

Abstract
Fibroblast growth factor 23 (FGF23) is a bone-derived hormone involved in the control of phosphate (P) homeostasis and vitamin D metabolism. Despite advances, however, molecular details of this gene’s regulation remain uncertain. In this report, we created mouse strains in which four epigenetically marked FGF23 regulatory regions were individually deleted from the mouse genome using CRISPR/Cas9 gene-editing technology, and the consequences of these mutations were then assessed on Fgf23 expression and regulation in vivo. An initial analysis confirmed that bone expression of Fgf23 and circulating intact FGF23 (iFGF23) were strongly influenced by both chronic dietary P treatment and acute injection of 1,25-dihydroxyvitamin D3 [1,25(OH)2D3]. However, further analysis revealed that bone Fgf23 expression and iFGF23 could be rapidly upregulated by dietary P within 3 and 6 hours, respectively; this acute upregulation was lost in the FGF23-PKO mouse containing an Fgf23 proximal enhancer deletion but not in the additional enhancer-deleted mice. Of note, prolonged dietary P treatment over several days led to normalization of FGF23 levels in the FGF23-PKO mouse, suggesting added complexity associated with P regulation of FGF23. Treatment with 1,25(OH)2D3 also revealed a similar loss of Fgf23 induction and blood iFGF23 levels in this mouse. Finally, normal lipopolysaccharide (LPS) induction of Fgf23 expression was also compromised in the FGF23-PKO mouse, a result that, together with our previous report, indicates that the action of LPS on Fgf23 expression is mediated by both proximal and distal Fgf23 enhancers. These in vivo data provide key functional insight into the genomic enhancers through which Fgf23 expression is mediated.
Fibroblast growth factor 23 (FGF23) is a newly discovered member of the endocrine subfamily of systemic FGF hormones that in humans include FGF19 and FGF21 (1, 2). This peptide factor is transcribed from its cognate gene largely in mature bone osteoblasts and osteocytes and is delivered into the circulation either as a full-length biologically active glycoprotein or as an inactive C-terminal cleavage product (3). Both the synthesis and processing of FGF23 represent regulatory steps that constitute primary determinants of the levels of intact functional FGF23 (iFGF23) in the blood. As a result, major efforts are presently under way to understand the dynamics and the mechanisms that characterize these processes. The primary function of FGF23 is to control systemic phosphate (P) levels, largely by regulating the concentrations and activities of the sodium-P cotransporters NPT2a and NPT2c that are expressed in the kidney and that control P diuresis (4–6). Indeed, a mechanistic action of FGF23 is to promote the internalization of these transporters from the apical membranes of epithelial cells lining renal proximal tubules, thereby restricting P reabsorption. Given this key function of FGF23, it should not be surprising that P represents a major systemic regulator of both cellular FGF23 expression and posttranscriptional processing (7).
P homeostasis is also modulated in the kidney and in bone by both 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] and PTH and in the intestine by 1,25(OH)2D3. Thus, additional physiological functions of FGF23 include the downregulation of PTH transcription from the parathyroid glands and decreased vitamin D hormone production by the kidney (3, 8–10). Accordingly, FGF23 has been shown to inhibit both the synthesis and secretion of PTH and to suppress CYP27B1 while inducing CYP24A1, the renal enzymes responsible for the synthesis and homeostatic maintenance of circulating 1,25(OH)2D3. Our recent studies have provided considerable molecular insight into how FGF23 coordinately regulates the genes for these two enzymes via unique kidney-specific genomic control modules (11–13). Because both PTH and 1,25(OH)2D3 also modulate P homeostasis and PTH represents a primary inducer of the Cyp27b1 gene (11, 14, 15), these activities of FGF23 constitute key negative feedback loops that serve to limit the activity of both hormones in the target tissues described previously. With these profound endocrine actions of FGF23 on P homeostasis and on both the synthesis and secretion of 1,25(OH)2D3 and PTH, it is not surprising that in addition to P, both PTH and 1,25(OH)2D3 also regulate FGF23 in normal healthy tissues (16, 17).
The complexity of FGF23 and its activation system as well as its involvement with many additional factors that participate in the regulation of P metabolism bring to light a myriad of genetic diseases of altered mineral homeostasis. Although the descriptions of most of these diseases predated the discovery of FGF23, they also facilitated the eventual identification of the hormone as a central mediator of P metabolism (7, 18, 19). As important, however, are the many diseases that cause dysregulated FGF23 expression secondary to the aberrant levels of numerous physiological factors that affect FGF23 expression and/or regulation. Chronic kidney disease (CKD)‒mineral and bone disorder represents a classic condition wherein progressive upregulation of FGF23 occurs, often together with increased serum P levels that are also subsequently associated with striking increases in PTH levels (20, 21). Of note, this disease may have highlighted the discovery of a central role for inflammation in the regulation of FGF23, setting the stage for the identification of bacterial LPS, IL-1β, TNFα, and other inflammatory agents as inducers of this phosphaturic hormone not only in bone but also in many additional tissues (22–26).
Previous knowledge of the signaling pathways and mechanisms of action of regulators such as the vitamin D hormone, PTH, and inflammatory mediators have led to specific insights into the mechanisms of FGF23 regulation. However, these studies have been conducted largely in vitro, frequently resulting in divergent observations. For example, one study suggests that vitamin D action may be mediated via an upstream, yet unconserved, Fgf23 vitamin D response element (VDRE), whereas a second study suggests the actions of the hormone occur via a more proximal element and may be secondary to vitamin D receptor (VDR) induction (27, 28). Regarding inflammation, a number of studies have suggested the presence of regulatory sequences that mediate the actions of TNFα and IL-1β and the iron induction of HIF-1α, although direct identification of the interactions of these factors with DNA sequences has not been forthcoming (22, 29). In contrast, little is known of how P operates to upregulate FGF23 expression, or indeed whether it does so at a transcriptional or posttranscriptional level. We therefore reasoned that an in vivo approach in mice in the context of the endogenous Fgf23 gene itself might advance our understanding of Fgf23 expression and regulation.
In this report, we used a clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) approach to delete from the mouse genome each of four potential epigenetically marked regulatory regions previously identified within proximity of the Fgf23 gene locus (30, 31). Each had the potential for FGF23 regulation, and our earlier studies had defined the involvement of one of these in mediating induction by LPS (31). We then examined the effect of these deletions on the induction of Fgf23 expression in bone and other tissues and on the blood levels of iFGF23 in the mouse as a consequence of dietary P, 1,25(OH)2D3, and LPS treatment. Our studies identified a primary enhancer region involved in Fgf23 expression and regulation by dietary P and 1,25(OH)2D3; this enhancer contributed to LPS induction of FGF23 as well. These studies provide an in vivo basis for further exploration into the molecular mechanisms through which Fgf23 expression is regulated.
Materials and Methods
Generation of enhancer-deleted mice
Fgf23 enhancer-deleted mice were generated using the CRISPR/Cas9 approach. RNA guides used for editing the mouse genome are listed in Fig. 1A and were optimized for the least number of potential off-target sites and fewest sites within coding exons using the Zhang Laboratory CRISPR Design tool (crispr.mit.edu). Guides were annealed, cloned into the pX458 vector (Addgene), and used to produce appropriate RNA as previously described (32, 33). Mutant mice were generated at the Genome Editing-Animal Models Facility of the University of Wisconsin Biotechnology Center using the isolated RNA guides and Cas9 protein as previously described (34). The resulting pups were genotyped with spanning primers (Table 1). The precise boundaries of the deleted segments were determined through DNA sequencing (Fig. 1A), and schematic locations of the deleted regions in the mutant strains are displayed in Fig. 1B.
Figure 1.

Generation of Fgf23-enhancer mutant mouse strains. (A) Putative Fgf23 enhancers epigenetically identified by chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) analyses were individually deleted to create the mouse strains indicated. Sequences of the guide RNAs used for the CRISPR/Cas9 genome editing method for each mutant are documented. Genomic locations of the deleted region in each mouse strain were determined by sequencing and are provided. FGF23-16KO mouse strain was previously generated (31). (B) Illustration of the Fgf23 gene locus in the mouse. Relative locations of the deleted regions in FGF23-PKO (PKO), FGF23-38KO (38KO), FGF23-IKO (IKO), and FGF23-16KO (16KO) mouse strains are displayed as black boxes. Exons and introns of Fgf23 gene are represented by gray and white boxes, respectively. Transcription start site is marked as an arrow. PAM, Protospacer adjacent motif; TSS, transcription start site.
Table 1.
Oligonucleotides for Genotyping
| Mutant Strains | Primers for Genotyping | Alleles (Size) |
|---|---|---|
| FGF23-PKO | TTGCTCCTGTCAAGTATGGAACA | WT (435 bp) |
| GTGAGAAGGAGGGCGTTTTGG | ||
| TTGCTCCTGTCAAGTATGGAACA | KO (284 bp) | |
| CAGGAAAAACAATGCCGCCAC | ||
| FGF23-38KO | CGACTGGGTTATGAGTTTCTTGG | WT (350 bp) |
| CCTGCTTAGGCTGTAGTGATGAG | ||
| ACAATAAGACTCCTCCCTTGGATG | KO (317 bp) | |
| CATTTTAAGGGCGAGGAAAGTGC | ||
| FGF23-IKO | GGTTGAAGATGAATGTTAGTCTAGTC | WT (556 bp) |
| GGCGAACAGTGTAGAAATGCAG | ||
| GGTTGAAGATGAATGTTAGTCTAGTC | KO (271 bp) | |
| GGCGAACAGTGTAGAAATGCAG | ||
| FGF23-16KOa | ACGCCCTTTAGCCCAGTAAT | WT (537 bp) |
| ATGGTGGGAAGGTGTGGTAA | ||
| AACAGTGTGAAGGGGTCCTG | KO (600 bp) | |
| TACCCACGAGGAACATGTGA |
Previously generated (31).
Animal study
Mutant mouse strains were maintained as heterozygotes through outbreeding with C57BL/6 mice (The Jackson Laboratory). All commercial diets were obtained from Envigo. After weaning, all mice were fed a diet of standard rodent chow (5008) unless specified otherwise. The same number of males and females were used for each experiment unless indicated otherwise. Sex was not considered a factor in the statistical analysis. To examine the effect of serum P and 1,25(OH)2D3 on Fgf23 expression in C57BL/6 mice, 4-week-old males were fed diets containing 0.1% (low; TD.09048), 0.6% (normal; TD.150838), or 1.65% (high; TD.160749) P for 2 weeks, subjected to a single intraperitoneal injection of either vehicle or 1,25(OH)2D3 [10 ng/g body weight (bw)], and then euthanized 6 hours later. To examine the involvement of the deleted Fgf23 regulatory regions in P response, 5-week-old mice of each strain were fed a P-deficient diet (0.02% P-containing diet; TD.86128) for 1 week and then transferred to a high-P diet for the indicated periods to maximize the overall response of the Fgf23 gene to P. Of note, all mice were fasted for 15 hours immediately before high-P diet transfer to ensure comparable dietary consumption. To examine whether the DNA regions deleted in the mutant mouse strains were involved in Fgf23 expression after chronic exposure to P, 4-week-old mice were fed 0.1%, 0.6%, or 1.65% P-containing diets for 2 weeks before euthanasia. To determine the DNA regions responsible for Fgf23 induction by 1,25(OH)2D3 and LPS, 8-week-old mice were intraperitoneally injected with either vehicle, 1,25(OH)2D3 (10 ng/g bw), or LPS (10 µg/g bw) (L6529; Sigma-Aldrich) and euthanized at the indicated times. Mice were exposed to a 12-hour light-dark cycle. All animal studies were reviewed and approved by the Research Animal Care and Use Committee of the University of Wisconsin-Madison.
Serum analyses
Blood was collected from anesthetized mice via cardiac puncture and serum, and EDTA-plasma was obtained as previously described (31). Serum P levels were determined using the QuantiChrom Phosphate Assay Kit (DIPI-500; BioAssay Systems) according to the manufacturer’s instructions. Levels of iFGF23 in EDTA-plasma were measured using Mouse/Rat FGF23 (intact) ELISA Kit (60-6800; Quidel) (35) as instructed.
Gene expression analysis
Tissues collected from euthanized mice were snap-frozen in liquid nitrogen and stored at −80°C. Total RNA was prepared from tissues using TRIzol Reagent (Life Technologies) and then subjected to reverse transcription using the High Capacity cDNA Reverse Transcription Kit following the manufacturer’s protocols as previously described (36, 37). Lumbar vertebra 5 (L5) was used to analyze gene expression in bone. Gene expression was assessed by TaqMan-mediated quantitative PCR on a StepOnePlus (Applied Biosystems). The following TaqMan primers (Applied Biosystems) were used: Fgf23 (Mm00445621), Dmp1 (Mm01208363_m1), Tnfsf11 (Mm00441906), Spp1 (Mm00436767_m1), Kl (Mm00502002), Il6 (Mm00446190), and Gapdh (4352339E).
Statistical analysis
All data were tested by GraphPad Prism 8 to determine whether they were normally distributed. Data that did not pass this normalization test were then normalized using the square root method. Using GraphPad Prism 8, normalized data were then subjected to two-way ANOVA and Benjamini and Hochberg assessment as a post hoc test for multiple comparisons to identify significant differences between experimental groups (P < 0.05).
De novo transcription factor motif analysis
To predict transcription factor binding motifs in the proximal regulatory region deleted in the FGF23-PKO mouse, the deleted sequence listed in Table 1 was subjected to genomic analysis using the Genomatix MatInspector program (38). The Matrix Library Version 11.0 was used with 0.75 core similarity and optimized matrix similarity.
Results
Dietary P intake and 1,25(OH)2D3 control Fgf23 expression in bone and iFGF23 levels in the circulation to maintain serum P
Although the general effects of dietary P, 1,25(OH)2D3, and mediators of inflammation on Fgf23 expression have been well established in vivo, progress toward understanding the mechanisms that underlie this regulation at the level of transcription has been limited largely to traditional cellular investigations in vitro (5, 16, 17, 39). We therefore initiated studies exclusively in vivo aimed at understanding both the features of Fgf23 induction and the broad genomic sites of action of these three agents at the Fgf23 gene. To initiate such studies, we first confirmed that exposure to low, normal, and high P contents in the diet administered continuously for 2 weeks resulted in a sequential rise in serum P concentration as seen in Fig. 2; of note, the increase in FGF23 mRNA expression relative to low dietary P levels occurred only in mice exposed to normal dietary P but did not occur in high dietary P relative to normal dietary P. We also noted a progressive increase in iFGF23 protein in each case as a function of P content in the diet. Recent studies suggest that this latter increase due to high serum P levels may be the result of processing alone (40). 1,25(OH)2D3, on the other hand, induces both osseous Fgf23 expression as well as iFGF23 levels under all dietary P regimens. These data also indicated that acute administration of 1,25(OH)2D3 after exposure to increasing dietary P levels prompted a further elevation in serum P limited to low and normal dietary P conditions. Because the effects of 1,25(OH)2D3 on FGF23 content in both bone and blood under conditions of high dietary P occurred in the absence of an increase in serum P, as is evident in Fig. 2, these data support the idea that the actions of 1,25(OH)2D3 are independent of its ability to upregulate P in the blood (41). These data also indicate that P exerts potential transcriptional as well as nontranscriptional mechanisms to upregulate FGF23, whereas the effects of 1,25(OH)2D3 are most likely exclusively transcriptional in nature.
Figure 2.
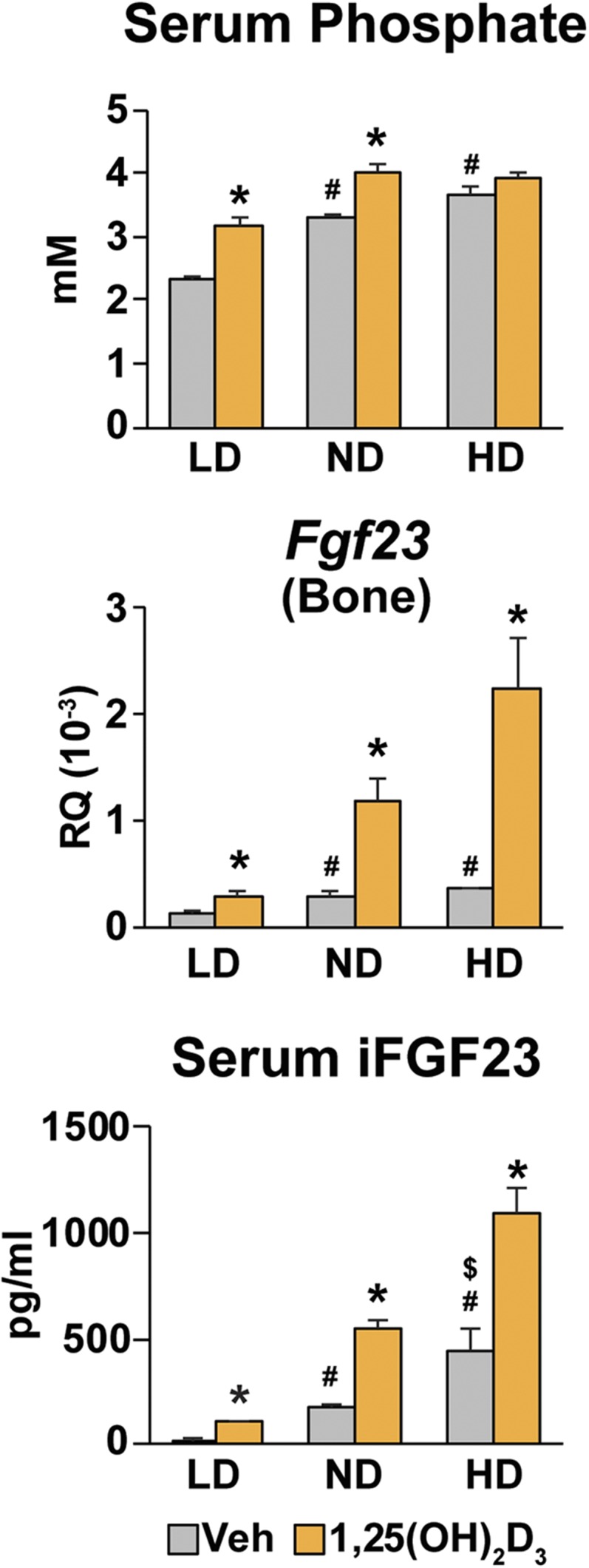
Regulation of Fgf23 expression and serum iFGF23 in response to P and 1,25(OH)2D3 in mice. C57BL/6 males at 4 wk of age were fed diets containing 0.1% P [low-phosphate diet (LD)], 0.6% P [normal diet (ND)], or 1.65% P [high-phosphate diet (HD)] until 6 wk of age and then treated with either vehicle (Veh) or 10 ng/g body weight of 1,25(OH)2D3 for 6 h. Serum P and iFGF23 levels and Fgf23 expression level in bone are presented. Gene expression represents relative quantitation (RQ) normalized to Gapdh (ΔΔCT). Each value is the average of six mice per group, and error bars represent SEM. *P < 0.05 compared with vehicle controls; #P < 0.05 compared with LD-fed vehicle controls; $P < 0.05 compared with ND-fed vehicle controls.
Increased dietary uptake of P induces Fgf23 expression via an Fgf23 proximal regulatory region‒mediated genomic mechanism
Although the effect of P on FGF23 processing and secretion is generally understood, direct evidence for a transcriptional mechanism of action of P on Fgf23 expression in bone and other tissues has not been established. This is due largely to the uncertainties of utilizing cell lines to explore the effects of P on Fgf23 transcription and to the inability of traditional methods of cell-based assays to identify regulatory regions that operate in the context of the intact Fgf23 gene (28, 42). As can be seen in Fig. 2, these uncertainties also extend to experiments in which P upregulation of Fgf23 via dietary studies in vivo used exposure times that ranged from days to weeks, placing the observed outcome far from any initial mechanism.
To establish whether P provokes a transcriptional upregulation of FGF23, we developed an approach to assess the temporal nature of Fgf23 response to P in mouse tissues in vivo as described in the “Materials and Methods” and then examined this response in the context of loss of enhancer function experiments in the Fgf23 enhancer-deleted mouse strains. The deletion locations, enhancer boundaries, and strain designations are indicated in Fig. 1. One of these mutant strains (FGF23-16KO) was used in an earlier study to identify the inflammation-mediating properties of a distal Fgf23 enhancer at 16 kb upstream of the Fgf23 transcription start site (TSS), thereby validating this general approach (31). To achieve the first objective, we initially conducted a temporal evaluation similar to that outlined in Fig. 2, wherein we explored earlier time points; these studies revealed that all of the effects of dietary P on serum P and Fgf23 expression were fully evident as early as 24 hours after exposure to different concentrations of dietary P (data not shown).
On the basis of this finding, we then fed FGF23-PKO, FGF23-16KO, FGF23-38KO, and FGF23-IKO mice and their wild-type littermate controls a P-deficient diet for 1 week to lower Fgf23 expression and then fed them for an additional 24 hours with either the same P-deficient diet (basal control) or one containing high dietary P (experimental test). Mice were euthanized, and serum P, bone FGF23 mRNA, and iFGF23 protein levels in the blood were examined. As can be seen in Fig. 3, each of these FGF23 parameters was upregulated at 24 hours in virtually all the wild-type littermate controls for each of the mutant strains. High-P diet‒fed FGF23-PKO mice showed significantly higher serum P levels than their wild-type counterparts, although the difference was not observed in the other mutant strains. Importantly, however, although serum P level was upregulated in the FGF23-PKO mouse, a corresponding upregulation of osseous FGF23 mRNA in these mice was almost completely abrogated, an effect that also resulted in a reduction in the levels of iFGF23 in the blood. It appears that reduced levels of both Fgf23 expression and iFGF23 may be responsible for the higher serum P levels in this mouse strain relative to those in wild-type controls. This dietary P–induced increase in FGF23 mRNA was also compromised in the FGF23-16KO mouse, although this effect was not statistically significant and was likely due to the reduced basal expression of FGF23 mRNA, as the fold-induction of FGF23 was unaffected. Perhaps more importantly, this reduction of the level of upregulation was not evident at the level of iFGF23 in the blood. Dietary P had no effect on FGF23 in either FGF23-38KO or FGF23-IKO mice where the regions that bind CTCF were deleted (31) and therefore may be involved at this locus in other genomic functions. These results suggest that the effects of dietary P on Fgf23 expression are not only relatively rapid, but they are also likely mediated via specific upstream DNA sequences excised in the FGF23-PKO mouse.
Figure 3.

Proximal regulatory region‒mediated genomic regulation of Fgf23 expression by increased dietary intake of P. FGF23-PKO (PKO), FGF23-16KO (16KO), FGF23-38KO (38KO), and FGF23-IKO (IKO) mice and their wild-type (WT) littermates were fed P-deficient diets (0.02% P) at 5 wk of age for 1 wk and then fed either a P-deficient diet (gray bars) or a high-P diet (red bars) for 24 h. Serum P and iFGF23 levels and Fgf23 expression level in bone are presented. Gene expression level represents relative quantitation (RQ) normalized to Gapdh (ΔΔCT). Each value is the average of six mice per group, and error bars represent SEM. *P < 0.05 compared with P-deficient diet‒fed controls; #P < 0.05 compared with WT littermates fed the same diet.
P-induced FGF23 mRNA expression is rapid and reminiscent of hormonal action
Given the initial finding that FGF23 induction was likely transcriptional in nature, we next explored simultaneously the temporal and transcriptional features of the FGF23 response to dietary P. To maximize response, we fed wild-type littermate controls and FGF23-PKO mice a P-deficient diet for 1 week, and then after a brief dietary restriction, the mice were transferred to a high-P diet, as described in the “Materials and Methods.” The mice were harvested at time points beginning at 3 hours, and the levels of serum P, bone FGF23 mRNA, and blood iFGF23 protein were assessed. As can be seen in Fig. 4A, an increase in serum P level was evident at the earliest time point in both control and FGF23-PKO mice. Of note, however, although serum P continued to rise at 6 hours in the FGF23-PKO mouse, it began to decline in wild-type mice; both declined by 24 hours. FGF23 mRNA levels in control mice, on the other hand, were also strongly increased by P at 3 hours, reaching maximum induction at 6 hours that was stable at 24 hours as well. However, no effect of dietary P was observed on bone Fgf23 expression at 3 or 6 hours in FGF23-PKO mice, although levels were modestly upregulated at 24 hours. iFGF23 levels in the blood were also increased in control mice; however, as was anticipated for a secondary process (protein synthesis following by secretion), this increase was not apparent until 6 hours and was further upregulated by 24 hours. In FGF23-PKO mice, however, iFGF23 remained unperturbed at 3 and 6 hours as was observed also for FGF23 mRNA, but it was then also modestly upregulated at 24 hours. This delayed upregulation of both FGF23 mRNA and protein in the FGF23-PKO mice indicates that in addition to an acute transcriptional mechanism of P action, both FGF23 mRNA and protein are also upregulated by dietary P through a chronic mechanism that appears secondary to blood P. This latter mechanism could be either transcriptional [via the induction of an additional systemic regulator(s) operating via a second Fgf23 enhancer] or posttranscriptional in nature.
Figure 4.

Primary and secondary responses of Fgf23 expression in response to dietary P intake. (A and C) FGF23-PKO and wild-type (WT) littermates were fed P-deficient diets at 5 wk of age for 1 wk and then a high-P diet for 0, 3, 6, or 24 h. Serum P and iFGF23 levels and expression levels of the indicated genes in the tissues described are presented. Gene expression level represents relative quantitation (RQ) normalized to Gapdh (ΔΔCT). Each value is the average of four to six mice per group, and error bars represent SEM. *P < 0.05 compared with 0-hour groups; #P < 0.05 compared with WT littermates fed the high-P diet for the same period. (B) FGF23-PKO and WT littermates at 4 wk of age were fed diets containing 0.1% P [low-phosphate diet (LD)], 0.6% P [normal diet (ND)], or 1.65% P [high-phosphate diet (HD)] until 6 wk of age. Serum P and iFGF23 levels and Fgf23 expression level in bone are presented. Gene expression level represents RQ normalized to Gapdh (ΔΔCT). Each value is the average of six to eight mice per group, and error bars represent SEM. #P < 0.05 compared with the same genotype mice fed an LD; $P < 0.05 compared with the same genotype mice fed an ND. No statistical difference was found between FGF23-PKO and WT littermates.
To determine whether FGF23 regulation reached equilibrium beyond 24 hours in the absence of the acute transactivation that drives Fgf23 expression in normal mice, we placed both wild-type and FGF23-PKO mice on low, normal, and high P diets for 2 weeks and then examined serum P levels and both osseous FGF23 mRNA and iFGF23 levels in both mouse strains. As can be seen in Fig. 4B, although normal P content in the diet increased all FGF23-related parameters above those seen under low–dietary P conditions, neither was affected by the absence of the acute transcriptional mechanism that led to rapid normalization of blood iFGF23 level. The absence of the acute mechanism also had no effect on the ability of high dietary P to raise FGF23 mRNA levels, although these levels were also not elevated above those seen in control mice on a normal P diet.
Finally, although increasing the P content in the diet elevated blood levels of iFGF23, the absence of the acute mechanism again had no effect on eventual iFGF23 levels in the blood. This finding suggests that P exhibits a multifaceted mechanism to upregulate FGF23 and that the traditional long-term treatment of mice under variable dietary P conditions may obscure an important acute mechanism of P action.
P upregulates the expression of additional genes involved in the control of bone metabolism
To further validate the early actions of P on gene transcription, we examined the possibility that dietary P might induce additional genes involved in bone mineral metabolism. Klotho (Kl) represents a coreceptor for FGF23 action and is a primary determinant of the selective activity of FGF23 in the kidney to promote P diuresis (43, 44). Dentin matrix protein1 (Dmp1), on the other hand, represents a known mediator of P regulation, and its deletion results in a disease phenotype of P metabolism in humans similar to that of FGF23 loss of function (19). Additional regulators involved in P include osteopontin (Spp1), one of many mineralization inhibitors (45), and Receptor Activator of Nuclear Factor κΒ Ligand (Tnfsf11), a key factor involved in osteoclast-mediated bone resorption (46). The results in Fig. 4C reveal that with the exception of Kl, an FGF23 coreceptor, each of these genes is acutely upregulated by dietary P in a manner not unlike that evident for FGF23. Kl, on the other hand, is slowly downregulated, a suppression that was noted previously (44). These findings suggest that the mechanism of P induction of FGF23 may be similar for additional bone genes related to P metabolism, thereby providing an advantage in future studies for identifying the specific transcription factors and perhaps the upstream signaling pathways responsible for the actions of serum P.
1,25(OH)2D3 induction of Fgf23 is mediated via a DNA segment located upstream of the Fgf23 gene promoter
The actions of 1,25(OH)2D3 have been well characterized and are mediated by the nuclear VDR and its heterodimer partner, the retinoid X receptor (47, 48). However, early studies of mouse FGF23 regulation by 1,25(OH)2D3 using a traditional cell-based transfection approach suggested that the actions of this hormone were mediated by the VDR largely via a single VDRE located approximately 1 kb upstream of the Fgf23 gene (27). In silico studies, on the other hand, have suggested the presence of multiple VDREs located both upstream and downstream of the human FGF23 gene; several of these exhibited considerable homology with that of the mouse gene, whereas others revealed functional capabilities when transfected into cell lines in vitro (49). Most recent analyses, however, have focused on the more proximal region of the mouse Fgf23 gene, where both the involvement of a NURR1-like element and the possibility of a secondary mechanism have been suggested (28).
Given these diverse observations, we first used the mutant mouse models that we had created to narrow the general genomic location of the element(s) responsible for the transcriptional actions of 1,25(OH)2D3 on Fgf23 expression and to assess the effects of the hormone on serum P and iFGF23. The results in Fig. 5A reveal that although 1,25(OH)2D3 strongly induced both Fgf23 expression in wild-type littermate controls from all the mutant strains of mice, the induction of osseous FGF23 mRNA and iFGF23 in the blood was almost completely lost in FGF23-PKO mice. Thus, the activity of 1,25(OH)2D3 appears to be largely mediated via this proximal segment of DNA and not through segments deleted in the FGF23-38KO, FGF23-16KO, or FGF23-IKO mutant mice. Because the deletion in the FGF23-PKO mouse is large, however, these results do not resolve whether or which of the active VDREs identified within this region could represent the active element(s) involved.
Figure 5.

Proximal regulatory region‒mediated genomic regulation of Fgf23 expression by 1,25(OH)2D3. (A) FGF23-PKO (PKO), FGF23-16KO (16KO), FGF23-38KO (38KO), and FGF23-IKO (IKO) mice and their wild-type (WT) littermates were treated with either vehicle (Veh) or 10 ng/g bw of 1,25(OH)2D3 for 6 h. Serum P and iFGF23 levels and Fgf23 expression level in bone are presented. Gene expression level represents relative quantitation (RQ) normalized to Gapdh (ΔΔCT). Each value is the average of six to eight mice per group, and error bars represent SEM. *P < 0.05 compared with vehicle controls; #P < 0.05 compared with WT littermates that received the same treatments. (B) FGF23-PKO and WT littermates were treated with 10 ng/g bw 1,25(OH)2D3 for 0, 1, 3, 6, 12, and 24 h. Serum P and iFGF23 levels and expression levels of the indicated genes in bone are presented. Gene expression level represents RQ normalized to Gapdh (ΔΔCT). Each value is the average of four to eight mice per group, and error bars represent SEM. *P < 0.05 compared with 0-h groups; #P < 0.05 compared with WT littermates that received the same treatments.
We then contrasted the temporal response of bone Fgf23 with that of Cyp24a1, a classic vitamin D target gene, in both wild-type control and FGF23-PKO mice, assessing the temporal effects of the hormone on serum P as well. As can be seen in Fig. 5B, although the temporal induction profiles by 1,25(OH)2D3 were similar for both Cyp24a1 and Fgf23, they were not identical. Accordingly, the induction of Cyp24a1 was highly robust with the induction of FGF23 mRNA, peaking at 6 and 12 hours, respectively. However, as seen in Fig. 5B, these responses provided no clear evidence for an indirect induction of FGF23 by 1,25(OH)2D3in vivo. Of note, 1,25(OH)2D3 also induced a significant increase in blood P levels (Fig. 5B), presumably through the rapid induction of intestinal P uptake. It remains possible that this increase in serum P could influence Fgf23 upregulation. In total, these results suggest that the actions of 1,25(OH)2D3 on Fgf23 expression are both direct and transcriptionally mediated. Further dissection together with chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) analyses is necessary to resolve the precise locations of both P and 1,25(OH)2D3 actions and to identify the VDRE sequence(s) essential for 1,25(OH)2D3‒mediated activation of FGF23.
Transcriptional upregulation of FGF23 by inflammatory mediators in multiple tissues is co-mediated via the proximal enhancer in the Fgf23 gene
Inflammatory mediators that include LPS, IL-1β, and TNFα, which are present during diseases of inflammation such as CKD, appear to induce Fgf23 expression in bone as well as in a variety of additional tissues (22). Indeed, we showed in recent studies of Fgf23 induction that the actions of these mediators to induce FGF23 were partially compromised in FGF23-16KO mice, although we also noted a baseline reduction (31). The presence of DNA sequence motifs in this enhancer reinforced the possibility that factors active in the induction of inflammation, such as nuclear factor κB (NF-κB), HIF-1α, and/or STAT proteins, could be involved in vivo. Importantly, earlier studies in primary bone cells and cell lines in vitro had also indicated that inflammatory activity might be mediated via these factors through elements located within a segment near the Fgf23 promoter (22, 29).
We therefore assessed whether the proximal region in the FGF23-PKO mouse as well as those in FGF23-38KO and FGF23-IKO mice might also contribute to Fgf23 upregulation in vivo. As can be seen in Fig. 6, although LPS induced control IL-6 expression in multiple tissues of both wild-type controls and in virtually all mutant mouse strains tested, normal induction of Fgf23 expression and upregulation of iFGF23 protein in the blood by LPS observed in wild-type FGF23-PKO littermate controls was significantly reduced in FGF23-PKO mice (Fig. 6A). LPS induction of FGF23 production was unaffected in the enhancer-deleted mice derived from FGF23-38KO and FGF23-IKO strains of mice (Fig. 6B and 6C). These experiments confirm that the proximal enhancer region of Fgf23 is indeed involved in mediating inflammatory response in vivo, suggesting that this region as well as the distal enhancer located 16 kb upstream of the Fgf23 gene are likely involved (31). This finding identifies an additional mechanism through which high levels of FGF23 may be produced during diseases characterized by inflammation.
Figure 6.

Proximal regulatory region is involved in Fgf23 expression induced by LPS. (A) FGF23-PKO (PKO), (B) FGF23-38KO (38KO), and (C) FGF23-IKO (IKO) mice and their wild-type (WT) littermates were treated with either vehicle (Veh) or 10 mg/kg bw of LPS for 6 h. Levels of serum iFGF23 and Fgf23 and Il6 expression in the indicated tissues are presented. Gene expression level represents relative quantitation (RQ) normalized to Gapdh (ΔΔCT). Each value is the average of six to eight mice per group, and error bars represent SEM. *P < 0.05 compared with vehicle controls; #P < 0.05 compared with WT littermates that received the same treatments. Fgf23 levels were undetectable by RT-PCR. UD, undetectable.
The proximal regulatory region of Fgf23 includes potential binding motifs for the transcription factors involved in regulation of Fgf23 expression in response to P, 1,25(OH)2D3, and inflammatory stimuli
The 4 kb of sequence deleted upstream of the Fgf23 TSS in the FGF23-PKO mouse appears to mediate the induction of FGF23 by P and 1,25(OH)2D3 and contribute to induction by the inflammatory mediator LPS. This segment is composed of abundant repetitive sequences that appear to be more predominant in the distal region of this enhancer, as indicated in Fig. 7A. Importantly, a total of 1265 motifs for a variety of putative transcription factors were identified within this 4-kb sequence using the Genomatix MatInspector program (data not shown), including those for the transcription factors involved in FGF23 regulation, such as VDR [1,25(OH)2D3 response], NF-κB, and nuclear factor of activated T cells (NFAT; inflammatory response) (27, 28, 49–51), as seen in Fig. 7. Although mammalian transcription factors for P activation have yet to be identified, motifs for the E-box transcription factor μE3, a transcription factor belonging to a large family of regulators that may mediate P induction of NaPi2a (52, 53), are also seen. The presence of each of these motifs within a genomic segment now established as functionally active in vivo provides the impetus for further dissection and eventual determination of the involvement of these putative transcription factors in Fgf23 expression and for the identification of their specific DNA sites of action.
Figure 7.

Putative transcription factor binding motifs in proximal enhancer region. (A) Repetitive sequences defined by the University of California, Santa Cruz Genome Browser (genome.uscs.edu) are marked in the proximal regulatory region as gray boxes. Locations of potential binding motifs of MITF/TFE3, VDR/RXR, NFAT, and NF-κB are presented. Transcription start site is shown by an arrow. (B) Putative transcription factor binding motifs were predicted by MatInspector as described in the “Materials and Methods.” Matrix names, matrix similarity (Matrix sim), sequences, and locations of the motifs for MITF/TFE3, VDR/RXR, NFAT, and NF-κB are presented. MIT, microphthalmia transcription factor; RXR, retinoid X receptor; TSS, transcription start site.
Discussion
FGF23 is modulated at both transcriptional and posttranscriptional levels by a multitude of local and systemic regulators (19). In this report, we explored the genomic sites of action of P and 1,25(OH)2D3, two major physiological regulators, as well as LPS, a recently discovered FGF23 inducer linked to inflammation. The effects of each of these physiological and pathological agents are not limited to the transcriptional control of FGF23 but can frequently extend to posttranscriptional mechanisms as well. Accordingly, we showed through loss of enhancer function studies in mice that the actions of each of these three inducers are mediated via elements located within a segment immediately upstream of the Fgf23 gene promoter. Although LPS is also active within a more distal region −16 kb upstream, none of these regulators is active at sites located at −38 kb or within a downstream intron at +7 kb. The Fgf23 proximal regulatory domain that we examined via deletion extends upstream of the Fgf23 TSS from −115 to −4110 bp and is highly conserved. It contains potential binding site motifs for a number of transcription factors that are known mediators of 1,25(OH)2D3, inflammation, and other modulators of Fgf23 expression. On the basis of our findings as well as those of others, it is evident that the Fgf23 gene is regulated by at least two separate functional enhancers. Future analyses will enable a refinement of the sites of actions of P, 1,25(OH)2D3, and inflammation and provide crucial insights into the molecular mechanisms through which they operate.
The mechanisms of P regulation of transcription in mammalian cells are highly complex and have in most cases eluded molecular evaluation. Studies in yeast have identified a single classic P regulatory paradigm involving the PHO regulon, where a complex signaling pathway transduces P status to some 20 genes via P sensors, protein kinases, and PHO4, a key transcription factor of P regulation (54, 55). This factor is highly regulated by the yeast P transduction pathway, binds to DNA, and functions much like that of numerous mammalian transcriptional modulators to regulate the PHO regulon. This regulation occurs through the modulation of chromatin structure, analogous to that which occurs in higher organisms. Analogues of Pho4 do not appear to be expressed in mammalian cells, however. In mice and humans, numerous dietary/nutritional studies have revealed the broad effects of altered serum P content on a myriad of biochemical and cellular processes in tissues, including bone, and on the regulation of genes. These effects are frequently measured over time frames that disallow the elucidation of mechanisms.
With regard to the regulation of FGF23, much effort has gone into the study of P induction of this gene using both measurements of FGF23 mRNA and analyses of cloned Fgf23 promoter constructs to identify P-sensitive regions (3). None of these attempts has resulted in an understanding of the transcriptional control by P or of the identification of mechanism(s) that could provide an experimental basis for further effort. In the present studies, however, we used the well-established upregulatory response of FGF23 to dietary P and developed a straightforward method with which to determine the temporal characteristics of inducible response to dietary P. As was demonstrated, the effect of dietary P on Fgf23 expression and blood iFGF23 levels was acute, occurring within 3 and 6 hours, respectively. The physiological consequence of this rapid action to upregulate FGF23 was also self-evident, as a decrease in serum P levels became apparent by 6 hours in wild-type mice but not in FGF23-PKO mice. It was also fully dependent on a genomic segment present upstream of the Fgf23 gene that suggested a transcriptional underpinning. Deletion of this segment did not prevent a delayed secondary upregulation of Fgf23 expression that was first evident within 24 hours and that led to complete normalization of both FGF23 mRNA in bone and protein levels in the blood a few days later. Although the acute action is clearly transcriptional in nature, the mechanism of the latter chronic action is uncertain; this process could occur either through transcriptional or posttranscriptional/posttranslational mechanisms. The presence of this secondary mechanism may have obscured our understanding of P regulation of FGF23 in vivo. Of note, this rapid effect of P also appears to affect other genes, most prominently Dmp1. Further dissection of both the acute effects of P within the segment located upstream of the Fgf23 promoter and the potential localization of the chronic upregulation of FGF23 to other regions of the Fgf23 gene will allow us to identify important DNA sequences, transcription factors, and perhaps upstream signaling components involved in P action.
Deletion of the proximal enhancer segment in the FGF23-PKO mouse also led to a loss of FGF23 response to 1,25(OH)2D3. This observation is important because it is consistent with several previous localization studies of FGF23 action by the vitamin D hormone using traditional methods in cell culture. However, the work here does not resolve which or whether either of these elements is involved. However, these earlier studies diverge with respect to the localization of functional sites of action, with one describing the presence of a VDRE approximately −1038 nt upstream of the TSS and the second at a classic NURR1-like nuclear receptor response element at −247 nt that could mediate response to 1,25(OH)2D3 (27, 28). Although components of the canonical vitamin D signaling pathway that include the VDR as well as target VDREs are known, Saini et al. (49) also suggested that this effect may be a secondary one mediated by direct upregulation of the VDR by additional transcription factors active at the Fgf23 gene.
Clearly, evidence for a direct effect of 1,25(OH)2D3 at FGF23 will require an assessment of the presence of the VDR at one or more of these putative sites, an effort that, while successful at numerous vitamin D‒regulated genes using ChIP-seq analysis in an osteocyte cell line, failed to provide evidence of VDR occupancy at sites within the FGF23 locus (30). Earlier studies also identified additional distal putative VDREs located outside the deleted proximal enhancer segment at Fgf23 (49). However, although several of these elements are located within the deletions created in the FGF23-38KO and FGF23-IKO mice, it is worth noting that there were no apparent consequences to the induction of Fgf23 in these mice; the remaining locations of the potential VDREs were not explored because they do not retain the characteristic epigenetic signatures of enhancers.
1,25(OH)2D3 treatment is normally accompanied by a transient increase in blood P level. This upregulation of P by 1,25(OH)2D3 suggests that the actions of the hormone could be mediated or synergistically enhanced by serum P. Occurrence of the former seems unlikely, however, because while a chronically administered diet high in P upregulates iFGF23 but not serum P or Fgf23 (compared with a normal P diet), all three parameters are increased upon further acute cotreatment with 1,25(OH)2D3. This suggests that the effects of the vitamin D hormone are mechanistically independent of P. Nevertheless, assessment of this question requires further dissection of the proximal enhancer region of Fgf23 in vivo to determine whether the sites of action of P and 1,25(OH)2D3 are distinct.
The induction of FGF23 by inflammatory mediators represents a more recent discovery (22, 50). Indeed, these factors are known to be associated with FGF23 upregulation in human diseases such as CKD and inflammatory bowel disease (19). This effector of FGF23 upregulation is of clear importance because it provokes elaboration of FGF23 from numerous tissues rather than exclusively from bone, and the consequence of this upregulation on blood levels of iFGF23 is profound. Importantly, this induction also leads to levels of FGF23 that appear to bypass the requirement for Klotho as a coreceptor of the FGF23 receptor FGFR1c in the kidney and parathyroid glands, which enables a low affinity interaction of FGF23 with other members of the FGFR family in the cardiovascular system, the heart, and likely elsewhere. These interactions are largely pathological in nature and result in increased mortality in humans (56–58). Inflammation, like that of P, also affects FGF23 processing (22, 23). Thus, the levels of iFGF23 protein in the blood can far surpass the upregulation observed at the levels of transcription.
The results provided from this study in the FGF23-PKO mouse suggest that the induction of FGF23 is mediated by sequences located directly upstream of the Fgf23 promoter. Thus, this regulatory region, together with the enhancer at −16 kb that was explored previously in the FGF23-16KO mouse, participates collectively in the transcriptional regulation of Fgf23 (31). Importantly, this proximal region, like that of the distal −16 kb region, contains numerous sequence motifs capable of binding transcription factors known to mediate the actions of inflammatory factors, including those for NF-κB, NFAT, and STAT family members. This observation is consistent with previous studies using traditional in vitro approaches, which provided evidence via the use of transactivation inhibitors that NF-κB, NFAT, and/or HIF-1α may be directly involved (22, 50, 51). However, further dissection as well as ChIP or ChIP-seq analysis at the Fgf23 locus will be required to identify the actual transcription factors in question and to show which of the sequences present in these regions are direct targets of these factors. It is also possible that these two regions may interact either additively or synergistically to facilitate the induction of FGF23, thereby providing an additional mechanism through which FGF23 can be strongly upregulated via inflammation. An interaction such as this could well contribute to the high levels of FGF23 that are evident in inflammatory states such as CKD and inflammatory bowel disease.
CKD is characterized by uremia, alterations in circulating hormones involved in mineral metabolism such as PTH upregulation and vitamin D metabolite suppressions, and in striking disturbances in P metabolism (21, 59). All of these have the potential to affect FGF23 expression, as demonstrated in this report, which begins to rise early in the development of CKD. The initial effect seems likely to be inflammation, whereby the sensitivity of the gene to inflammatory mediators has the potential to be extreme owing in part to the actions of two enhancers that act in multiple tissues. It is interesting that chronic challenge of high dietary P on FGF23 regulation is driven largely by homeostatic FGF23 processing rather than transcription (40). However, it is also curious that dietary P overload for 8 weeks dose dependently induced inflammation in a CKD rat model and that serum P level in patients with advanced CKD is associated with proinflammatory cytokines such as IL-6 (60, 61). Thus, hyperphosphatemia caused by a failure of renal P excretion in later stages of CKD may upregulate FGF23 through both transcriptional and posttranslational processing mechanisms (23). Because high FGF23 levels are associated with cardiovascular disease and increased mortality in patients with CKD (58, 62, 63), further efforts to understand both the primary and secondary mechanisms of P action are warranted.
In conclusion, we used an invivo approach to show that FGF23 regulation by P and 1,25(OH)2D3 is mediated via a proximal enhancer region, whereas inflammation is mediated via this as well as a more distal region. Although further studies are necessary to dissect sites of action and the molecular mechanisms that will likely illuminate the underlying mechanisms responsible for P regulation, the current studies establish unequivocal functions for these regions within the mouse Fgf23 gene, thereby setting the stage for future analyses.
Acknowledgments
We thank Dustin Irving and Douglas Jacobson for the animal husbandry associated with this study.
Financial Support: This study was supported by National Institutes of Health Grants AR064424 and DK118174 (to J.W.P.).
Glossary
Abbreviations:
- 1,25(OH)2D3
1,25-dihydroxyvitamin D3
- bw
body weight
- Cas9
clustered regularly interspaced short palindromic repeats–-associated protein-9 nuclease
- ChIP-seq
chromatin immunoprecipitation followed by DNA sequencing
- CKD
chronic kidney disease
- CRISPR
clustered regularly interspaced short palindromic repeats
- FGF23
fibroblast growth factor 23
- iFGF23
intact fibroblast growth factor 23
- LPS
lipopolysaccharide
- NFAT
nuclear factor of activated T cells
- NF-κB
nuclear factor κB
- P
phosphate
- TSS
transcription start site
- VDR
vitamin D receptor
- VDRE
vitamin D response element
Additional Information
Disclosure Summary: The authors have nothing to disclose.
Data Availability: All data generated or analyzed during this study are included in this published article or in the data repositories listed in References.
References and Notes
- 1. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98(11):6500–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75(1):503–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012;8(5):276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280(4):2543–2549. [DOI] [PubMed] [Google Scholar]
- 5. Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16(2):221–232. [DOI] [PubMed] [Google Scholar]
- 6. Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18(7):1227–1234. [DOI] [PubMed] [Google Scholar]
- 7. White KE, Hum JM, Econs MJ. Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis. Curr Osteoporos Rep. 2014;12(3):252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117(12):4003–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krajisnik T, Björklund P, Marsell R, Ljunggren O, Akerström G, Jonsson KB, Westin G, Larsson TE. Fibroblast growth factor-23 regulates parathyroid hormone and 1α-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195(1):125–131. [DOI] [PubMed] [Google Scholar]
- 10. López I, Rodríguez-Ortiz ME, Almadén Y, Guerrero F, de Oca AM, Pineda C, Shalhoub V, Rodríguez M, Aguilera-Tejero E. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 2011;80(5):475–482. [DOI] [PubMed] [Google Scholar]
- 11. Meyer MB, Benkusky NA, Kaufmann M, Lee SM, Onal M, Jones G, Pike JW. A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J Biol Chem. 2017;292(42):17541–17558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer MB, Benkusky NA, Kaufmann M, Lee SM, Redfield RR, Jones G, Pike JW. Targeted genomic deletions identify diverse enhancer functions and generate a kidney-specific, endocrine-deficient Cyp27b1 pseudo-null mouse. J Biol Chem. 2019;294(24):9518–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyer MB, Lee SM, Carlson AH, Benkusky NA, Kaufmann M, Jones G, Pike JW. A chromatin-based mechanism controls differential regulation of the cytochrome P450 gene Cyp24a1 in renal and non-renal tissues. J Biol Chem. 2019;294(39):14467–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(6Suppl)1689S–1696S. [DOI] [PubMed] [Google Scholar]
- 15. Haussler MR, Whitfield GK, Haussler CA, Sabir MS, Khan Z, Sandoval R, Jurutka PW. 1,25-Dihydroxyvitamin D and Klotho: a tale of two renal hormones coming of age. Vitam Horm. 2016;100:165–230. [DOI] [PubMed] [Google Scholar]
- 16. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–435. [DOI] [PubMed] [Google Scholar]
- 17. Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T, Fukumoto S, Yamashita T. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289(5):F1088–F1095. [DOI] [PubMed] [Google Scholar]
- 18. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079–2086. [DOI] [PubMed] [Google Scholar]
- 19. Bär L, Stournaras C, Lang F, Föller M. Regulation of fibroblast growth factor 23 (FGF23) in health and disease. FEBS Lett. 2019;593(15):1879–1900. [DOI] [PubMed] [Google Scholar]
- 20. Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, Fukagawa M. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44(2):250–256. [DOI] [PubMed] [Google Scholar]
- 21. Quarles LD. Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res. 2012;318(9):1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, Zumbrennen-Bullough KB, Sun CC, Lin HY, Babitt JL, Wolf M. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89(1):135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. David V, Francis C, Babitt JL. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol. 2017;312(1):F1–F8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Jüppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutiérrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Group. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305(23):2432–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Isakova T, Wahl P, Vargas GS, Gutiérrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–1315. [DOI] [PubMed] [Google Scholar]
- 28. Kaneko I, Saini RK, Griffin KP, Whitfield GK, Haussler MR, Jurutka PW. FGF23 gene regulation by 1,25-dihydroxyvitamin D: opposing effects in adipocytes and osteocytes. J Endocrinol. 2015;226(3):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Q, Doucet M, Tomlinson RE, Han X, Quarles LD, Collins MT, Clemens TL. The hypoxia-inducible factor-1α activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. 2016;4(1):16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. St. John HC, Bishop KA, Meyer MB, Benkusky NA, Leng N, Kendziorski C, Bonewald LF, Pike JW. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014;28(7):1150–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Onal M, Carlson AH, Thostenson JD, Benkusky NA, Meyer MB, Lee SM, Pike JW. A novel distal enhancer mediates inflammation-, PTH-, and early onset murine kidney disease-induced expression of the mouse Fgf23 Gene. JBMR Plus. 2018;2(1):31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meyer MB, Benkusky NA, Onal M, Pike JW. Selective regulation of Mmp13 by 1,25(OH)2D3, PTH, and Osterix through distal enhancers. J Steroid Biochem Mol Biol. 2016;164:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meyer M, de Angelis MH, Wurst W, Kühn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci USA. 2010;107(34):15022–15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. RRID:AB_2813726, https://scicrunch.org/resolver/AB_2813726.
- 36. Lee SM, Bishop KA, Goellner JJ, O’Brien CA, Pike JW. Mouse and human BAC transgenes recapitulate tissue-specific expression of the vitamin D receptor in mice and rescue the VDR-null phenotype. Endocrinology. 2014;155(6):2064–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Onal M, St. John HC, Danielson AL, Pike JW. Deletion of the distal Tnfsf11 RL-D2 enhancer that contributes to PTH-mediated RANKL expression in osteoblast lineage cells results in a high bone mass phenotype in mice. J Bone Miner Res. 2016;31(2):416–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21(13):2933–2942. [DOI] [PubMed] [Google Scholar]
- 39. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, Nangaku M, Abe M, Matsuhisa M, Kato S, Matsumoto T, Fukumoto S. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci. 2019;116(23):11418–11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36(6):971–977. [DOI] [PubMed] [Google Scholar]
- 42. Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab. 2005;288(6):E1101–E1109. [DOI] [PubMed] [Google Scholar]
- 43. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. [DOI] [PubMed] [Google Scholar]
- 44. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem. 2006;281(10):6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, Lafage-Proust MH, Dresselaers T, Feng JQ, Bonewald LF, Meyer MB, Pike JW, Bouillon R, Carmeliet G. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest. 2012;122(5):1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. [DOI] [PubMed] [Google Scholar]
- 47. Pike JW, Meyer MB, Benkusky NA, Lee SM, St. John H, Carlson A, Onal M, Shamsuzzaman S. Genomic determinants of vitamin D-regulated gene expression. Vitam Horm. 2016;100:21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pike JW, Meyer MB, Lee SM, Onal M, Benkusky NA. The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J Clin Invest. 2017;127(4):1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saini RK, Kaneko I, Jurutka PW, Forster R, Hsieh A, Hsieh JC, Haussler MR, Whitfield GK. 1,25-dihydroxyvitamin D3 regulation of fibroblast growth factor-23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcif Tissue Int. 2013;92(4):339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ito N, Wijenayaka AR, Prideaux M, Kogawa M, Ormsby RT, Evdokiou A, Bonewald LF, Findlay DM, Atkins GJ. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cell Endocrinol. 2015;399:208–218. [DOI] [PubMed] [Google Scholar]
- 51. Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23 [published correction appears in J Biol Chem. 2015;290(33):20101]. J Biol Chem. 2015;290(16):10447–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kido S, Miyamoto K, Mizobuchi H, Taketani Y, Ohkido I, Ogawa N, Kaneko Y, Harashima S, Takeda E. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. 1999;274(40):28256–28263. [DOI] [PubMed] [Google Scholar]
- 53. Taketani Y, Segawa H, Chikamori M, Morita K, Tanaka K, Kido S, Yamamoto H, Iemori Y, Tatsumi S, Tsugawa N, Okano T, Kobayashi T, Miyamoto K, Takeda E. Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3: identification of a vitamin D-responsive element in the human NAPi-3 gene. J Biol Chem. 1998;273(23):14575–14581. [DOI] [PubMed] [Google Scholar]
- 54. Bergwitz C, Jüppner H. Phosphate sensing. Adv Chronic Kidney Dis. 2011;18(2):132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kang HJ, Jeong SJ, Kim KN, Baek IJ, Chang M, Kang CM, Park YS, Yun CW. A novel protein, Pho92, has a conserved YTH domain and regulates phosphate metabolism by decreasing the mRNA stability of PHO4 in Saccharomyces cerevisiae. Biochem J. 2014;457(3):391–400. [DOI] [PubMed] [Google Scholar]
- 56. Smith K, deFilippi C, Isakova T, Gutiérrez OM, Laliberte K, Seliger S, Kelley W, Duh SH, Hise M, Christenson R, Wolf M, Januzzi J. Fibroblast growth factor 23, high-sensitivity cardiac troponin, and left ventricular hypertrophy in CKD. Am J Kidney Dis. 2013;61(1):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, Kiss I, Rosivall L, Kosa J, Lakatos P, Kovesdy CP, Mucsi I. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011;22(5):956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St. John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-o M, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chelluboina B, Vemuganti R. Chronic kidney disease in the pathogenesis of acute ischemic stroke. J Cereb Blood Flow Metab. 2019;39(10):1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamada S, Tokumoto M, Tatsumoto N, Taniguchi M, Noguchi H, Nakano T, Masutani K, Ooboshi H, Tsuruya K, Kitazono T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am J Physiol Renal Physiol. 2014;306(12):F1418–F1428. [DOI] [PubMed] [Google Scholar]
- 61. Navarro-González JF, Mora-Fernández C, Muros M, Herrera H, García J. Mineral metabolism and inflammation in chronic kidney disease patients: a cross-sectional study. Clin J Am Soc Nephrol. 2009;4(10):1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marthi A, Donovan K, Haynes R, Wheeler DC, Baigent C, Rooney CM, Landray MJ, Moe SM, Yang J, Holland L, di Giuseppe R, Bouma-de Krijger A, Mihaylova B, Herrington WG. Fibroblast growth factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J Am Soc Nephrol. 2018;29(7):2015–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mehta R, Cai X, Lee J, Scialla JJ, Bansal N, Sondheimer JH, Chen J, Hamm LL, Ricardo AC, Navaneethan SD, Deo R, Rahman M, Feldman HI, Go AS, Isakova T, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators. Association of fibroblast growth factor 23 with atrial fibrillation in chronic kidney disease, from the Chronic Renal Insufficiency Cohort Study. JAMA Cardiol. 2016;1(5):548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]