

Summary
Disarming pathogens by targeting virulence factors is a promising alternative to classic antibiotics. Many virulence factors in Gram‐negative bacteria are secreted via the autotransporter (AT) pathway, also known as Type 5 secretion. These factors are secreted with the assistance of two membrane‐based protein complexes: Sec and Bam. To identify inhibitors of the AT pathway, we used transcriptomics analysis to develop a fluorescence‐based high‐throughput assay that reports on the stress induced by the model AT hemoglobin protease (Hbp) when its secretion across the outer membrane is inhibited. Screening a library of 1600 fragments yielded the compound VUF15259 that provokes cell envelope stress and secretion inhibition of the ATs Hbp and Antigen‐43. VUF15259 also impairs β‐barrel folding activity of various outer membrane proteins. Furthermore, we found that mutants that are compromised in outer membrane protein biogenesis are more susceptible to VUF15259. Finally, VUF15259 induces the release of vesicles that appear to assemble in short chains. Taken together, VUF15259 is the first reported compound that inhibits AT secretion and our data are mostly consistent with VUF15259 interfering with the Bam‐complex as potential mode of action. The validation of the presented assay incites its use to screen larger compound libraries with drug‐like compounds.
A promising new category of antibiotics are molecules that interfere with virulence factors. Many virulence factors are transported across the bacterial cell envelope into the host by the autotransporter (AT) pathway. We developed a high‐throughput assay that reports on compromised AT secretion. Screening a compound library resulted in an inhibitor of AT secretion that also interfered with biogenesis of β‐barrel outer membrane proteins.
Introduction
Antibiotic resistance in clinically relevant pathogens continues to emerge and spread. Despite increasing awareness, the progress in addressing this challenge appears insufficient. In particular, the lack of antibiotics with a novel mechanism of action in the drug development pipeline necessitates the development of new therapeutic strategies (Boucher et al., 2013). A promising new class of antibiotics are anti‐virulence drugs, designed to specifically target virulence factor functions (Clatworthy et al., 2007; Baron, 2010). Virulence factors allow pathogens to replicate and disseminate within a host, for example by evading the host immune system (Heras et al., 2015). Disarming pathogens of their virulence factors may permit the immune system to clear the infection or increase the susceptibility of the pathogens toward conventional antibiotics. It has been hypothesized that anti‐virulence drugs result in a lower selective pressure to develop resistance because they do not target vital bacterial functions and are less harmful to the resident microbial flora (Allen et al., 2014).
In diderm bacteria, most virulence factors are secreted across the cell envelope by multiple secretion systems (SS), classified as Type 1SS‐9SS (also known as Type I‐IX) (Mecsas, 2016). The cell envelope is comprised of both an inner membrane and an outer membrane, separated by the periplasmic space. The T1SS, T3SS, T4SS and T6SS transport proteins across these membranes in a coordinated one‐step mechanism, while substrates of the T2SS and the T5SS traverse the cell envelope in two consecutive steps and feature a periplasmic intermediate. The T3SS, T4SS and T6SS can also transport proteins across the host cell membrane, delivering secreted proteins directly into the host cytosol. Especially for T3SS, several promising inhibitors were discovered using cell‐based high‐throughput screening (HTS) approaches (Tsou et al., 2013). A similar strategy was used in this study to identify inhibitors of the T5SS, also known as the autotransporter (AT) pathway.
The AT pathway is the most widespread mechanism in Gram‐negative bacteria to secrete virulence factors by pathogens that cause human diseases, such as meningitis, peritonitis and whooping cough (Henderson and Nataro, 2001). The mechanism of secretion and basic structure of most ATs is conserved (van Ulsen et al., 2013), making the T5SS an attractive target for the development of anti‐virulence drugs. AT secretion is a relatively autonomous secretion system in which the secreted AT effector domain (the passenger) is flanked by an N‐terminal signal peptide that mediates translocation across the inner membrane via the protein conducting Sec‐translocon and by the β‐domain at the C‐terminus that forms a β‐barrel in the outer membrane (Fig. 1). The β‐domain mediates translocation of the AT passenger across the outer membrane in an intricate, not fully understood mechanism that also involves the Bam‐complex (Sauri et al., 2009), the primary function of which is the insertion of β‐barrel proteins into the outer membrane (Sauri et al., 2009). Upon translocation to the cell surface, the AT passenger folds into a conserved β‐helical conformation, which is thought to provide part of the driving force for the translocation process (van Ulsen et al., 2013; Fan et al., 2016). At the cell surface, most ATs are proteolytically cleaved from the β‐domain to perform their function as released proteins. Alternatively, the passenger remains attached to the β‐domain to serve for instance as an adhesion (Fan et al., 2016).
Figure 1.
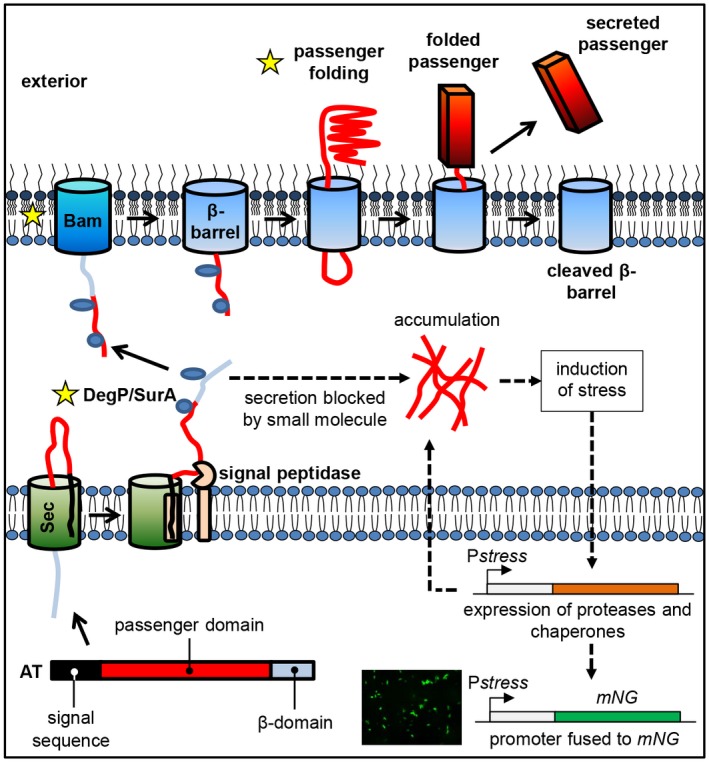
The T5SS with potential targets of inhibitors and the principle of the reporter assay. Organization of protein domains in ATs (signal sequence, passenger domain and β‐domain). AT secretion involves (A) transfer from the cytoplasm across the inner membrane into the periplasm via the Sec translocon, and (B) translocation across the outer membrane to the exterior of the cell via the Bam‐complex. Late stage targets for T5SS inhibitors are marked with a star and include Skp, DegP, SurA, the Bam‐complex and passenger folding. Blocking secretion leads to AT accumulation in the periplasm, inducing cell envelope stress that triggers the expression of proteases and chaperones to relieve the stress. Stress is monitored by expressing the fluorescent protein mNeonGreen (mNG) under control of a stress‐regulated promoter. [Colour figure can be viewed at https://wileyonlinelibrary.com]
When considering the complete AT pathway as a drug target, it is important to note that the system makes use of generic components such as the Sec and Bam machineries as well as chaperones and targeting factors in the cytoplasm and periplasm. In addition, AT‐specific elements, such as the folding of the AT passenger at the cell surface and the interplay between the β‐domain and the Bam‐complex are critical for the secretion process. Hence, screening for compounds that impair the secretion of ATs may yield hits that interfere at different levels in the biogenesis pathway, some of which could be unknown (Fig. 1). Compounds that target essential proteins with a more generic function may be bactericidal at higher concentrations and represent more classical antibiotics whereas AT‐specific inhibitors may address virulence rather than growth and survival per se. Here, we aimed to develop an HTS assay to select inhibitors that affect the more specific late stages in the secretion pathway, i.e. the targeting to and translocation across the bacterial outer membrane.
Previously, we have described a late stage translocation intermediate of the model secreted AT hemoglobin protease (Hbp) that is blocked during transfer across the outer membrane due to the formation of a disulfide bond between two engineered cysteine residues in the passenger domain and, as a consequence, induces cell envelope stress (Jong et al., 2007). In the current study, we characterized this stress response in more detail using RNA sequencing and used the data to develop a fluorescence‐based HTS assay that reports on impaired outer membrane translocation of ATs. As proof‐of‐concept a fragment library, consisting of small organic molecules, was screened that yielded an inhibitor (VUF15259, Rac‐2‐(4‐aminopiperidin‐1‐yl)‐1‐(3,4‐dichlorophenyl)ethan‐1‐ol hydrochloride hydrate) of Hbp secretion and assembly of β‐barrel outer membrane proteins (OMPs).
Results
Development of an HTS assay to identify AT secretion inhibitors
To develop an HTS assay for compounds that inhibit late stages of AT secretion, we decided to monitor the consequences of accumulation of the model AT Hbp in the periplasm at the transcriptome level. We have shown previously that secretion of the paired cysteine mutant Hbp110C/348C across the outer membrane is impaired due to the formation of a disulfide bond in the oxidizing periplasm. The jammed intermediate is exposed to both the periplasm and the cell surface and induces cell envelope stress (Jong et al., 2007). As a result, the expression of the periplasmic protease and chaperone DegP is upregulated, which degrades the intermediates and improves bacterial growth under these conditions (Jong et al., 2007). We reasoned that genes that are induced under this stress condition could be used for the construction of sensitive and robust reporter systems to monitor inhibition of AT secretion.
To characterize the cellular responses to Hbp stalling in more detail, a transcriptomic analysis was carried to compare responses upon expression of Hbp and the stalled secretion mutant Hbp110C/348C. Expression of the constructs was induced in E. coli strain MC4100 for 30 min and total RNA was isolated and analyzed (Table 1). About 56 genes were found to be differentially expressed upon production of Hbp110C/348C compared to Hbp, with a p‐value > 0.01. The majority (66%) of the induced genes belonged to the Sigma E (σE) regulon that directs a major cell envelope stress response involved in maintaining outer membrane homeostasis (Ruiz and Silhavy, 2005). Many of the upregulated genes encode quality control factors, such as the periplasmic chaperones Skp and DegP, and transcriptional regulators of the stress response (Dartigalongue et al., 2001). As mentioned above, upregulation of degP expression under these conditions has been reported earlier (Jong et al., 2007). In addition, enhanced expression of genes that encode subunits of the Bam‐complex was observed. These genes are also part of the σE regulon (Bury‐Moné et al., 2009). Importantly, the Bam‐complex is essential for Hbp secretion (Bury‐Moné et al., 2009). Additional up‐regulated genes of interest relate to the phage shock stress response (Psp) and the Cpx cell envelope stress response. The Cpx regulon shows substantial overlap with the σE regulon and both are activated by unfolded periplasmic proteins. However, there are also specific inducing cues for each pathway. While σE is activated by high temperatures and aberrant LPS, Cpx is activated by alkaline pH and changes in inner and outer membrane lipid composition (Ruiz and Silhavy, 2005). The Psp stress response is induced when the integrity of the inner membrane is compromised. For instance, it responds to a collapse of the proton motive force and defects in protein translocation (Darwin, 2005). Among the repressed genes we found are those encoding the major OMPs OmpF, OmpC and OmpA. This is probably due to small regulatory RNAs (snRNA), induced by the σE response, that bind to the 5′ mRNA region and halt de novo synthesis (Gogol et al., 2011). Taken together, the differential gene expression analysis revealed that expression of Hbp110C/348C leads to activation of the σE, Cpx and Psp stress responses.
Table 1.
Significantly up‐regulated and down‐regulated genes upon expression of Hbp110C/348C compared to expression of wild‐type Hbp.
| Gene ID | Gene a | Description/function | Log2 fold change b | P‐value |
|---|---|---|---|---|
| BWG_1136 | pspA | Phage shock protein A | 2.2 | 1.08E‐29 |
| BWG_0154 | degP | Periplasmic protease | 2.1 | 2.59E‐25 |
| BWG_1137 | pspB | Phage shock protein B | 2.1 | 7.93E‐17 |
| BWG_2181 | yfeK | Uncharacterized protein | 1.7 | 3.27E‐06 |
| BWG_2766 | htrG | Uncharacterized signal transduction protein | 1.6 | 3.68E‐06 |
| BWG_1138 | pspC | Phage shock protein C | 1.6 | 6.04E‐04 |
| BWG_3583 | cpxP | Repressor CpxP; regulator Cpx response | 1.5 | 2.77E‐06 |
| BWG_2336 | rseA | Anti‐sigma factor, regulation of σE activity | 1.4 | 1.70E‐06 |
| BWG_2356 | raiA | Translation inhibitor and ribosome stability factor | 1.4 | 1.70E‐09 |
| BWG_3754 | plsB | Membrane bound glycerol‐3‐phosphate acyltransferase | 1.3 | 7.51E‐10 |
| BWG_2337 | rpoE | RNA polymerase sigmaE (σE) factor | 1.3 | 5.98E‐04 |
| BWG_1139 | pspD | Phage shock protein D | 1.3 | 9.15E‐06 |
| BWG_3234 | eptB | Phosphoethanolamine transferase | 1.2 | 2.46E‐09 |
| BWG_2194 | yfeY | Predicted outer membrane lipoprotein | 1.2 | 4.64E‐07 |
| BWG_2334 | rseC | Regulation of σE activity | 1.1 | 1.74E‐06 |
| BWG_0061 | yabI | Conserved inner membrane protein | 1.1 | 4.61E‐08 |
| BWG_2680 | yggN | Hypothetical protein, potentially involved in biofilm formation | 1.1 | 2.38E‐05 |
| BWG_1619 | yeaY | Predicted lipoprotein | 1.1 | 7.66E‐05 |
| BWG_3216 | yhjJ | Periplasmic zinc‐dependent peptidase | 1.1 | 3.97E‐06 |
| BWG_2335 | rseB | Regulation of σE activity | 1.1 | 6.00E‐08 |
| BWG_1688 | cutC | Copper homeostasis protein | 1.1 | 5.78E‐10 |
| BWG_0261 | sbmA | Inner membrane transporter | 1 | 2.51E‐08 |
| BWG_1618 | fadD | Fatty acyl coA synthetase | 1 | 1.01E‐05 |
| BWG_3038 | fkpA | Periplasmic peptidyl prolyl isomerase | 1 | 4.40E‐04 |
| BWG_0897 | mdoG | Regulation periplasmic glucan biosynthesis | 0.9 | 3.10E‐07 |
| BWG_2182 | yfeS | Uncharacterized protein | 0.9 | 2.18E‐05 |
| BWG_2854 | yraP | Potentially regulating the DegP/Skp folding pathway | 0.8 | 8.50E‐04 |
| BWG_2112 | sixA | Phosphohistidine phosphatase | 0.8 | 1.30E‐06 |
| BWG_1451 | pdxY | Pyridoxamine kinase | 0.8 | 5.42E‐04 |
| BWG_0169 | bamA | Outer membrane protein assembly factor YaeT, component of the Bam‐complex | 0.7 | 1.33E‐03 |
| BWG_0168 | rseP | Inner membrane protease, regulation of σE activity | 0.7 | 5.78E‐05 |
| BWG_2951 | mreB | Rod shape‐determining protein | 0.7 | 5.48E‐06 |
| BWG_3404 | yieF | Catalyzes reduction of quinones | 0.7 | 5.23E‐04 |
| BWG_2258 | yfgC | Metalloprotease | 0.7 | 1.26E‐05 |
| BWG_0262 | yaiW | Putative DNA‐binding transcriptional regulator | 0.7 | 3.36E‐05 |
| BWG_1140 | pspE | Phage shock protein E | 0.7 | 7.66E‐04 |
| BWG_2950 | mreC | Rod shape‐determining protein | 0.6 | 3.53E‐05 |
| BWG_3152 | rpoH | RNA polymerase factor sigma 32 | 0.6 | 8.52E‐04 |
| BWG_3745 | trpB | Tryptophan synthase subunit beta | 0.6 | 1.38E‐03 |
| BWG_1242 | aldA | Aldehyde dehydrogenase A | 0.6 | 9.23E‐05 |
| BWG_1670 | yebA | Murein endopeptidase | 0.5 | 7.59E‐04 |
| BWG_2241 | bamC | Lipoprotein, component of the Bam‐complex | 0.5 | 2.15E‐04 |
| BWG_2555 | recB | Exodeoxyribonuclease V essential for recombination | 0.5 | 8.49E‐04 |
| BWG_0171 | lpxD | Lipid A biosynthesis | 0.5 | 7.08E‐04 |
| BWG_0170 | skp | Periplasmic chaperone, part of the DegP/Skp folding pathway | 0.5 | 3.70E‐04 |
| BWG_2355 | bamD | Lipoprotein, component of the Bam‐complex | 0.5 | 2.84E‐04 |
| BWG_1044 | chaA | Sodium exporter | 0.5 | 1.25E‐03 |
| BWG_1089 | trpA | Tryptophan synthase subunit alpha | 0.5 | 4.03E‐04 |
| BWG_2276 | bamB | Lipoprotein, component of the Bam‐complex | 0.4 | 7.81E‐04 |
| BWG_0871 | efeB | Peroxidase | ‐0.7 | 1.44E‐04 |
| BWG_0870 | efeO | Involved in iron uptake | ‐0.8 | 2.42E‐05 |
| BWG_0809 | ompA | Outer membrane protein A | ‐1 | 3.82E‐04 |
| BWG_0667 | ompX | Outer membrane protein X | ‐1 | 3.55E‐05 |
| BWG_1989 | ompC | Outer membrane protein C | ‐1.4 | 5.30E‐05 |
| BWG_0781 | ompF | Outer membrane protein F | ‐2.3 | 1.59E‐49 |
Genes previously reported to be regulated by the σE, Cpx or Psp stress response are indicated in blue, red and light green respectively. Genes whose expression is controlled by both σE and Cpx are shown in yellow. The genes in dark green are not known to belong to a cell envelope stress response (Bury‐Moné et al., 2009).
The log (base 2) changes indicate the ratios of gene signal intensities of the E. coli strain TOP10F’ harboring pEH3‐Hbp110C/348C to the reference strain TOP10F’ containing pEH3‐Hbp wild‐type.
We reasoned that small molecules that inhibit outer membrane translocation of Hbp will induce similar responses as Hbp110C/348C. Therefore, in order to identify compounds that target AT biogenesis, we set up a stress‐based assay that visualizes E. coli cells affected in Hbp secretion. This was done by placing a fluorescent protein under control of a stress‐regulated promoter that is strongly induced upon accumulation of Hbp in the periplasm (Fig. 1). The promoter of rpoE was selected because it responds to impaired Hbp secretion according to the transcriptomic analysis (Table 1) and RpoE is the key regulator of the corresponding stress response. Although the Psp stress response was also strongly activated, the cues for this response are less clearly defined (Jovanovic et al., 2006) and may reflect indirect effects of periplasmic Hbp accumulation on the integrity of the inner membrane.
The rpoE promoter was fused to the gene encoding the fluorescent reporter protein mNeonGreen (mNG). mNG has a shorter maturation time than GFP as well as a higher brightness and quantum yield (Shaner et al., 2013). These features allow for rapid detection of stress response induction and a higher signal to noise ratio. To test whether impaired Hbp secretion can be reliably detected using this PrpoE‐mNG reporter construct, it was introduced in E. coli TOP10F’ cells harboring pEH3‐Hbp110C/348C or pEH3‐Hbp and expression of the Hbp derivatives was induced with IPTG. As shown in Fig. 2A, fluorescence from the reporter construct was increased approximately threefold upon expression of the translocation intermediate Hbp110C/348C compared to Hbp. Of note, it was found that expression of Hbp already slightly induced σE stress as compared to cells carrying an empty pEH3 vector, which is most likely caused by saturation of the translocation machinery under the conditions used.
Figure 2.
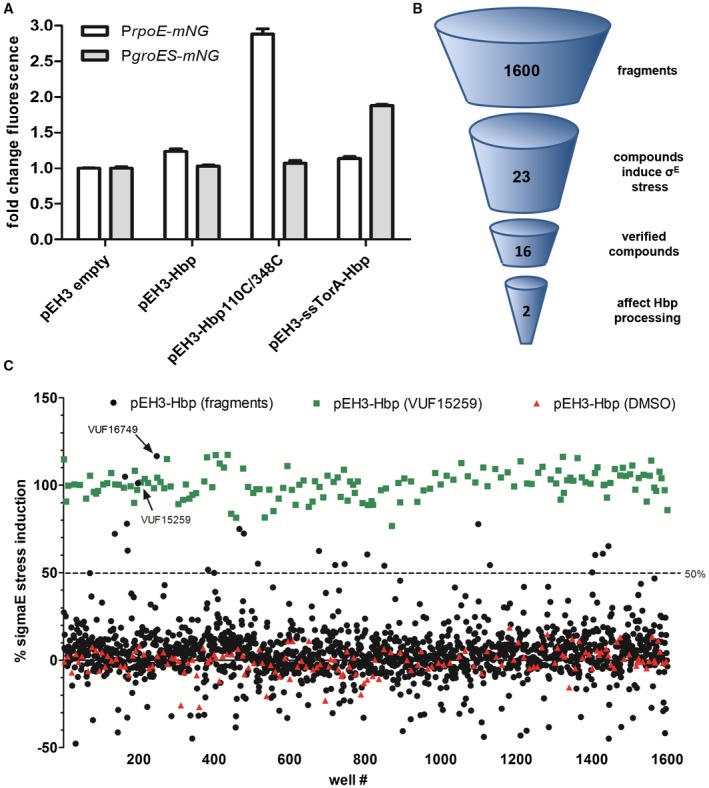
Development of stress‐based assay and summary of fragment screen. A. Cell envelope stress and cytosolic stress were determined using PrpoE‐mNG and PgroES‐mNG reporter constructs respectively. Hbp species were co‐expressed from the pEH3 plasmid in E. coli TOP10F’ bacteria grown in a 96‐well plate. Hbp expression was induced with IPTG and after 3 h of incubation mNG fluorescence and OD660 were measured. Fluorescence intensities were corrected for growth and the fold increase in fluorescence was calculated compared to the empty vector control (pEH3). Error bars represent the standard deviation of triplicate samples. B. In total, 1600 fragments were screened for σE stress induction. 23 compounds induced σE stress in the primary screen whereas secondary screening verified 16 compounds as hits. An orthogonal assay showed that two compounds, VUF15259 and VUF16749, impaired secretion of Hbp. C. Plot of σE stress induction of each compound compared to cells expressing Hbp incubated in 200 µM VUF15259 (positive control, green) and cells expressing Hbp incubated in 1% DMSO (negative control, red). The positive control was set to 100%. Compounds were selected as hits with a stress induction of ≥ 50%, indicated by a dashed line. Compound VUF15259 and VUF16749 is indicated with an arrow. [Colour figure can be viewed at https://wileyonlinelibrary.com]
To confirm the specificity of the PrpoE‐mNG reporter construct in the detection of stress caused by periplasmic Hbp accumulation, a second reporter assay was designed to monitor Hbp accumulation in the cytosol. This is expected to induce the heat shock response, which includes the chaperone pair GroEL/ES that catalyzes ATP‐dependent protein folding (Arsène et al., 2000). Therefore, a PgroES‐mNG fusion was constructed similar to the cell envelope stress reporter construct. To prove that PgroES‐mNG responds to accumulation of Hbp in the cytosol, an Hbp mutant was expressed in which the Hbp signal sequence (ss) was replaced with that of TMAO‐reductase (TorA) ss. We have previously shown that the resulting ssTorA‐Hbp fusion protein accumulates in the cytosol in insoluble form (Jong et al., 2017). Co‐expression of ssTorA‐Hbp and the PgroES‐mNG reporter induced a twofold increase in fluorescence intensity, confirming that heat shock stress was induced (Fig. 2A). To further confirm the specificity of the PrpoE‐mNG and PgroES‐mNG reporters, fluorescence was monitored upon expression of Hbp110C/348C and ssTorA‐Hbp. Indeed, expression of Hbp110C/348C and ssTorA‐Hbp almost exclusively induced the σE and Heat shock reporters respectively. Taken together, the data show that our PrpoE‐mNG reporter‐based assay detects cells that accumulate Hbp in the periplasm.
Screening of a fragment‐based library
To examine the suitability of our reporter assay to identify compounds that affect Hbp secretion, we conducted a screen on the proprietary VUF library of 1600 fragments (De Kloe et al., 2011; Verheij et al., 2011). Fragment‐based drug discovery is an approach that uses very small organic compounds (‘fragments’) as starting points for subsequent hit optimization (Erlanson et al., 2016). The fragments in the library cover a diverse chemical space and the majority of the compounds comply to the criteria defined in the ‘Rule of 3’, in which the molecular mass is < 300 Dalton, the number of hydrogen bond donors is ≤ 3 and the number of hydrogen bond acceptors is ≤ 3 (Congreve et al., 2003).
To test the suitability of the VUF fragment library, the compounds were pre‐screened using a dot blot analysis of supernatants of Hbp secreting cells (data not shown). Although the assay was tedious and difficult to reproduce, compound VUF15259 consistently affected Hbp secretion. In addition, VUF15259 activated σE stress in our reporter assay (see below), showing that this compound was suitable as a positive control for the HTS stress assay.
To establish whether the assay is robust for HTS, we first determined the Z′ factor. This statistical parameter reflects on both the signal dynamic range between the positive and negative controls and the data variation associated with the signal measurements (Zhang et al., 1999). E. coli TOP10F′ cells carrying the PrpoE‐mNG reporter construct and the pEH3‐Hbp plasmid were induced for Hbp expression and either incubated in DMSO (negative control) or compound VUF15259 (positive control). To calculate the Z′ factor, the mNG fluorescence in a 96‐well plate was measured in time and corrected for growth. After several optimization experiments, we obtained a reliable Z′ factor of 0.8 after three h of growth, showing that the assay was sufficiently robust for HTS in 96‐well plates.
To identify more inhibitors of the Hbp secretion pathway, all 1600 VUF fragments were tested for their ability to induce σE stress in cells harboring the wild‐type pEH3‐Hbp plasmid in combination with the PrpoE‐mNG reporter construct. Hbp expression was induced with IPTG and compounds were added simultaneously to a final concentration of 200 µM. A high concentration of compound was used in the primary screen because the affinity of fragments is generally low (Joseph‐McCarthy et al., 2014). After 3 h of incubation, the optical density (OD660) and mNG fluorescence were measured. Compounds were selected as hits when their values were higher than 50% with the positive control set at 100%. This selection protocol resulted in 23 hit compounds (Fig. 2B,C). After primary screening, the assay was repeated in triplicate for the 23 compounds. Replicates were considered verified when their mean value reached higher than 50% stress, yielding 16 hits, including VUF15259 (data not shown).
The 16 hits were tested in an orthogonal assay to validate the σE stress response biochemically and to test whether the detected stress correlates with inhibition of Hbp secretion. As mentioned earlier, the paired cysteine mutant Hbp110C/348C accumulates in the periplasm in the so‐called Hbp pro‐form, in which the Hbp passenger domain is still connected to the Hbp β‐domain (Jong et al., 2007). The Hbp pro‐form is subject to degradation by the periplasmic protease DegP and can only be detected in bacteria that express the proteolytically inactive degP::S210A mutant (Jong et al., 2007). Likewise, Hbp species that accumulate due to the presence of secretion blocking compounds are expected to be degraded by DegP. Therefore, we used E. coli MC1061 degP::S210A to examine the effects of the compound hits on Hbp processing and secretion. Bacteria treated with the 16 hits were analyzed by SDS‐PAGE and Western blotting. In the absence of any compound, mature Hbp passenger domain (110 kDa) was detected as expected, but also a small fraction of pro‐Hbp (142 kDa) indicating that secretion is slightly saturated under these expression conditions (Fig. 3A). On the contrary, Hbp110C/348C exclusively accumulated as 142 kDa pro‐form as expected. Only 2 of the 16 compounds, VUF16749 (Fig. 3B) and VUF15259 (Fig. 3C), induced an increase in Hbp pro‐form relative to the mature Hbp passenger, indicating interference with Hbp secretion. Both VUF15259 and VUF16749 are racemic compounds with some overlapping pharmacophore features. We proceeded with VUF15259 because it showed the strongest effect on Hbp processing. To further analyze compound VUF15259, a larger batch was re‐synthesized and the structure of the compound was confirmed (Figs S4‐S10).
Figure 3.
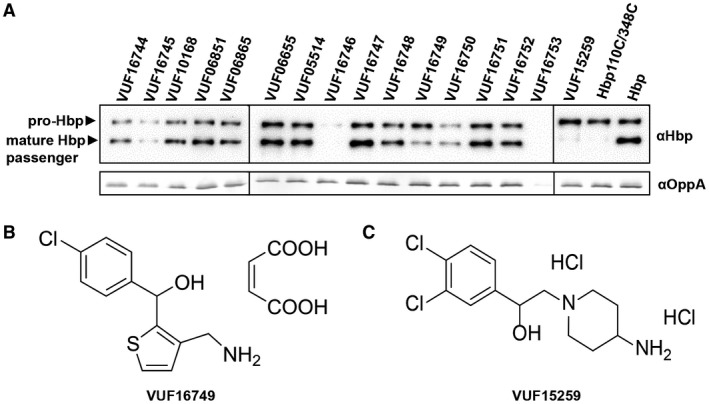
Hbp expression and processing in response to compound hits. A. E. coli MC1061 degP::S210A cells were grown in 96‐well plates and induced for Hbp(‐derivative) expression in the presence or absence (last two lanes) of 200 µM of the indicated VUF compounds. Subsequently, cells were collected and separated from spent medium by centrifugation and analyzed by SDS‐PAGE and Western blotting. The periplasmic protein OppA, part of the oligopeptide permease Opp, was used as loading control. B. Structure of VUF16749. C. Structure of VUF15259.
VUF15259 inhibits Hbp secretion
To study the effect of VUF15259 in more detail, the biogenesis of Hbp was tested in MC1061 degP::S210 upon incubation with different concentrations of the compound. In the absence of VUF15259, Hbp is processed, as shown by the presence of the 110 kDa mature Hbp passenger and the 28 kDa Hbp β‐domain (Fig. 4A). Addition of VUF15259 led to a dose‐dependent accumulation of pro‐Hbp and a concomitant decrease in mature Hbp passenger and Hbp β‐domain (Fig. 4A). Consistently, a dose‐dependent reduction in the amount of secreted passenger domain in the spent medium was observed (Fig. 4B), confirming that VUF15259 inhibits Hbp secretion. Analysis of extracellular stress using the PrpoE‐mNG reporter construct showed a dose‐dependent induction of σE stress (Fig. 4C). This induction of cell envelope stress was confirmed by examination of the levels of the stress‐regulated protease DegP (Fig. 4A). We also found a dose‐dependent upregulation of the Psp stress response marker PspA (Fig. 4A), which is consistent with the transcriptomic analysis of Hbp110C/348C expressing cells (Table 1). Taken together, the data demonstrate that VUF15259 impairs Hbp secretion, leading to accumulation of unprocessed Hbp in the periplasm and subsequent upregulation of cell envelope stress.
Figure 4.
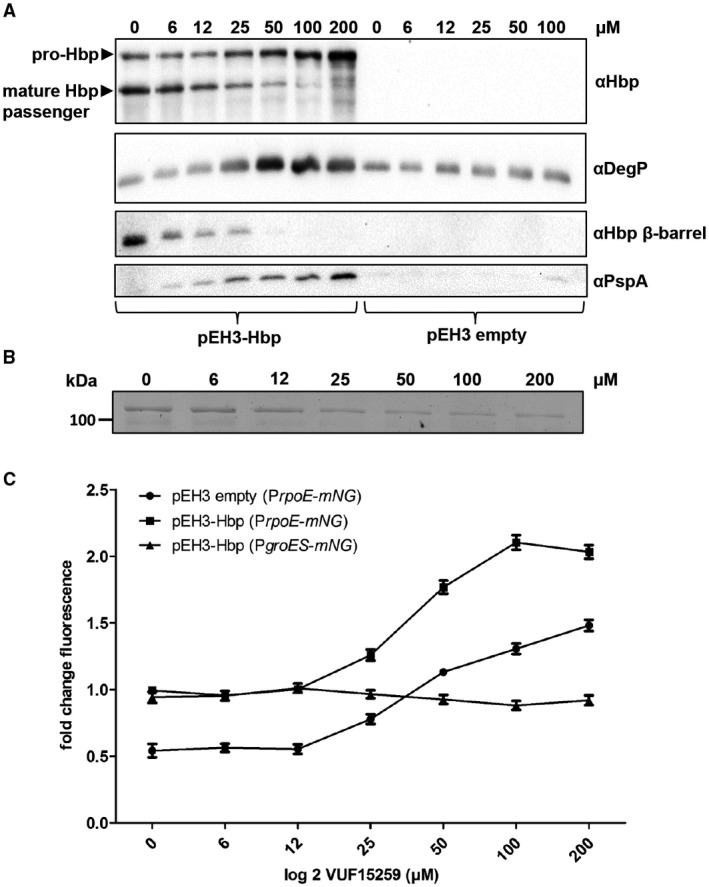
VUF15259 stress induction and inhibition of Hbp secretion. E. coli TOP10F’ bacteria were grown in a 96‐well plate and Hbp was expressed from the pEH3 plasmid. Cells were exposed for 3 h to an increasing concentration of VUF15259 as indicated in the Figure. A. Whole cell lysates were analyzed with SDS‐PAGE and Western blotting. The whole cell lysate of empty vector (pEH3) E. coli cells treated with VUF15259 were also analyzed. B. The spent medium was TCA precipitated and analyzed by SDS‐PAGE and Coomassie staining. C. Stress was monitored using PrpoE‐mNG and PgroES‐mNG on the pUA66 plasmid. Error bars represent the standard deviation of triplicate samples.
VUF15259 could either function as a virulence inhibitor by specifically targeting Hbp biogenesis or as a more classic antibiotic by interfering with a common step in protein secretion. Since VUF15259 also induces σE stress in cells that do not express Hbp, albeit at a lower level (Fig. 4C), the latter explanation appears likely.
VUF15259 inhibits the T5SS but not the T3SS
The mechanism of secretion of ATs is conserved and requires the assistance of the periplasmic chaperones SurA and DegP, as well as the Bam‐complex in the outer membrane (van Ulsen et al., 2013). To examine a potential generic effect of VUF15259 on the T5SS, the influence on the biogenesis of another AT, Antigen‐43 (Ag‐43), was analyzed. Ag‐43 is an adhesin involved in biofilm formation (Henderson and Nataro, 2001). Successful surface display of Ag‐43 in E. coli causes auto‐aggregation and sedimentation, which can be easily determined by measuring the OD660. Indeed, expression of Ag‐43 resulted in sedimentation of mock‐treated E. coli cells whereas VUF15259‐treated cells showed hardly any change in OD660, indicating that VUF15259 prevents surface display of Ag‐43 (Fig. 5).
Figure 5.
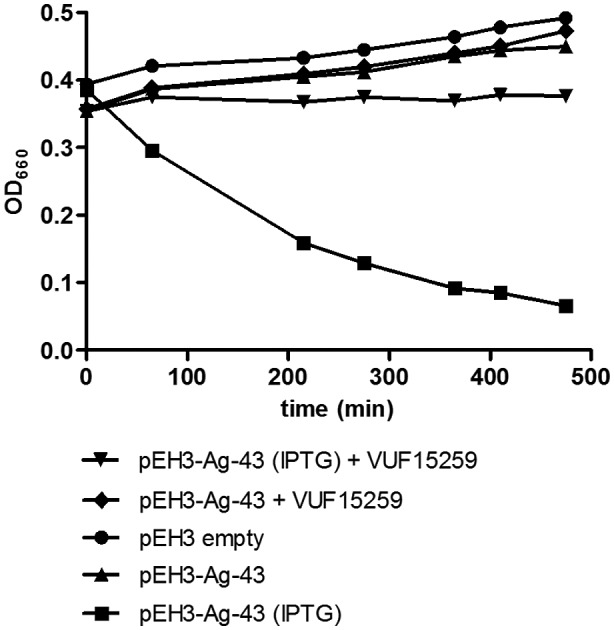
VUF15259 inhibits surface display of Ag‐43. Ag‐43 was expressed from the pEH3 plasmid in E. coli TOP10F’. Cells were grown in 96‐well plates and Ag‐43 expression was induced with IPTG. After 3 h of growth, the cultures were transferred to a cuvette and left standing. OD660 was measured in time in the top of the cuvette.
To investigate whether VUF15259 interferes with protein secretion in general, its effect on T3SS‐mediated secretion was tested in Salmonella typhimurium. In VUF15259‐treated S. typhimurium, secretion via the T3SS was not changed compared to the mock‐treated control (Fig. S1A), while Hbp secretion was affected in this species (Fig. S1B,C). This indicated that VUF15259 does not interfere with protein secretion in general.
VUF15259 interferes with Bam‐related processes
To obtain insight in the target of VUF15259, we studied the effect of the compound on DegP, SurA and the Bam‐complex, which are involved in AT secretion but primarily function in insertion of β‐barrel OMPs (Konovalova et al., 2017). We reasoned that if VUF15259 interferes with this latter process, mutant strains that are compromised in OMP assembly might be particularly sensitive to the addition of this compound.
To test this hypothesis, the effect of VUF15259 on growth was examined in strains in which surA, bamB or degP has been inactivated. VUF1529 moderately affected growth in the parental strains TOP10F, MC4100, MC1061 and KS272 (Fig. 6). However, bacteria became more susceptible to VUF15259 in the absence of surA or bamB. Enhanced susceptibility was also observed in a protease‐deficient degP::S210A mutant background, while a particularly strong effect was observed upon complete knock out of degP. These synergistic effects suggest that the compound interferes with OMP biogenesis.
Figure 6.
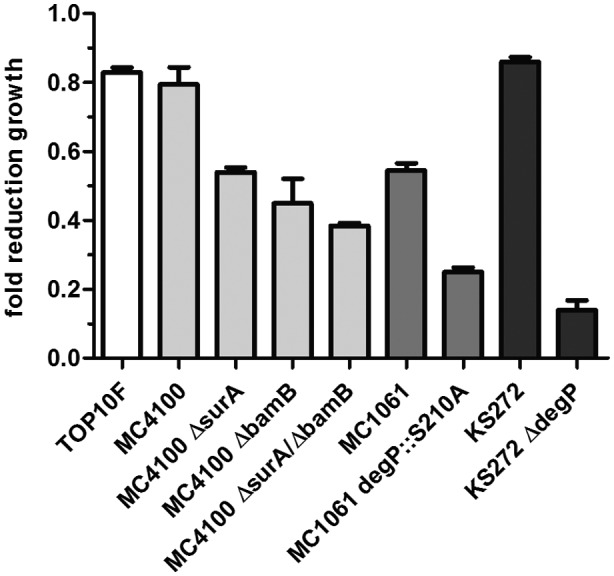
VUF15259 affects growth, in particular of strains with a compromised OMP assembly pathway. Bacteria were grown in a 96‐well plate overnight with 100 µM VUF15259 or DMSO in Thermostar shakers at 37°C shaken at 600 RPM with a start OD660 of 0.001. The OD660 was measured after overnight growth and the fold reduction in OD660 was calculated between bacteria treated with compound and bacteria treated with DMSO only. Error bars represent the standard deviation of triplicate samples.
To analyze more directly whether VUF15259 affects OMP, biogenesis E. coli cells were grown in presence and absence of VUF15259, lysed and cell envelopes were collected for analysis by SDS‐PAGE and Western blotting to examine the level of OMPs. Importantly, this analysis is complicated by the fact that σE stress downregulates OMP synthesis and upregulates expression of subunits of the Bam‐complex (Bury‐Moné et al., 2009). Hence, secondary effects of the periplasmic Hbp accumulation and/or σE stress may obscure the primary effects of the VUF15259 compound. To address this issue, we took controls in which the σE stress was induced to a similar extent by expressing σE directly in the absence of compound (Fig. 7A). Under these conditions, we did observe an approximately 1.5‐fold decrease in the amounts of OmpF/C/A compared to cells carrying an empty pEH3 vector (Fig. 7B), but unexpectedly we did not see a change in levels of BamA (Fig. 7C). The levels of the inner membrane protein LepB were largely unaffected (Fig. 7C). Exposure to VUF15259 decreased OmpF/C/A levels ~ 3‐fold (Fig. 7B) and, strikingly, BamA levels ~ 5‐fold compared to bacteria expressing the σE factor (Fig. 7C). The levels of the associated Bam lipoproteins BamB, BamC and BamD were unaffected (Fig. 7D). Furthermore, we observed a ~ 4‐fold reduction in the amount of fully assembled Bam‐complex in the cell envelope of compound treated cells analyzed by Blue Native PAGE (Fig. 7E), consistent with the decreased BamA level (Fig. 7C).
Figure 7.
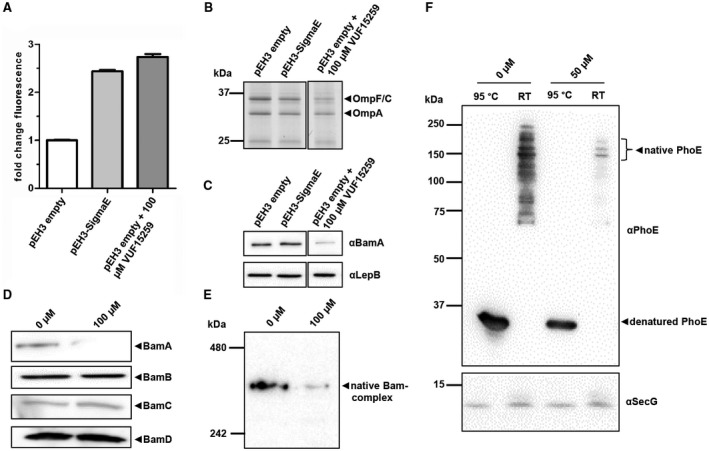
VUF15259 interferes with targeting or insertion of β‐barrel type OMPs. (A) E. coli TOP10F’ cells, carrying the pEH3 vector expressing σE factor and TOP10F’ cells, carrying the empty pEH3 vector, were treated with VUF15259 or with 1% DMSO. Cells were grown in 96‐well plates for 3 h and the σE stress was measured using the PrpoE‐mNG reporter construct. The fold mNG fluorescence is depicted compared to the DMSO‐treated empty vector cells, with the error bars representing the standard deviation of triplicate samples. Bacteria in the 96‐well plate were collected and separated from medium by centrifugation. Cell envelopes were isolated using ultracentrifugation and analyzed by (B) SDS‐PAGE and (C) Western blotting analysis using antibodies against BamA and LepB. For the empty vector, control cells also an antibody staining against the whole Bam‐complex was performed analyzed under (D) denaturing conditions by SDS‐PAGE, and under (E) native conditions by Blue Native PAGE. (F) TOP10F’ cells, expressing phoE from the pEH3 plasmid, were grown in 96‐well plates and treated with VUF15259 for 3 h. Cell envelopes were isolated and analyzed by a semi‐native PAGE and Western blotting. To examine heat‐modifiability, samples were either incubated at room temperate (RT) or at 95°C for 10 min. The inner membrane protein SecG was used as loading control.
When comparing the effect of VUF15259 on the biogenesis of Hbp and OMPs, respectively, it is important to note that Hbp expression is induced from an expression plasmid in the presence of the compound. In contrast, OMPs are constitutively expressed and may therefore seem less affected due to a pool of fully assembled OMP present at the time of compound addition. To better compare effects on Hbp and OMP biogenesis, we expressed the β‐barrel OMP PhoE from the pEH3 plasmid and analyzed its membrane insertion and folding in the presence and absence of VUF15259. Cell envelopes were isolated and the expression, folding and assembly of PhoE was analyzed using a heat‐modifiability assay, which is based on the observation that correctly folded β‐barrels retain their native conformation in the presence of SDS unless they are completely denatured upon heating (Noinaj et al., 2015). This effect can be monitored using semi‐native SDS‐PAGE, in which the heat‐denatured β‐barrel runs at a position that is different from that of its non‐heated native form. For PhoE, the native conformation is a trimer (Jansen et al., 2000). Importantly, expression of phoE is not regulated by σE and its membrane insertion is dependent on BamA (Gogol et al., 2011). As shown in Fig. 7F, levels of PhoE in the cell envelope were decreased when cells were incubated with VUF15259. With and without compound PhoE remained heat‐modifiable, suggesting that VUF15259 impairs membrane targeting or insertion of PhoE, rather than folding and assembly into its native form. Collectively, our results indicate that VUF15259 interferes with the targeting or insertion of β‐barrel‐type OMPs.
VUF15259 does not affect in vitro folding of OmpA
To explore the effect of VUF15259 on the Bam‐complex in more detail, we performed an in vitro reconstitution of assembly of the β‐barrel OmpA into proteoliposomes that contain the purified Bam‐complex using a procedure described before by Van der Does et al. (2003) and Roman‐Hernandez et al. (2014). In brief, all five bam genes were expressed in a single E. coli strain and the Bam‐complex was purified from a detergent‐solubilized enriched outer membrane fraction using a C‐terminal His6‐tag on BamE. Blue native PAGE and SDS‐PAGE analysis showed that the purified Bam‐complex was fully assembled, containing all five Bam proteins and forming a stable complex under the native conditions used (Fig. S2A,B). The purified Bam‐complex was incorporated into liposomes using a detergent dilution strategy (Hagan et al., 2010; Roman‐Hernandez et al., 2014). OmpA folds efficiently into the liposomes only in the presence of the Bam‐complex, indicated by the appearance of the 30 kDa band representing folded OmpA (Fig. S2C). Pre‐incubating the proteoliposomes with 100 µM VUF15259 did neither affect the insertion nor the folding of OmpA indicating that under these conditions, VUF15259 is unable to interfere with this Bam‐mediated process.
VUF15259 affects the integrity of the outer membrane and induces vesicle formation
The effect of VUF15259 on OMP biogenesis prompted us to investigate its impact on the barrier function of the outer membrane. We probed the permeability of the outer membrane by monitoring the uptake of N‐phenyl‐1‐naphthylamine (NPN). NPN is a fluorescent dye that only fluoresces in the hydrophobic membrane environment. For this to occur, it needs to penetrate the outer membrane. E. coli cells were either incubated in the presence of different concentrations of VUF15259 or only treated with DMSO. Subsequently, NPN was added and fluorescence was measured. Polymyxin B (PMB) was used as a positive control because it binds to lipopolysaccharide and destabilizes the outer membrane, causing increased membrane permeability. Addition of VUF15259 resulted in an up to ~ 4‐fold increase in fluorescence intensity, compared to a ~ 6‐fold increase when cells were treated with 8 µg ml–1 PMB (Fig. 8A).
Figure 8.
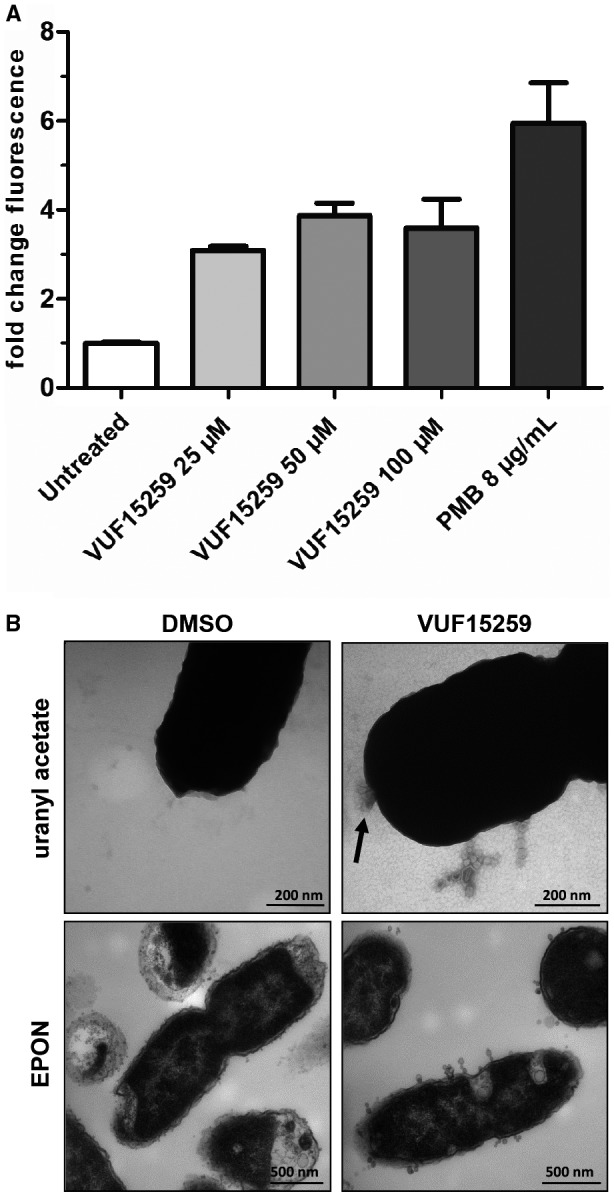
VUF15259 compromises outer membrane integrity and increases vesicle release. A. E. coli TOP10F’ cells were grown in a 96‐well plate and treated with compound VUF15259 and uptake of NPN was measured after 3 h. As a positive control, PMB was added and immediately afterwards uptake of NPN was determined. The fold difference in fluorescence is shown compared to control (DMSO treated) cells with the error bars representing the standard deviation of triplicate samples. B. E. coli TOP10F’ cells were grown to steady‐state and transferred to a 96‐well plate for the treatment with 100 µM VUF15259 or DMSO as control. After 3 h of growth, cells were collected, washed and analyzed by TEM using EPON sectioning and negative staining (uranyl acetate). Scale bars are indicated in the figure. The arrow indicates a potential membrane disruption.
To examine the effect of VUF15259 on cell envelope morphology in more detail, VUF15259‐ treated E. coli cells were analyzed using transmission electron microscopy, with either EPON embedding and sectioning or negative staining of intact bacteria with uranyl acetate. Compared to untreated cells, VUF15259‐treated cells showed round protrusions at the cell surface. These putative outer membrane vesicles (OMVs) mostly remained attached to the cell envelope forming chain‐like structures (Fig. 8B). The cell envelope also seemed to be damaged at specific sites, as indicated by an arrow. Combined, the data show that VUF15259 alters the hydrophobic barrier function of the outer membrane and increases vesicle release.
Discussion
We present a robust fluorescence‐based HTS assay for the screening of inhibitors of virulence factor secretion via the T5SS or AT pathway. Of note, the inhibitors may block specific and generic features and machineries that are required for the T5SS. The stress reporter assay is based on transcriptomics analysis that shows a strong σE cell envelope stress response when a translocation intermediate of the model AT Hbp is expressed that is stuck in the outer membrane and, as a consequence, partly exposed to the periplasm (Jong et al., 2007). In addition, the Cpx and Psp cell envelope stress responses were triggered. While the σE and Cpx responses partly overlap and are known to report on periplasmic protein accumulation, the Psp response is linked to defects in the inner membrane and the energy state of the cell (Darwin, 2005; Ruiz and Silhavy, 2005). At present, the cues for this latter response are unclear. It may be related to an imbalance in periplasmic chaperones needed for clearance of for instance YidC, a peripheral subunit of the Sec‐translocon that contacts Hbp prior to release in the periplasm (Jong et al., 2010). Functional depletion of YidC is known to trigger the Psp response (Wang et al., 2010).
Screening of a fragment library resulted in the small molecule VUF15259 that was verified to induce cell envelope stress by impairing secretion of the ATs Hbp and Ag‐43. In concept, the VUF15259 compound could impair Hbp secretion by impeding different steps in the AT secretion pathway, as shown in Fig. 1. First, the compound might directly inhibit Hbp folding at the surface of the cell, a process known to be required for efficient translocation (Soprova et al., 2010). However, this mode of action would be inconsistent with the observed induction of σE stress in the absence of Hbp expression, the generic effect on outer membrane integrity and the effect on OMP assembly. Second, the compound could interfere with targeting of the AT to the outer membrane, for instance by acting on the periplasmic chaperones SurA, DegP and Skp, which are also involved in OMP biogenesis (Rollauer et al., 2015). DegP and Skp appear unlikely targets because they are dispensable for Hbp biogenesis and secretion (Sauri et al., 2009). In addition, VUF15259 affected bacterial growth in a surA knockout strain, indicating that SurA is also not a direct target of VUF15259. Third, the compound could interfere with Bam‐mediated insertion and translocation of ATs (Sauri et al., 2009). We think that our data are most consistent with this last scenario.
First, VUF15259 showed an increased bactericidal effect in E. coli strains in which β‐barrel OMP assembly was already compromised due to deletions of surA, degP or bamB. Particularly, a deletion of the protease DegP resulted in severe additive growth defects, most likely a result of toxic accumulation of misfolded OMPs in the periplasm. Second, VUF15259 impaired the levels of various β‐barrel OMPs (OmpA, OmpC, OmpF and PhoE) that are known to require the Bam‐complex for insertion (Selkrig et al., 2014). This effect can be partly explained by the σE stress response itself, which inhibits synthesis of OMPs through the σE‐regulated snRNAs MicA and RybB (Gogol et al., 2011). However, synthesis of PhoE is not regulated by these snRNAs. In addition, when the σE stress response was directly induced to the same level by over‐expressing σE, OMP levels were only slightly decreased. In contrast, in VUF15259‐treated E. coli cells, OMP levels were severely affected, suggesting a more direct effect of VUF15259 on OMP assembly. Third, VUF15259 decreased the level of BamA, the β‐barrel subunit of the Bam‐complex. Consistently, a lower level of the fully assembled Bam‐complex was detected. This cannot be explained by the induced σE as it rather leads to an upregulation of Bam expression (Bury‐Moné et al., 2009). In fact, we indirectly confirmed this effect in our transcriptomics analysis of the consequences of periplasmic Hbp accumulation (Table 1). Fourth, VUF15259 was shown to alter membrane permeability, which has also been reported for E. coli strains with defects in the Bam‐complex (Mahoney et al., 2016; Storek et al., 2018). Hence, VUF15259 may directly or indirectly affect the insertion or stability of BamA. However, even though BamA is essential, we only observed a modest effect on growth of compound VUF15259 in a wild‐type E. coli strain. It should be noted that under laboratory conditions, bacteria only need very low levels of BamA and only prolonged depletion times show growth defects in a conditional bamA strain (Li et al., 2009). This could also explain why VUF15259 does not show strong bactericidal effects. VUF15259 did also not impair in vitro folding of the β‐barrel OmpA into Bam‐complex containing proteoliposomes. This result should be interpreted with caution though since the orientation of the functional Bam‐complex is inverted in proteoliposomes as compared to intact bacteria. If the compound binds to surface exposed parts of BamA in whole bacteria, it might not reach this target in the lumen of proteoliposomes. Also, the use of pre‐assembled Bam‐complex in the proteoliposomes will obscure potential effects of VUF15259 on assembly of the Bam‐complex. Moreover, Bam‐mediated insertion of OmpA does not require SurA, implying that this step cannot be interrogated by the assay performed (Kahne, 2013).
Interference with the essential and relatively accessible Bam‐complex is currently being explored as an attractive antibiotic target. Two studies showed that small peptides can interfere with Bam functioning by competing for binding of BamA to BamD. A peptide consisting of the C‐terminal 96 amino acids of BamA that binds to BamD compromised in vitro folding of BamA in Bam‐complex containing proteoliposomes (Hagan et al., 2015). Similarly, a peptide that contains the putative BamA‐binding region of Pseudomonas aeruginosa BamD increased membrane permeability (Mori et al., 2012). Recently, the monoclonal antibody MAB1 was reported to bind to an extracellular loop of BamA affecting growth of E. coli with truncated LPS (Storek et al., 2018). In addition, MAB1 showed upregulation of σE stress, impaired β‐barrel folding activity and increased membrane permeability. Interestingly, the effects described are reminiscent of those induced by VUF15259 supporting a potentially interference with Bam‐complex activity.
In addition to the effects on Hbp secretion and β‐barrel OMP biogenesis, the VUF15259 compound induces the formation of membrane vesicles that stay attached to the cell envelope and seem to form short chains. Judged from the morphology and size, these are presumably outer membrane vesicles (OMVs). The increased vesiculation could be induced by σE stress, for instance through a reduction in the levels of the OMPs and the lipoprotein Pal that are known to be downregulated through snRNA (McBroom et al., 2006). Disruption of the Tol‐Pal system that bridges the inner and outer membranes is known to increase OMV release (Walburger et al., 2002; Gogol et al., 2011). Also, strains that lack OmpA produce more OMVs (Deatherage et al., 2009). Yet, it remains unclear why chains of OMVs are formed, rather than OMVs that are individually released. This phenotype has not been reported before to our knowledge.
In conclusion, we report on the development of a simple and robust fluorescence‐based reporter assay that is suitable for HTS screening of Hbp secretion inhibitors. Fragment screening yielded a compound (VUF15259) that inhibits secretion of ATs and interferes with β‐barrel OMP insertion into the outer membrane. Our data are consistent with but do not prove that the compound affects Bam‐complex functioning. Future studies will focus on identifying the target of VUF15259. Although the effects of VUF15259 are appealing, its potential for use in a therapeutic context is currently limited as the compound shows low affinity. Importantly, the validation of the assay presented here incites its use to screen larger compound libraries with drug‐like compounds. This will probably lead to the identification of different classes of compounds. The assay may lead to specific inhibitors of AT secretion, which can function as anti‐virulence drugs or more classical bactericidal compounds that interfere with the biogenesis of β‐barrel proteins in general.
Experimental procedures
Strains and media
The E. coli strains that were used in this study are listed in Table S1. E. coli bacteria were grown either in Luria Broth medium (LB) supplemented with 0.2% glucose or in minimal M9 medium with 0.2% glucose and 0.2% casamino acids (Difco, Detroit, MI, USA), as indicated (Miller, 1992). For selective growth and transformations, chloramphenicol (30 μg ml–1), streptomycin (100 μg ml–1), kanamycin (50 μg ml–1) and tetracycline (6.25 μg ml–1) were added to the medium, where appropriate.
Reagents, enzymes and sera
Rapid DNA Dephosphorylation and Ligation Kit was from Roche Applied Science. Roche or New England Biolabs (NEB) provided restriction enzymes. Phusion High Fidelity DNA polymerase was obtained from NEB. GeneJET Plasmid Miniprep Kit was from Thermo Scientific. The pre‐stained Precision Plus SDS‐PAGE protein marker was obtained from Biorad. QIAquick Gel Extraction Kit, QIAquick RNA Purification kit and QIAquick PCR Purification Kit were purchased from Qiagen. Sigma–Aldrich provided all other reagents, primers and chemicals. Experiments in 96‐well plates were performed in µClear Chimney black clear‐bottom plates TC sterile from Greiner. Plates were sealed with non‐sterile clear multi‐well plate sealers from Greiner. Antisera against BamA and PspA were provided by J. Tommassen (Utrecht University, the Netherlands). Antisera against PhoE and DegP were from H. de Cock (Utrecht University, the Netherlands) and J. Beckwith (Harvard Medical School, USA) respectively. Antisera against the native Bam‐complex, recognizing all Bam subunits was from T. den Blaauwen (University of Amsterdam, the Netherlands). Antisera against LepB, SecG, Hbp passenger domain and Hbp β‐domain were from our own collection. HRP‐conjugated affinity purified anti‐rabbit IgG from Rockland was used as the secondary antibody.
RNA extraction and RNA sequencing
Wild‐type Hbp and Hbp110C/348C were expressed from the pEH3 plasmid under lac promoter control in E. coli strain MC4100. Wild‐type Hbp will be referred to as Hbp in the remainder of this work. Bacteria were grown overnight in M9 medium at 30°C in triplicate. The overnight cultures were used to inoculate 20 ml M9 medium in 100 ml culture flasks at 30°C to an OD660 of 0.3. Subsequently, protein expression was induced with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) for 30 min and total RNA was extracted using the Qiagen RNA purification kit according to the manufacturer's instructions. The RNA integrity (RNA Integrity Score ≥ 8) and quantity was determined on the Agilent 2100 Bioanalyzer (Agilent; Palo Alto, CA, USA). To deplete ribosomal RNA, Invitrogen's RiboMinusTM Prokaryotic kit was used according to manufacturer's instructions. Briefly, 2 µg of total RNA samples was hybridized with prokaryotic rRNA sequence‐specific 5′‐biotin labeled oligonucleotide probes to selectively deplete large rRNA molecules from total RNA. Then, these rRNA‐hybridized, biotinylated probes were removed from the sample with streptavidin‐coated magnetic beads. The resulting RNA samples were concentrated using the RiboMinusTM concentrate module according to the manufacturer's protocol. The final RiboMinusTM RNA samples were subjected to thermal mRNA fragmentation using Elute, Prime and fragment Mix from the Illumina TruSeqTM RNA sample preparation kit v2 (Low‐Throughput protocol). The fragmented mRNA samples were subjected to cDNA synthesis using the Illumina TruSeqTM RNA sample preparation kit (Low‐Throughput protocol) according to manufacturer's protocol. Briefly, cDNA was synthesized from enriched and fragmented RNA using SuperScript III Reverse Transcriptase (Invitrogen) and SRA RT primer (Illumina). The cDNA was further converted into double‐stranded DNA using the reagents supplied in the kit, and the resulting dsDNA was used for library preparation. To this end, cDNA fragments were end‐repaired and phosphorylated, followed by adenylation of 3′ends and adapter ligation. A number of 12 cycles of PCR amplification cycles were then performed, and the library was finally purified with AMPure beads (Beckman Coulter) as per the manufacturer's instructions. A small aliquot (1 µl) was analyzed on the Invitrogen Qubit and Agilent Bioanalyzer. The bar‐coded cDNA libraries were pooled together in equal concentrations in one pool before sequencing on Illumina HiSeq2000 using the TruSeq SR Cluster Generation Kit v3 and TruSeq SBS Kit v3. Data were processed with the Illumina Pipeline Software v1.82.
Sequence reads of the different conditions were mapped against the reference sequence of E. coli MC4100 as paired end data using SMALT (parameter:‐y 0.5; ‐i 1000 ‐r 0). With SAMtools, the output was transformed to a bam file (Li et al., 2009). We obtained the read counts for each transcript and condition with bedtools (parameter ‐D) (Anders and Huber, 2010). The read counts were used in DESeq to first generate a distance matrix. Next, we performed a differential expression analysis, following the description of the DESeq paper (Anders and Huber, 2010).
Plasmid construction
Plasmids used in this study are listed in Table S2 and primers in Table S3. To construct the pUA66 plasmid derivatives carrying the stress promoters PrpoE and PgroES fused to mNG, the promoter sequences were amplified by PCR using genomic DNA of the E. coli strain MG1655 as template. The primers were flanked by XhoI and BamHI restriction sites. The resulting PCR fragments were cloned into the XhoI/BamHI sites of pUA66 (already containing gfpmut2), creating pUA66‐RpoE and pUA66‐GroES in which gfpmut2 was under control of the indicated stress promoters. In order to construct fusions of the rpoE and groES promoter sequences to mNG, the gene encoding mNG was amplified by PCR using pUC57‐mNG as template (kindly provided by Tanneke den Blaauwen, University of Amsterdam, the Netherlands). The resulting PCR product carrying flanking XbaI sites was cloned into pUA66‐RpoE and pUA66‐GroES using the XbaI/XbaI ligation site. This resulted in replacement of gfpmut2 by mNG, yielding plasmids pUA66‐RpoE‐NeonGreen and pUA66‐GroES‐NeonGreen. Note that an optimized version of mNG was constructed in which C/G nucleotides were substituted to A/T nucleotides in the 5′ UTR of mNG to enhance expression. To construct the pEH3 plasmid carrying the rpoE gene under lac promoter control, the gene was amplified by PCR using genomic DNA of E. coli strain MG1655 as a template. The primers contained BamHI and KpnI restriction sites and the resulting PCR fragment was cloned into the BamHI/KpnI site of the pEH3 plasmid. The pEH3 plasmid carrying ag‐43 under lac promoter control was constructed similar to pEH3‐RpoE, but using the restriction sites XbaI and EcoRI. The bam genes were PCR amplified from genomic E. coli MC4100 DNA and subcloned into pCDF‐Duet and pET‐Duet. The genes bamA and bamB were cloned into multiple cloning site 2 (MCS2), using the NdeI/BglII site, and MCS1, using the NcoI/HindIII site, of pCDF, respectively, creating the pCDF‐BamAB plasmid. In the pET vector, bamC was cloned into MCS1 using the NcoI/HindIII site. In the same plasmid, bamD and bamE‐His6 (polycistronic) were cloned in MCS2 using the NdeI/BglII and BglII/XhoI sites, respectively, creating the pET‐BamCDE plasmid. To make plasmids that express ompA (22‐346) and His6‐tagged surA (21‐408) without their signal peptides, the genes were amplified from genomic E. coli MC4100 DNA and subcloned into pET22b using the NdeI and XhoI sites, creating pET22b‐OmpA and pET22b‐SurA‐His6. The pET22b plasmid already contained a His6‐tag sequence after the XhoI site which allowed incorporation of a His6‐tag in surA at the C‐terminus. The sequence of all plasmids was confirmed by DNA sequencing (Macrogen Europe).
Growth and stress measurements in 96‐well plates
Growth and fluorescence experiments were performed in 96‐well plates as follows. Bacteria were grown in M9 medium to mid‐log phase in regular culture flasks at 37°C. Then, the culture was diluted to an OD660 of 0.1 and protein expression was induced with 80 μM IPTG. After 5 min of incubation, 50 μL culture aliquots were transferred to a 96‐well plate and compound or DMSO was added in 50 μL M9 medium. Compounds were dissolved in DMSO and 0.5% DMSO was used as final concentration in the bacterial cultures in the wells, unless stated otherwise. After sealing the plate, growth was continued at 37°C in Thermostar (BMG Labtech) shakers at 600 RPM. Fluorescence (excitation 485 nm, emission 535 nm) and OD660 were measured after 3 h of incubation using the Clariostar plate reader (BMG Labtech). Fluorescence intensities were corrected for growth.
For compound screening, the following adjustments were implemented. Library compounds and the positive control VUF15259 were pre‐plated into 96‐well plates at a concentration of 400 µM in 50 µl M9 medium containing 2% DMSO. E. coli TOP10F’ bacteria harboring pEH3‐Hbp were grown to steady‐state at an OD660 of 0.1 in a culture flask. Expression of Hbp was induced with 80 µM IPTG and after 5 min of incubation 50 µl of bacterial culture was transferred to the 96‐well plates using robotics and growth was continued as described above. A positive and negative control was included in each row of the 96‐well plates, containing 1% DMSO as final concentration in the well. The positive control was set to reflect 100% stress induction. Compounds that induced ≥ 50% stress or more were selected as hits. Hit compounds were resynthesized to verify the structure, purity and activity. Compounds that affected growth resulted in negative fluorescent values compared to the negative control. As this screen aims to find secretion inhibitors, these growth inhibiting compounds were not further analyzed.
Antigen‐43 aggregation assay
E. coli TOP10F’ cells, carrying the pEH3‐Ag‐43 or empty pEH3 plasmid as control, were grown in a 96‐well plate with 10 wells per condition. Ag‐43 protein expression was subsequently induced with 40 µM IPTG for 3 h at 37°C in the Thermostar shaker at 600 RPM. To analyze cell aggregation, the cultures in the wells of each condition were combined, adjusted to an OD660 of 0.8 and left to stand at 4°C in a 1 ml cuvette. Cell aggregation was monitored by measuring the OD660 of the upper part of the cuvette of each condition at 30 min intervals.
Cell envelope protein analysis
E. coli TOP10F’ cells, harboring the pEH3‐PhoE plasmid, were grown in LB medium at 37°C to mid‐log phase and diluted to an OD660 of 0.05 in 5 ml LB medium in a round bottom tube. Expression of PhoE from the pEH3 vector was induced with 40 µM IPTG. Subsequently, cells were exposed to 100 µM compound VUF15259 or DMSO as a control and incubated at 37°C with shaking for 2.5 h. Next, cells were centrifuged at 5,000 ×g for 10 min and taken up in ice‐cold resuspension buffer (10 mM Tris‐HCl, 3 mM EDTA, pH 8.0). EDTA‐free Protease Inhibitor Cocktail (cOmpleteTM, Roche) was added and the cells were lysed by tip sonication on ice. Unbroken cells were pelleted by centrifugation at 5,000 ×g at 4°C for 10 min. The supernatant was used to isolate the cell envelopes by ultra‐centrifugation at 200,000 ×g at 4°C for 60 min. The collected cell envelopes were resuspended in solubilization buffer, containing 50 mM Tris‐HCl, 150 mM NaCl, pH 8.0 (TBS) with 1% (w/v) n‐Dodecyl‐β‐D‐Maltoside (Anatrace), followed by overnight incubation at 4°C. The solubilized cell envelopes were analyzed by 11% semi‐native PAGE (Noinaj et al., 2015) or 11% SDS‐PAGE, Coomassie staining and Western blotting. Quantification of protein bands was performed in ImageJ. Blue native PAGE analysis was performed according to the manufacturer's instructions (Thermo Scientific).
Electron microscopy
E. coli TOP10F’ cells were grown to steady‐state exponential phase. The bacteria were transferred to a 96‐well plate with a start OD660 of 0.025 in 100 µL. Next, compound VUF15259 was added to a concentration of 50, 100 and 200 µM. As controls, non‐treated cells and cells incubated with DMSO were used. For each condition, 6 wells were used. After sealing the plate, the bacteria were grown in the Synergy H1 plate reader (Biotek) at 37°C with 2 mm linear shaking for 3 h. Subsequently, the cells of each condition were collected and pelleted at 5,000 ×g for 10 min. Cells were washed in phosphate‐buffered saline (PBS) and finally resuspended in 1 ml 4% paraformaldehyde and 0.5% glutaraldehyde (EM grade) for 2 h at room temperature and post‐fixed with 1% OsO4. Subsequently, the samples were dehydrated in an alcohol series and embedded in Epon (LX‐112 resin Ladd research, Williston, VT, USA). Ultrathin (80 nm) epon sections were collected on Formvar‐coated grids and counterstained with uranyl acetate and lead citrate. In addition, intact aldehyde fixed cells were negatively stained with uranyl acetate and collected on Formvar‐coated grids. Grids were examined using a FEI Tecnai‐12 G2 Spirit Biotwin electron microscope (Fei, Eindhoven, the Netherlands).
NPN uptake assay
Outer membrane permeability was determined by measuring the uptake of NPN. NPN was dissolved in acetone to a concentration of 500 µM. E. coli TOP10F’ cells were grown in 96‐well plates and incubated with twofold increasing concentrations of VUF15259 for 3 h followed by the addition of 20 μM NPN. Directly after adding NPN, fluorescence (excitation 350 nm, emission 420 nm) was measured every min for 10 min in the Synergy H1 plate reader. In a separate well, after 3 h of growth, 10 µg ml–1 PMB was added as a positive control and fluorescence was measured directly after exposing the cells to PMB.
Synthesis of compounds
THF, DCM and diethylether were dried using a PureSolv Micro Multi Unit solvent purification system from Inert. 2‐Bromo‐1‐(3,4‐dichlorophenyl)ethan‐1‐one was from Fluorochem. 2‐Bromo‐1‐(3,4‐difluorophenyl)ethan‐1‐one was from Apollo. 2‐Bromo‐1‐(3,4‐dimethylphenyl)ethan‐1‐one, 2‐bromo‐1‐(4‐methylphenyl)ethan‐1‐one and 2‐bromo‐1‐(4‐fluorophenyl)ethan‐1‐one were from Combi Blocks. 1‐([1,1′‐Biphenyl]‐4‐yl)‐2‐bromoethan‐1‐one and 2‐bromo‐1‐(naphthalen‐2‐yl)ethan‐1‐one were from Acros. VUF16749 was obtained from our in‐house library. All other solvents and chemicals were acquired from Sigma–Aldrich and were used as received. ChemBioDraw Ultra 16 was used to generate systematic names for all molecules. All reactions were performed under a nitrogen atmosphere. TLC analyzes were carried out with alumina silica plates (Merck F254) using staining and/or UV visualization. Column purifications were performed manually using Silicycle Ultra Pure silica gel or automatically using Biotage equipment. The NMR spectra (1H, 13C and 2D) were recorded on a Bruker 300 (300 MHz), Bruker 400 (400 MHz) or a Bruker 500 (500 MHz) spectrometer. Chemical shifts are reported in ppm (δ) and the residual solvent was used as internal standard. A Bruker microTOF mass spectrometer using ESI in positive ion mode was used to record HRMS spectra. Elemental Analyzes were carried out by Mikroanalytisches Labor Pascher (Germany). A Shimadzu LC‐20AD liquid chromatograph pump system linked to a Shimadzu SPD‐M20A diode array detector with MS detection using a Shimadzu LC‐MS‐2010EV mass spectrometer was used to perform LC‐MS analyzes. An Xbridge (C18) 5 µm column (50 mm, 4.6 mm) was used. The solvents that were used were the following: solvent B (acetonitrile with 0.1% formic acid) and solvent A (water with 0.1% formic acid), flow rate of 1.0 ml min–1, start 5% B, linear gradient to 90% B in 4.5 min, then 1.5 min at 90% B, then linear gradient to 5% B in 0.5 min, then 1.5 min at 5% B; total run time of 8 min. All compounds have a purity of ≥ 95% (unless specified otherwise), calculated as the percentage peak area of the analyzed compound by UV detection at 230 nm. A schematic depiction of the synthesis route for VUF15259 (Fig. S3) and spectral data for VUF15259 (Figs S4‐S10) can be found in the supplementary information.
Tert‐butyl (1‐(2‐(3,4‐dichlorophenyl)‐2‐oxoethyl)piperidin‐4‐yl)carbamate (4, VUF15343). 2‐Bromo‐1‐(3,4‐dichlorophenyl)ethan‐1‐one 2 (1.30 g, 4.85 mmol) and 4‐(Boc‐amino)piperidine 3 (2.275 g, 11.36 mmol) were dissolved in DCM (70 ml). Et3N (1.35 ml, 9.70 mmol) was added and the mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo. Column chromatography (EtOAc/heptane 0/100 to 40/60) afforded the product as a yellow solid (1.55 g, 82%).1H NMR (CDCl3, 300 MHz) 8.09 (d, 1H, J = 1.9 Hz), 7.82 (dd, 1H, J = 8.3, 1.9 Hz), 7.55 (d, 1H, J = 8.3 Hz), 4.53‐4.44 (br, 1H), 3.87 (s, 2H), 3.64‐3.46 (m, 1H), 3.08‐2.95 (m, 2H), 2.58‐2.40 (m, 2H), 2.04‐1.94 (m, 2H), 1.77‐1.50 (m, 2H), 1.44 (s, 9H). LC‐MS: tR = 3.5 min, UV purity (251 nm): > 98%, MS [M+1]+ 387.1. Rac‐tert‐butyl (1‐(2‐(3,4‐dichlorophenyl)‐2‐hydroxyethyl)piperidin‐4‐yl)carbamate (5, VUF15345). Ketone 4 (1.55 g, 4.00 mmol) was dissolved in DCM (110 ml). NaBH4 (554 mg, 14.65 mmol) was added and the mixture was stirred for 2 h at room temperature. MeOH (5 ml) was added dropwise (caution: gas evolution). DCM (50 ml) was added and the mixture was washed with water (50 ml). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. This afforded the product as an off‐white solid (1.40 g, 90%). 1H NMR (CDCl3, 300 MHz) 7.48 (d, 1H, J = 2.0 Hz), 7.40 (d, 1H, J = 8.2 Hz), 7.18 (dd, 1H, J = 8.2, 2.0 Hz), 4.75 (app br d, 1H, J = 10.6 Hz), 4.54‐4.42 (m, 1H), 3.61‐3.44 (m, 1H), 3.20‐3.08 (m, 1H), 2.94‐2.79 (m, 1H), 2.62‐2.38 (m, 3H), 2.33‐2.19 (m, 1H), 2.09‐1.92 (m, 3H), 1.67‐1.49 (m, 2H), 1.44 (s, 9H). LC‐MS: tR = 3.5 min, UV purity (210 nm, low epsilon): > 95%, MS [M + 1]+ 389.1. Rac‐2‐(4‐aminopiperidin‐1‐yl)‐1‐(3,4‐dichlorophenyl)ethan‐1‐ol hydrochloride hydrate (1, VUF15259). Boc‐protected amine 5 (1.40 g, 3.60 mmol) was stirred at room temperature in 2.0 M HCl in dioxane (72 ml). A white precipitate formed. After 1 h, the mixture was evaporated in vacuo to give an off‐white solid. The precipitate was dissolved in hot EtOH (35 ml). Cooling, filtering and washing delivered the product (0.64 g, 48%) as a white solid. 1H NMR: analysis shows multiple double sets of peaks, which may be attributed to slowly interconverting conformers. Extensive 2D NMR analysis shows connectivity that supports this theory. Where a signal identity can be reasonably attributed to overlapping peaks resulting from this phenomenon the peak is designated as a ‘m op’ (=‘multiplet as a result of overlapping peaks’). Recording in DMSO‐d6 at elevated temperatures mostly leads to coalescence of peaks (Fig. S11). 1H NMR (DMSO‐d6, 500 MHz) 10.52 (br, 1H), 8.63‐8.47 (m op, 3H), 7.73‐7.65 (m op, 2H), 7.48‐7.39 (m op, 1H), 6.55‐6.46 (m op, 1H), 5.28‐5.17 (m op, 1H), 3.75‐3.54 (m op, 2H), 3.52‐3.03 (m, 5H + water), 2.22‐1.92 (m, 4H). 13C NMR analysis shows multiple sets of peaks (one with large signals and one with small signals), which may be attributed to slowly interconverting conformers. Only the major peaks are listed. 13C NMR (DMSO‐d6, 125 MHz) 143.5, 131.5, 131.1, 130.6, 128.5, 126.8, 66.3, 62.2, 51.7, 50.3, 45.6, 27.4, 27.3. Elem Anal calc. for C13H18Cl2N2O.1.8HCl.1.0H2O: C 41.88%, H 5.89%, N 7.51%, Cl 36.13%.; found: C 42.01%, H 5.75%, N 7.54%, Cl 36.1%. LC‐MS: tR = 2.1 min, UV purity (230 nm, low epsilon): > 98% excl. solvent front, MS purity (TIC): > 95%. HR‐MS calc. for C13H19Cl2N2O+: 289.0869, found: 289.0864.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MS, JL, WSJ and PvU contributed to the conception or design of the study. MS, AA, SMdM, SK, GJS, BvdB, SW, SW, NNvdW, and MW were involved in the acquisition, analysis or interpretation of the data. MS, and JL contributed to the writing of the manuscript.
Supporting information
Acknowledgements
We thank Hans Custers for synthetic efforts, and Iwan de Esch, Wilbert Bitter, Peter van Ulsen, Alexander Speer, Tanneke den Blaauwen, Corinne M. Ten Hagen‐Jongman, Henry F. Vischer and Frank J. Bruggeman for valuable input in the project and critical reading of the manuscript.
References
- Allen, R.C. , Popat, R. , Diggle, S.P. and Brown, S.P. (2014) Targeting virulence: can we make evolution‐proof drugs? Nature Reviews. Microbiology, 12(4), 300–308. [DOI] [PubMed] [Google Scholar]
- Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biology, 11(106), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsène, F. , Tomoyasu, T. and Bukau, B. (2000) The heat shock response of Escherichia coli . International Journal of Food Microbiology, 55(13), 3–9. [DOI] [PubMed] [Google Scholar]
- Baron, C. (2010) Antivirulence drugs to target bacterial secretion systems. Current Opinion in Microbiology, 13(1), 100–105. [DOI] [PubMed] [Google Scholar]
- Boucher, H.W. , Talbot, G.H. , Benjamin, D.K. , Bradley, J. , Guidos, R.J. , Jones, R.N. , et al. (2013) 10 x ’20 progress‐development of new drugs active against gram‐negative bacilli: an update. From the infectious diseases society of America. Clinical Infectious Diseases, 56(12), 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bury‐Moné, S. , Nomane, Y. , Reymond, N. , Barbet, R. , Jacquet, E. , Imbeaud, S. , et al. (2009) Global analysis of extracytoplasmic stress signaling in Escherichia coli . PLoS Genetics, 5(9), e1000651 10.1371/journal.pgen.1000651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy, A.E. , Pierson, E. and Hung, D.T. (2007) Targeting virulence: a new paradigm for antimicrobial therapy. Nature Chemical Biology, 3(9), 541–548. [DOI] [PubMed] [Google Scholar]
- Congreve, M. , Carr, R. , Murray, C. and Jhoti, H. (2003) A “Rule of Three” for fragment‐based lead discovery? Drug Discovery Today, 8(19), 876–877. [DOI] [PubMed] [Google Scholar]
- Dartigalongue, C. , Missiakas, D. and Raina, S. (2001) Characterization of the Escherichia coli sigma E Regulon. Journal of Biological Chemistry, 276(24), 20866–20875. [DOI] [PubMed] [Google Scholar]
- Darwin, A.J. (2005) The phage‐shock‐protein response. Molecular Microbiology, 57(3), 621–628. [DOI] [PubMed] [Google Scholar]
- Deatherage, B.L. , Lara, J.C. , Bergsbaken, T. , Rassoulian Barrett, S.L. , Lara, S. and Cookson, B.T. (2009) Biogenesis of bacterial membrane vesicles. Molecular Microbiology, 72(6), 1395–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson, D.A. , Fesik, S.W. , Hubbard, R.E. , Jahnke, W. and Jhoti, H. (2016) Twenty years on: the impact of fragments on drug discovery. Nature Reviews Drug Discovery, 15(9), 605–619. [DOI] [PubMed] [Google Scholar]
- Fan, E. , Chauhan, N. , Gupta Udatha, D.B.R.K. , Leo, J.C. and Linke, D. (2016) Type V secretion systems in bacteria. Microbiology Spectrum. 10.1128/microbiolspec.VMBF-0009-2015 [DOI] [PubMed] [Google Scholar]
- Gogol, E.B. , Rhodius, V.A. , Papenfort, K. , Vogel, J. and Gross, C.A. (2011) Small RNAs endow a transcriptional activator with essential repressor functions for single‐tier control of a global stress regulon. Proceedings of the National Academy of Sciences of the United States of America, 108(31), 12875–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan, C.L. , Kim, S. and Kahne, D. (2010) Reconstitution of outer membrane protein assembly from purified components. Science, 328(5980), 890–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan, C.L. , Wzorek, J.S. and Kahne, D. (2015) Inhibition of the β‐barrel assembly machine by a peptide that binds BamD. Proceedings of the National Academy of Sciences of the United States of America, 112(7), 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, I.A.N.R. and Nataro, J.P. (2001) Virulence functions of autotransporter proteins. Infection and Immunity, 69(3), 1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heras, B.B. , Scanlon, M.J. and Martin, J.L. (2015) Targeting virulence not viability in the search for future antibacterials. British Journal of Clinical Pharmacology, 79(2), 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, C. , Heutink, M. , Tommassen, J. and De Cock, H. (2000) The assembly pathway of outer membrane protein PhoE of Escherichia coli . European Journal of Biochemistry, 267(12), 3792–3800. [DOI] [PubMed] [Google Scholar]
- Jong, W.S.P. , Ten Hagen‐Jongman, C.M. , Den Blaauwen, T. , Jan Slotboom, D. , Tame, J.R.H. , Wickström, D. , et al. (2007) Limited tolerance towards folded elements during secretion of the autotransporter Hbp. Molecular Microbiology, 63(5), 1524–1536. [DOI] [PubMed] [Google Scholar]
- Jong, W.S.P. , ten Hagen‐Jongman, C.M. , Ruijter, E. , Orru, R.V.A. , Genevaux, P. and Luirink, J. (2010) YidC is involved in the biogenesis of the secreted autotransporter hemoglobin protease. The Journal of Biological Chemistry, 285(51), 39682–39690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong, W.S.P. , Vikström, D. , Houben, D. , Berg van Saparoea, H.B. , Gier, J.W. and Luirink, J. (2017) Application of an E. coli signal sequence as a versatile inclusion body tag. Microbial Cell Factories, 16(1). 10.1186/s12934-017-0662-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph‐McCarthy, D. , Campbell, A.J. , Kern, G. and Moustakas, D. (2014) Fragment‐based lead discovery and design. Journal of Chemical Information and Modeling, 54(3), 693–704. [DOI] [PubMed] [Google Scholar]
- Jovanovic, G. , Lloyd, L.J. , Stumpf, M.P.H. , Mayhew, A.J. and Buck, M. (2006) Induction and function of the phage shock protein extracytoplasmic stress response in Escherichia coli . Journal of Biological Chemistry, 281(30), 21147–21161. [DOI] [PubMed] [Google Scholar]
- Kahne, D. (2013) Bam lipoproteins assemble BamA in vitro . Biochemistry, 76, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kloe, G.E. , Kool, J. , Van Elk, R. , Van Muijlwijk‐Koezen, J.E. , Smit, A.B. , Lingeman, H. , et al. (2011) Online parallel fragment screening and rapid hit exploration for nicotinic acetylcholine receptors. MedChemComm, 2(7), 590–595. [Google Scholar]
- Konovalova, A. , Kahne, D.E. and Silhavy, T.J. (2017) Outer membrane biogenesis. Annual Review of Microbiology, 71(1), 539–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney, T.F. , Ricci, D.P. and Silhavy, T.J. (2016) Classifying β‐barrel assembly substrates by manipulating essential bam complex members. Journal of Bacteriology, 198(14), 1948–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBroom, A.J. , Johnson, A.P. , Vemulapalli, S. and Kuehn, M.J. (2006) Outer membrane vesicle production by Escherichia coli is independent of membrane instability. Journal of Bacteriology, 188(15), 5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecsas, J. (2016) Bacterial secretion systems – an overview. Microbiology Spectrum, 4(1), 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.H. (1992) A Short Course in Bacterial Genetics – A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. USA: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Mori, N. , Ishii, Y. , Tateda, K. , Kimura, S. , Kouyama, Y. , Inoko, H. , et al. (2012) A peptide based on homologous sequences of the β‐barrel assembly machinery component bamd potentiates antibiotic susceptibility of Pseudomonas aeruginosa . Journal of Antimicrobial Chemotherapy, 67(9), 2173–2181. [DOI] [PubMed] [Google Scholar]
- Noinaj, N. Kuszak, A.J. and Buchanan, S.K. (2015) Heat modifiability of outer membrane proteins from gram negative bacteria. Methods in Molecular Biology, 1329, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollauer, S.E. , Sooreshjani, M.A. , Noinaj, N. , Buchanan, S.K. and Buchanan, S.K. (2015) Outer membrane protein biogenesis in Gram‐negative bacteria. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 10.1098/rstb.2015.0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman‐Hernandez, G. , Peterson, J.H. and Bernstein, H.D. (2014) Reconstitution of bacterial autotransporter assembly using purified components. ELife. 10.7554/eLife.04234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz, N. and Silhavy, T.J. (2005) Sensing external stress: watchdogs of the Escherichia coli cell envelope. Current Opinion in Microbiology, 8(2), 122–126. [DOI] [PubMed] [Google Scholar]
- Sauri, A. , Soprova, Z. , Wickström, D. , de Gier, J.W. , Van der Schors, R.C. , Smit, A.B. , et al. (2009) The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology, 155(12), 3982–3991. [DOI] [PubMed] [Google Scholar]
- Selkrig, J. , Leyton, D.L. , Webb, C.T. and Lithgow, T. (2014) Assembly of β‐barrel proteins into bacterial outer membranes. Biochimica et Biophysica Acta – Molecular Cell Research, 1843(8), 1542–1550. [DOI] [PubMed] [Google Scholar]
- Shaner, N.C. , Lambert, G.G. , Chammas, A. , Ni, Y. , Cranfill, P.J. , Baird, M.A. , et al. (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum . Nature Methods, 10(5), 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soprova, Z. , Sauri, A. , van Ulsen, P. , Tame, J.R.H. , den Blaauwen, T. , Jong, W.S.P. and Luirink, J. (2010) A conserved aromatic residue in the autochaperone domain of the autotransporter Hbp is critical for initiation of outer membrane translocation. The Journal of Biological Chemistry, 285(49), 38224–38233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storek, K.M. , Auerbach, M.R. , Shi, H. , Garcia, N.K. , Sun, D. , Nickerson, N.N. , et al. (2018) Monoclonal antibody targeting the β‐barrel assembly machine of Escherichia coli is bactericidal. Proceedings of the National Academy of Sciences of the United States of America, 115(14), 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou, L.K. , Dossa, P.D. and Hang, H.C. (2013) Small molecules aimed at type III secretion systems to inhibit bacterial virulence. MedChemComm, 4(1), 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Does, C. , De Keyzer, J. , Van der Laan, M. and Driessen, A.J.M. (2003) Reconstitution of purified bacterial preprotein translocase in liposomes. Methods in Enzymology, 372(2000), 86–98. [DOI] [PubMed] [Google Scholar]
- van Ulsen, P. , Rahman, S.U. , Jong, W.S.P. , Daleke‐Schermerhorn, M.H. and Luirink, J. (2013) Type V secretion: from biogenesis to biotechnology. Biochimica et Biophysica Acta, 1843, 1592–1611. [DOI] [PubMed] [Google Scholar]
- Verheij, M.H.P. , De Graaf, C. , De Kloe, G.E. , Nijmeijer, S. , Vischer, H.F. , Smits, R.A. , et al. (2011) Fragment library screening reveals remarkable similarities between the G protein‐coupled receptor histamine H4and the ion channel serotonin 5‐HT3A. Bioorganic and Medicinal Chemistry Letters, 21(18), 5460–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walburger, A. , Lazdunski, C. and Corda, Y. (2002) The Tol/Pal system function requires an interaction between the C‐terminal domain of TolA and the N‐terminal domain of TolB. Molecular Microbiology, 44(3), 695–708. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Kuhn, A. and Dalbey, R.E. (2010) Global change of gene expression and cell physiology in YidC‐depleted Escherichia coli . Journal of Bacteriology, 192(8), 2193–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Chung, T.D.Y. and Oldenburg, K.R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening, 4(2), 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials