

Abstract
Essentials.
Blood coagulation is driven by vitamin K (VK)‐dependent proteases.
We have identified and characterized ‘sirtilin’ as an additional VK‐dependent protease.
Sirtilins emerged early in the evolution of the coagulation system of vertebrates.
Ubiquitous occurrence might indicate an important functional role of sirtilins.
Summary
Background
Vitamin K (VK)‐dependent proteases are major players in blood coagulation, including both the initiation and the regulation of the cascade. Five different members of this protease family have been described, comprising the following coagulation factors: factor VII, FIX, FX, protein C (PC), and prothrombin (FII). FVII, FIX, FX and PC share a typical domain architecture, with an N‐terminal γ‐carboxyglutamate (Gla) domain, two epidermal growth factor‐like (EGF) domains, and a C‐terminal trypsin‐like serine protease (SP) domain.
Objectives
We have identified uncharacterized proteins in snake genomes showing the typical Gla–EGF1–EGF2–SP domain architecture but relatively low sequence conservation compared to known VK‐dependent proteases. On the basis of sequence analysis, we hypothesized that these proteins are functional members of the VK‐dependent protease family.
Methods/results
Using phylogenetic analyses, we confirmed the so‐called ‘sirtilins’ as an additional VK‐dependent protease class. These proteases were found in several vertebrates, including jawless fish, cartilaginous fish, bony fish, reptiles, birds, and marsupials, but not in other mammals. The recombinant zymogen form of Thamnophis sirtalis sirtilin was produced by in vitro renaturation, and was activated with human activated FXI. The activated form of sirtilin proteolytically cleaved peptide and protein substrates, including prothrombin. Mass spectrometry‐based substrate profiling of sirtilin revealed a narrower sequence specificity than those of FIX and FX.
Conclusions
The ubiquitous occurrence of sirtilins in many vertebrate classes might indicate an important functional role. Understanding the detailed functions of sirtilins might contribute to a deeper understanding of the evolution and function of the vertebrate coagulation system.
Keywords: blood coagulation factors, hemostasis, mass spectrometry, phylogeny, sequence analysis, trypsin‐like serine protease
Introduction
Vitamin K (VK)‐dependent proteases are major players in blood coagulation 1. A subgroup of VK‐dependent proteases comprises the following coagulation factors: factor VII, FIX, FX, and protein C (PC). These are referred to as Gla–EGF1–EGF2–SP proteases throughout this article. They share a common domain structure comprising an N‐terminal γ‐carboxyglutamate (Gla) domain followed by two epidermal growth factor‐like (EGF) domains, i.e. EGF1 and EGF2, and a C‐terminal trypsin‐like serine protease (SP) domain 2. The minimal catalytically active component comprises the EGF2 and SP domains 3. Thrombin, which is another related VK‐dependent protease, has two Kringle domains instead of the EGF domains. VK‐dependent proteases are synthesized as zymogens, and their activation requires proteolytic cleavage of the activation peptide. The newly formed N‐terminus of the SP domain inserts into its activation pocket, and thereby induces and stabilizes its active conformation 4.
The VK‐dependent proteases have highly specific functions in blood coagulation 1, 5, 6. This is shown by unique activation patterns and tightly regulated substrate specificities. Indeed, cofactors are crucial for the interaction between Gla–EGF1–EGF2–SP proteases and their substrates. The complex of activated FVII (FVIIa) and tissue factor (TF) initiates the TF pathway (also called the extrinsic pathway), cleaving and thus activating FX to form activated FX (FXa). FXa, together with its activated cofactor activated FV (FVa), forms the prothrombinase complex, converting prothrombin (FII) to active thrombin (activated FII [FIIa]). The alternative contact activation pathway (also called the intrinsic pathway) leads to the proteolytic activation of FIX to activated FIX (FIXa). FIXa, together with its cofactor activated FVIII, is a highly efficient activator of FX. FX represents a junction point in the blood coagulation system, acting as a target molecule of a positive feedback loop driven by the intrinsic pathway 5. Thereby, the downstream effector thrombin activates FXI, whose active form (activated FXI [FXIa]), in turn, activates FIX and thus boosts FXa and finally thrombin accumulation. Thrombin further proteolytically activates both the procoagulatory cofactors FV and FVIII of FIXa and FXa, respectively, and the inhibitory PC. Activated PC, together with its cofactor protein S, inactivates the upstream cofactors by proteolytic cleavage, and thus triggers a regulatory negative feedback loop. The intrinsic and the extrinsic pathways converge in the activation of fibrinogen by thrombin 7. This results in accumulation and polymerization of fibrin at the site of vessel injury, the final step of hemostasis. Efficient fibrin formation in humans depends largely on the positive feedback loop triggered by FIX and FVIII. Indeed, dysfunction of FIX or FVIII causes the bleeding disorders hemophilia B and hemophilia A, respectively 8.
Interestingly, birds, reptiles, amphibians, fishes and lower vertebrates have only FIX, and lack the other proteases of the intrinsic pathway 9, 10, 11. In the lamprey, for instance, FIX and its upstream activators are completely missing 10, 12. This fact indicates an earlier appearance of the extrinsic pathway in evolution than of the intrinsic pathway. Consequently, the absence of the latter also implies functional differences in the coagulation systems between these organisms. The VK‐dependent proteases probably emerged from a common ancestor via multiple gene duplication events 9, 11, 13. This resulted in early divergence of PC, FVII and FX in vertebrate evolution, which is, for example, reflected by the situation found in the lamprey 10, 12. FIX diverged from FX later in evolution, as exemplified by cartilaginous fishes and bony fishes 10, 12. This process probably also triggered the emergence of the intrinsic coagulation pathway in mammals, including plasma kallikrein, activated FXII, and FXIa, the upstream activator of FIX.
Here, we describe an additional class of VK‐dependent coagulation proteases, which we found to exist in organisms ranging from jawless fish to marsupials. These proteases appeared in evolution even before the divergence of FVII and FX, shedding new light on the emergence and function of the vertebrate blood coagulation system.
Materials and methods
Enzymes
Human FXa (Molecular Innovations, Novi, MI, USA), human FXIa (Coachrom, Enzersdorf, Austria) and human prothrombin (Coachrom) were purchased from commercial vendors.
Sequence analysis
Multiple sequence alignments were calculated with clustal omega 14, and emboss needle 15 was used for pairwise sequence alignments. Phylogeny analysis was conducted with mega (v.7.0.26 16). The phylogenetic tree was calculated with the maximum likelihood method, by use of an LG amino acid substitution model with a discrete gamma distribution (1.0246) and invariable sites (7.24%). Positions in alignments with < 95% site coverage were excluded from phylogenetic analyses. The database accession numbers of used sequences are shown in Table S1.
Protein expression and purification
Expression, renaturation and purification of human FIX (Cys134–Thr461) were performed as described previously 17. FIX zymogen was concentrated up to 3 mg mL−1 and stored at − 20 °C. Procedures described for FIX 17 were adapted for expression, isolation of inclusion bodies (IBs), renaturation and purification of Thamnophis sirtalis sirtilin (Glu129–Ser489). IBs were reduced, solubilized (1 g wet weight in 10 mL of 50 mmol L−1 Tris‐HCl, pH 4.5, 8.0 mol L−1 guanidine‐HCl, 50 mmol L−1 NaCl, and 20 mmol L−1 EDTA), and renatured by rapid dilution in 0.5 mol L−1 arginine, 50 mmol L−1 Tris‐HCl (pH 8.5), 150 mmol L−1 NaCl, 20 mmol L−1 CaCl2, 3 mmol L−1 cysteine and 0.3 mmol L−1 cystine at 16 °C in six pulses (final dilution of 1 : 100). The renaturation reaction mixture was concentrated and dialyzed 1 : 100 against ion exchange chromatography (IEC) buffer A (20 mmol L−1 Tris‐HCl, pH 8.0, 50 mmol L−1 NaCl, and 5 mmol L−1 CaCl2). The sample was applied to a HiPrep 16‐10 Q‐Sepharose FF column (GE Healthcare, Chicago, IL, USA), washed with five column volumes of IEC buffer A and three column volumes of 10% IEC buffer B (20 mmol L−1 Tris/HCl, pH 8.0, 1.0 mol L−1 NaCl, CaCl2, 5 mmol L−1 CaCl2). The sirtilin zymogen was eluted with 20% IEC buffer B. The protein was concentrated up to 3 mg mL−1 and subjected to gel permeation chromatography (GPC) in 10 mmol L−1 HEPES (pH 7.5), 150 mmol L−1 NaCl and 2 mmol L−1 CaCl2 by use of a Superdex 75 column (GE Healthcare). The protein was concentrated to ~ 2.0 mg mL−1 and stored at − 20 °C. Sirtilin was activated in GPC buffer by addition of 5 μg mL−1 chymostatin and 25 μg mL−1 FXIa (~ 200 units per mg) for 24 h at 25 °C. FXIa was separated with two consecutive GPC steps. Sirtilin‐a was concentrated up to 1 mg mL−1 and stored at 4 °C.
Enzyme kinetics
The enzyme activity was measured with the 7‐amido‐4‐methylcoumarin (AMC) P1′‐conjugated peptide substrates Pefafluor‐IXa (methylsulfonyl‐d‐cyclohexylglycyl‐Gly‐Arg‐AMC), IEGR‐AMC (t‐butyloxycarbonyl‐Ile‐Glu‐Gly‐Arg‐AMC) or GPR‐AMC (tosyl‐Gly‐Pro‐Arg‐AMC) in buffer C (50 mmol L−1 HEPES, pH 7.4, 150 mmol L−1 NaCl, and 5 mmol L−1 CaCl2) or buffer D (50 mmol L−1 HEPES, pH 7.4, 150 mmol L−1 NaCl, 5 mmol L−1 CaCl2, and 0.1% PEG8000) at 25 °C in a fluorescence microplate reader (excitation, 380 nm; emission, 460 nm). Initial reaction velocities were determined with linear regression analyses determining molar velocity values on the basis of AMC standard curves.
Michaelis–Menten kinetics were determined in buffer D, which was supplemented with 0.01% Triton X‐100 for sirtilin‐a and FXa. Assays with Pefafluor‐IXa (dissolved in H2O) were conducted with 2.4 nmol L−1, 0.14 nmol L−1 and 320 nmol L−1 sirtilin‐a, FXa, and FIXa, respectively. Assays with IEGR‐AMC (dissolved in dimethylsulfoxide, 1% assay concentration) were conducted with 117 nmol L−1, 1.4 nmol L−1 and 6.4 μmol L−1 sirtilin‐a, FXa, and FIXa, respectively. Progress curves (Fig. S1) were evaluated with graphpad prism (v.5.0; GraphPad Software, San Diego, CA, USA) on the basis of initial reaction velocities. Pefafluor‐IXa turnover by FIXa was fitted with a classical Michaelis–Menten model. Other reactions were fitted with a substrate inhibition model estimating the formation of a ternary non‐productive enzyme–substrate complex: v = V max × [S]/(K M + [S] × {1 + [S]/K I}) 18.
Sirtilin activation assays
Activation of the zymogen forms (27.4 μmol L−1) of FIX and sirtilin was performed in buffer C supplemented with 2 μg mL−1 chymostatin at 25 °C. The activation reactions were initiated by addition of 0.16 μmol L−1 FXIa. Activity of the samples was assayed in buffer C supplemented with 0.01% Triton X‐100. Activity assays were performed after 30 min of incubation at 25 °C in the presence of 30% ethylene glycol 19, with 2 mmol L−1 Pefafluor‐IXa as substrate. Recombinant human furin 20 was used to test the activation of sirtilin and of the control protein FXS195AΔ38AP 21 by furin in buffer C.
Substrate profiling
Proteomic identification of protease cleavage sites (PICS) assays were conducted as described previously 22 with peptide libraries from whole proteome extracts of Escherichia coli BL21 23. The proteome (2.2 mg mL−1) was dissolved in 100 mmol L−1 HEPES/NaOH (pH 7.5), and digested overnight at 37 °C with 22 μg mL−1 endoproteinase GluC or 44 μg mL−1 legumain 24, 25, with the pH adjusted to 5.5 for legumain. Peptide libraries (1 mg mL−1) were digested with sirtilin‐a (0.61 μmol L−1), FXa (0.43 μmol L−1) or FIXa (5.8 μmol L−1) in buffer E (50 mmol L−1 Tris, pH 8.0, 150 mmol L−1 NaCl, and 5 mmol L−1 CaCl2) overnight at 25 °C. Peptides were duplex stable isotope‐labeled by reductive dimethylation, with CH2O for controls and 13CD2O for protease‐treated samples. Dimethylation was performed for 2 h at room temperature, and quenched with 100 mmol L−1 Tris‐HCl (pH 7.5) for 1 h. The protease‐treated samples were mixed with the controls, and purified with C18 StageTips 26.
Peptides were analyzed on an HPLC‐coupled Q‐TOF mass spectrometer (ImpactII; Bruker, ?Billerica, MA, USA) as described previously 22. Peptides were identified and quantified from the mass spectra with maxquant v1.6.0.16 27. IceLogos 28 were calculated for (legumain/GluC) 75/112, 143/148 and 323/332 cleavage windows of sirtilin‐a, FIXa, and FXa, respectively. Detailed settings for mass spectrometry measurements and data evaluation are shown in Data S1.
Activation of prothrombin and FX by sirtilin
Activation of prothrombin (6.9 μmol L−1) by sirtilin‐a (2.3 μmol L−1) and FXa (0.077 μmol L−1 in the presence of 0.01% Triton X‐100) was investigated in buffer E at 30 °C. The activity of thrombin was measured with the substrate GPR‐AMC (100 μmol L−1), with the samples diluted 1 : 5000 in activity assays with buffer C supplemented with 0.01% Triton X‐100. Activation trials with FX (8.5 μmol L−1) were conducted in buffer E in the presence of 2.3 μmol L−1 sirtilin‐a at 30 °C. The samples were diluted 1 : 500 in the activity assays with buffer C supplemented with 0.01% Triton X‐100 and 1 mmol L−1 diisopropylfluorophosphate (DIFP). To inactivate sirtilin‐a, the reactions were incubated for 20 min at 25 °C before addition of Pefafluor‐IXa (0.2 mmol L−1).
Results
‘Sirtilins’ – a novel class of VK‐dependent proteases
As a result of extensive database analyses 29, we have identified so far uncharacterized Gla–EGF1–EGF2–SP‐type sequences (Table S1) from the snakes T. sirtalis (garter snake), Python bivittatus (Burmese python), and Protobothrops mucrosquamatus (Asian pitviper). These predicted proteins showed overall average sequence identities with the human coagulation factors PC, FVII, FIX and FX of 33.3%, 34.8%, 37.9%, and 37.8%, respectively. A multiple sequence alignment with human FIX and human FX verified a typical domain architecture including an N‐terminal Gla‐domain, two EGF‐like domains (EGF1 and EGF2), and a C‐terminal trypsin‐like protease domain (Fig. 1A–C). The EGF2 and catalytic domains of the snake‐derived proteins are interspersed by putative activation peptides of ~ 70 amino acids, which are longer than those of human FIX (35 amino acids) and human FX (52 amino acids; Fig. 1C). The activation peptide is flanked by putative FXIa cleavage motifs, which are highly similar between the new snake proteins (TR↓S184 and TR↓I254, T. sirtalis; absolute sequence numbering is used throughout with initiation methionine +1 30, if not otherwise stated) and human FIX (TR↓A192 and TR↓V227). In FIX, these sites are alternatively cleaved by the FVIIa–TF complex 1. The N‐terminal cleavage site, however, lacks the typical furin cleavage motif RRKR↓ as found for FX (Fig. 1A–C). On the basis of the sequence alignments, additional functional features were identified, including the conserved interdomain disulfide bridge between the EGF2 and catalytic domains, the N‐terminal insertion sequence (↓IIGG) of the catalytic domain, and the catalytic triad (Fig. 1C).
Figure 1.

Domain architecture and sequence alignments of FIX/FX‐like proteins. (A, B) Schematic representation of the domain structure of (A) factor IX and (B) FX showing the Gla (yellow), EGF1 (light green), EGF2 (green) and trypsin‐like protease (S1, cyan) domains. The activation peptides are shown in pink, and their protease cleavage sites are marked with arrowheads. The connecting interdomain disulfide bridge between the EGF2 and S1 domains is marked with orange dots. Typical proteolytic activation sites are indicated; in addition to the intrinsic FXase FIXa–FVIIIa, FVIIa–TF can activate FX (extrinsic FXase). (C) Multiple sequence alignments of human FIX and FX, and of new vitamin K‐dependent proteases from Thamnophis sirtalis, Protobothrops mucrosquamatus, and Python bivittatus (database accession codes are given in Table S1). The absolute sequence numbering of human FIX is given in the alignment. The position of the domains is marked in the coloring scheme of (A). Identical amino acids are underlined in light gray (60%), dark gray (80%), and black (100%). Functionally important residues are colored orange (interdomain disulfide bridge), magenta (protease cleavage sites), and cyan (catalytic triad). The sequence numbers of the catalytic triad are also shown in chymotrypsin numbering (white with asterisk). EGF, epidermal growth factor‐like; FVIIa, activated FVII; FVIIIa, activated FVIII; FXIa, activated FXI; Gla, γ‐carboxyglutamate; TF, tissue factor.
Analyses of molecular markers of serine protease evolution 31, 32, including the AGY codons of Ser195 and Ser214 and of tyrosine at position 225 (chymotrypsin numbering), revealed typical similarities of sitilins to FVII, FIX, FX, and PC (Fig. S2).
Prompted by the relatively low sequence identity of the snake‐derived Gla–EGF1–EGF2–SP proteases with human VK‐dependent proteases, we performed pairwise sequence comparisons between coagulation factor homologs of different organisms (Fig. S3). Interestingly, we found additional homologs of the newly identified snake‐derived proteases in Gallus gallus (birds) and in Phascolarctos cinereus (marsupials), but not in Homo sapiens. The sequences of these proteases showed identities between the tested species of 58.1–63.0% (Fig. S3). This finding suggests the existence of a novel class of VK‐dependent proteases, which we called ‘sirtilins’ according to the source organism T. sirtalis (see below). We also subjected Gla–EGF1–EGF2–SP proteases of jawless fishes (Lethenteron japonicum, lamprey), cartilaginous fishes (Callorhinchus milii, elephant shark), bony fishes (Danio rerio, zebrafish), P. mucrosquamatus, G. gallus, P. cinereus and H. sapiens to phylogenetic analyses. Except for H. sapiens, all of these organisms did indeed show a sirtilin homolog, clustering in a unique clade (Fig. 2). Interestingly, we also found a homologue in L. japonicum that was previously denoted as FVII‐B 12. Apparently, sirtilins emerged early in the evolution of the vertebrate coagulation system, which might indicate an important functional role.
Figure 2.
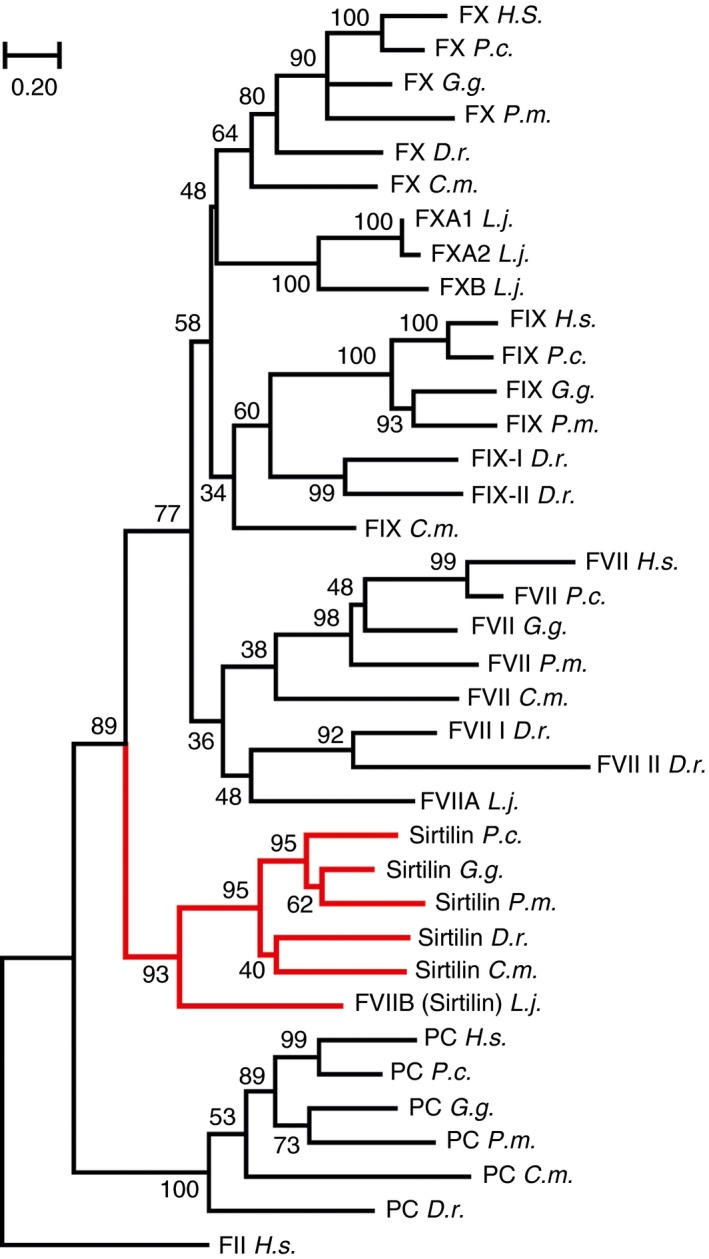
Phylogenetic analysis of sirtilins and related vitamin K‐dependent proteases. A neighbor‐joining tree of the highest log likelihood (− 12 192.26) is shown, with branch lengths representing the number of amino acid substitutions per site. The reliability of the branches is given at the nodes as a percentage of 1000 bootstrap replicates. Human prothrombin (FII) was used as the outgroup. Homologs of sirtilin are highlighted in red. Sequences of the catalytic domains of Homo sapiens (H.s.), Phascolarctos cinereus (P.c.), Gallus gallus (G.g.), Protobothrops mucrosquamatus (P.m.), Danio rerio (D.r.), Callorhinchus milii (C.m.) and Lethenteron japonicum (L.j.) were analyzed as shown in Table S1.
Preparation of T. sirtalis sirtilin
Snakes are poikilothermic organisms, and might have evolved proteins with broader stability and activity optima. Considering the advantages of heat stress‐adapted proteins in biochemical studies, the snake sirtilins were chosen for further characterization.
VK‐dependent proteases have been successfully prepared by in vitro folding in previous studies (e.g. 33, 34, 35). We have adapted the preparation procedure of recombinant FIX 17 to sirtilin comprising the catalytically active assembly of the EGF2 and catalytic domains (amino acids 129–489). Although the tested sirtilins of P. mucrosquamatus, P. bivittatus and T. sirtalis were expressed equally well as IBs, the highest in vitro folding efficiency was observed for the T. sirtalis homolog. Thus, T. sirtalis sirtilin was exclusively used for further analyses. The zymogen was purified with IEC and GPC, giving yields of 1–2 mg of purified protein per gram of IBs (Fig. 3). The sirtilin zymogen was activated with human FXIa (see below). FXIa and the low molecular weight cleavage products were separated from activated sirtilin (sirtilin‐a) with GPC (Fig. 3).
Figure 3.
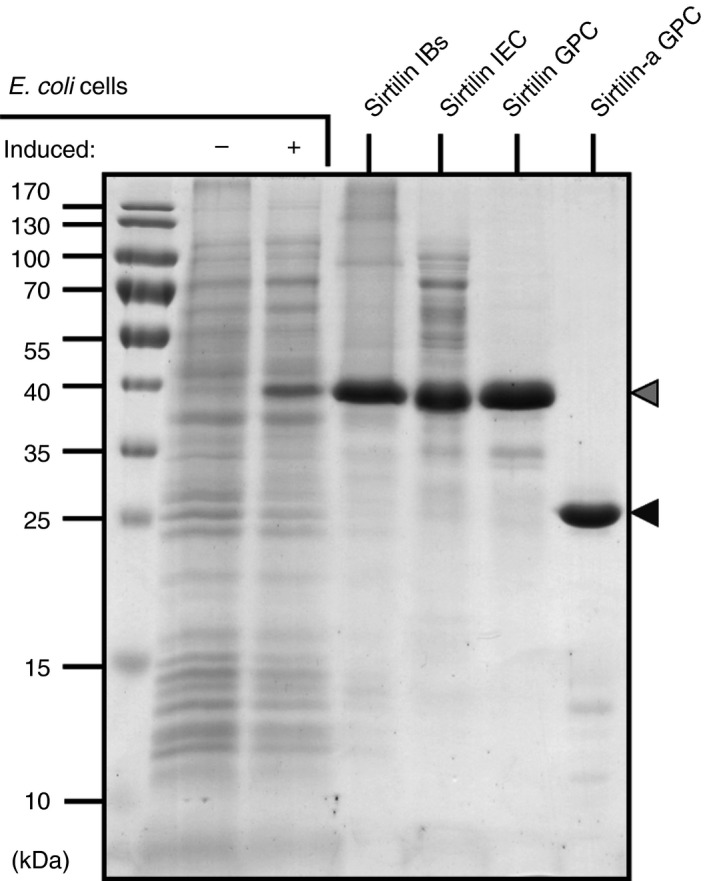
Purification of sirtilin: SDS‐PAGE analyses showing expression and purification of sirtilin. The sirtilin zymogen (gray) and activated sirtilin (sirtilin‐a, black) are marked with arrowheads. IB, inclusion body; IEC, ion exchange chromatography; GPC, gel permeation chromatography.
Activation of sirtilin by FXIa
To test whether sirtilins are activated in an FIX‐like manner (see above), we performed activation assays with FXIa (Fig. 4). Stepwise proteolytic cleavage of sirtilin to at least three intermediate forms was observed in the presence of FXIa (Fig. 4A). In contrast, FIX was converted by two sequential cleavage events to FIXaα and FIXaαβ (Fig. 4B 36). This observation is well explained by the additional lysine and arginine residues found in the activation peptide of sirtilin forming potential FXIa cleavage sites (Fig. 1C). Activation of the zymogen finally results in a disulfide‐linked heterodimer comprising the EGF2 and catalytic domains of ∼33 kDa, as shown for FIX and FX 36, 37. Under reducing conditions, the ~ 6‐kDa EGF2 domain is released and thus separated by SDS‐PAGE. Only the catalytic domains of FIXa and sirtilin‐a with theoretical molecular weights of ~ 26 kDa are observed on the gel (Fig. 4A,B).
Figure 4.
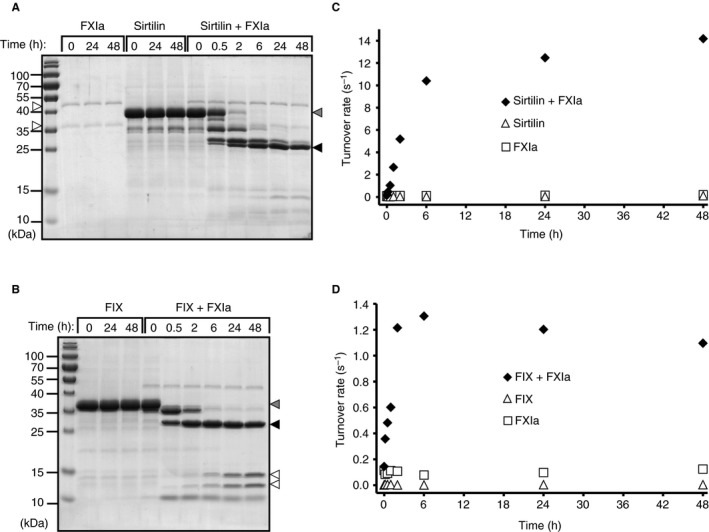
Activation of sirtilin by activated factor XI (FXIa). (A) SDS‐PAGE analysis of FXIa‐dependent cleavage of sirtilin (sirtilin + FXIa) and control assays (FXIa, sirtilin) under reducing conditions. Arrowheads indicate the light and heavy chains of FXIa (white), sirtilin zymogen (gray), and activated sirtilin (sirtilin‐a, black). (B) SDS‐PAGE analysis of FXIa‐dependent activation of FIX (FIX + FXIa) and the control assays without FXIa (FIX) under reducing conditions. Arrowheads indicate the FIX zymogen (gray), FIXαβ (black) and degradation products of FIX (white) upon prolonged FXIa exposure. (C) Gain of the sirtilin‐specific turnover rate (moles of AMC per second per mole of sirtilin; substrate: Pefafluor‐IXa) upon addition of FXIa. (D) Gain of the FIX‐specific turnover rate (moles of AMC per second per mole of FIX; substrate: Pefafluor‐IXa) upon addition of FXIa.
Activation of trypsin‐like proteases requires precise cleavage and subsequent insertion of the new N‐terminus into the activation pocket of the catalytic domain 4. To probe the productive activation of sirtilin, the accumulated catalytic activity was monitored with Pefafluor‐IXa (Fig. 4C). For sirtilin and FIX, a rapid increase in activity was observed (Fig. 4C,D), turning into a stationary phase after several hours. FIX showed slight, continuous degradation (bands between 10 kDa and 15 kDa; Fig. 4B) and a decrease in activity after 6 h (Fig. 4D), which was absent for sirtilin. In the control reactions, the proteins and the activity remained stable over time.
For sirtilin and FIX, the increase in activity upon FXIa cleavage excellently correlated with the accumulation of the 26‐kDa band, confirming that this entity is part of the catalytically active component. Interestingly, the maximal turnover rate (activity per mole of protease) of sirtilin‐a (~ 14 s−1) was higher than that of FIXa (~ 1.3 s−1) (Fig. 4C,D), indicating higher specific activity of sirtilin‐a.
The amino acid sequence of sirtilin lacks a proper furin consensus cleavage motif as is found for FX (see above). Nonetheless, the N‐terminal FXIa cleavage site (TR↓A; see above) is part of a paired basic motif (KRSTR↓A) that is found, for example, in some furin substrates (e.g. 38, 39). Indeed, this sequence was cleaved by recombinant furin (Fig. S4), although with very low efficiency as compared with recombinant FX.
Kinetic characterization of sirtilin
We characterized the kinetic properties of sirtilin‐a in comparison with FIXa and FXa by using fluorogenic peptide substrates. Unlike IEGR‐AMC, Pefaflour‐IXa interacts in a non‐canonical manner with trypsin‐like proteases, inserting the P3 d ‐cyclo‐hexyl‐glycyl side chain into the P4 pocket 40. With both substrates, we observed similar K M values, in the mmol L−1 range, for sirtilin‐a (Table 1; Fig. S5A,B). The k cat values, however, were substantially different, being 30.1 ± 4.3 s−1 and 0.50 ± 0.07 s−1 for Pefaflour‐IXa and IEGR‐AMC, respectively (Table 1; Fig. S5A,B). This behavior is more similar to that of FIXa than to that of FXa, whereby the latter showed large differences in K M for the two substrates (0.27 ± 0.02 mmol L−1 for Pefaflour‐IXa, and 1.8 ± 0.2 mmol L−1 for IEGR‐AMC; Table 1; Fig. S5C,D). Interestingly, FXa showed the lowest differences in k cat between Pefaflour‐IXa and IEGR‐AMC, the k cat values being 259 ± 9 s−1 and 72.5 ± 7.2 s−1, respectively (Table 1; Fig. S5E,F). The catalytic efficiency of sirtilin‐a (8600 ± 2700 s−1 mol−1 L) for Pefaflour‐IXa was approximately two orders of magnitude higher than that of FIXa (65 ± 5 s−1 mol−1 L) and two orders of magnitude lower than that of FXa (960 000 ± 100 000 s−1 mol−1 L). Similar differences between the proteases were observed for IEGR‐AMC, with the k cat/K M values being 161 ± 49 s−1 mol−1 L, 2.5 ± 1.4 s−1 mol−1 L and 40 300 ± 8500 s−1 mol−1 L for sirtilin‐a, FXIa, and FX, respectively (Table 1).
Table 1.
Kinetic parameters of sirtilin‐a in comparison with activated factor IX (FIXa) and activated FX (FXa)
| K M (mmol L−1) | k cat (s−1) | K I * (mmol L−1) | k cat/K M (s−1 mol−1 L) | |
|---|---|---|---|---|
| Pefafluor‐IXa | ||||
| Sirtilin‐a | 3.5 ± 0.6 | 30.1 ± 4.3 | 2.0 ± 0.4 | 8600 ± 2700 |
| FIXa | 2.0 ± 0.1 | 0.130 ± 0.003 | – | 65 ± 5 |
| FXa | 0.27 ± 0.02 | 259 ± 9 | 3.7 ± 0.3 | 960 000 ± 100 000 |
| IEGR‐AMC | ||||
| Sirtilin‐a | 3.1 ± 0.5 | 0.50 ± 0.07 | 14.9 ± 8.2 | 161 ± 49 |
| FIXa | 4.4 ± 1.2 | 0.011 ± 0.003 | 5.5 ± 2.7 | 2.5 ± 1.4 |
| FXa | 1.8 ± 0.2 | 72.5 ± 7.2 | 4.0 ± 0.8 | 40 300 ± 8500 |
IEGR‐AMC, t‐butyloxycarbonyl‐Ile‐Glu‐Gly‐Arg‐AMC; Pefafluor‐IXa, methylsulfonyl‐d‐cyclohexylglycyl‐Gly‐Arg‐AMC. *Substrate inhibition model.
Substrate profiling of sirtilin‐a
We applied a mass spectrometry‐based method (PICS 22, 23) to characterize the sequence specificity of sirtilin‐a. The identified cleavage motifs of sirtilin‐a were visualized as ‘IceLogos’ 28 indicating the preferred consensus sequence. Similar consensus sequences were obtained for sirtilin‐a, FIXa and FXa at the non‐primed side (Fig. 5A–F). The typical cleavage motifs of these proteases show a strong preference for arginine at P1 and a comparable weaker preference for glycine and small hydrophobic amino acids at P2. In addition, the tested proteases share a preference for hydrophobic amino acids such as leucine and valine at P3. Beyond this common cleavage motif, however, sirtilin‐a showed unique preferences at the non‐primed side (Fig. 5A,B). At P1′, serine, threonine and valine were overrepresented. Alanine, glycine and aromatic amino acids are frequently found at P2′ in peptides cleaved by sirtilin‐a. In contrast, FIXa (Fig. 5C,D) preferred leucine at P2′, and FXa was, in general, less specific at this position (Fig. 5E,F).
Figure 5.
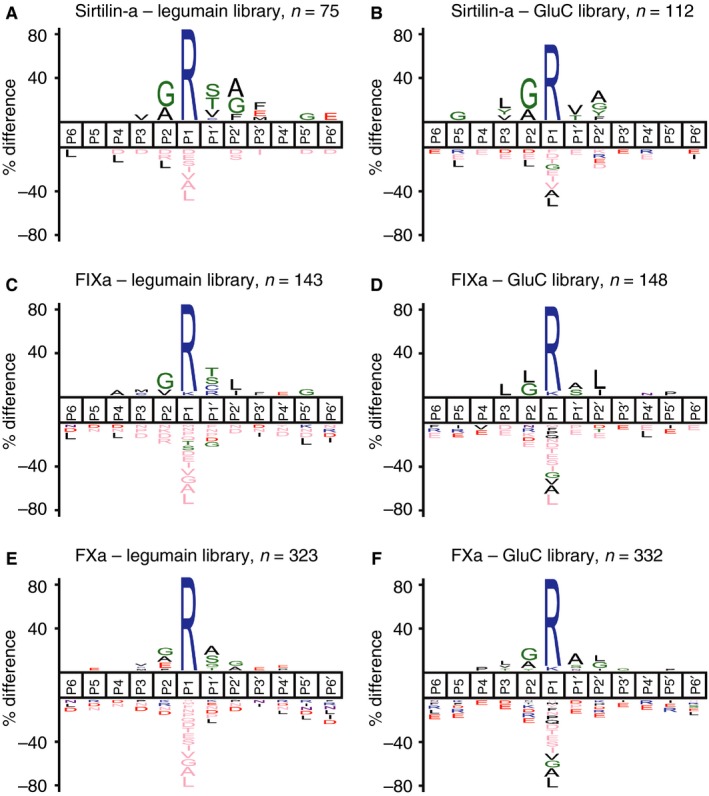
Substrate profiles of sirtilin‐a, FIXa and FXa. (A) Sirtilin‐a – legumain library. (B) Sirtilin‐a – GluC library. (C) FIXa – legumain library. (D) FIXa – GluC library. (E) FXa – legumain library. (F) FIXa – GluC library. Semispecific peptides with an at least four‐fold increase in intensity upon protease treatment were used for reconstruction of the cleavage sites displayed as IceLogos. The number of identified cleavage windows (n) is given at the respective IceLogos. The IceLogos show the percentage difference of a specific amino acid relative to its natural abundance (P = 0.05) at a certain substrate position from P6 to P6′. FIXa, activated FIX; FXa, activated FX.
Activation of prothrombin and FX by sirtilin‐a
The homology of sirtilin with FIX and FX, and the similar sequence specificity, suggest overlapping functions of these proteases in blood coagulation. Therefore, we investigated the ability of sirtilin‐a to cleave and activate the two macromolecular substrates prothrombin and/or FX.
To detect the activity of thrombin selectively, we used the specific substrate GPR‐AMC. Cleavage of thrombin is indicated immediately after addition of sirtilin‐a by the formation of a double band at ~ 37 kDa (Fig. 6A). These cleavage products were also generated by FXa cleavage (Fig. 6B). In the absence of FVa and phospholipids, the Arg271 cleavage site of prothrombin was preferred over Arg320 by FXa, generating the fragments F1.2 and Prethrombin‐2 (P2) (Fig. 6C 37). The preference for Arg271 was even more pronounced for sirtilin‐a, as indicated by slow cleavage of Arg320 at the P2 fragment and thus by slow thrombin (α‐thrombin) formation (Fig. 6A). Even after 24 h, the B fragment of thrombin at 35 kDa was visible as a faint band only. For FXa, however, significant generation of thrombin occurred more rapidly, as indicated by accumulation of the B fragment after 30 min (Fig. 6A). F1.2 is susceptible to cleavage by thrombin itself at Arg155, generating the fragments F1 and F2 (Fig. 6C). This secondary cleavage at Arg155 was observed immediately after addition of FXa, again indicating rapid generation of thrombin (Fig. 6B). In the presence of sirtilin, however, F1 and F2 became visible after 1 h, indicating slower processing of F1.2 and thus slower thrombin formation (Fig. 6A). These observations are in excellent agreement with the accumulation of the thrombin‐specific activity over time (Fig. 6D,E). Incubation of prothrombin with FXa generated turnover of the fluorogenic substrate by thrombin of ~ 1431 h−1 after 4.5 h (Fig. 6E). The same amount of sirtilin‐a resulted in a comparable lower thrombin‐specific turnover of ~ 1.7 h−1 (Fig. 6D).
Figure 6.
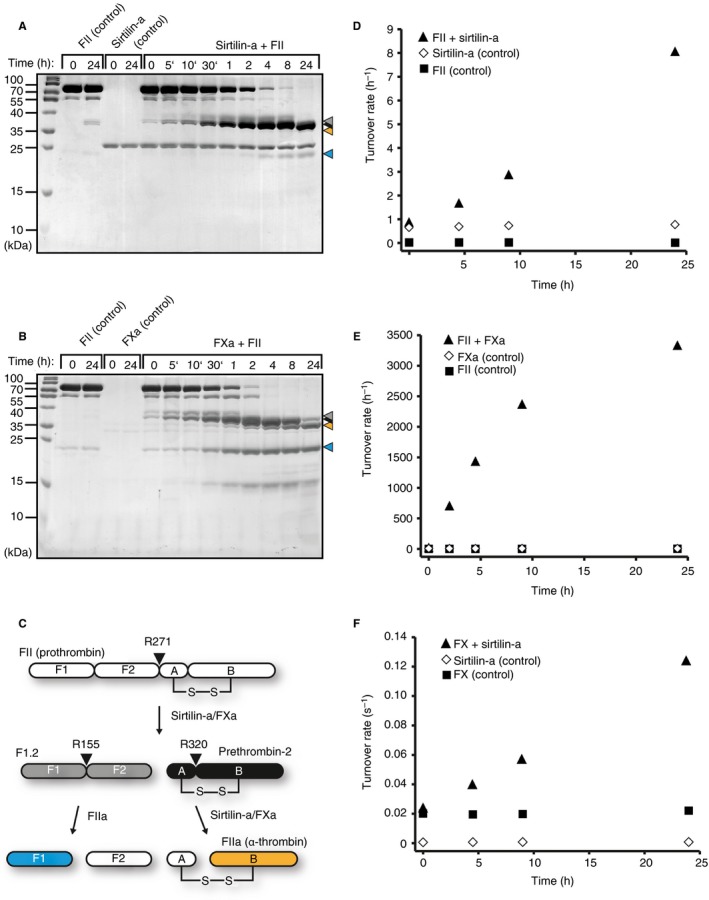
Activation of prothrombin and factor X by sirtilin‐a. (A, B) SDS‐PAGE analysis of limited proteolysis reactions with (A) sirtilin‐a or (B) activated FX (FXa) and prothrombin (FII), including controls with the individual proteins. The cleavage products of prothrombin are marked with arrowheads. Coloring is according to (C). (C) Schematic representation of prothrombin cleavage. Prothrombin cleavage fragments are indicated according to 37. (D, E) Increase in thrombin‐specific turnover with incubation time in the presence of (D) sirtilin‐a or (E) FXa. The turnover rate is given as the ratio between the thrombin‐specific turnover of GPR‐AMC (mol AMC h–1) per mole of sirtilin‐a (2.35 μmol L−1) and FXa (0.077 μmol L−1). (F) Increase in FXa‐specific turnover as a function of incubation time. Turnover rate refers to the ratio between the FXa‐specific turnover of the fluorogenic substrate (mol AMC s–1) per mole of FX zymogen.
In blood coagulation, FX is activated by FIXa by proteolysis at Arg194. We tested whether sirtilin can also activate FX. To specifically detect the activity of newly formed FXa in the presence of sirtilin‐a, we selectively inhibited the latter with DIFP (Fig. S6). We did indeed observe a constant increase in turnover upon incubation of FX with sirtilin‐a, indicating productive conversion of FX to FXa (Fig. 6F). The activity of the control reactions was constant over time. Cleavage of FX by sirtilin‐a, however, was very slow, as indicated by the turnover rate of ~ 0.12 s−1 after 24 h of incubation (the k cat of FXa was 259 s−1 under substrate saturation; Table 1). The activating cleavage site of FX (TR↓IV) is very different from the consensus cleavage motif of sirtilin‐a (see above), which might explain the slow activation kinetics.
Discussion
We have identified sirtilins as a novel class of VK‐dependent proteases that were found in fishes, reptiles, birds, and marsupials, but not in higher mammals. Sirtilins showed the typical domain architecture of the Gla–EGF1–EGF2–SP family members. The catalytically active component of T. sirtalis sirtilin, comprising the EGF2 and trypsin‐like protease domains, was prepared by in vitro folding.
As expected from the sequence homology of sirtilin with other VK‐dependent proteases, we found stringent specificity for arginine at P1. In comparison with FIX and FX, additional preferences were identified, especially at the primed and non‐primed positions, suggesting a unique substrate specificity of sirtilin. Substrates with P3 d‐amino acids interact with trypsin‐like proteases in a ‘non‐canonical’ manner, binding with the P3 side chain to the S4 pocket 40. In the case of FIXa and FXa, this resulted in combined K M and k cat effects as compared with a canonical substrate. For sirtilin, however, both substrates showed very similar K M values. Nonetheless, an approximately 60‐fold increase in k cat was observed, indicating a strong effect on the catalytic mechanism rather than on substrate binding. Limited proteolysis studies with the protein substrate prothrombin provided further clues regarding the substrate specificity of sirtilin at the primed positions. Sirtilin cleaved prothrombin preferentially at Arg271 (R271↓TA) with the optimal amino acids at P1′ and P2′ as compared with Arg320 (R320↓IV). This cleavage preference was less pronounced for FXa, which showed lower specificity, especially at the primed positions. In the presence of the cofactor FVa, however, the prothrombinase complex (FVa–FXa) selectively binds the ‘closed’ conformation of prothrombin 41 and thus triggers cleavage at Arg320. The extended and comparatively high sequence specificity of sirtilin indicates increased importance of direct protease–substrate interactions. This finding might hint at a cofactor‐independent function of sirtilin. Alternatively, exosites might contribute to enzyme–substrate interactions of sirtilin, thereby conformationally selecting the open prothrombin form.
A characteristic feature of T. sirtalis sirtilin is the enlarged activation peptide as compared with the other Gla–EGF1–EGF2–SP family members. Interestingly, it is readily cleaved at several sites by FXIa, resulting in the activation of the protease. The activation scheme of the coagulation proteases determines the exact sequence of the coagulation cascade, and thus is substantially connected to their function. Strikingly, FXIa is found only in mammals, so an alternative physiological activator of sirtilin has to come into play in lower vertebrates. Because it is found in many organisms lacking the intrinsic pathway, sirtilin might fulfil a function as an ancient intrinsic coagulation factor. The intrinsic pathway in higher mammals is initiated by autoactivation of FXII 42. This process is triggered by platelet‐derived polyphosphates in vivo and by negatively charged surfaces in vitro. Although it is tempting to speculate about a similar autoactivation mechanism of sirtilin, this has not been observed so far.
In conclusion, a new member of the VK‐dependent proteases was identified, and a deeper understanding of the functions of sirtilin could provide novel insights into blood coagulation of vertebrates as well as into the evolution of coagulation factors.
Addendum
S. O. Dahms designed and performed research, analyzed data, and wrote the manuscript. F. Demir and P. Huesgen designed the PICS experiments, performed mass spectrometry measurements, and analyzed data. K. Thorn planned research and analyzed data. H. Brandstetter planned research, analyzed data, and wrote the manuscript. All authors edited the final manuscript.
Disclosure of Conflict of Interests
S. O. Dahms and H. Brandstetter received funding from Novo Nordisk. K. Thorn is an employee of Novo Nordisk. The other authors state that they have no conflict of interest.
Supporting information
Fig. S1. Progress curves of substrate hydrolysis.
Fig. S2. Analysis of molecular markers in sirtilins.
Fig. S3. Pairwise alignments of VK‐dependent proteases.
Fig. S4. Cleavage of sirtilin by furin.
Fig. S5. Enzyme kinetics of sirtilin‐a in comparison with FIXa and FXa.
Fig. S6. Inhibitor profile of sirtilin‐a in comparison with FIXa and FXa and selective inhibition by DIPF.
Table S1. Sequences used for alignments and phylogenetic analyses.
Data S1. Supplementary data.
Acknowledgements
The authors thank E. Dall for critical reading of the manuscript.
Dahms SO, Demir F, Huesgen PF, Thorn K, Brandstetter H. Sirtilins – the new old members of the vitamin K‐dependent coagulation factor family. J Thromb Haemost 2019; 17: 470–81.
Manuscript handled by: R. Camire
Final decision: P. H. Reitsma, 4 January 2019
References
- 1. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 2013; 93: 327–58. [DOI] [PubMed] [Google Scholar]
- 2. Stenflo J. Contributions of Gla and EGF‐like domains to the function of vitamin K‐dependent coagulation factors. Crit Rev Eukaryot Gene Expr 1999; 9: 59–88. [PubMed] [Google Scholar]
- 3. Zogg T, Brandstetter H. Complex assemblies of factors IX and X regulate the initiation, maintenance, and shutdown of blood coagulation. Prog Mol Biol Transl Sci 2011; 99: 51–103. [DOI] [PubMed] [Google Scholar]
- 4. Huber R, Bode W. Structural basis of activation and action of trypsin. Acc Chem Res 1978; 11: 114–22. [Google Scholar]
- 5. Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 1991; 30: 10363–70. [DOI] [PubMed] [Google Scholar]
- 6. Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006; 26: 41–8. [DOI] [PubMed] [Google Scholar]
- 7. Laurens N, Koolwijk P, de Maat MP. Fibrin structure and wound healing. J Thromb Haemost 2006; 4: 932–9. [DOI] [PubMed] [Google Scholar]
- 8. Bolton‐Maggs PH, Pasi KJ. Haemophilias A and B. Lancet 2003; 361: 1801–9. [DOI] [PubMed] [Google Scholar]
- 9. Ponczek MB, Bijak MZ, Nowak PZ. Evolution of thrombin and other hemostatic proteases by survey of protochordate, hemichordate, and echinoderm genomes. J Mol Evol 2012; 74: 319–31. [DOI] [PubMed] [Google Scholar]
- 10. Kimura A, Ikeo K, Nonaka M. Evolutionary origin of the vertebrate blood complement and coagulation systems inferred from liver EST analysis of lamprey. Dev Comp Immunol 2009; 33: 77–87. [DOI] [PubMed] [Google Scholar]
- 11. Doolittle RF. Step‐by‐step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol 2009; 74: 35–40. [DOI] [PubMed] [Google Scholar]
- 12. Doolittle RF. Bioinformatic characterization of genes and proteins involved in blood clotting in lampreys. J Mol Evol 2015; 81: 121–30. [DOI] [PubMed] [Google Scholar]
- 13. Davidson CJ, Hirt RP, Lal K, Snell P, Elgar G, Tuddenham EG, McVey JH. Molecular evolution of the vertebrate blood coagulation network. Thromb Haemost 2003; 89: 420–8. [PubMed] [Google Scholar]
- 14. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 2000; 16: 276–7. [DOI] [PubMed] [Google Scholar]
- 16. Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol Biol Evol 2016; 33: 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kristensen LH, Olsen OH, Blouse GE, Brandstetter H. Releasing the brakes in coagulation factor IXa by co‐operative maturation of the substrate‐binding site. Biochem J 2016; 473: 2395–411. [DOI] [PubMed] [Google Scholar]
- 18. Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. New York: Wiley‐VCH, 2000. [Google Scholar]
- 19. Sichler K, Kopetzki E, Huber R, Bode W, Hopfner KP, Brandstetter H. Physiological fIXa activation involves a cooperative conformational rearrangement of the 99‐loop. J Biol Chem 2003; 278: 4121–6. [DOI] [PubMed] [Google Scholar]
- 20. Dahms SO, Hardes K, Becker GL, Steinmetzer T, Brandstetter H, Than ME. X‐ray structures of human furin in complex with competitive inhibitors. ACS Chem Biol 2014; 9: 1113–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Griessner A, Zogg T, Brandstetter H. The activation peptide of coagulation factor IX and X serves as a high affinity receptor to cationic ligands. Thromb Haemost 2013; 110: 620–2. [Google Scholar]
- 22. Biniossek ML, Niemer M, Maksimchuk K, Mayer B, Fuchs J, Huesgen PF, McCafferty DG, Turk B, Fritz G, Mayer J, Haecker G, Mach L, Schilling O. Identification of protease specificity by combining proteome‐derived peptide libraries and quantitative proteomics. Mol Cell Proteomics 2016; 15: 2515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schilling O, Huesgen PF, Barre O, Auf dem Keller U, Overall CM. Characterization of the prime and non‐prime active site specificities of proteases by proteome‐derived peptide libraries and tandem mass spectrometry. Nat Protoc 2011; 6: 111–20. [DOI] [PubMed] [Google Scholar]
- 24. Dall E, Brandstetter H. Activation of legumain involves proteolytic and conformational events, resulting in a context‐ and substrate‐dependent activity profile. Acta Crystallogr F Struct Biol Cryst Commun 2012; 68: 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dall E, Brandstetter H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc Natl Acad Sci USA 2013; 110: 10940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rappsilber J, Mann M, Ishihama Y. Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2007; 2: 1896–906. [DOI] [PubMed] [Google Scholar]
- 27. Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 2016; 11: 2301–19. [DOI] [PubMed] [Google Scholar]
- 28. Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat Methods 2009; 6: 786–7. [DOI] [PubMed] [Google Scholar]
- 29. NCBI Resource Coordinators . Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2018; 46: D8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goodeve AC, Reitsma PH, McVey JH; Working Group on Nomenclature of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis . Nomenclature of genetic variants in hemostasis. J Thromb Haemost 2011; 9: 852–5. [DOI] [PubMed] [Google Scholar]
- 31. Krem MM, Di Cera E. Molecular markers of serine protease evolution. EMBO J 2001; 20: 3036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brenner S. The molecular evolution of genes and proteins: a tale of two serines. Nature 1988; 334: 528–30. [DOI] [PubMed] [Google Scholar]
- 33. Zogg T, Brandstetter H. Structural basis of the cofactor‐ and substrate‐assisted activation of human coagulation factor IXa. Structure 2009; 17: 1669–78. [DOI] [PubMed] [Google Scholar]
- 34. Hopfner KP, Brandstetter H, Karcher A, Kopetzki E, Huber R, Engh RA, Bode W. Converting blood coagulation factor IXa into factor Xa: dramatic increase in amidolytic activity identifies important active site determinants. EMBO J 1997; 16: 6626–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sichler K, Banner DW, D'Arcy A, Hopfner KP, Huber R, Bode W, Kresse GB, Kopetzki E, Brandstetter H. Crystal structures of uninhibited factor VIIa link its cofactor and substrate‐assisted activation to specific interactions. J Mol Biol 2002; 322: 591–603. [DOI] [PubMed] [Google Scholar]
- 36. Zogg T, Brandstetter H. Activation mechanisms of coagulation factor IX. Biol Chem 2009; 390: 391–400. [DOI] [PubMed] [Google Scholar]
- 37. Krishnaswamy S. The transition of prothrombin to thrombin. J Thromb Haemost 2013; 11(Suppl. 1): 265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin‐dependent proteolysis. J Biol Chem 2004; 279: 18111–14. [DOI] [PubMed] [Google Scholar]
- 39. McColl BK, Paavonen K, Karnezis T, Harris NC, Davydova N, Rothacker J, Nice EC, Harder KW, Roufail S, Hibbs ML, Rogers PA, Alitalo K, Stacker SA, Achen MG. Proprotein convertases promote processing of VEGF‐D, a critical step for binding the angiogenic receptor VEGFR‐2. FASEB J 2007; 21: 1088–98. [DOI] [PubMed] [Google Scholar]
- 40. Stubbs MT, Bode W. A player of many parts: the spotlight falls on thrombin's structure. Thromb Res 1993; 69: 1–58. [DOI] [PubMed] [Google Scholar]
- 41. Chinnaraj M, Chen Z, Pelc LA, Grese Z, Bystranowska D, Di Cera E, Pozzi N. Structure of prothrombin in the closed form reveals new details on the mechanism of activation. Sci Rep 2018; 8: 2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood 2012; 120: 4296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Progress curves of substrate hydrolysis.
Fig. S2. Analysis of molecular markers in sirtilins.
Fig. S3. Pairwise alignments of VK‐dependent proteases.
Fig. S4. Cleavage of sirtilin by furin.
Fig. S5. Enzyme kinetics of sirtilin‐a in comparison with FIXa and FXa.
Fig. S6. Inhibitor profile of sirtilin‐a in comparison with FIXa and FXa and selective inhibition by DIPF.
Table S1. Sequences used for alignments and phylogenetic analyses.
Data S1. Supplementary data.