

Summary
Maize (Zea mays mays) oil is a rich source of polyunsaturated fatty acids (FAs) and energy, making it a valuable resource for human food, animal feed, and bio‐energy. Although this trait has been studied via conventional genome‐wide association study (GWAS), the single nucleotide polymorphism (SNP)‐trait associations generated by GWAS may miss the underlying associations when traits are based on many genes, each with small effects that can be overshadowed by genetic background and environmental variation. Detecting these SNPs statistically is also limited by the levels set for false discovery rate. A complementary pathways analysis that emphasizes the cumulative aspects of SNP‐trait associations, rather than just the significance of single SNPs, was performed to understand the balance of lipid metabolism, conversion, and catabolism in this study. This pathway analysis indicated that acyl‐lipid pathways, including biosynthesis of wax esters, sphingolipids, phospholipids and flavonoids, along with FA and triacylglycerol (TAG) biosynthesis, were important for increasing oil and FA content. The allelic variation found among the genes involved in many degradation pathways, and many biosynthesis pathways leading from FAs and carbon partitioning pathways, was critical for determining final FA content, changing FA ratios and, ultimately, to final oil content. The pathways and pathway networks identified in this study, and especially the acyl‐lipid associated pathways identified beyond what had been found with GWAS alone, provide a real opportunity to precisely and efficiently manipulate high‐oil maize genetic improvement.
Keywords: maize, lipid metabolism, pathway analysis, genome‐wide association study
Significance Statement
This pathway analysis provided insights into the genetic basis of lipid metabolism, beyond what could be detected via GWAS alone, and indicated specific opportunities to precisely and efficiently manipulate high‐oil maize genetic improvement.
Introduction
Oil content and composition are important determinants of maize (Zea mays mays) kernel quality (Watson, 1987). Maize oil is rich in high‐energy lipids in the form of triacylglycerols (TAGs), which include unsaturated fatty acids (FAs, e.g. oleic acid and linoleic acid), making maize oil a valuable resource for human food, animal feed, and bio‐energy. Different ratios of each FA may be more beneficial to different end uses. For instance, palm oil is well known for its high level of saturated FA and has many advantages for the food industry thanks to its high oxidative stability and high melting point, making it a good alternative to trans‐fats. Oils with higher oleic‐acid content are healthier because oleic acid can reduce blood pressure, inflammation, and oxidative damage, and may help prevent cancer. Therefore, understanding the genetic architecture of lipid metabolism in maize enables work on this key target for maize breeding and biotechnology‐assisted improvement. Maize kernel oil concentration and FA composition are highly heritable traits but are quantitative in nature and controlled by multiple genes.
Long‐term artificial selection in high‐oil maize populations has provided unique genetic resources for dissection of the genetic architecture of oil biosynthesis in maize kernels (Dudley and Lambert, 2004; Lambert et al., 2004; Song and Chen, 2004). In 1896, C. G. Hopkins started a selection experiment on the percent oil and protein of maize kernels using the open‐pollinated cultivar ‘Burr's White’ at the University of Illinois (Dudley and Lambert, 2004). He analyzed 163 ears for oil and protein content and selected the 24 ears highest in protein and another 24 for high oil, and the 12 ears lowest in protein and another 12 for low oil. Therefore, he initiated the Illinois High Oil (IHO), Illinois Low Oil (ILO), Illinois High Protein (IHP), and Illinois Low Protein (ILP) strains. After 100 generations of continued artificial selection, the kernel oil concentration of IHO increased from the initial 4.69 to 20.37%; in the ILO, after 85 generations it had decreased from 4.69 to 0.05% (below which germination was compromised; Dudley and Lambert, 2004). It is remarkable that 100 generations of selection had not eliminated the genetic variability in this population, and an upper limit has not been reached in the IHO (Dudley and Lambert, 2004). This indicates that many genes, each with a small effect, have been the targets of continual selection over the course of the experiment, and may continue to be so for some time to come.
Quantitative trait loci (QTL) for oil concentration and FA composition in maize have been identified from multiple studies of high‐oil maize lines (Sughrour and Rocheford, 1994; Alrefai et al., 1995; Laurie et al., 2004; Clark et al., 2006). These studies found several QTL with a range of small to large effects on different FA composition traits, many with additive effects. Using a recombinant inbred line (RIL) population, epistatic effects were found as well, and a gene under a QTL affecting maize seed oil and oleic‐acid content that encodes an acyl‐CoA:diacylglycerol acyltransferase (DGAT1‐2), which catalyzes the final step of oil synthesis, was identified by Yang et al. (2010). A phenylalanine insertion in DGAT1‐2 increased oil and oleic‐acid content (Zheng et al., 2008). Our previous research combining linkage and association analyses allowed fine mapping of a QTL influencing levels of palmitic acid to a 90‐kb region containing a candidate gene, Zea mays fatb (Zmfatb), which encodes an acyl‐acyl carrier protein (ACP) thioesterase. An 11‐bp insertion in the last exon of this gene decreases palmitic acid content and concentration, leading to an optimization of the ratio of saturated to unsaturated FAs, while having no effect on total oil content (Li et al., 2011). Using near isogenic lines (NIL), Zhang et al. (2012) fine mapped a major QTL for embryo to endosperm ratio (EER) and kernel oil concentration, and identified ZmGE2, which encodes a cytochrome p450 protein. A 247‐bp transposable element (TE) insertion in the 3′‐untranslated region was associated with increased EER and kernel oil, and was a selection target during long‐term selection for high EER in a high‐oil population (Zhang et al., 2012).
Fine mapping with recombinant inbred lines (RILs) and map‐based cloning are highly accurate, but the process is time‐consuming and challenging. The genomic regions identified often span very large physical distances containing many repetitive sequences. Association mapping can shorten the research period while simultaneously analyzing greater allele numbers and greatly improving mapping resolution, often to the single gene level (Remington et al., 2001; Yu and Buckler, 2006). Beló et al. (2008) used GWAS to identify loci with major effects on oleic‐acid concentration in maize kernels and, with relatively few (8590) SNPs, identified Zmfad2 as responsible for the differences in the oleic‐acid content. Recently, we conducted a GWAS with 1.03 million SNPs characterized in a panel of 368 maize inbred lines and identified 74 loci significantly associated with kernel oil concentration and FA composition, which we subsequently examined by expression QTL mapping, linkage mapping and coexpression analysis (Li et al., 2013). Most gene characterizations that have come from GWAS have discovered that the association is not actually in the causal gene; identifying the correct gene can be quite an arduous process.
Metabolic pathway analysis combined with GWAS focuses on the cumulative effects of many genes grouped according to their shared biological function. This promising approach can give clues to the genetic basis of a trait by finding biological insights missed when focusing on only one or a few genes that are most significantly associated (Tang et al., 2015). Although originally developed to study differences in gene expression data for medically important diseases (Subramanian et al., 2005), pathway analysis has also been used with association mapping data, first in human disease studies (Wang et al., 2007) and then plants (Tang et al., 2015). In addition, biologically relevant pathways can be used to determine additional candidate genes for further study or to interpret large data sets produced by other high‐throughput approaches, including RNA sequencing, proteomics, and metabolomics. This is being done in maize, in targeted pathways such as carotenoid biosynthesis (Owens et al., 2014); in functionally related networks (Baute et al., 2016; Walley et al., 2016); and in protein interaction networks (Musungu et al., 2015).
Tang et al. (2015) took the pathway‐based approach using the results of an aflatoxin GWAS study in maize to jointly consider all genetic sequences associated with aflatoxin accumulation resistance, regardless of magnitude of allele effects. GWAS alone identified significant associations but none led to plausible resistance mechanisms; 17 high ranking pathways representing mechanisms to prevent fungal growth and production of deleterious aflatoxin were identified via the pathway approach (Tang et al., 2015). Many of these pathways were consistent with known resistance mechanisms, primarily centered on the production of the bioactive plant hormone methyl‐jasmonate, known to orchestrate plant responses to several biotic stresses. The interaction of production pathways of different lipids makes it difficult to find genes that account for all phenotypic variation of one FA alone. Levels of one FA are generally linked to levels of others, as production of one may start with, and therefore decrease, another (Li‐Beisson et al., 2013). To better understand lipid metabolism in maize and identify key points for genetic or genomic manipulation, this study was performed to elucidate the biochemical pathways, metabolic networks, and associated genes that contribute to oil‐related traits by accounting for linkage disequilibrium and association statistics among SNPs from a large‐scale GWAS study.
Results
GWAS and tagSNPs
SNP‐trait associations calculated for oil concentration and 9 FA‐related traits were reported in Li et al. (2013) and Table S1. The GWAS was run with TASSEL on 560 000 SNPs genotyped on each of the 368 inbred maize lines in this study. In total, 139 SNPs associated at P < 1.8 × 10−6 were identified for oil concentration, for which R 2 values ranged between 6.61 × 10−6 to 0.18 (Li et al., 2013). A pathway analysis was run using the cumulative association information from all SNPs in the GWAS, by identifying tagSNPs and linked genes per linkage block and assigning them to common metabolic pathways. In total, 259 206 unique tagSNPs were used to locate 25 404 genes, of which 3106 mapped to the 313 MaizeCyc pathways that had five or more genes. This information, along with the number of tagSNPs and genes mapped to the significant pathways, is shown in Table S2.
Most significant pathways for increasing oil concentration
Three pathways were significantly associated with increased oil concentration at P < 0.01, and an additional 16 at P < 0.05 (Table 1). These include seven pathways directly involved in lipid metabolism (Table 1), and some of these are shown in Figure 1. Linoleate biosynthesis I (PWY‐5995) was the most significant (P = 3.51 × 10−4) pathway identified in this study. Linoleate is a polyunsaturated FA that accounts for more than 50% of the oil content in the maize grains of this panel (Li et al., 2013). Most of the oleoyl‐CoA converted from oleoyl‐ACP is incorporated into lipids, forming phosphatidylglycerols (PGs), diglycerides, and phosphatidylcholines (PCs). Further desaturation of the oleoyl groups to linoleoyl groups in the endoplasmic reticulum (ER) occurs while being incorporated into lipids (Figure 2). This step is catalyzed by the enzyme acyl‐lipid ω‐6 desaturase (EC1.14.19.f, or cytochrome b5), encoded by the FAD2 gene, which was also significantly associated with oil concentration in our previous GWAS results (Li et al., 2013). This gene was the second most important in the calculation of the running enrichment score (RES) for PWY‐5995; all genes in the pathway can be seen in Figure 2, along with their relative contribution to oil concentration (genes are sorted and denoted by the hash marks along the top of the RES graph). The most important gene in the pathway (according to enrichment score), which had long‐chain fatty acid‐CoA ligase activity (MaizeCyc v2.1; Monaco et al., 2013), was not identified in the GWAS by Li et al. (2013).
Table 1.
Pathways associated with increased oil concentration and with enrichment scores that were significant at P < 0.05. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study
| MaizeCyc ID | Pathway name | P | FDR |
|---|---|---|---|
| PWY‐5995 | Linoleate biosynthesis I (plants) | 0.00035 | 0.10553 |
| PWY‐5121 | Superpathway of geranylgeranyldiphosphate biosynthesis II (via MEP) | 0.00273 | 0.40943 |
| PWY‐5143 | Fatty acid activation | 0.00684 | 0.61996 |
| PWY‐5912 | 2′‐Deoxymugineic acid phytosiderophore biosynthesis | 0.01065 | 0.61996 |
| PWY‐5687 | Pyrimidine ribonucleotides interconversion | 0.01489 | 0.61996 |
| NONMEVIPP‐PWY | Methylerythritol phosphate pathway | 0.01522 | 0.61996 |
| PWY‐5123 | Trans, trans‐farnesyl diphosphate biosynthesis | 0.01543 | 0.61996 |
| PWY‐4081 | Glutathione redox reactions I | 0.01879 | 0.61996 |
| PWY0‐163 | Salvage pathways of pyrimidine ribonucleotides | 0.02114 | 0.61996 |
| PWY‐5366 | Palmitoleate biosynthesis II | 0.02588 | 0.61996 |
| PWY‐5142 | Acyl‐ACP thioesterase pathway | 0.02588 | 0.61996 |
| UDPNACETYLGALSYN‐PWY | UDP‐N‐acetyl‐D‐glucosamine biosynthesis II | 0.02665 | 0.61996 |
| TRIGLSYN‐PWY | Triacylglycerol biosynthesis | 0.02689 | 0.61996 |
| PWY2OL‐4 | Linalool biosynthesis | 0.03074 | 0.61996 |
| SO4ASSIM‐PWY | Sulfate reduction I (assimilatory) | 0.03532 | 0.61996 |
| PWY‐5340 | Sulfate activation for sulfonation | 0.03532 | 0.61996 |
| COA‐PWY‐1 | Coenzyme A biosynthesis | 0.04194 | 0.61996 |
| PWY‐5885 | Wax esters biosynthesis II | 0.04270 | 0.61996 |
| PWY‐5278 | Sulfite oxidation III | 0.04559 | 0.61996 |
Figure 1.
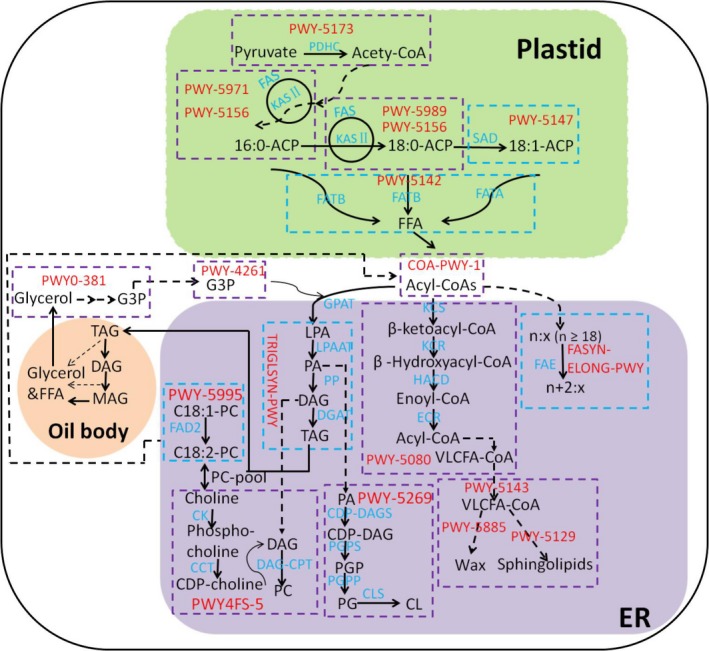
The simplified lipid metabolic pathway in maize. The reactions corresponding to significant pathways detected by pathway analysis are in purple and blue dotted line boxes. The colored boxes also denote pathways that were missed (purple) and detected (blue) by GWAS alone (Li et al., 2013). Plastid and endoplasmic reticulum (ER) are shaded with green and purple, respectively. PDHC, pyruvate dehydrogenase complex; FAS, fatty acid synthase; KAS, ketoacyl‐ACP synthase; SAD, stearoyl‐ACP desaturase; ACP, acyl carrier protein; FAT, acyl‐ACP thioesterase; FFA, free fatty acid; FAE, fatty acid elongase. KCS, ketoacyl‐CoA synthase; KCR, ketoacyl‐CoA reductase; HACD, hydroxyacyl‐CoA dehydrase; ECR, enoyl‐CoA reductase; VLCFA, very long‐chain fatty acid; G3P, glycerol 3‐phosphate; GPAT, glycerol 3‐phosphate acyltransferase; LPA, lysophosphatidic acid; PA, phosphatidic acid; LPAAT, lysophosphatidic acid acyltransferase; DAG, diacylglycerol; PP, PA phosphatase; TAG, triacylglycerol; DGAT, acyl‐CoA: diacylglycerol acyltransferase; MAG, monoacylglycerol; FAD2, oleate desaturase; CK, choline kinase; CCT, choline‐phosphate cytidylyltransferase; DAG‐CPT, diacylglycerol cholinephosphotransferase; PC, phosphatidylcholine; CDP‐DAG, CDP‐diacylglycerol; CDP‐DAGS, CDP‐DAG synthase; PGPS, phosphatidylglycerophosphate synthase; PGP, phosphatidylglycerol phosphate; PGPP, PGP phosphatase; PG, phosphatidylglycerol; CL, cardiolipin; CLS, cardiolipin synthase. Pathway names: PWY‐5173, superpathway of acetyl‐CoA biosynthesis; PWY‐5971, palmitate biosynthesis II (bacteria and plants); PWY‐5156, superpathway of fatty acid biosynthesis II (plant); PWY‐5989, stearate biosynthesis II (plants); PWY‐5147, oleate biosynthesis I (plants); PWY‐5142, acyl‐ACP thioesterase pathway; COA‐PWY‐1, coenzyme A biosynthesis; PWY‐4261, glycerol degradation IV; PWY‐5080, very long‐chain fatty acid biosynthesis; PWY‐5143, fatty acid activation; PWY‐5885, wax esters biosynthesis II; PWY‐5129, sphingolipid biosynthesis (plants); TRIGLSYN‐PWY, triacylglycerol biosynthesis; FASYN‐ELONG‐PWY, fatty acid elongation – saturated; PWY0‐381, glycerol degradation I; PWY‐5995, linoleate biosynthesis I (plants); PWY4FS‐5, superpathway of phosphatidylcholine biosynthesis; PWY‐5269, cardiolipin biosynthesis II.
Figure 2.
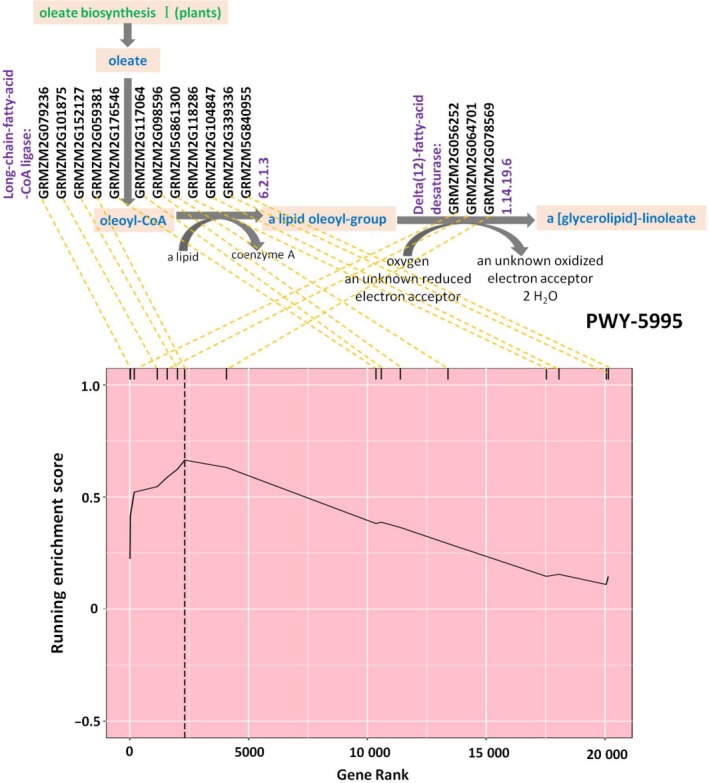
Graphs of pathway reactions and the running enrichment score (RES) calculation for PWY‐5995, linoleate biosynthesis I. Genes were ranked in descending order (left to right) by their effect scores and paired using the yellow dotted line with genes in the pathway reaction, denoted by hash marks at the top of the RES graph. The pathway enrichment score that coincided with the maximum running enrichment score is marked by the black vertical dashed line.
Another pathway with enrichment scores significant at the P < 0.05 is the TAG biosynthesis pathway (TRIGLSYN‐PWY, Figure 1 and Table 1). The major lipid in maize grains reserve is TAG, a glycerol backbone onto which three FAs are sequentially esterified in higher plants. The assembly of TAG occurs in the ER by four consecutive reactions. DGAT, encoding diacylglycerol acyltransferase, catalyzes the final step of TAG synthesis, which was associated with oil concentration in our previous GWAS result (Li et al., 2013). Over‐expression of DGAT increases oil and oleic‐acid concentration by up to 41 and 107% in maize kernels, respectively (Zheng et al., 2008).
Most significant pathways for increasing FA
Pathway analyses were run on associations calculated for nine FA concentration traits, including the concentration levels of palmitic (C16:0), stearic (C18:0), oleic (C18:1), linoleic (C18:2), and linolenic (C18:3) acids, and the ratios of palmitic to stearic (C16:0/C18:0), stearic to oleic (C18:0/C18:1), oleic to linoleic (C18:1/C18:2), and linoleic to linolenic (C18:2/C18:3). Of the 313 MaizeCyc pathways analyzed, 28 that increased the concentration of one or more FA were identified, and had enrichment scores that were significant at P < 0.01 (Table 2). Of these, 10 are directly involved in lipid metabolism, including known biosynthesis pathways for palmitate, stearate, oleate, and linoleate (Li‐Beisson et al., 2010). An extended list for pathways with enrichment scores significant at P < 0.05 is included in Table S3. In addition to the direct biosynthesis pathways, others that are known to be involved in lipid metabolism were identified (Figure 1). Pathway PWY‐5147, oleate biosynthesis I, identified at P < 0.01 in two traits, is central to lipid metabolism and has two sequential reactions that begin with stearoyl‐ACP. Based on the ranks of the effect values, 15 of the 19 genes in this pathway contributed to the enrichment score (Table S4). Two of these genes (GRMZM5G829544 and GRMZM2G079308) had also been identified via GWAS (Li et al., 2013). The key enzyme in this pathway is stearoyl‐ACP 9‐desaturase (SAD), which introduces the first double bond at the delta (9) position of stearoyl‐ACP to produce oleoyl‐ACP. This enzyme is responsible for the conversion of saturated FAs to unsaturated FAs in the synthesis of vegetable oils (Dyer and Mullen, 2001; Han et al., 2017). This example, and all pathway results, illustrates the effectiveness of the pathway analysis for interpreting GWAS results beyond what is seen simply with single point association data (Tables S1 and S4).
Table 2.
Pathways associated with increased FA concentration and with enrichment scores that were significant at P < 0.01. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study
| MaizeCyc ID | Lead traita | Other traitb | Pathway name | Lead trait P‐value | Lead trait FDR | Other trait FDR |
|---|---|---|---|---|---|---|
| COA‐PWY‐1 | C16:0 | Coenzyme A biosynthesis | 0.00399 | 0.15680 | ||
| PWY‐5035 | C16:0 | Gibberellin biosynthesis III (early C‐13 hydroxylation) | 0.00585 | 0.18314 | ||
| PWY‐5147 | C16:0 | C18:1 | Oleate biosynthesis I (plants) | 0.00138 | 0.15680 | 0.22442 |
| PWY‐5995 | C16:0 | Linoleate biosynthesis I (plants) | 0.00378 | 0.15680 | ||
| FASYN‐ELONG‐PWY | C18:0 | Fatty acid elongation – saturated | 0.00364 | 0.15615 | ||
| PWY‐5142 | C18:0 | Acyl‐ACP thioesterase pathway | 0.00449 | 0.15615 | ||
| PWY‐5156 | C18:0 | C16:0 | Superpathway of fatty acid biosynthesis II (plant) | 0.00146 | 0.07626 | 0.15680 |
| PWY‐5366 | C18:0 | Palmitoleate biosynthesis II | 0.00449 | 0.15615 | ||
| PWY‐5367 | C18:0 | C16:0 | Petroselinate biosynthesis | 0.00009 | 0.00926 | 0.15680 |
| PWY‐5971 | C18:0 | C16:0 | Palmitate biosynthesis II (bacteria and plants) | 0.00012 | 0.00926 | 0.15680 |
| PWY‐5973 | C18:0 | C16:0 | Cis‐vaccenate biosynthesis | 0.00009 | 0.00926 | 0.15680 |
| PWY‐5989 | C18:0 | C16:0 | Stearate biosynthesis II (plants) | 0.00012 | 0.00926 | 0.15680 |
| PWY‐6151 | C18:0 | S‐adenosyl‐l‐methionine cycle I | 0.00113 | 0.07093 | ||
| ARO‐PWY | C18:1 | Chorismate biosynthesis I | 0.00112 | 0.17456 | ||
| PWY‐4081 | C18:1 | Glutathione redox reactions I | 0.00904 | 0.41883 | ||
| PWY‐5912 | C18:1 | C16:0 | 2′‐Deoxymugineic acid phytosiderophore biosynthesis | 0.00306 | 0.22442 | 0.18314 |
| PWY‐6457 | C18:1 | Trans‐cinnamoyl‐CoA biosynthesis | 0.00937 | 0.41883 | ||
| PWY‐6628 | C18:1 | Superpathway of phenylalanine biosynthesis | 0.00200 | 0.20860 | ||
| PWY‐6629 | C18:1 | Superpathway of tryptophan biosynthesis | 0.00098 | 0.17456 | ||
| PWY‐5340 | C18:2 | Sulfate activation for sulfonation | 0.00462 | 0.48231 | ||
| PWY‐5687 | C18:2 | Pyrimidine ribonucleotides interconversion | 0.00693 | 0.54214 | ||
| SO4ASSIM‐PWY | C18:2 | Sulfate reduction I (assimilatory) | 0.00462 | 0.48231 | ||
| UDPNACETYLGALSYN‐PWY | C18:2 | UDP‐N‐acetyl‐d‐glucosamine biosynthesis II | 0.00322 | 0.48231 | ||
| PWY1F‐353 | C18:3 | Glycine betaine biosynthesis III (plants) | 0.00469 | 0.36720 | ||
| PWY‐3282 | C18:3 | Ammonia assimilation cycle II | 0.00125 | 0.36720 | ||
| PWY‐381 | C18:3 | Nitrate reduction II (assimilatory) | 0.00426 | 0.36720 | ||
| PWY‐5934 | C18:3 | Fe(III)‐reduction and Fe(II) transport | 0.00594 | 0.37199 | ||
| PYRIDNUCSYN‐PWY‐1 | C18:3 | NAD biosynthesis I (from aspartate, plastidic) | 0.00314 | 0.36720 |
The pathways with the most significant P‐value among FA concentration traits.
Additional FA concentration trait with significant P‐value (P < 0.01).
The identified pathways often influenced more than one of the FAs, which was expected because FAs are frequently modified or converted from one into another (Tables 2 and S3). The correlation between the levels of all FAs is strongly significant (P < 0.001, Li et al., 2013), and the same pathways are frequently identified when running the analysis on different FAs at P < 0.05 (Tables 2 and S3). In addition, single pathways often affect the levels of multiple FAs. For example, the coenzyme A biosynthesis pathway (COA‐PWY‐1) had a high enrichment score for C16:0 and C18:2; FA elongation – saturated (FASYN‐ELONG‐PWY) had a high enrichment score for C16:0 and C18:0; and the acyl‐ACP thioesterase pathway (PWY‐5142) had a high enrichment score for C16:0, C18:0 and C18:1 (Figure 1; Tables 2 and S3).
Coenzyme A (CoA) functions as an acyl carrier and carbonyl‐activating group in numerous central biochemical transformations, including the tricarboxylic acid cycle (TCA, also known as the citric acid cycle (CAC) or Krebs cycle) and FA metabolism (Rubio et al., 2006). The biosynthesis of CoA from (R)‐pantothenate (Figure 3) is an essential universal pathway in both prokaryotes and eukaryotes. In the present analysis, eight genes in the CoA biosynthesis pathway (COA‐PWY‐1) contributed the most to the enrichment score. The steps in the pathway mediated by the products of these genes are indicated in Figure 3 as well. Therefore, it can be seen where the allelic variation in this panel was critical in the biosynthesis of CoA for determining the level of C16:0 and C18:2 in maize kernels. The FA elongation pathway (FASYN‐ELONG‐PWY) includes the reactions that constitute one turn of a cycle that lengthens the chain of an acyl‐ACP molecule by two carbons. The products of this pathway are saturated FAs such as C12:0, C14:0, C16:0 and C18:0 (Li‐Beisson et al., 2010), and the importance of this pathway was confirmed by our results (Table S3). None of the genes in these two pathways was identified in the GWAS results of Li et al. (2013). In this study's analysis, however, the cumulative effect of many genes in the pathway allowed it to be identified as associated with multiple measured traits. The pathway for acyl‐ACP thioesterase (PWY‐5142) is encoded by nine genes, and releases FA from acyl‐ACPs. Two of these nine genes, GRMZM2G079308 (FATB.a) and GRMZM5G829544 (FATB.b), contributed to increasing the RES. These two genes were identified in previous GWAS results (Li et al., 2013; Table S1). The main products of acyl‐ACP thioesterase B (FATB) are C16:0 and to a lesser extent C18:0 in all plants (Salas and Ohlrogge, 2002; Li‐Beisson et al., 2013).
Figure 3.
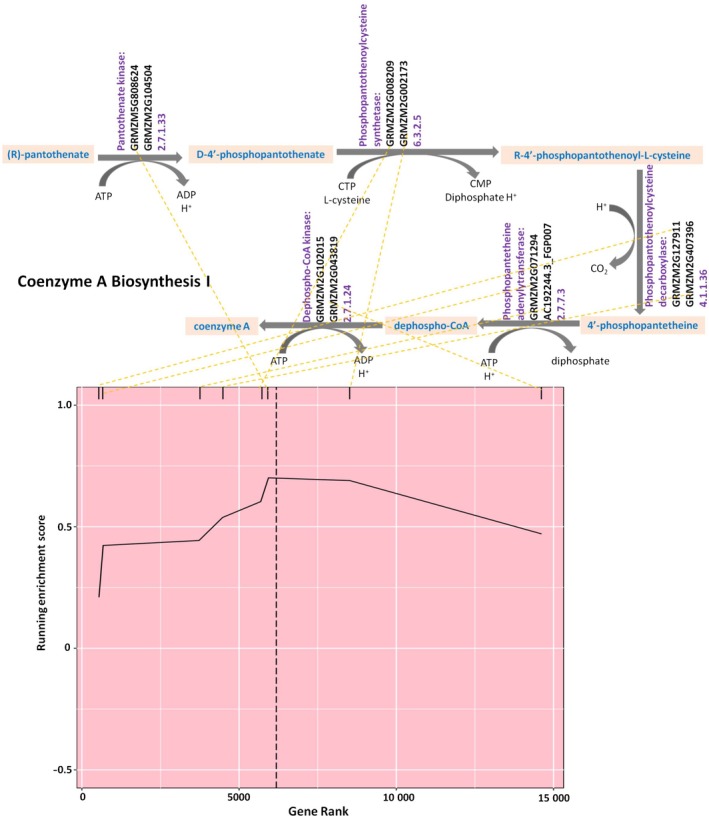
Graphs of pathway reactions and the running enrichment score (RES) calculation for COA‐PWY‐1, coenzyme A biosynthesis. Genes were ranked in descending order (left to right) by their effect scores and paired using the yellow dotted line with genes in the pathway reaction, denoted by hash marks at the top of the RES graph. The pathway enrichment score that coincided with the maximum running enrichment score is marked by the black vertical line.
In addition to FA biosynthesis pathways, acyl‐lipid pathways were also identified in this analysis of increased FA traits. These included wax esters biosynthesis II (PWY‐5885) for C18:1; sphingolipid biosynthesis (PWY‐5129) and phospholipid biosynthesis II (PHOSLIPSYN2‐PWY) for C18:2; flavonoid biosynthesis (PWY1F‐FLAVSYN) for C18:2 and C18:3; and the superpathway of phosphatidylcholine biosynthesis (PWY4FS‐5) for C18:3. The wax esters biosynthesis pathway (PWY‐5885) is carried out by several unrelated acyltransferases, including wax ester synthase (WS)/DGAT. Wax esters biosynthesis uses very long‐chain fatty acids (VLCFA) (elongated from small to medium length FAs), which are esterified to a short chain alcohol (King et al., 2007). Flavonoids are secondary metabolites formed from phenylpropanoid and FA derivatives and have an important function, acting as UV‐B protectors, signal molecules in legume−rhizobium bacteria interactions, and in response to biotic stress (Peters et al., 1986; Ryan et al., 2001). Pathways involved in hormone synthesis, amino acid degradation, and energy production also contributed to the increase in FA levels in this analysis (Figure 1; Tables 2 and S3).
Most significant pathways for decreasing oil concentration and FA
The pathways associated with a decrease in oil and FA concentration can be found in Table S5. Among the 86 significantly (P < 0.05) associated pathways, 11 were related to lipid metabolism. These were mainly lipid degradation pathways, and 23 were related to degradation of primary metabolites in general (Figure 4; Table S5). For instance, glycerol degradation IV (PWY‐4261) was the most significantly enriched in the analysis of C18:0 (P < 0.01) and C18:1 (P < 0.05, Figure 1; Table S5). During seed germination, TAG is broken down to FAs and glycerol, which are converted to sucrose via the glyoxylate cycle to support seedling growth (Li‐Beisson et al., 2013). In the glycerol degradation pathway (PWY‐4261), glycerol is phosphorylated to glycerol‐3‐phosphate by glycerol kinase, and then converted to dihydroxyacetone phosphate by glycerol‐3‐phosphate dehydrogenase, acs7. The product of glycerol degradation, dihydroxyacetone phosphate, can be converted to sugars via gluconeogenesis (Friso et al., 2010). Therefore, there may be less glycerol‐3‐phosphate available for TAG biosynthesis and subsequent production of oil. The acs7 gene, as well as 12 others, together increased the enrichment score of PWY‐4261; these genes were not identified in the GWAS results of Li et al. (2013).
Figure 4.
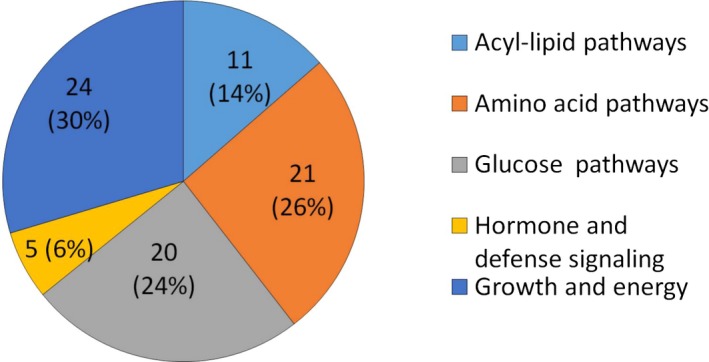
Pathway annotation category for the 86 significantly associated pathways for decreasing oil content and FA.
Another pathway reducing lipids is heptadecane biosynthesis (PWY‐6622), which was found to be enriched in the analysis of C16:0 and C18:0 (P < 0.05, Table S5). The alkanes are produced from FAs in three steps – activation of the FA by an ACP, reduction of the activated FA to an aldehyde, and decarbonylation of the aldehyde by gl1, resulting in an alkane that is one carbon shorter than the original FAs. Therefore, the increase in metabolites derived from FAs would decrease the level of FAs, and this was shown in our results. The list of pathways that decrease oil and FA concentration was dominated by carbon partitioning pathways, such as amino acid and glucose related pathways, which made up over 50% of total pathways with decreasing effect values (Figure 4; Table S5). Examples of significant substrate consumption pathways include pyruvate fermentation to ethanol (PWY‐5486) and gluconeogenesis I (GLUCONEO‐PWY) (Table S5). Gluconeogenesis is the generation of glucose from non‐sugar carbon substrates such as pyruvate and glycerol. The process is essentially the reversal of the glycolysis pathway. In addition to the important role played by carbon partitioning in lipid metabolism, pathways involved in hormone production, cell growth, or energy production decrease lipid levels as well (Figure 4).
Discussion
The candidate genes identified by previous GWAS of oil‐related traits were mostly involved in FA and TAG biosynthesis (Li et al., 2013; Table S1). Following biosynthesis, FAs can then be subjected to elongation, desaturation and eventual export from the plastid, ultimately giving rise to distinct acyl‐lipid species, including phosphatidylinositols (PIs), phosphatidylcholines (PCs), phosphatidylethanolamines (PEs), phosphatidylglycerols (PGs), monogalactosyldiacylglycerols (MGDGs), digalactosyldiacylglycerols (DGDGs), sulfoquinovosyldiacylglycerols (SQDGs) (Li‐Beisson et al., 2013). However, the large number of intermediates and the interaction between lipid pathways render the characterization of lipid metabolism quite challenging (De Abreu et al., 2018). Pathway analysis of oil‐related traits provided a complementary method to detect the mechanisms of lipid metabolism in maize kernels and emphasized multiple aspects of SNP‐trait associations rather than just significance, as in conventional GWAS.
Our pathway analysis results indicate that acyl‐lipid pathways, including wax esters biosynthesis, sphingolipid biosynthesis, phospholipid biosynthesis and flavonoid biosynthesis, were as important as FA and TAG biosynthesis pathways for increasing oil and FA concentration (Tables 1, S1 and S3), and for changing FA ratios (Tables S6). Due to the nature of statistical tests, it is possible that one or more of the pathways identified by this analysis was a false positive result, and not actually involved in oil biosynthesis. However, resampling with replacement and false discovery rate protection were both done to reduce the incidence of false positives. It is therefore likely that detected pathways which are not known lipid biosynthesis pathways do contribute to overall levels of lipids in maize kernels, in a less direct manner. Further experiments with molecular biology tools and/or CRISPR/Cas9 would be required to provide validation of involvement of all pathways in oil biosynthesis.
Lipid metabolism in the maize kernel
By combining pathway analysis results with knowledge about lipid metabolism in the model species Arabidopsis (Li‐Beisson et al., 2013), we can propose further modifications that can be used to achieve optimum levels of FAs and lipids in maize for different end uses. The acyl‐lipid metabolic pathways in maize (Figure 1) and Arabidopsis generate an acetyl‐CoA pool from pyruvate through the action of the plastidial pyruvate dehydrogenase complex (PDHC). FAs are synthesized in the plastid by a Type II fatty acid synthase complex (FAS) adding two carbon units to the extending FA chain in serial reactions. Resulting FAs are typically 16 or 18 carbons long and attached to an ACP. Stearic acid (18:0‐ACP) can be desaturated by SAD. Long‐chain acyl groups are then hydrolyzed by FAT that release FAs, which are ultimately activated to CoA esters and exported to the ER. In the ER, they are assembled into phosphatidic acid (PA) in the acyl‐CoA‐dependent Kennedy pathway with the enzymes glycerol‐3‐phosphate acyltransferases (GPATs) and lysophosphatidic acid acyltransferases (LPAATs), and acyl‐CoA. This PA can then be dephosphorylated de novo by PA phosphatases (PP) to create diacylglycerol (DAG), which is then available for acyltransferase reaction. DGAT transfers acyl‐CoAs to the sn‐3 position of DAG to produce TAG. Phosphatidylcholine (PC) is created via the CDP‐choline pathway using CDP‐choline:diacylglycerol cholinephosphotransferase (DAG‐CPT); and cardiolipin (CL) is created via the action of enzymes phosphatidylglycerol phosphate (PGP) synthase (PGPS), PGP phosphatase (PGPP), and CL synthase (CLS).
The first step in wax and sphingolipid biosynthesis is an elongation cycle converting C16:0 and C18:0 fatty acyl‐CoAs to generate VLCFA wax and sphingolipid precursors between 20 and 34 carbons in length. The first step involves condensation of malonyl‐CoA with an acyl‐CoA catalyzed by a β‐ketoacyl‐CoA synthase (KCS). The resulting β‐ketoacyl‐CoA is reduced by a β–ketoacyl‐CoA reductase (KCR) and the resulting β‐hydroxyacyl‐CoA then undergoes dehydration by a β–hydroxyacyl‐CoA dehydratase (HCD). In the final step, the enoyl‐CoA is reduced to an acyl‐CoA by enoyl‐CoA reductase (ECR). This cycle results in an acyl chain extended by two carbons, and the cycle can be repeated. FA elongation is catalyzed by an ER‐associated, multienzyme complex known as fatty acid elongase (FAE). Therefore, the levels of specific FAs, and total oil, depend on the synthesis of these lipids from precursors, which are frequently other lipids, and the degradation of these lipids (often into other lipids, but also many other compounds). There will be many more pathways associated with the amount of each FA, ratios of FAs, and total oil than simply the biosynthesis pathways that create them, and these pathways will be a key to achieve target levels of any specific FA or lipid.
Practical modification of lipid metabolism guided by pathway analysis
The GWAS by Li et al. (2013) identified 74 loci significantly associated with kernel oil concentration and fatty acid composition. Many of these genes are in pathways that were identified in the current study (Table S1). However, many of those genes are not in pathways in the MaizeCyc database, so they could not be identified by pathway analysis. Conversely, many of the pathways identified in the current study contained no genes identified by GWAS, as no single gene was associated at significance levels exceeding the GWAS threshold, but the cumulative RES of multiple genes in these pathways allowed them to be identified. We therefore see that the two analyses are complementary for identifying more mechanisms and candidate genes for complex traits.
We have shown that identification of biosynthesis pathways for FA derivatives greatly broaden understanding of mechanisms of lipid metabolism and/or catabolism. Because starch, oil and protein are the three main components in maize kernels, the ability to manipulate oil and FA concentrations now allows breeders to create specialty maize for different breeding targets. There is a tradeoff between the lipid and starch components in maize grain (Schwender et al., 2015). Increasing lipid content also increases plastidic fatty acid synthesis and glycolytic flux, while decreasing glycolytic intermediates. In addition, the lipid/starch tradeoff is most likely mediated by allosteric feedback regulation of phosphofructokinase and ADP‐glucose pyrophosphorylase. Our pathway results provide hints as to the mechanism of this lipid/starch tradeoff. Allelic variation exists within the genotypes of this GWAS panel that should allow precise manipulation of FA content within the FA biosynthesis and carbon partitioning pathways. Genes at critical branching points of pathways with the strongest associations to the desired FA can be targeted via allele mining or genome editing to create lines with the exact desired levels of these FA.
TAG is the predominant form of storage lipids in seeds of oil crops. It is clear from this pathway analysis that the synthesis and assembly of TAG in plants involves a metabolic network of FA fluxes through multiple subcellular compartments containing alternative pathways to produce different lipid compositions. Therefore, systems biology offers a powerful and effective tool to assemble multiple genes involved in lipid metabolic engineering. For example, genes for competing pathways can be disrupted to direct metabolic flux toward a desired route. From Figure 1, we see that FAD2 encodes the enzyme that converts 18:1 into 18:2. Genome editing could be used to ‘knock out’ or reduce FAD2 activity for enhancement of oleic acid. This approach has been demonstrated by silencing the soybean FAD2 gene family to create a high oleic‐acid seed trait (Haun et al., 2014). In addition, DGAT1‐2 is responsible for the TAG content (Figure 1). We could reduce FAD2 activity and increase DGAT1‐2 activity for enhancement of oleic acid and TAG simultaneously to design a maize line with high oleic acid and oil content.
While yield improvement has long been the primary goal of conventional maize breeding, other nutritional or industrial traits are becoming more important. This pathway analysis of lipid metabolism provides information to identify which pathways should be tweaked at one or a few key genes each, to create lines with the precise levels or ratios of lipids desired. This information may soon be used, along with the development and application of synthetic biology tools to design and assemble large DNA constructs, and/or gene editing, to bring target lipid synthesis traits together with excellent agronomic and yield performance in the creation of superior high‐oil maize lines.
Experimental procedures
GWAS dataset
The GWAS analysis results of Li et al. (2013), which were used as input data for the pathway analysis are described briefly as follows. A GWAS analysis was run on a panel of 368 diverse inbred lines including 23 high‐oil lines. All entries were genotyped using RNA sequencing. About 560 000 polymorphisms with minor allele frequency (MAF) ≥ 0.05 were selected for analysis. Linkage disequilibrium (LD) was calculated between each pair of SNPs in TASSEL (Bradbury et al., 2007). Overall LD decay was rapid, reaching 500 bp (r 2 = 0.1) in the 368 lines.
All 368 lines were phenotyped for oil concentration and composition in replicated field experiments in four environments in China. Levels of total oil and 10 individual FAs in the kernels were measured according to Yang et al. (2010) and reported in Li et al. (2013), along with ratios of different FAs. The pathway analyses presented here were run on oil concentration, the FA concentration of palmitic (C16:0), stearic (C18:0), oleic (C18:1), linoleic (C18:2), and linolenic (C18:3) acids, and the ratios of palmitic to stearic (C16:0/C18:0), stearic to oleic (C18:0/C18:1), oleic to linoleic (C18:1/C18:2), and linoleic to linolenic (C18:2/C18:3), 10 traits in all.
Pathway analysis
The resulting SNP‐trait association data generated by TASSEL (Bradbury et al., 2007) were implemented in the pathway analysis according to Tang et al. (2015) and included the SNP‐trait association values for significance (P), correlation (R 2 or proportion of the phenotypic variation accounted for), and effect values along with the calculated LD values for D′, and R 2, and P between each marker SNP and its closest neighboring SNPs (50 upstream and 50 downstream). SNPs were then assigned to genes using a tagSNP approach outlined by Tang et al. (2015) in their decision tree. Briefly, if LD analysis showed no linkage between a reference SNP and other SNPs, then the reference SNP was the tagSNP. If LD analysis found linkage between the reference SNP and a block of other SNPs in either upstream and downstream directions, then selection of the tagSNP from the reference SNP or the SNPs in the linkage block was based on the SNP‐trait association effect values (majority sign, positive or negative, and magnitude), P‐values, and distance between the reference SNP and closest SNP in the linkage block. The purpose of the tagSNP was to reduce the dimensionality of the dataset to SNPs with the greatest effects on the trait under analysis. The gene(s) causing the SNP‐trait association was then assumed to be within 1 Kb of the SNP. The reason for using only 1 kb is because many cis‐acting elements that regulate gene expression reside within the gene or in immediate regions upstream and downstream of the gene coding region. If a gene was located, then the SNP‐trait association effect value was transferred to the gene and used for gene‐set enrichment analysis (Tang et al., 2015). Gene sets were defined as the genes according to membership in pathways and superpathways found in the MaizeCyc database (MaizeCyc v2.1; Monaco et al., 2013). Only pathways with five or more genes (313 pathways in total) were considered to reduce bias from a small sample size. Genes were ranked by their effects (negative to positive), and a running sum statistics similar to a weighted Kolmogorov−Smirnov statistics was calculated. The weighting factor for the running sum statistics was the absolute value of the SNP‐trait association effect. The enrichment score for the pathway was the maximum positive deviation of the running sum statistics from zero. Significance of the enrichment score was determined by permutation analysis (1000 random permutations of the effect values). The P‐values for the pathway enrichment scores were then corrected for false discovery rate (FDR) using the QVALUE package in R (Storey and Tibshirani, 2003, R package version 2.2.2. http://github.com/jdstorey/qvalue).
Conflict of Interest
The use of trade name, commercial product or corporation in this publication is for the information and convenience of the reader and does not imply an official recommendation, endorsement or approval by the US Department of Agriculture or the Agricultural Research Service for any product or service to the exclusion of others that may be suitable. USDA is an equal opportunity provider and employer. The authors declare that they have no competing interests.
Supporting information
Table S1. Summary of significantly associated genes from the GWAS study of Li et al. (2013), and those that were also identified by pathway analysis. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/).
Table S2. Summary of significantly associated SNPs from the GWAS study of Li et al. (2013) with R 2 values, and the number of tagSNPs and genes from the present study.
Table S3. Pathways associated with increased FA content and with enrichment scores that were significant at 0.01 < P < 0.05. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study.
Table S4. Number of genes which contributed to the enrichment score in significant pathways at P < 0.01, and which are shown as hatch marks at the top of the running enrichment score graphs. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/).
Table S5. Pathways associated with decreased oil and FA concentration with enrichment scores significant at P < 0.05. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study.
Table S6. Pathways associated with effects on the ratio of different FA traits with enrichment scores significant at P < 0.01. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study. (https://www.maizegdb.org/metabolic_pathways/).
Acknowledgements
The authors thank Drs Natalia de Leon and Tim Setter for reviewing the manuscript and providing excellent suggestions for improvement. We are grateful to the National Key Research and Development Program of China (2016YFD0100503) and the National Natural Science Foundation of China (31771797) to HL, and USDA‐ARS project number 6064‐21000‐013‐00D to MW.
References
- Alrefai, R. , Berke, T.G. and Rocheford, T.R. (1995) Quantitative trait locus analysis of fatty acid concentration in maize. Genome, 38, 894–901. 10.1139/g95-118. [DOI] [PubMed] [Google Scholar]
- Baute, J.D. , Herman, F. , Coppens, J. , De Block, B. , Slabbinck, B. , Dell'Aqcua, M. , Pè, M.E. , Maere, S. , Nelissen, H. and Inzé, D. (2016) Combined large‐scale phenotyping and transcriptomics in maize reveals a robust growth regulatory network. Plant Physiol. 170, 1848–1867. 10.1104/pp.15.01883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beló, A. , Zheng, P.Z. , Luck, S. , Shen, B. , Meyer, D.J. , Li, B. , Tingey, S. and Rafalski, A. (2008) Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genomics, 279, 1–10. 10.1007/s00438-007-0289-y. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z.W. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Clark, D. , Dudley, J.W. , Rocheford, T.R. and LeDeaux, J.R. (2006) Genetic analysis of corn kernel chemical composition in the random mated 10 generation of the cross of generation 70 of IHO × ILO. Crop Sci. 46, 807–819. 10.2135/cropsci2005.06-0153. [DOI] [Google Scholar]
- De Abreu, E. , Lima, F. , Li, K. , Wen, W. , Yan, J. , Nikoloski, Z. , Willmitzer, L. and Brotman, Y. (2018) Unraveling lipid metabolism in maize with time‐resolved multi‐omics data. Plant J. 93, 1102–1115. 10.1104/pp.15.01883. [DOI] [PubMed] [Google Scholar]
- Dudley, J.W. and Lambert, R.J. (2004) 100 generation of selection for oil and protein in corn. Plant Breed. Rev. 24, 79–110. 10.1002/9780470650240.ch5. [DOI] [Google Scholar]
- Dyer, J.M. and Mullen, R.T. (2001) Immunocytological localization of two plant fatty acid desaturases in the endoplasmic reticulum. FEBS Lett. 494, 44–47. 10.1016/s0014-5793(01)02315-8. [DOI] [PubMed] [Google Scholar]
- Friso, G. , Majeran, W. , Huang, M. , Sun, Q. and van Wijk, K.J. (2010) Reconstruction of metabolic pathways, protein expression, and homeostasis machineries across maize bundle sheath and mesophyll chloroplasts: large‐ quantitative proteomics using the first maize genome assembly. Plant Physiol. 152, 1219–1250. 10.1104/pp.109.152694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, Y. , Xu, G. , Du, H. , Hu, J. , Liu, Z. , Li, H. , Li, J. and Yang, X. (2017) Natural variations in stearoyl‐acp desaturase genes affect the conversion of stearic to oleic acid in maize kernel. Theor. Appl. Genet. 130, 151–161. 10.1007/s00122-016-2800-5. [DOI] [PubMed] [Google Scholar]
- Haun, W. , Coffman, A. , Clasen, B.M. et al. (2014) Improved soybean oil quality by targeted mutagenesis of the fatty acid desaturase 2 gene family. Plant Biotechnol. J. 12, 934–940. 10.1111/pbi.12201. [DOI] [PubMed] [Google Scholar]
- King, A. , Nam, J.W. , Han, J. , Hilliard, J. and Jaworski, J.G. (2007) Cuticular wax biosynthesis in petunia petals: cloning and characterization of an alcohol‐acyltransferase that synthesizes wax‐esters. Planta, 226, 381–394. 10.1007/s00425-007-0489-z. [DOI] [PubMed] [Google Scholar]
- Lambert, R.J. , Alexander, D.E. and Mejaya, I.J. (2004) Single kernel selection for increased grain oil in maize synthetics and high‐oil hybrid development. Plant Breed. Rev. 24, 153–175. 10.1002/9780470650240. [DOI] [Google Scholar]
- Laurie, C.C. , Chasalow, S.D. , LeDeaux, J.R. , McCarroll, R. , Bush, D. , Hauge, B. , Lai, J.S. , Clark, D. , Rocheford, T.R. and Dubley, J.W. (2004) The genetic architecture of response to long‐term artificial selection for oil concentration in the maize kernel. Genetics, 168, 2141–2155. 10.1534/genetics.104.029686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Li, H. , Li, Q. et al. (2011) An 11‐bp insertion in Zea mays fatb reduces the palmitic acid content of fatty acids in maize grain. PLoS One, 6, e24699 10.1371/journal.pone.0024699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Peng, Z.Y. , Yang, X.H. et al. (2013) Genome‐wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. 10.1038/ng.2484. [DOI] [PubMed] [Google Scholar]
- Li‐Beisson, Y. , Shorrosh, B. , Beisson, F. et al. (2010) Acyl‐lipid metabolism. Arabidopsis Book, 11, e0161 10.1199/tab.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li‐Beisson, Y. , Shorrosh, B. , Beisson, F. et al. (2013) Acyl‐lipid metabolism. Arabidopsis Book, 11, e0161 10.1199/tab.0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco, M.K. , Sen, T.Z. , Dharmawardhana, P.D. et al. (2013) Maize metabolic network construction and transcriptome analysis. Plant Genome, 6, 1–12. 10.3835/plantgenome2012.09.0025. [DOI] [Google Scholar]
- Musungu, B. , Bhatnagar, D. , Brown, R.L. , Fakhoury, A.M. and Geisler, M. (2015) A predicted protein interactome identifies conserved global networks and disease resistance subnetworks in maize. Front. Genet. 6, 201 10.3389/fgene.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, B.F. , Lipka, A.E. , Magallanes‐Lundback, M. et al. (2014) A foundation for provitamin A biofortification of maize: genome‐wide association and genomic prediction models of carotenoid levels. Genetics, 198, 1699–1716. 10.1534/genetics.114.169979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, N.K. , Frost, J.W. and Long, S.R. (1986) A plant flavone, luteolin, induces expression of Rhizobium meliloti nodulation genes. Science, 233, 977–980. 10.1126/science.3738520. [DOI] [PubMed] [Google Scholar]
- Remington, D.L. , Thornsberry, J.M. , Matsuoka, Y. , Wilson, L.M. , Whitt, S.R. , Doebley, J. , Kresovich, S. , Goodman, M.M. and Buckler, E.S. (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA, 98, 11479–11484. 10.1073/pnas.201394398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio, S. , Larson, T.R. , Gonzalez‐Guzman, M. , Alejandro, S. , Graham, I.A. , Serrano, R. and Rodriguez, P.L. (2006) An Arabidopsis mutant impaired in coenzyme A biosynthesis is sugar dependent for seedling establishment. Plant Physiol. 140, 830–843. 10.1104/pp.105.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, K.G. , Swinny, E.E. , Winefield, C. and Markham, K.R. (2001) Flavonoids and UV photoprotection in Arabidopsis mutants. Z. Naturforsch. C. 56, 745–754. 10.1515/znc-2001-9-1013. [DOI] [PubMed] [Google Scholar]
- Salas, J.J. and Ohlrogge, J.B. (2002) Characterization of substrate specificity of plant FatA and FatB acyl‐ACP thioesterases. Arch. Biochem. Biophys. 403, 25–34. 10.1016/s0003-9861(02)00017-6. [DOI] [PubMed] [Google Scholar]
- Schwender, J. , Hebbelmann, I. , Heinzel, N. et al. (2015) Quantitative multilevel analysis of central metabolism in developing oilseeds of oilseed rape during in vitro culture. Plant Physiol. 168, 828–848. 10.1104/pp.15.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, T.M. and Chen, S.J. (2004) Long term selection for oil concentration in five maize populations. Maydica, 49, 9–14. https://journals-crea.4science.it/index.php/maydica/issue/archive. [Google Scholar]
- Storey, J.D. and Tibshirani, R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U S A. 100, 9440‐9445. 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V.K. et al. (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sughrour, J.R. and Rocheford, T.R. (1994) Restriction fragment length polymorphism differences among Illinois long‐term selection oil trains. Theor. Appl. Genet. 87, 916–924. 10.1007/bf00225785. [DOI] [PubMed] [Google Scholar]
- Tang, J.D. , Perkins, A. , Williams, W.P. and Warburton, M.L. (2015) Using genome‐wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics, 16, 673 10.1186/s12864-015-1874-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley, J.W. , Sartor, R.C. , Shen, Z. et al. (2016) Integration of omic networks in a developmental atlas of maize. Science, 353, 814–818. 10.1126/science.aag1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Bucan, M. (2007) Pathway‐based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 81, 1278–1283. 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, S.A. (1987) Structure and composition In Corn: Chemistry and Technology (Watson S.A. and Ramstad P.E., eds.). St. Paul, MN: American Association of Cereal Chemists, Inc., pp. 53–82. https://my.aaccnet.org/ItemDetail?iProductCode=27330 [Google Scholar]
- Yang, X.H. , Guo, Y.Q. , Yan, J.B. , Zhang, J. , Song, T.M. , Rocheford, T. and Li, J.S. (2010) Major and minor QTL and epistasis contribute to fatty acid composition and oil content in high‐oil maize. Theor. Appl. Genet. 120, 665–678. 10.1007/s00122-009-1184-1. [DOI] [PubMed] [Google Scholar]
- Yu, J.M. and Buckler, E.S. (2006) Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 17, 155–160. 10.1016/j.copbio.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Zhang, P. , Allen, W.B. , Nagasawa, N. et al. (2012) A transposable element insertion within ZmGE2 gene is associated with increase in embryo to endosperm ratio in maize. Theor. Appl. Genet. 125, 1463–1471. 10.1007/s00122-012-1926-3. [DOI] [PubMed] [Google Scholar]
- Zheng, P. , Allen, W.B. , Roesler, K. et al. (2008) A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat. Genet. 40, 367–372. 10.1038/ng.85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of significantly associated genes from the GWAS study of Li et al. (2013), and those that were also identified by pathway analysis. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/).
Table S2. Summary of significantly associated SNPs from the GWAS study of Li et al. (2013) with R 2 values, and the number of tagSNPs and genes from the present study.
Table S3. Pathways associated with increased FA content and with enrichment scores that were significant at 0.01 < P < 0.05. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study.
Table S4. Number of genes which contributed to the enrichment score in significant pathways at P < 0.01, and which are shown as hatch marks at the top of the running enrichment score graphs. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/).
Table S5. Pathways associated with decreased oil and FA concentration with enrichment scores significant at P < 0.05. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study.
Table S6. Pathways associated with effects on the ratio of different FA traits with enrichment scores significant at P < 0.01. Pathway identifier (ID) and name are drawn from the MaizeCyc database (https://www.maizegdb.org/metabolic_pathways/), and P and FDR were calculated in this study. (https://www.maizegdb.org/metabolic_pathways/).