sytsma K. J., Treutlein J., and Wink M.. 2004. Salvia (Lamiaceae) is not monophyletic: implications for the systematics, radiation, and ecological specializations of Salvia and tribe Mentheae. American Journal of Botany 91: 1115–1125. [DOI] [PubMed] [Google Scholar]
- Walker, J. B. , Drew B. T., and Sytsma K. J.. 2015. Unravelling species relationships and diversification within the iconic California Floristic Province sages (Salvia subgenus Audibertia, Lamiaceae). Systematic Botany 40: 826–844. [Google Scholar]
- Wang, M. , Zhao H. X., Wang L., Wang T., Yang R. W., Wang X. L., Zhou Y. H., et al. 2013. Potential use of DNA barcoding for the identification of Salvia based on cpDNA and nrDNA sequences. Gene 528: 206–215. [DOI] [PubMed] [Google Scholar]
- Wegner, W. , Worner G., Harmon R. S., and Jicha B. R.. 2011. Magmatic history and evolution of the Central American Land Bridge in Panama since Cretaceous times. Bulletin of the Geological Society of America 123: 703–724. [Google Scholar]
- Wei, Y. K. , Wang Q., and Huang Y. B.. 2015. Species diversity and distribution in Salvia . Biodiversity Science 23: 3–10. [Google Scholar]
- Wen, J. , and Ickert‐Bond S. M.. 2009. Evolution of the Madrean‐Tethyan disjunctions and the North and South American amphitropical disjunctions in plants. Journal of Systematics and Evolution 47: 331–348. [Google Scholar]
- Wen, J. , Nie Z. L., and Ickert‐Bond S. M.. 2016. Intercontinental disjunctions between eastern Asia and western North America in vascular plants highlight the biogeographic importance of the Bering land bridge from late Cretaceous to Neogene. Journal of Systematics and Evolution 54: 469–490. [Google Scholar]
- Wester, P. , and Claßen‐Bockhoff R.. 2006. Bird pollination in South African Salvia species. Flora ‐ Morphology, Distribution, Functional Ecology of Plants 201: 396–406. [Google Scholar]
- Wester, P. , and Claßen‐Bockhoff R.. 2007. Floral diversity and pollen transfer mechanisms in bird‐pollinated Salvia species. Annals of Botany 100: 401–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester, P. , and Claßen‐Bockhoff R.. 2011. Pollination syndromes of New World Salvia species with special reference to bird pollination. Annals of the Missouri Botanical Garden 98: 101–155. [Google Scholar]
- Will, M. , and Claßen‐Bockhoff R.. 2014. Why Africa matters: evolution of Old World Salvia (Lamiaceae) in Africa. Annals of Botany 114: 61–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, M. , and Claßen‐Bockhoff R.. 2017. Time to split Salvia s.l. (Lamiaceae) ‐ New insights from Old World Salvia phylogeny. Molecular Phylogenetics and Evolution 109: 33–58. [DOI] [PubMed] [Google Scholar]
- Will, M. , Schmalz N., and Claßen‐Bockhoff R.. 2015. Towards a new classification of Salvia s.l.: (re)establishing the genus Pleudia Raf. Turkish Journal of Botany 39: 693–707. [Google Scholar]
- de Winter, B. 1971. Floristic relationships between the northern and southern arid areas in Africa. Mitteilungen der Botanischen Staatssammlung München 10: 424–437. [Google Scholar]
- Wolfe, J. 1975. Some aspects of plant geography of the Northern Hemisphere during the late Cretaceous and Tertiary. Annals of the Missouri Botanical Garden 62: 264–279. [Google Scholar]
- Yang, Z. 2006. Computational molecular evolution. Oxford Press, London, UK. [Google Scholar]
- Zachos, J. , Pagani M., Sloan L., Thomas E., and Billups K.. 2001. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292: 686–693. [DOI] [PubMed] [Google Scholar]
- Zanne, A. E. , Tank D. C., Cornwell W. K., Eastman J. M., Smith S. A., FitzJohn R. G., McGlinn D. J., et al. 2014. Three keys to the radiation of angiosperms into freezing environments. Nature 506: 89–92. [DOI] [PubMed] [Google Scholar]
- Zohary, M. 1973. Geobotanical foundations of the Middle East, vols. I and II. Gustav Fischer Verlag, Stuttgart, Germany; Swets & Zeitlinger, Amsterdam, Netherlands. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Species names and GenBank information for nrDNA data (ETS and ITS). Species with *** were sampled for AHE and the data are deposited in data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8m40rb0.
Appendix S2. Species names, subgeneric or clade designations, vouchers, GenBank accessions, biogeographical area scoring (see Appendix S4 for area names), biome scoring (see Appendix S5 for biome names), and pollinator scoring. The “Salvia aegyptiaca clade” is informally listed here with subgen. Zhumeria. Species in the informal “Heterosphace” are listed here under two subgroups, New World “Salviastrum” or Old World “Verticillata”.
Appendix S3. Terminals, proportion of extant species sampled for each clade for BAMM analyses of Salvia. The “Salvia aegyptiaca clade” is informally listed here with subgen. Zhumeria. The informal “Heterosphace” are listed here under New World “heterosphaceA” or Old World “heterosphaceB”.
Appendix S4. BioGeoBEARS area: details regarding model development, temporal stratification, and dispersal probabilities among the geographical regions through time for BioGeoBEARS analyses of Salvia. See Appendix S2 for species coding.
Appendix S5. BioGeoBEARS biome: details regarding model development, temporal stratification, and dispersal probabilities among biomes through time for BioGeoBEARS analyses of Salvia. See Appendix S2 for species coding.
Appendix S6. RAxML tree of anchored hybrid enrichment genomic data based on 316 concatenated nuclear genes across 35 species of Salvia and five outgroup genera in the tribe Mentheae. Bootstrap support values are provided next to each node. Asterisks indicate clades that are designated as monophyletic in the BEAST analyses of 528 species based on nrDNA. Edges are colored by subgenus (see Table 1). (The informal “Heterosphace” is used here and includes New World “Salviastrum” and Old World “Verticillata” and their relatives.) See Fig. 2 for ASTRAL II coalescent bootstrap tree based on anchored hybrid enrichment genomic data.
Appendix S7. RAxML bootstrap tree of 528 taxa of Salvia and two outgroup genera based on analysis of nrDNA ITS/ETS only (provided no topological constraints). Phylogram showing all species tips, their major subgeneric clades designations, and RAxML bootstrap values for each branch. Relationships among major clades are not completely congruent with those based on anchored hybrid enrichment genomic data (see Fig. 2, Appendix S6).
Appendix S8. BEAST maximum clade credibility tree (yule model) using Salvia nrDNA and rooted with a monophyletic outgroup of Lepechinia and Melissa. Major clades that were well supported based on anchored hybrid enrichment genomic data (see Fig. 2) were used as topological constraints in BEAST. This tree is same as shown in Fig. 4 but with species terminals and 95% nodal age intervals provided.
Appendix S9. The PCoA 1 and 2 distribution of 1000 PP BEAST trees from Salvia nrDNA and the 10 identified grove tree sets (and the MCC tree from Fig. 3 and Appendix S8) from the Treespace analysis (Kendall, M., and C. Colijn. 2016. Mapping phylogenetic trees to reveal distinct patterns of evolution. Molecular Biology and Evolution 33: 2735–2743).
Data Availability Statement
Species, vouchers, and GenBank accessions for targeted sequences are provided in Appendix S1. AHE raw sequences and final alignments are available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.8m40rb0 (Kriebel et al., 2019). Species data for area, biome, and pollinator are provided in Appendix S2. The R scripts, aligned data, trees, and data necessary to reproduce the analyses in this paper are deposited in Dryad or in [Link], [Link], [Link].