

Abstract
Objective
Some features of subjects with Prader‐Willi syndrome (PWS) resemble those seen in growth hormone deficiency (GHD). Children with PWS are treated with growth hormone (GH), which has substantially changed their phenotype. Currently, young adults with PWS must discontinue GH after attainment of adult height when they do not fulfil the criteria of adult GHD. Limited information is available about the prevalence of GHD in adults with PWS. This study aimed to investigate the GH/insulin‐like growth factor (IGF‐I) axis and the prevalence of GHD in previously GH‐treated young adults with PWS.
Design
Cross‐sectional study in 60 young adults with PWS.
Measurements
Serum IGF‐I and IGFBP‐3 levels, GH peak during combined growth hormone‐releasing hormone (GHRH)‐arginine stimulation test.
Results
Serum IGF‐I was <−2 standard deviation scores (SDS) in 2 (3%) patients, and IGFBP‐3 was within the normal range in all but one patient. Median (IQR) GH peak was 17.8 μg/L (12.2; 29.7) [~53.4 mU/L] and below 9 μg/L in 9 (15%) patients. Not one patient fulfilled the criteria for adult GHD (GH peak < 9 μg/L and IGF‐I < −2 SDS), also when BMI‐dependent criteria were used. A higher BMI and a higher fat mass percentage were significantly associated with a lower GH peak. There was no significant difference in GH peak between patients with a deletion or a maternal uniparental disomy (mUPD).
Conclusions
In a large group of previously GH‐treated young adults with PWS, approximately 1 in 7 exhibited a GH peak <9 μg/L during a GHRH‐arginine test. However, none of the patients fulfilled the consensus criteria for adult GHD.
Keywords: arginine, growth hormone, growth hormone deficiency, growth hormone‐releasing hormone, IGF‐I, Prader‐Willi syndrome
1. INTRODUCTION
Prader‐Willi syndrome (PWS) is a rare disorder resulting from the lack of expression of the PWS region (q11‐q13) on the paternally derived chromosome 15. This is mostly caused by a paternal deletion or maternal uniparental disomy (mUPD) and in some cases by an imprinting centre defect or paternal chromosomal translocation.1, 2 Clinical findings change with age, with infancy being characterized by muscular hypotonia and failure to thrive, while short stature, psychomotor delay, hyperphagia and obesity are more prominent during childhood and adulthood.1, 3
Some features of people with PWS resemble those seen in growth hormone deficiency (GHD), such as short stature and an abnormal body composition with a low lean body mass (LBM) and an increased fat mass (FM). Long‐term continuous growth hormone (GH) treatment in children with PWS improves body composition, linear growth, physical strength, cognition and adaptive functioning, substantially changing the phenotype of children with PWS.4, 5, 6, 7, 8, 9 Currently, young adults with PWS have to stop GH treatment after attainment of adult height, when they do not fulfil the criteria of adult GHD. Studies have shown that GH treatment is beneficial for adults with PWS, with a sustained improvement in FM and LBM when GH is continued after attainment of adult height, and a deterioration of body composition when GH treatment is discontinued.10, 11
Reduced serum insulin‐like growth factor (IGF‐I) levels and a reduced GH response to provocative stimuli were found in a varying percentage of children and adults with PWS.3, 12, 13, 14, 15, 16, 17, 18 Most studies in adults with PWS have investigated GHD in adults who were not treated with GH during childhood and the prevalence of GHD varied dependent on the diagnostic test and the chosen cut‐off points, which were or were not corrected for BMI. This study aimed to assess the GH response to a growth hormone releasing hormone (GHRH)‐arginine stimulation test in a large sample of young adults with PWS who had attained adult height and were previously treated with GH during childhood.
2. METHODS
2.1. Patients
Inclusion criteria for the present study were (i) genetically confirmed diagnosis of PWS by a positive methylation test, (ii) GH treatment during childhood for at least two years and (iii) having attained adult height, defined as a height velocity <0.5 cm per 6 months and epiphyseal closure as demonstrated by a radiograph of the left hand and wrist. Exclusion criteria were (i) medication to reduce weight or fat or (ii) noncooperative behaviour. Sixty subjects were included, who were all on a diet and exercise programme. Written informed consent was obtained from patients and their parents or legal representatives. The study protocol was approved by the Medical Ethics Committee of the Erasmus University Medical Centre, Rotterdam.
2.2. Design
All participants were followed at the Dutch PWS Reference Centre. They were treated with biosynthetic GH (Pfizer Inc) during childhood, administered subcutaneously once daily at bedtime in a dose of 1.0 mg/m2/d (≈0.035 mg/kg/d). The GH dose was regularly adjusted based on calculated body surface area and serum IGF‐I levels. At attainment of adult height, GH treatment was discontinued for at least six weeks prior to performing a standard GH stimulation test with growth hormone‐releasing hormone (GHRH) and arginine.19
2.3. Anthropometry
Standing height was measured using a Harpenden Stadiometer, and weight was measured on a calibrated electric scale (Servo Balance KA‐20‐150S; Servo Berkel Prior). Height, weight and body mass index (BMI) were expressed as standard deviation scores (SDS) using growth analyser 4.0 (available at http://www.growthanalyser.org), adjusting for age (18 years) and sex according to Dutch reference values.20, 21
2.4. Body composition
Body composition was assessed by dual‐energy X‐ray absorptiometry (DXA; Lunar Prodigy; GE Healthcare), within four months of the GHRH‐arginine stimulation test in 41 participants. Total fat mass (FM; kg) and lean body mass (LBM; kg) were assessed. All scans were made on the same machine, with daily quality assurance. The intra‐assay coefficient of variation (CV) for fat tissue was 0.41%‐0.88% and for LBM 1.57%‐4.49%.22 FM was also expressed as percentage of total body weight (FM%). LBM was calculated as fat‐free mass minus bone mineral content. FM% SDS and LBM SDS were calculated according to age‐ and sex‐matched Dutch reference values.23
2.5. GHRH‐arginine test
Growth hormone stimulation tests started at 8.30 am after overnight fasting, with the patients recumbent. After an indwelling catheter had been placed, each participant received GHRH (1 μg/kg as intravenous bolus at 0 minute, with a maximum dose of 100 μg) and arginine (0.5 g/kg during 30 minutes, with a maximum dose of 50 g). Blood samples for GH determination were drawn at 0, 15, 30, 45, 60 and 90 minutes after the intravenous bolus of GHRH. Levels of GH, IGF‐I and IGFBP‐3 were measured using the IDS‐iSYS immunoassay system, which is based on chemoluminescence. The intra‐assay variations were <6.4%, <7.5% and <5.1%, respectively. Levels of IGF‐I and IGFBP‐3 were expressed as SDS, adjusting for age and gender.24, 25
2.6. Statistical analysis
Statistical analyses were performed with spss 24.0 (SPSS Inc). Data were expressed as median (IQR). The GH response to GHRH and arginine was assessed by the evaluation of the highest GH plasma concentration, that is the GH peak. GHD in adults was defined as a GH peak level after GHRH‐arginine test <9 μg/L in combination with a serum IGF‐I SDS level <–2.0 adjusted for age and gender.3, 19 We also applied the BMI‐dependent cut‐off points for the GHRH‐arginine test: a GH peak of <11.5 μg/L if BMI is <25 kg/m2, a GH peak of <8.0 μg/L if BMI is 25‐30 kg/m2, and a GH peak of <4.2 μg/L if BMI >30 kg/m2.26
Pearson's correlation coefficients were used to assess relationships between IGF‐I and GH peak and anthropometric measurements and body composition variables. Student's t tests were used to calculate differences between groups. P values <0.05 were considered statistically significant.
3. RESULTS
3.1. Baseline characteristics
Sixty young adults with PWS (27 males, 33 females) with a median (IQR) age of 17.9 (16.3; 19.6) years were included in the current evaluation of GH response to a GHRH‐arginine stimulation test (Table 1). GH treatment during childhood was started at a median (IQR) age of 6.6 (4.0; 8.8) years. Twenty‐nine young adults had a deletion (48.3%), 25 a maternal uniparental disomy (mUPD; 41.7%) and five a paternal translocation (8.3%). One patient refused further investigation of the genetic subtype.
Table 1.
Clinical characteristics at adult height and during childhood
| Adult height | During childhooda | |
|---|---|---|
| Age (y) | 17.9 (16.3; 19.6) | 6.6 (4.0; 8.8) |
| Male/Female (n) | 27/33 | 27/33 |
| Genetic subtype | ||
| Deletion/mUPD/translocation/unknown | 29/25/5/1 | 29/25/5/1 |
| Height for age (SDS) | −1.0 (−1.7; −0.3) | −2.2 (−3.0; −1.8) |
| BMI | 24.2 (21.1; 27.9) | 17.9 (16.3; 19.4) |
| BMI for age (SDS) | 1.1 (−0.2; 1.9) | −0.6 (−1.1; 0.1) |
| FM% | 40.5 (35.7; 47.5) | 34.2 (28.9; 38.3) |
| FM% SDS | 2.3 (1.8; 2.6) | 2.3 (2.1; 2.6) |
| LBM SDS | −2.3 (−3.1; −1.2) | −2.5 (−2.8; −2.0) |
Data expressed as median (IQR). FM and LBM at adult height were assessed in 41 individuals and in 38 individuals during childhood.
Abbreviations: BMI, body mass index; FM%, fat mass percentage; LBM, lean body mass; mUPD, maternal uniparental disomy; SDS, standard deviation scores.
GH naïve before start of GH treatment.
Twenty‐nine patients were receiving sex steroid replacement therapy (SSRT), and seven had spontaneous oestrogen or testosterone levels within the normal range. The remaining 24 subjects were considered hypogonadal, but were not receiving SSRT at time of evaluation.
3.2. GH response to GHRH‐arginine test
Table 2 and Figure 1 show the results of the GHRH‐arginine tests and serum IGF‐I and IGFBP‐3 levels. Median (IQR) GH peak was 17.8 μg/L (12.2; 29.7) [~53.4 mU/L]. Median (IQR) serum IGF‐I was −0.4 (−1.1; 0.4) SDS and IGFBP‐3 1.6 (1.0; 2.2) SDS.
Table 2.
GH response to GHRH‐arginine stimulation test
| At adult height | During childhooda | |
|---|---|---|
| IGF‐I (nmol/L) | 32.4 (21.3; 39.6) | 7.5 (5.2; 12.7) |
| IGF‐I SDS | −0.4 (−1.1; 0.4) | −1.7 (−2.2; −0.9) |
| IGFBP‐3 (mg/L) | 4.7 (4.0; 5.7) | 1.2 (0.9; 1.5) |
| IGFBP‐3 SDS | 1.6 (1.0; 2.2) | −2.0 (−3.1; −1.4) |
| GH peak (μg/L) | 17.8 (12.2; 29.7) | — |
| Time to GH peak (min) | 45.0 (30.0; 60.0) | — |
Data expressed as median (IQR).
Abbreviations: GH, growth hormone; IGFBP‐3, Insulin‐like growth factor‐binding protein 3; IGF‐I, Insulin‐like growth factor; SDS, standard deviation scores.
GH naïve before start of GH treatment.
Figure 1.
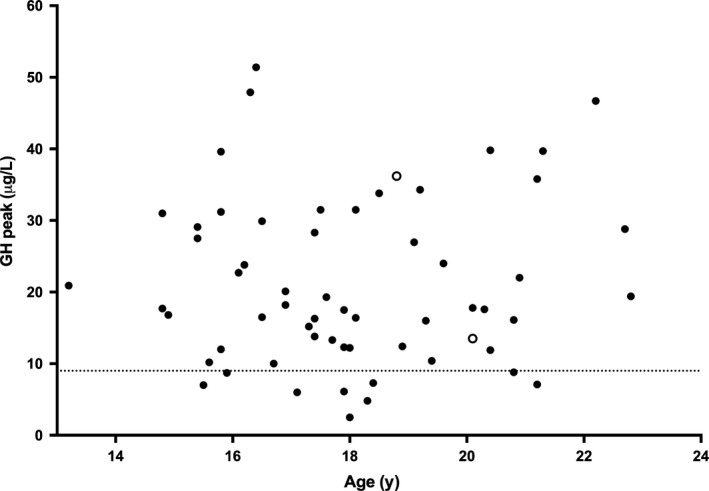
Growth hormone (GH) Peak and IGF‐I SDS in 60 young adults with PWS. GH peak during GHRH‐arginine test according to age in young adults with PWS. The dashed line shows the cut‐off value for GHD (9 μg/L). The open circles represent patients with an IGF‐I SDS <−2 SDS, the black circles >−2 SDS. GHD, growth hormone deficiency; GHRH, growth hormone‐releasing hormone; IGF‐I, Insulin‐like growth factor; SDS, standard deviation scores
Serum IGF‐I was <−2 SDS in two (3%) patients, and IGFBP‐3 was within the normal range in all but one participant. Nine participants had a peak GH level <9 μg/L (15%) during the GHRH‐arginine test. None of these patients also had an IGF‐I level <−2.0 SDS. When BMI‐dependent criteria were used, nine participants had a GH peak below the cut‐off (15%). Again, none of these patients also had an IGF‐I <−2.0 SDS. Thus, not one of our patients fulfilled the criteria of adult GHD.
Growth hormone peak was not significantly different between participants with a deletion and those with a mUPD (P = 0.99) or between males and females (P = 0.41). There was also no significant difference in GH peak between hypogonadal patients who were not receiving SSRT and patients who were receiving SSRT or had spontaneous oestrogen and testosterone levels within the normal range at the time of the GHRH‐arginine stimulation test (P = 0.67).
3.3. Correlation analyses
A higher BMI SDS and a higher FM% SDS were significantly associated with a lower GH peak (r = −0.46, P < 0.01 and r = −0.43, P < 0.01, resp.). The GH peak did neither correlate with age or waist‐hip ratio (both P > 0.13), nor with IGF‐I SDS (r = 0.01, P = 0.93). There was no significant correlation between IGF‐I SDS and BMI SDS or FM% SDS (r = −0.14, P = 0.94 and r = 0.008, P = 0.96, resp.).
3.4. IGF‐I at start of GH treatment during childhood
Clinical characteristics and median serum IGF‐I and IGFBP‐3 levels of the young adults at start of GH treatment during childhood are shown in Tables 1 and 2, respectively. Before starting GH treatment at a median age of 6.6 years, 18 children (30%) had an IGF‐I <−2 SDS. Serum IGF‐I was unknown in eight children (13.3%). Of the 18 children with an IGF‐I <−2 SDS at start of GH, only two had an IGF‐I <−2 SDS after cessation of GH. None of them had a GH peak <9 μg/L during the GHRH‐arginine test. So, none of the young adults with an IGF‐I <−2 at start of GH during childhood fulfilled the criteria of adult GHD after cessation of GH treatment at attainment of adult height, also when BMI‐dependent cut‐off values were used.
Before start of GH treatment, there was no significant difference in height, BMI, FM% and LBM SDS between children with an IGF‐I <−2 SDS and >−2 SDS (all P > 0.34).
4. DISCUSSION
This study investigated the GH/IGF‐I axis and the prevalence of GHD in a large group of 60 young adults with PWS, who were treated with GH for more than 10 years during childhood until attainment of adult height. Against expectations, our data show that none of the patients had adult GHD according to the consensus criteria, also when BMI was taken into account. Results were variable, with some patients having a low serum IGF‐I and normal GH peak, and others a normal serum IGF‐I and low GH peak. In most countries, adults with PWS cannot be treated with GH after attainment of adult height, if they do not fulfil the consensus criteria for adult GHD.
Patients with PWS have several clinical features that resemble those seen in GHD, including short stature and an abnormal body composition with an increased fat mass and decreased muscle mass.1 Both children and adults with PWS respond favourably to GH treatment, with a complete normalization of stature in children, an increase in serum IGF‐I and IGFBP‐3 levels and a significant improvement in body composition, without safety concerns.4, 10, 11 It was generally believed that young adults with PWS would have GHD and could continue GH treatment after attainment of adult height. However, completely unexpected, none of the 60 young adults fulfilled the current consensus criteria for the diagnosis of adult GHD. Only two of our patients (3%) had an IGF‐I level <−2 SDS, and none of them had a GH peak <9 μg/L. Thus, a decreased IGF‐I level was not accompanied by a low GH peak during provocative testing. On the other hand, nine (15%) patients had a low GH peak, but completely normal serum IGF‐I levels.
The reported prevalence of GHD in children and adults with PWS varies. According to a review by Burman et al,12 40%‐100% of children with PWS fulfilled the criteria for GHD, depending on the stimulation test used. Three recent studies investigated the prevalence of GHD in mostly GH‐naïve adult PWS patients, aged 16‐43 years, by performing GHRH‐arginine tests.3, 16, 27 Two of these studies, in 15 and 41 adult patients, also reported a low prevalence of GHD, with only one (8%) and six (15%) of the participants fulfilling the diagnostic criteria for GHD according to BMI‐dependent cut‐off points, respectively.3, 27 In contrast, Grugni et al16 described severe GHD in 16 out of 37 adults with PWS (43.5%). However, mean BMI of their study group was 45.2 kg/m2, which is much higher than the median BMI of 24.2 kg/m2 in our study and the reported median BMI of approximately 27 kg/m2 in the other two studies.3, 27 Even though BMI‐dependent cut‐off points were used, a mean BMI of 45.2 kg/m2 is so far above the upper limit of 30 kg/m2 that there is an increased likelihood of a blunted GH response to a GHRH‐arginine test. Conceivably, the current BMI‐dependent cut‐off points may not be suitable for patients with very severe obesity.28 Also, in contrast to the adults investigated in the aforementioned studies, all young adults participating in our study were treated with GH from a median age of 6.6 years until attainment of adult height.
The GHRH‐arginine test is reported as reliable as the insulin tolerance test (ITT) for (re)testing patients treated with GH during childhood.19, 29 Given the increased risk of severe side effects during an ITT and the higher burden of this test, we chose to perform GHRH‐arginine instead of insulin tolerance tests. There is discussion about the appropriate cut‐off values for GH peak during GHRH‐arginine tests. A cut‐off value of 15.9 μg/L for severe adult GHD has been proposed for retesting late adolescents with childhood‐onset GHD. However, due to the small number of overweight patients, a separate cut‐off for adolescents with a high BMI was not obtained.30 One‐third of our patients had a BMI >25 kg/m2, and the cut‐off values, as proposed by Dreismann et al,26 are therefore not applicable to our patients with PWS. We decided to use the BMI‐adjusted adult cut‐off values instead.
Two studies in young children with PWS described a low GH peak after clonidine or arginine in 68%‐85%.17, 31 One of these studies in 27 children with PWS also performed a combined test (GHRH‐arginine or GHRH‐pyridostigmine test), which showed a much lower prevalence of GHD (15%) than the clonidine and arginine tests.31 As studies in adults with PWS have described a higher prevalence of GHD after combined stimulation tests, the authors of both studies suggested that GHD in PWS might be due to an evolving process, with pituitary GH reserve decreasing with age.17, 31 Although we did not perform GH stimulation tests during childhood before starting GH treatment, our data show that, of the 18 children with an IGF‐I <−2 SDS at start of GH treatment, all but two patients had normal serum IGF‐I levels (>−2 SDS) after cessation of GH at adult height attainment. This finding contradicts the hypothesis that GHD might become more prevalent with age.
Our data are in line with those of most non‐PWS patients with idiopathic childhood‐onset GHD, who have a normal GH response when retested as adults.32, 33, 34 The reasons why are unknown, but may include a variable GH response to stimulation, GHD being a transient condition or GHD being partial, preventing normal growth during childhood, but not causing symptoms in adulthood. We cannot, however, exclude that GH treatment stimulates GH secretory cells in the pituitary and that this GH effect persists, even after GH treatment is discontinued. This hypothesis is supported by a recent RCT, which showed that one year of placebo in previously GH‐treated young adults with PWS did not deteriorate cognitive functioning and might suggest that the neurotropic effects of long‐term GH during childhood last into adulthood.35
During the transition period, the body undergoes physical and psychological changes that conclude the shift from childhood to adulthood. GH seems to play an important role in this transition, as it favourably influences body composition, lipid profile and peak bone density in adolescents with GHD.36 In PWS, studies have shown a deterioration of body composition after cessation of GH treatment, while continuing GH after attainment of adult height maintained the improved FM and LBM obtained during childhood, supporting the presence of clinical GHD.10, 11 However, as most adults with PWS do not fulfil the consensus criteria for adult GHD, they currently cannot be treated with GH after attainment of adult height.
In conclusion, in a large group of previously GH‐treated patients with PWS, GH peak levels during a GHRH‐arginine test were low in 15% of patients, but not one patient fulfilled the criteria for adult GHD. These results are against expectations, as several studies have shown that GH treatment has positive effects on body composition and health profile in adults with PWS.
CONFLICT OF INTEREST
Investigator‐initiated study for which AHK received an independent research grant from Pfizer. The other authors have nothing to disclose.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
ACKNOWLEDGEMENTS
We express our gratitude to all children and parents for their enthusiastic participation in this study and thank Mariëlle van Eekelen for all her work. We thank all collaborating paediatric endocrinologists, paediatricians and other healthcare providers.
Donze SH, Damen L, van Alfen‐van der Velden JAEM, et al. Prevalence of growth hormone (GH) deficiency in previously GH‐treated young adults with Prader‐Willi syndrome. Clin Endocrinol (Oxf). 2019;91:118–123. 10.1111/cen.13988
Funding information
This study was an investigator‐initiated study, supported by an independent research grant from Pfizer. Pfizer was not involved in conception or design of the study, nor in collection, analysis or interpretation of data, writing the manuscript or decision to submit the manuscript for publication.
Data Availability Statement: The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader‐Willi syndrome. Genet Med. 2012;14(1):10‐26. [DOI] [PubMed] [Google Scholar]
- 2. Goldstone AP, Holland AJ, Hauffa BP, Hokken‐Koelega AC, Tauber M. Recommendations for the diagnosis and management of Prader‐Willi syndrome. J Clin Endocrinol Metab. 2008;93(11):4183‐4197. [DOI] [PubMed] [Google Scholar]
- 3. van Nieuwpoort IC, Sinnema M, Castelijns JA, Twisk JW, Curfs LM, Drent ML. The GH/IGF‐I axis and pituitary function and size in adults with Prader‐Willi syndrome. Horm Res Paediatr. 2011;75(6):403‐411. [DOI] [PubMed] [Google Scholar]
- 4. Bakker NE, Kuppens RJ, Siemensma E, et al. Eight years of growth hormone treatment in children with Prader‐Willi syndrome: maintaining the positive effects. J Clin Endocrinol Metab. 2013;98(10):4013‐4022. [DOI] [PubMed] [Google Scholar]
- 5. Festen D, Wevers M, Lindgren AC, et al. Mental and motor development before and during growth hormone treatment in infants and toddlers with Prader‐Willi syndrome. Clin Endocrinol. 2008;68(6):919‐925. [DOI] [PubMed] [Google Scholar]
- 6. Siemensma E, Tummers‐de Lind van Wijngaarden R, Festen D, et al. Beneficial effects of growth hormone treatment on cognition in children with Prader‐Willi syndrome: a randomized controlled trial and longitudinal study. J Clin Endocrinol Metab. 2012;97(7):2307‐2314. [DOI] [PubMed] [Google Scholar]
- 7. Lo ST, Festen D, Tummers‐de Lind van Wijngaarden R, Collin P, Hokken‐Koelega A. Beneficial effects of long‐term growth hormone treatment on adaptive functioning in infants with Prader‐Willi Syndrome. Am J Intellect Dev Disabil. 2015;120(4):315‐327. [DOI] [PubMed] [Google Scholar]
- 8. Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB. Long‐term growth hormone therapy changes the natural history of body composition and motor function in children with Prader‐Willi syndrome. J Clin Endocrinol Metab. 2010;95(3):1131‐1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deal CL, Tony M, Hoybye C, et al. GrowthHormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader‐Willi syndrome. J Clin Endocrinol Metab. 2013;98(6):E1072‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuppens RJ, Bakker NE, Siemensma E, et al. Beneficial effects of GH in young adults with Prader‐Willi syndrome: a 2‐year crossover trial. J Clin Endocrinol Metab. 2016;101(11):4110‐4116. [DOI] [PubMed] [Google Scholar]
- 11. Sode‐Carlsen R, Farholt S, Rabben KF, et al. Growth hormone treatment in adults with Prader‐Willi syndrome: the Scandinavian study. Endocrine. 2012;41(2):191‐199. [DOI] [PubMed] [Google Scholar]
- 12. Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader‐Willi syndrome: a review with special reference to GH. Endocr Rev. 2001;22(6):787‐799. [DOI] [PubMed] [Google Scholar]
- 13. Corrias A, Bellone J, Beccaria L, et al. GH/IGF‐I axis in Prader‐Willi syndrome: evaluation of IGF‐I levels and of the somatotroph responsiveness to various provocative stimuli. Genetic Obesity Study Group of Italian Society of Pediatric Endocrinology and Diabetology. J Endocrinol Invest. 2000;23(2):84‐89. [DOI] [PubMed] [Google Scholar]
- 14. Grugni G, Marzullo P, Ragusa L, et al. Impairment of GH responsiveness to combined GH‐releasing hormone and arginine administration in adult patients with Prader‐Willi syndrome. Clin Endocrinol. 2006;65(4):492‐499. [DOI] [PubMed] [Google Scholar]
- 15. Diene G, Mimoun E, Feigerlova E, et al. Endocrine disorders in children with Prader‐Willi syndrome–data from 142 children of the French database. Horm Res Paediatr. 2010;74(2):121‐128. [DOI] [PubMed] [Google Scholar]
- 16. Grugni G, Giardino D, Crino A, et al. Growth hormone secretion among adult patients with Prader‐Willi syndrome due to different genetic subtypes. J Endocrinol Invest. 2011;34(7):493‐497. [DOI] [PubMed] [Google Scholar]
- 17. Cohen M, Harrington J, Narang I, Hamilton J. Growth hormone secretion decreases with age in paediatric Prader‐Willi syndrome. Clin Endocrinol. 2015;83(2):212‐215. [DOI] [PubMed] [Google Scholar]
- 18. Hoybye C, Frystyk J, Thoren M. The growth hormone‐insulin‐like growth factor axis in adult patients with Prader Willi syndrome. Growth Horm IGF Res. 2003;13(5):269‐274. [DOI] [PubMed] [Google Scholar]
- 19. Aimaretti G, Corneli G, Razzore P, et al. Comparison between insulin‐induced hypoglycemia and growth hormone (GH)‐releasing hormone + arginine as provocative tests for the diagnosis of GH deficiency in adults. J Clin Endocrinol Metab. 1998;83(5):1615‐1618. [DOI] [PubMed] [Google Scholar]
- 20. Fredriks AM, van Buuren S, Burgmeijer R, et al. Continuing positive secular growth change in The Netherlands 1955–1997. Pediatr Res. 2000;47(3):316‐323. [DOI] [PubMed] [Google Scholar]
- 21. Fredriks AM, van Buuren S, Wit JM, Verloove‐Vanhorick SP. Body index measurements in 1996–7 compared with 1980. Arch Dis Child. 2000;82(2):107‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Y, Franks PW, Brookshire T, Antonio TP. The intra‐ and inter‐instrument reliability of DXA based on ex vivo soft tissue measurements. Obes Res. 2004;12(12):1925‐1929. [DOI] [PubMed] [Google Scholar]
- 23. Boot AM, Bouquet J, de Ridder MA, Krenning EP, de Muinck Keizer‐Schrama SM. Determinants of body composition measured by dual‐energy X‐ray absorptiometry in Dutch children and adolescents. Am J Clin Nutr. 1997;66(2):232‐238. [DOI] [PubMed] [Google Scholar]
- 24. Bidlingmaier M, Friedrich N, Emeny RT, et al. Reference intervals for insulin‐like growth factor‐1 (igf‐i) from birth to senescence: results from a multicenter study using a new automated chemiluminescence IGF‐I immunoassay conforming to recent international recommendations. J Clin Endocrinol Metab. 2014;99(5):1712‐1721. [DOI] [PubMed] [Google Scholar]
- 25. Rikken B, van Doorn J, Ringeling A, Van den Brande JL, Massa G, Wit JM. Plasma levels of insulin‐like growth factor (IGF)‐I, IGF‐II and IGF‐binding protein‐3 in the evaluation of childhood growth hormone deficiency. Horm Res. 1998;50(3):166‐176. [DOI] [PubMed] [Google Scholar]
- 26. Corneli G, Di Somma C, Baldelli R, et al. The cut‐off limits of the GH response to GH‐releasing hormone‐arginine test related to body mass index. Eur J Endocrinol. 2005;153(2):257‐264. [DOI] [PubMed] [Google Scholar]
- 27. Sode‐Carlsen R, Farholt S, Rabben KF, et al. Body composition, endocrine and metabolic profiles in adults with Prader‐Willi syndrome. Growth Hormon IGF Res. 2010;20(3):179‐184. [DOI] [PubMed] [Google Scholar]
- 28. Popovic V. Approach to testing growth hormone (GH) secretion in obese subjects. J Clin Endocrinol Metab. 2013;98(5):1789‐1796. [DOI] [PubMed] [Google Scholar]
- 29. Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(6):1587‐1609. [DOI] [PubMed] [Google Scholar]
- 30. Dreismann L, Schweizer R, Blumenstock G, Weber K, Binder G. Evaluation of the GHRH‐arginine retest for young adolescents with childhood‐onset GH deficiency. Growth Horm IGF Res. 2016;27:28‐32. [DOI] [PubMed] [Google Scholar]
- 31. Di Giorgio G, Grugni G, Fintini D, et al. Growth hormone response to standard provocative stimuli and combined tests in very young children with Prader‐Willi syndrome. Horm Res Paediatr. 2014;81(3):189‐195. [DOI] [PubMed] [Google Scholar]
- 32. Molitch ME. Growth hormone treatment in adults with growth hormone deficiency: the transition. J Endocrinol Invest. 2011;34(2):150‐154. [DOI] [PubMed] [Google Scholar]
- 33. Meazza C, Gertosio C, Pagani S, et al. Is retesting in growth hormone deficient children really useful? Minerva Endocrinol. 2017;42(4):325‐330. [DOI] [PubMed] [Google Scholar]
- 34. Gasco V, Corneli G, Beccuti G, et al. Retesting the childhood‐onset GH‐deficient patient. Eur J Endocrinol. 2008;159(Suppl 1):S45‐52. [DOI] [PubMed] [Google Scholar]
- 35. Kuppens RJ, Mahabier EF, Bakker NE, Siemensma EP, Donze SH, Hokken‐Koelega AC. Effect of cessation of GH treatment on cognition during transition phase in Prader‐Willi syndrome: results of a 2‐year crossover GH trial. Orphanet J Rare Dis. 2016;11(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Balercia G, Giovannini L, Paggi F, et al. Growth hormone deficiency in the transition period: body composition and gonad function. J Endocrinol Invest. 2011;34(9):709‐715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.