

Abstract
Neuroinflammation and oligodendroglial cytoplasmic α‐synuclein (α‐syn) inclusions (GCIs) are important neuropathological characteristics of multiple system atrophy (MSA). GCIs are known to interfere with oligodendroglial maturation and consequently result in myelin loss. The neuroinflammatory phenotype in the context of MSA, however, remains poorly understood. Here, we demonstrate MSA‐associated neuroinflammation being restricted to myeloid cells and tightly linked to oligodendroglial α‐syncleinopathy. In human putaminal post‐mortem tissue of MSA patients, neuroinflammation was observed in white matter regions only. This locally restricted neuroinflammation coincided with elevated numbers of α‐syn inclusions, while gray matter with less α‐synucleinopathy remained unaffected. In order to analyze the temporal pattern of neuroinflammation, a transgenic mouse model overexpressing human α‐syn under the control of an oligodendrocyte‐specific myelin basic protein (MBP) promoter (MBP29‐hα‐syn mice) was assessed in a pre‐symptomatic and symptomatic disease stage. Strikingly, we detected an increased neuroinflammation in regions with a high α‐syn load, the corpus callosum and the striatum, of MBP29‐hα‐syn mice, already at a pre‐symptomatic stage. Furthermore, this inflammatory response was restricted to myeloid cells being highly proliferative and showing an activated, phagocytic phenotype. In contrast, severe astrogliosis was observed only in gray matter regions of MSA patients as well as MBP29‐hα‐syn mice. To further characterize the influence of oligodendrocytes on initiation of the myeloid immune response, we performed RNA sequencing analysis of α‐syn overexpressing primary oligodendrocytes. A distinct gene expression profile including upregulation of cytokines important for myeloid cell attraction and proliferation was detected in α‐syn overexpressing oligodendrocytes. Additionally, microdissected tissue of MBP29‐hα‐syn mice exhibited a similar cellular gene expression profile in white matter regions even pre‐symptomatically. Collectively, these results imply an early crosstalk between neuroinflammation and oligodendrocytes containing α‐syn inclusions leading to an immune response locally restricted to white matter regions in MSA.
Keywords: multiple system atrophy, neuroinflammation, oligodendrocytes, α‐synuclein, white matter
Abbreviations
- α‐syn
α‐synuclein
- ctx
cortex
- cc
corpus callosum
- gm
gray matter
- MBP‐hα‐syn tg mice line 29
MBP29‐hα‐syn mice
- MC
medium change
- MSA
multiple system atrophy
- RNA‐Seq
RNA sequencing
- ROI
region of interest
- str
striatum
- Virus‐TD
virus transduction
- wm
white matter
Introduction
Multiple system atrophy (MSA) is a fast progressing, atypical parkinsonian disorder in which patients commonly present a severe autonomic dysfunction and poor levodopa responsiveness 26, 49, 69. In addition, MSA patients show two variants, the parkinsonian MSA (MSA‐P) characterized by motor symptoms like bradykinesia and postural instability due to the striatonigral degeneration and the cerebellar MSA (MSA‐C) with ataxia linked to olivopontocerebellar atrophy 49. The structural hallmark of MSA is the accumulation of oligodendroglial cytoplasmic inclusions (GCIs) composed of α‐synuclein (α‐syn) aggregates categorizing MSA as an α‐synucleinopathy 17, 63. Additional neuropathological characteristics are profound demyelination and neuronal loss correlating with disease duration and GCI density 45, 52. In order to study underlying molecular mechanisms of MSA, transgenic mouse models were generated expressing α‐syn under the control of oligodendrocyte‐specific promotors. These models demonstrate different behavioral and structural phenotypes mimicking important aspects of MSA 43, 76, 81, 88, 92. Shults et al described a transgenic mouse line overexpressing human α‐syn controlled by a myelin basic protein (MBP) promotor (line 29, MBP29‐hα‐syn mice). These transgenic mice develop a severe behavioral phenotype after 2–3 months of age and die prematurely after 4–6 months 76.
An important feature of MSA pathology is a marked neuroinflammation comprising micro‐ and astrogliosis associated with increased pro‐inflammatory cytokine levels 41, 42, 78. As the resident innate myeloid immune cells of the central nervous system (CNS), microglia constantly survey the brain parenchyma, building the first line of defense against pathogens 60. Upon activation due to protein aggregates, microglia change both their morphology as well as gene expression profile as observed in MSA, Parkinson’s (PD) and Alzheimer’s disease (AD) 67, 89. A direct impact of α‐syn on microglial activation and induction of pro‐inflammatory immune responses via uptake of α‐syn or binding on pathogen recognition receptors (PRRs) was observed in different in vitro as well as in vivo studies 8, 37, 82, 83, 93. Microglial‐specific positron emission tomography (PET) using the benzodiazepine receptor ligand [11C]‐PK11195 in MSA patients suggested a regionally distinct neuroinflammatory activity, for example, in the putamen and the substantia nigra 24. To date it is not yet known whether infiltrating monocytes or macrophages contribute to neuroinflammatory processes in MSA as well. The classical markers commonly used to identify resting and activated myeloid cells, such as IBA1, CD11b and CD68, do not allow exact distinction between resident microglia and infiltrated myeloid cells 14, 28, 32, 38, 48. In contrast, recruitment of peripheral myeloid cells as well as lymphocytes is described in human post‐mortem brain tissue of PD patients as well as in respective in vitro and in vivo models 31, 36, 77. Besides myeloid cells, astrocytes are also activated by oligodendroglial α‐syn leading to morphological changes and cytokine production 50, 70. In addition, elevated RNA levels of pro‐inflammatory cytokines, such as tumor necrosis factor‐α (TNF‐α), interleukin‐1β (IL‐1β) and C‐C motif chemokine ligand 2 (CCL2) were detected in the striatum and the substantia nigra of MSA patients 74.
Given that a profound neuroinflammation is observed in MSA and its mouse models the underlying spatial and temporal pattern is still not well understood in the context of α‐syn inclusions in MSA. Therefore, we hypothesized that oligodendroglial α‐synucleinopathy activates neuroinflammatory responses spatially restricted and early in the disease. In order to understand the spatial pattern of resident neuroinflammation in MSA‐P, we analyzed myeloid as well as astrocytic markers in white and gray matter regions in human post‐mortem tissue of MSA‐P patients containing different levels of GCIs. In addition to the rather end stage human post‐mortem analysis, we investigated the kinetic and the cellular phenotype of the neuroinflammatory response in the transgenic MSA mouse model, MBP29‐hα‐syn mice. Finally, to better understand the interaction of oligodendroglia and neuroinflammatory processes, we transcriptionally analyzed α‐syn overexpressing primary oligodendrocytes. To our knowledge this is the first study that directly contrasts the white and the gray matter‐specific inflammatory response and emphasizes an early pro‐inflammatory impact of oligodendroglial α‐synucleinopathy in MSA‐P.
Materials and Methods
Human post‐mortem samples
Human post‐mortem samples of MSA‐P patients and controls were obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam (open access: http://www.brainbank.nl). For research purposes tissue samples were collected from donors who agreed to a brain autopsy and the use of the material in conjunction with the clinical history 39. Five MSA‐P patients clinically and neuropathologically diagnosed according to current consensus guidelines and five age‐ and gender‐matched controls without neurological diseases or α‐syn pathology were examined (Table S1). Sections of paraffin‐embedded putamen were sliced in 5 µm thickness and stained according to standard protocols for Luxol Fast Blue/Periodic Acid Schiff (LFB/PAS) and immunohistochemistry. Antibodies used for antigen detection are listed in Table S2. Before chromogenic detection was started, pretreatment with citrate buffer was performed for antigen retrieval. Hematoxylin was used for counterstaining of the nuclei. The density of α‐syn+ inclusions as well as CD68+ and GFAP+ cells in at least five outlined white matter striae and adjacent gray matter was quantified using a stereology software (Stereo Investigator, MicroBrightField bioscience). KI67+ cells were quantified in the entire putamen using the Stereo Investigator software. The optical density of LFB/PAS was analyzed in overview images of the putaminal region. Mean gray value was calculated using the ImageJ software. CNP staining was used to quantify the area of myelinated striae. Therefore, the putaminal area was outlined and a threshold was set. Afterward, images of the putaminal areas were analyzed using the particle analysis plugin of ImageJ 75 determining the area of white matter striae.
Animals and tissue processing
Transgenic mice overexpressing human α‐syn under the control of a MBP promoter (MBP‐hα‐syn tg mice line 29, hereafter named: MBP29‐hα‐syn mice) and non‐transgenic control littermates were maintained under standard animal housing conditions with a 12‐h dark‐light cycle and free access to food and water 30, 76. Mice were bred on a B6D2F1 background and heterozygous male animals were analyzed in comparison to male age‐matched non‐transgenic littermates (controls). The MBP29‐hα‐syn mouse line is a strong expresser line with high levels of human α‐syn in the forebrain. These mice show severe neurological symptoms as well as premature death after 4–6 months of age 76 (see also Figure 3A). According to the European (2010/63/EU) and NIH (National Institute of Health) guidelines for the human treatment of animals, mice were euthanized under anesthesia and transcardially perfused with 0.9% sodium chloride solution. Forebrains were microdissected and one hemisphere was fixed in 4% paraformaldehyde overnight. After transfer into 30% sucrose, hemispheres were coronally sectioned (40 µm) for free‐floating histological analysis using a sliding microtome and kept in cryoprotection solution at −20°C until staining. For gene expression analysis the cortex (ctx), the corpus callosum (cc) and the striatum (str) were microdissected and stored at −80°C until tissue homogenization was performed.
Histology
Coronal brain sections of MBP29‐hα‐syn and control mice were stained as previously described 53. Primary antibodies used for specific antigen detection are listed in Table S2. For staining of α‐syn, CD68 and PCNA a pretreatment with citrate buffer (Dako) for 30 min at 80°C was necessary for antigen retrieval. Chromogenic detection of IBA1 and GFAP was performed using a donkey anti‐rabbit‐biotin and a donkey anti‐goat‐biotin antibody for labeling of the primary antibody, respectively (further information see Table S3). Subsequently, for coupling the peroxidase to the specific antigen, tissue sections were incubated using an ABC complex (Vectastain Elite ABC HRP Kit). Visualization was achieved using a 3,3′‐diaminobenzidine (DAB) peroxidase (HRP) substrate kit supplemented with Nickel according to manufacturer’s instructions (Vector laboratories). Cell density was calculated by framing the region of interest (ROI) and quantifying the cell number within the ROI using the Stereo Investigator software (MicroBrightField Bioscience). Alexa488‐, Alexa568‐ and Alexa647‐labeled antibodies were used for fluorescence staining (detailed information see Table S3). Nuclei were visualized using DAPI (Sigma, 1:10,000). Human α‐syn expression in distinct brain regions (cc, ctx and str) was analyzed performing Z stacks followed by colocalization analysis using the ZEN software (Zeiss). α‐syn+ pixels were measured in the ROI after thresholding. PCNA+ and IBA1+ cells were quantified within the ctx, the cc and the str using the cell counter plugin of ImageJ 75. CD68+ voxels were quantified using the 3D object counter plugin of ImageJ 9.
Quantitative real‐time PCR (qPCR)
RNA was isolated using QIAzol Lysis Reagent (Qiagen) for tissue homogenization. After addition of chloroform, phase separation was performed by centrifugation. RNA was purified using an RNeasy mini kit (Qiagen) according to manufacturer’s instructions and concentration was determined using Nano‐Drop technology (PeqLab). GoScriptTM Reverse Transcription System (Promega) was used for cDNA generation and gene transcription was quantified in a Light Cycler 480 (Roche) using the SSo Fast EvaGreen Supermix (Biorad). The following primers were used:
| Target genes | forward | reverse | references |
|---|---|---|---|
| Pu.1 | CAGAAGGGCAACCGCAAGAA | GCCGCTGAACTGGTAGGTGA | Gomez‐Nicola et al. 2013 29 |
| Trem2 | CTGGAGGACCCTCTAGATGAC | CCACAGGATGAAACCTGCCT | NM_001272078.1 |
| Tlr2 | ATGTCGTTCAAGGAGGTGCG | CCGGTGATGCAATTCGGATG | NM_011905.3 |
| Tlr4 | GAGTCAGAATGAGGACTGGGTGAG | GGAATAAAGTCTCTGTAGTGAAGGCAG | Henn et al. 2009 33 |
| Cxcl10 | GCTCAAGTGGCTGGGATG | GAGGCAAAGGAGGGTGTGG | Sutthiwarotamakun et al. 2011 85 |
| Ccl2 | CAGATGCAGTTAACGCCCCA | GCTTCTTTGGGACACCTGCT | NM_011333.3 |
| Housekeeping genes | forward | reverse | references |
|---|---|---|---|
| β‐Actin | GCCTTCCTTCTTGGGTATGGAA | CAGCTCAGTAACAGTCCGCC | Süß et al. 2015 84 |
| 18S rRNA | GGACCAGAGCGAAAGCATTTG | GCCAGTCGGCATCGTTTATG | Pernot et al. 2010 65 |
| Hprt | GTCATGTCGACCCTCAGTCC | GCAAGTCTTTCAGTCCTGTCC | Trias et al. 2016 86 |
| Pgk1 | GTCGTGATGAGGGTGGACTT | AACCGACTTGGCTCCATTGT | Trias et al. 2016 86 |
Lentiviral vectors
Third‐generation lentiviral vectors containing the coding sequence for human α‐syn or green fluorescent protein (GFP) were generated as previously described 90. The lentiviral back bone encodes the elongation factor 1‐α (EF1α) promotor driving a constitutive expression of human α‐syn and/or an internal ribosomal entry site (IRES) followed by GFP. For virus production HEK293T cells were transfected with the coding vector of EF1α‐hu‐α‐syn‐IRES‐GFP (α‐syn) or EF1α‐IRES‐GFP (control) and three packaging vectors: pMDL, pRevRSV and pVSV‐G. Viral titers of EF1α‐hu‐α‐syn‐IRES‐GFP and EF1α‐IRES‐GFP lentiviral constructs were determined via biological titration in HEK293T cells and flow cytometry analysis.
Primary OPC culture
Primary cultures were obtained from mixed glial cultures derived from neonatal Wistar rats at the postnatal ages P0 to P3 (modified according to 54). In brief, mixed glial cultures were grown in poly‐D‐lysine‐coated flasks with Dulbecco’s minimum eagle medium supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin at 37°C and 5% CO2 for 10–14 days. For removal of primary microglia, flasks were placed on an orbital shaker with 200 rpm at 37°C for 60 min, followed by medium change (MC). Primary OPCs were collected by additional shaking in freshly added medium at 200 rpm for 16 h. Cells were seeded on poly‐ornithine‐coated plates and cultured in SATO medium supplemented with 10 ng/mL platelet‐derived growth factor‐aa (PDGF‐aa) and 10 ng/mL fibroblast growth factor‐2 (FGF‐2) for proliferation and in SATO medium supplemented with 40 ng/mL 3,3′,5‐Triiodo‐L‐thyronine (T3) for differentiation 10. One day after separation of primary OPCs, cells were lentivirally transduced (virus‐TD) with either the EF1α‐hu‐α‐syn‐IRES‐GFP or the EF1α‐IRES‐GFP vector overnight. After viral transduction, medium was changed and primary OPCs were differentiated for 4 days and harvested for transcriptomic analysis.
Library preparation and transcriptome analysis
Library preparation and sequencing of primary OPCs transduced with EF1α‐hu‐α‐syn‐IRES‐GFP (n = 4) or EF1α‐IRES‐GFP lentiviral vectors (n = 3) was performed as described previously 19. Briefly, RNA quality was determined and a corresponding library was prepared. Sequencing of the pooled libraries was performed with an average depth of 86 million 101‐bp single‐end reads. Reads were aligned against the Rattus norvegicus genome Rnor5.0 using STAR, with default parameters absolute read counts were determined and reads encoding rRNA, tRNA, mt rRNA and mt tRNA were discarded 2, 18. The statistical analysis of the raw counts and differential gene expression analysis was performed using DESeq2 R package version 1.6.3. Significance was assessed at a false discovery rate (FDR) of 0.05 using the Benjamini‐Hochberg method with at least 1.5‐fold difference 6. Functional enrichment analysis of the 81 upregulated genes (>1.5‐fold change, FDR <0.05) was performed using Metascape 87.
Statistics
Quantitative datasets were analyzed using GraphPad Prism® and are represented in mean ± standard error of the mean (SEM). Statistical analysis was performed using two‐way analysis of variation (ANOVA) in combination with Tukey’s multiple comparison post hoc test (mouse CD68) and in combination with Sidak’s multiple comparison post hoc test (IBA1, PCNA, mouse/human GFAP, human CD68 and RNA expression analysis). Human α‐syn levels in MBP29‐hα‐syn mice were statistically analyzed using a one‐way ANOVA (Friedman test) and post hoc Dunn’s multiple comparison testing. Statistical analysis of LFB/PAS, CNP and KI67 staining was evaluated using Mann‐Whitney U test (non‐parametric, unpaired). Human α‐syn+ inclusions in human post‐mortem brain tissue were statistically analyzed using the Wilcox t test (non‐parametric, paired). The relationship between human α‐syn+ inclusions in white and gray matter of the MSA patients and disease duration as well as the number of CD68+ cells was analyzed using the non‐parametric Spearman correlation. Correlation coefficient (r sp) and P‐values were calculated using GraphPad Prism® and a line was fitted using the linear regression tool. P‐values < 0.05 were considered statistically significant.
Results
Severe and topographically restricted inflammation in human putaminal white matter regions of MSA‐P patients
In order to analyze the link between α‐synucleinopathy and neuroinflammation in MSA, post‐mortem putaminal samples of five MSA‐P patients and five age‐ and gender‐matched controls were obtained from the NBB (Figure 1A; Table S1). We focused on MSA‐P to ensure best possible homogeneity in terms of clinical and neuropathological diagnosis. Histological staining of LFB/PAS (Figure 1A,B) confirmed a reduction of myelin staining intensity in the putamen of MSA‐P patients (see also 19). Additionally, analysis of CNP staining (Figure 1C) revealed a reduced area of striatal white matter fibers in MSA‐P patients. Statistical analysis of LFB/PAS and CNP did not reach significance (P = 0.056), however, an apparent trend towards decreased levels was observed. The presence of α‐syn inclusions was mainly observed in putaminal white matter and less pronounced in adjacent gray matter (Figure 1D, bottom panel; P = 0.063), whereas no α‐syn inclusions were observed in controls (Figure 1D, top panel). This supported the definite diagnosis of the parkinsonian subtype of MSA (MSA‐P) and formed the basis to compare areas with high and low α‐syn load. Additionally, a negative correlation of α‐syn inclusions and disease duration was observed (Figure S1A; white matter: r sp = −0.80, P = 0.133; gray matter: r sp = −1.00, P = 0.017).
Figure 1.
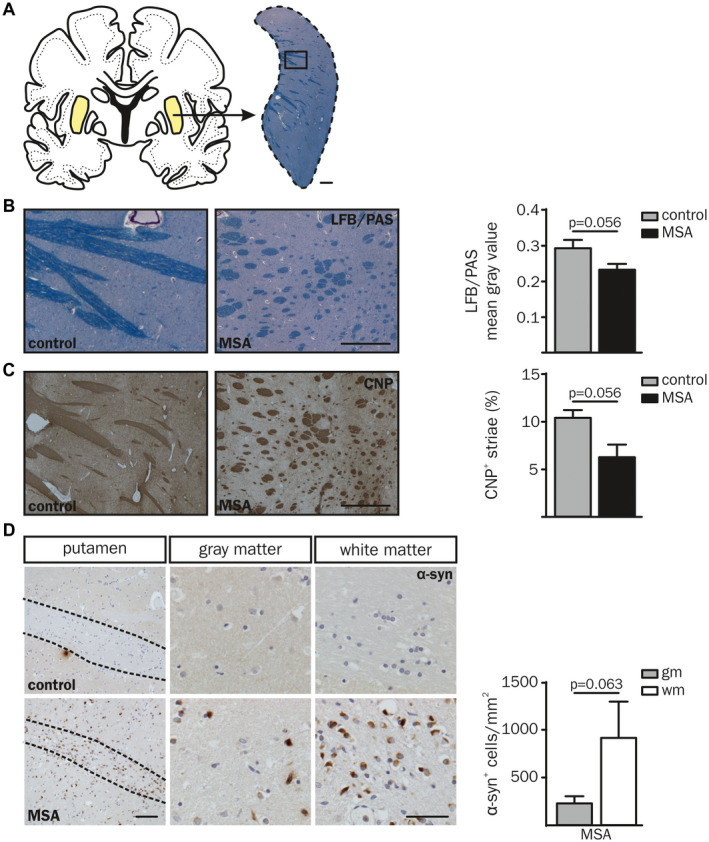
Neuropathological characteristics of putaminal white (wm) and gray matter (gm) in post‐mortem brain tissue of MSA‐P patients compared to controls. (A, B) Luxol Fast Blue/Periodic Acid‐Schiff (LFB/PAS) and (C) CNP staining was examined in 5 µm cryosections of human post‐mortem brain tissue of five MSA‐P patients compared to age‐ and gender‐matched controls (Table S1). (A) Overview of a human coronal brain section. The putamen is displayed in higher magnification and stained with LFB/PAS (right, scale bar: 1000 µm), boxed region is magnified in (B). Representative images of a MSA‐P patient (right; 67 yrs, male) compared to a control (left; 55 yrs, male) and quantification of the mean gray value of LFB/PAS staining (B) and CNP+ area (%) (C). Scale bars: 500 and 1000 µm, respectively. (D) The overview image of α‐synuclein (α‐syn) staining (left column) displays a representative putaminal white matter tract of a MSA‐P patient and a corresponding control (both 55 yrs, male). Scale bar: 100 µm. A higher magnification of putaminal white and gray matter region is depicted in the middle and right column, respectively (scale bar: 50 µm). White matter striae are framed with dashed lines. Putaminal white matter of MSA‐P patients showed increased levels of pathological α‐syn accumulation compared to gray matter (α‐syn+ cells per mm2). No α‐syn inclusions were present in controls (top panel). Data represent mean ± SEM (n = 5 patients per group). Statistical analyses were performed using Mann‐Whitney U test for LFB/PAS (P = 0.056) and CNP (P = 0.056). Wilcox t test was used for statistical analysis of α‐syn (P = 0.063). Abbreviation: wm: white matter, gm: gray matter.
Cellular CD68 expression is a well‐established marker to identify infiltrated and resident activated myeloid cells in human post‐mortem brain tissue 14, 38, 48. An increased number of CD68+ cells was observed in the putaminal white matter of MSA‐P compared to both the adjacent gray matter (2.3‐fold, P = 0.007) and the white matter of controls (6.7‐fold, P = 0.002) (Figure 2A). Intriguingly, the number of CD68+ cells was not different in putaminal gray matter of MSA‐P patients and controls indicating a distinct immune response associated to the putaminal white matter in MSA. However, the number of α‐syn+ inclusions did not correlate with the number of CD68+ cells indicating a myeloid immune response triggered by oligodendroglial α‐synucleinopathy rather than α‐syn itself (Figure S1B; white matter: r sp = 0.10, P = 0.950; gray matter: r sp = −0.20, P = 0.783).
Figure 2.
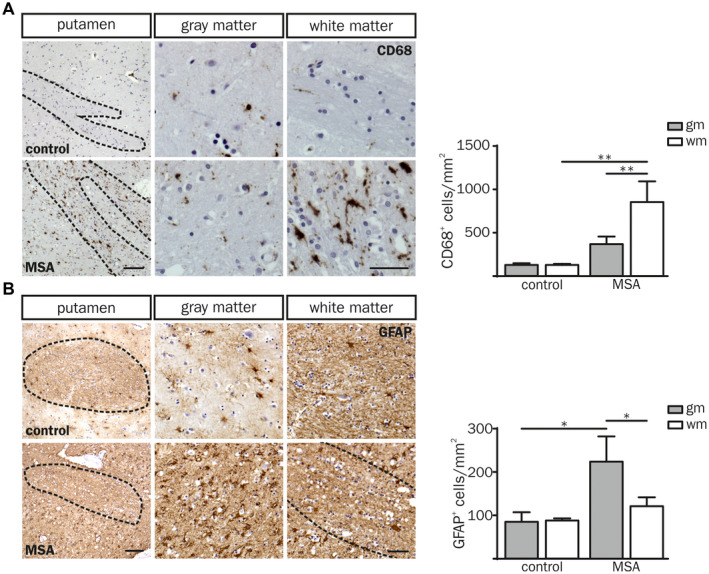
Neuroinflammatory response in putaminal post‐mortem brain tissue of MSA‐P patients compared to controls. (A, B) Overview images of a putaminal white matter tract (wm) with adjacent gray matter (gm; left column) derived from a control (55 yrs, male (CD68); 60 yrs, female (GFAP); top panel) and a MSA‐P (both 55 yrs, male; bottom panel). Scale bar: 100 µm. Higher magnification images of putaminal white and gray matter are depicted in the middle and right column, respectively. Dashed lines frame white matter striae. Scale bar: 50 µm. (A) The number of CD68+ cells was increased in the white matter of MSA‐P patients compared to controls (CD68+ cells per mm2). (B) Quantification of GFAP expressing cells per mm2 shows increased astrocyte numbers in the putaminal gray matter. Data represent mean ± SEM (n = 5 patients per group). Statistical analyses were performed using two‐way ANOVA. *P ≤ 0.05, **P ≤ 0.01. Abbreviation: wm: white matter, gm: gray matter.
In contrast to CD68+ myeloid cells, the number of GFAP+ astrocytes as well as the astroglial fiber density was increased in putaminal gray matter of MSA‐P patients only (Figure 2B). Taken together, we observed a regionally distinct neuroinflammatory pattern restricted to myeloid cells in the putaminal white matter and astrocytes in the gray matter of MSA‐P post‐mortem brain tissue.
Pre‐symptomatic neuroinflammation is spatially associated with α‐syn pathology in MBP29‐hα‐syn mice
Since post‐mortem brain samples reflect rather the end stage of MSA, we asked whether neuroinflammatory processes start early in the disease course. To investigate the temporal kinetic of neuroinflammation we analyzed a MSA mouse model mimicking prototypical features of MSA (MBP29‐hα‐syn mice). Due to high oligodendroglial expression of human α‐syn, MBP29‐hα‐syn mice show severe neurological symptoms 2–3 months after birth and die prematurely with a median age of 18.5 weeks (Figure 3A, Figure S2) 19, 76. MBP29‐hα‐syn mice at the postnatal age of 21 days (P21) represent a pre‐symptomatic disease stage without a motor phenotype (Figure 3A, left red arrow), whereas animals at P90 which already developed severe motor deficits, such as tremor and ataxia, represent an advanced disease stage (Figure 3A, right red arrow). This dual comparison allows a clear interpretation of the α‐syn‐related cellular phenotypes and reduces the influence of secondary pathological mechanisms. α‐syn expression was confirmed by co‐immunostaining of OLIG2 and human α‐syn (clone: 15G7) showing a 3.0 and 3.8‐fold increased area of human α‐syn protein expression in the corpus callosum (cc) compared to the cortex (ctx) and the striatum (str), respectively (Figure S2). Similarly to the human post‐mortem analysis, this observation formed the basis for analyzing neuroinflammatory responses as the consequence of oligodendroglial α‐synucleinopathy.
Figure 3.
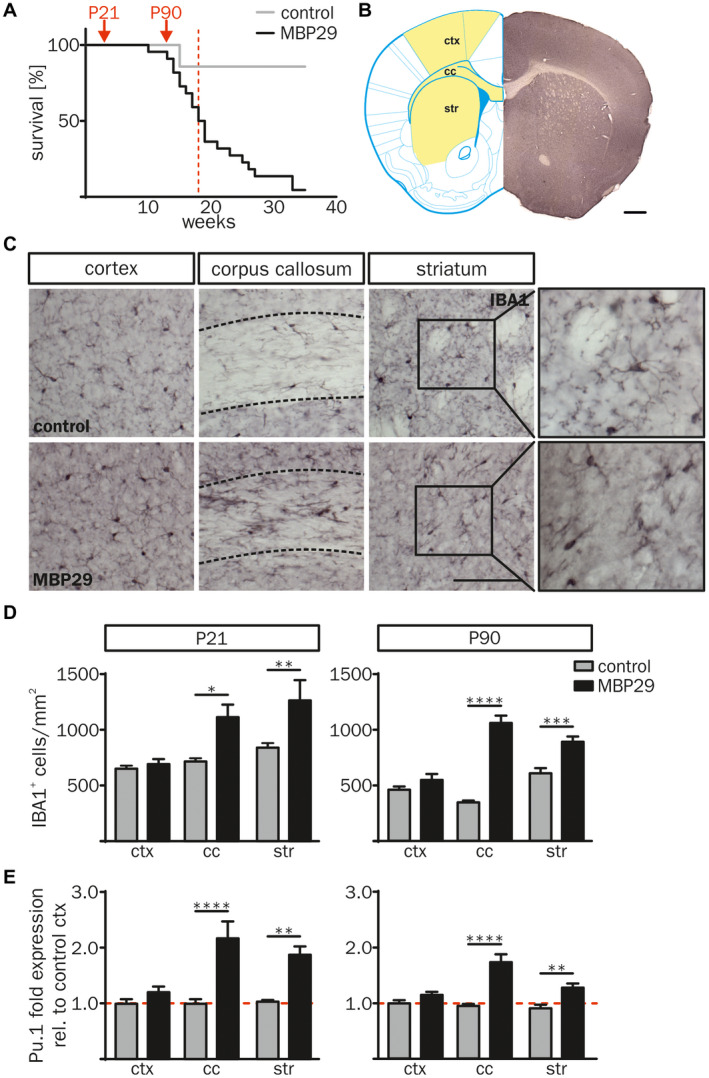
Increased number of IBA1+ cells in white matter regions of MBP29‐hα‐syn mice. (A) Kaplan‐Meier curve of MBP29‐hα‐syn mice (n = 22) compared to controls (n = 6). Red arrows indicate the time points of the combined structural and biochemical analysis of the animals at postnatal age of 21 d (P21) and 90 d (P90). Red dashed line highlights the median survival of 18.5 weeks of MBP29‐hα‐syn mice. (B) Coronal section of a non‐transgenic mouse at the age of P21 (right), transferred into a modified topographical map (left) adopted from Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates 4th Edition (Figure 22; coordinates: Interaural: 4.90 mm, Bregma: 1.09 mm; Copyright © Elsevier (2012) 64). Analyzed regions are highlighted in yellow (left) including the motor cortex M1 and M2 (ctx), the corpus callosum (cc) and the striatum (str). Scale bar: 500 µm. (C) Representative images of IBA1 immunostaining of the ctx, the cc and the str of control (top) and MBP29‐hα‐syn mice (bottom) at P90. Boxed regions show a higher magnification of representative IBA1+ cell morphologies in the striatum. Dashed lines frame the cc. Scale bar: 100 μm. (D) Quantification of IBA1+ myeloid cells of MBP29‐hα‐syn compared to control mice at the age of P21 (left) and P90 (right). The number of IBA1+ myeloid cells was increased in regions with white matter (cc, str), however, the ctx showed no change in IBA1+ cell numbers. (E) Gene expression analysis of the myeloid cell transcription factor Pu.1 in the ctx, the cc and the str of MBP29‐hα‐syn and control mice at P21 and P90 revealed a similar profile. Fold expression of Pu.1 was calculated relative to cortical expression levels of control mice. Data represent mean ± SEM (n = 5 animals per group). Statistical analyses were performed using two‐way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
Neuroinflammation in MBP29‐hα‐syn mice was first analyzed by quantifying the number of IBA1+ cells, a commonly used and robust marker labeling resting and activated myeloid cells 1. Areas of interest were chosen based on their high, medium and low content of α‐syn inclusions in oligodendrocytes, representing prototypical white (cc), mixed white and gray (str) as well as gray matter (ctx) regions, respectively (Figure 3B). The density of IBA1+ cells was increased in white matter regions (3.1‐fold in cc and 1.2‐fold in str) of MBP29‐hα‐syn mice at P90, whereas no difference was observed for the ctx (Figure 3C,D). Intriguingly, IBA1+ cell numbers in the cc and str of MBP29‐hα‐syn mice were already increased at the pre‐symptomatic stage (P21) by 1.5‐fold (Figure 3D). Confirming these results on a transcriptional level, the myeloid transcription factor Pu.1 was solely increased in the cc and the str of MBP29‐hα‐syn mice both at P21 and P90 (Figure 3E). The temporal pattern of myeloid cell numbers in MBP29‐hα‐syn mice suggests an early, pre‐symptomatic onset of neuroinflammation.
In contrast to the profound myeloid immune response in white matter regions, GFAP+ astrocyte numbers were increased in ctx and str only, matching our observations in human post‐mortem brain tissue (Figure 4). This distinct spatial pattern separating myeloid response and astrogliosis into white and gray matter regions, respectively, indicates that different triggers underlie myeloid inflammatory responses and astrocyte activation in MBP29‐hα‐syn mice being present at early disease stages.
Figure 4.
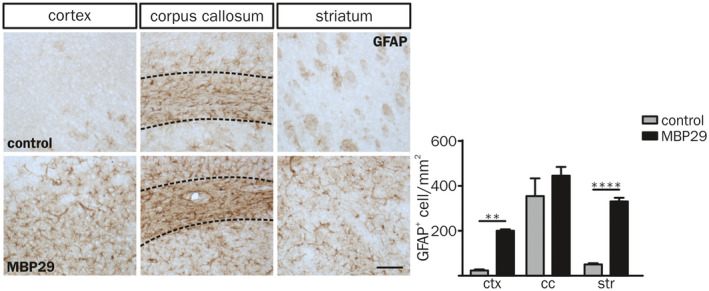
Astrogliosis in gray matter regions of MBP29‐hα‐syn mice. Characteristic GFAP staining (left) and quantification of GFAP+ cells per mm2 in the ctx, the cc and the str of control (top) and MBP29‐hα‐syn mice (bottom) at the age of P90 (Scale bar: 100 µm). The number of astrocytes is elevated in the ctx and the str of MBP29‐hα‐syn (n = 5) compared to control mice (n = 3). The cc is framed in dashed lines. Data represent mean ± SEM. Statistical analyses were performed using two‐way ANOVA. **P ≤ 0.01, ****P ≤ 0.0001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
Myeloid cells exhibit increased proliferation and activation in the white matter of MBP29‐hα‐syn mice
Next, we examined whether the increased number of IBA1+ cells may be due to the enhanced myeloid cell proliferation by co‐expression of the proliferating cell nuclear antigen (PCNA) and IBA1 (Figure 5A). Proliferation of IBA1+ cells was elevated in white matter regions (28.8‐fold in the cc and 54.8‐fold in the str) but also in the ctx (25.6‐fold) of MBP29‐hα‐syn mice at the pre‐symptomatic stage (P21). At P90 MBP29‐hα‐syn mice showed still a 4.7‐fold increased proliferation in the cc only (Figure 5A). Notably, the number of PCNA+ cells out of all IBA1+ cells decreased over time, for example, in the cc from 57.8% at P21 to 0.5% at P90 in MBP29‐hα‐syn and 2.2% at P21 to 0.1% at P90 in control mice.
Figure 5.
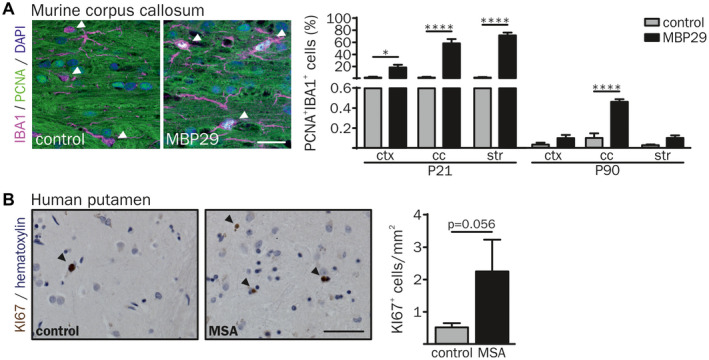
Increased proliferation of IBA1+ cells in the white matter of MBP29‐hα‐syn mice. (A) Proliferation of IBA1+ cells was examined by co‐labeling with the proliferation cell nuclear antigen (PCNA). Arrowheads indicate IBA1+ myeloid cells. At the age of P21 up to 70% of double‐labeled IBA1+/PCNA+ cells were identified in the cc and the str of MBP29‐hα‐syn mice. In contrast, at the age of P90 an increased proliferation of IBA1+ cells was present in the cc of MBP29‐hα‐syn mice only (n = 5 animals per group). Scale bar: 20 µm. (B) Representative image of proliferating KI67+ cells in the putamen of a control (77 yrs, female) and a MSA‐P (67 yrs, female) patient (left). Arrowheads indicate KI67+ cells. Quantification revealed an increased number of proliferating cells in MSA‐P patients compared to controls (right) (n = 5 patients per group). Scale bar: 50 µm. Data represent mean ± SEM. Statistical analyses were performed using two‐way ANOVA for PCNA and Mann‐Whitney U test (non‐parametric, unpaired) for KI67 quantification (KI67 statistics: P = 0.056). *P ≤ 0.05, ****P ≤ 0.0001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
We further asked whether a proliferating cell population is observed in the putamen of human MSA samples. Analysis of the proliferation marker KI67 revealed an increased density of proliferating cells from 0.5 to 2.3 cells per mm2 in the putamen of MSA‐P patients (P = 0.056) (Figure 5B).
Moreover, the morphology of IBA1+ cells showed an activated myeloid cellular phenotype including less ramified and short processes in white matter regions (cc and str) of MBP29‐hα‐syn mice only (Figure 3C) 71. To further characterize the α‐synucleinopathy‐related myeloid cell response, we examined the lysosomal‐associated protein CD68 as marker for phagocytic activity 91. A representative image of CD68 and IBA1 demonstrates increased immunoreactivity of both markers in the cc of MBP29‐hα‐syn mice (Figure 6A). Three dimensional (3D) determination of the number and size (mean volume and volume percent per region) of CD68+ lysosomal structures was performed using the 3D object counter plugin 9 of the ImageJ software 75 (Figure 6B). The analysis revealed a 24.1‐fold elevated number of CD68+ lysosomal structures occupying 40.6‐fold more volume in the cc of MBP29‐hα‐syn mice compared to controls at P90. Furthermore, CD68+ lysosomal structures were also increased in the str of MBP29‐hα‐syn mice, accounting a 6.2‐fold increase of the CD68+ volume compared to controls. At the pre‐symptomatic disease stage (P21) the mean volume of CD68+ lysosomal structures in the cc and the str was already enlarged up to 3.4‐fold in MBP29‐hα‐syn mice compared to controls; however, the occupied volume is less increased in MBP29‐hα‐syn mice at P21 compared to P90 (6.2‐fold in cc and 5.6‐fold in str). In addition, we analyzed the gene transcripts of Tlr2, Tlr4 and Trem2 linked to activation and phagocytic activity of myeloid cells (Figure 6C). Notably, the expression of these genes was highly upregulated in the cc and the str, but not in the ctx of MBP29‐hα‐syn mice at P21 and P90. Taken together, these findings indicate a pronounced, spatially and temporally defined neuroinflammatory response of myeloid cells with increased proliferative and phagocytic activity restricted to white matter regions in the transgenic MSA mouse model.
Figure 6.
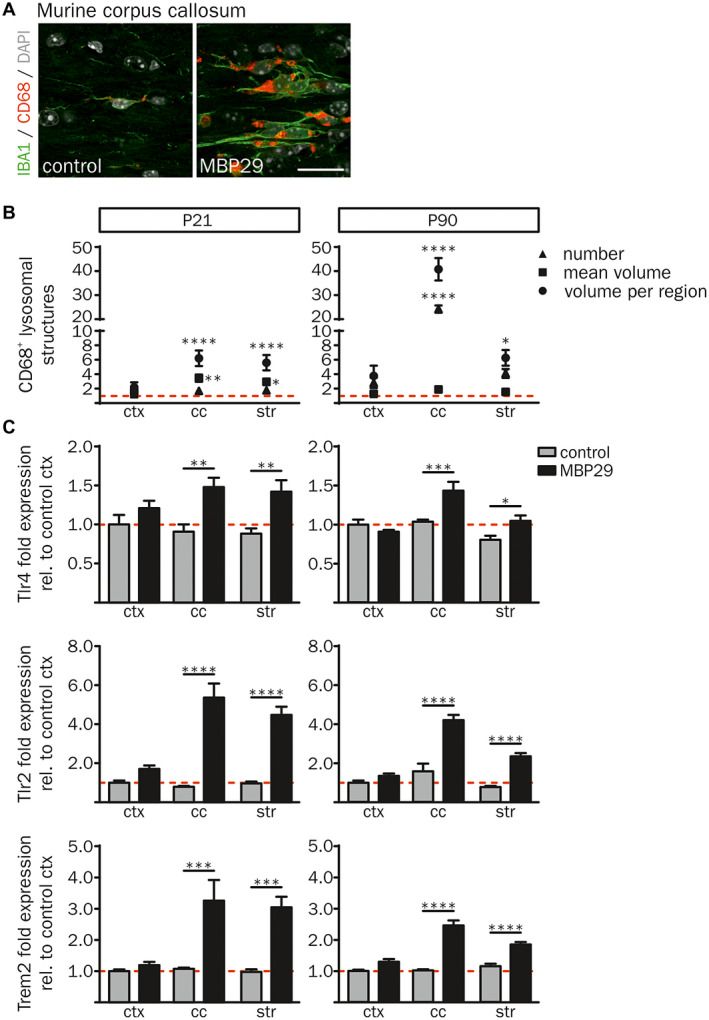
Activated, phagocytic phenotype of IBA1+ cells in MBP29‐hα‐syn mice. (A) Co‐immunofluorescence staining of IBA1 with the lysosomal‐associated protein CD68 revealed an increased expression of both markers in the cc of MBP29‐hα‐syn mice (right) in comparison to controls (left) at P21. Scale bar: 20 µm. (B) Quantification of the number of CD68+ lysosomal structures (triangle), their mean volume (square) as well as the percentage of their volume per region (circle) in MBP29‐hα‐syn mice relative to controls (red dashed line) revealed increased number and size of CD68+ lysosomal structures in transgenic animals at P21 (left graph) and P90 (right graph). (C) mRNA expression analysis relative to cortical gene expression of control mice. Fold expression of Tlr4 (top), Tlr2 (middle) and Trem2 (bottom) was highly increased in the cc and the str of MBP29‐hα‐syn mice at the age of P21 (left panel) and P90 (right panel). Data represent mean ± SEM (n = 5 animals per group). Statistical analyses were performed using two‐way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
α‐syn overexpressing oligodendrocytes show a pro‐inflammatory gene expression profile
Finally, we asked whether the distinct and early neuroinflammation is induced by α‐syn expressing oligodendrocytes. In order to investigate the role of oligodendroglial α‐synucleinopathy in initiating neuroinflammatory responses, primary oligodendrocytes were lentivirally transduced with an EF1α‐hu‐α‐syn‐IRES‐GFP or EF1α‐IRES‐GFP construct (see also Ref. 19). To detect genome‐wide differences in the transcriptional landscape of α‐syn overexpressing oligodendrocytes, we performed RNA sequencing on day 4 of maturation (Figure 7A). Analysis of the RNA expression profile revealed upregulation of 81 and downregulation of 16 genes in α‐syn overexpressing oligodendrocytes compared to GFP controls (>1.5 fold change, FDR 0.05) (Figure S3A). Further, GO analysis of the 81 upregulated genes revealed a significant enrichment for genes associated to pro‐inflammatory processes including cytokine signaling and production (Figure S3B). In particular transcripts important for chemotactic attraction and activation of myeloid cells, such as Cxcl10 and Ccl2, were upregulated in α‐syn overexpressing compared to GFP‐expressing primary oligodendrocytes (Figure 7B). Corroborating these results, an additional expression analysis of the transcripts of Cxcl10 and Ccl2 in MBP29‐hα‐syn mice showed an upregulation of their mRNA in areas of pronounced α‐syn pathology only, such as white matter regions at P21 and in the cc at P90 (Figure 7C). These data imply that α‐syn overexpression in oligodendrocytes triggers the expression of pro‐inflammatory cytokines inducing a distinct neuroinflammatory signature at a pre‐symptomatic disease stage.
Figure 7.
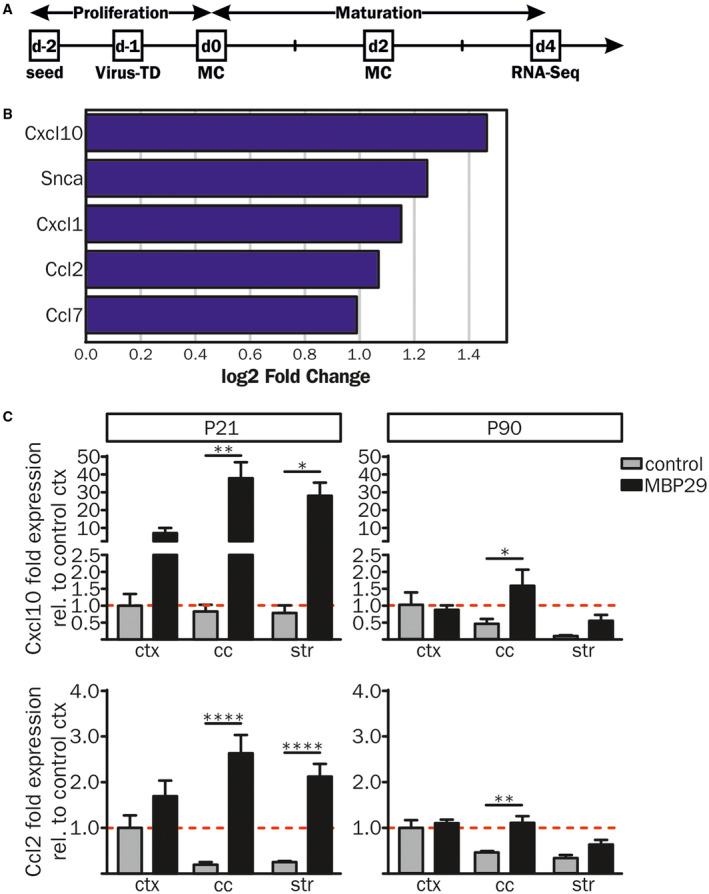
Pro‐inflammatory gene expression profiles of primary oligodendrocytes overexpressing human α‐syn. (A) Rat primary oligodendrocyte progenitor cells (OPCs) were lentivirally transduced (Virus‐TD) with EF1α‐hu‐α‐syn‐IRES‐GFP (n = 4) or EF1α‐IRES‐GFP (n = 3) constructs 1 day after cell seeding. OPCs were matured for 4 days prior to RNA sequencing (RNA‐Seq). (B) Compared to controls human α‐syn overexpressing OPCs showed an increased gene expression profile of pro‐inflammatory cytokines important for activation, migration and chemotactic attraction of immune cells. (C) RNA expression analysis of Cxcl10 and Ccl2 was performed via real‐time qPCR in the ctx, the cc and the str of MBP29‐hα‐syn and control mice at P21 (Cxcl10: n = 5 and n = 3; Ccl2: n = 5 and n = 4, respectively) and P90 (n = 4 and n = 3, respectively). Fold expression analysis was calculated relative to cortical expression levels of control mice and revealed increased mRNA levels in MBP29‐hα‐syn mice in the cc and the str at P21 (left panel). At P90 (right panel) Cxcl10 and Ccl2 expression levels were upregulated in the cc of MBP29‐hα‐syn mice only. Data represent mean ± SEM. Statistical analyses were performed using two‐way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. Abbreviations: MC: medium change, RNA‐Seq: RNA sequencing, Virus‐TD: virus transduction.
Discussion
Neuroinflammation has long been described as an important neuropathological feature in MSA while underlying mechanisms as well as its precise spatial and temporal pattern in this devastating neurodegenerative disease are poorly understood. Our study provides evidence for a tight topographical link between α‐synucleinopathy in oligodendrocytes and activation of neuroinflammatory responses in MSA already present at a pre‐symptomatic disease stage. In detail, we describe widespread α‐syn inclusions in putaminal white matter striae of post‐mortem tissue derived from MSA‐P patients being paralleled by a profound neuroinflammation restricted to myeloid cells. In contrast, the number of myeloid cells in the putaminal gray matter, as an area of minor α‐syn load, was unchanged. In addition to our findings in human post‐mortem samples, MBP29‐hα‐syn mice showed an early neuroinflammatory signature of myeloid cells in the cc and the str. Both regions show by far more pronounced α‐synucleinopathy than the ctx, in which no clear signs of myeloid response were detected even at later disease stages. Corroborating these histological analyses, expression of pro‐inflammatory cytokines was strongly increased both in α‐syn overexpressing oligodendrocytes in vitro and in areas with high α‐syn levels in MBP29‐hα‐syn mice.
In a first step, we analyzed neuroinflammation in post‐mortem tissue of MSA‐P patients, showing typical MSA pathology such as α‐syn inclusions within oligodendroglia and widespread reduction of myelin intensity. Statistical analysis of α‐syn located in white and gray matter regions of MSA‐P patients as well as LFB/PAS and CNP staining in the putamen of MSA‐P patients compared to controls revealed no significant difference (P = 0.056). However, an obvious trend towards increased α‐syn levels in the white matter of MSA‐P patients paralleled by reduced myelin intensity was observed leading to the conclusion that the high interindividual variability originates most likely from the limited sample size. Regions with varying levels of α‐synucleinopathy were investigated, allowing us to more clearly analyze the influence of oligodendroglial α‐syn inclusions on neuroinflammatory processes. Intriguingly, only in the white matter as a region with high α‐syn levels, prominent neuroinflammation was observed. In conjunction with our results, structural examination of the neuroinflammatory response revealed an increase of CD45+ myeloid cell numbers and gene expression of CD14 in the subcortical white matter of MSA post‐mortem brain tissue 61. A similar correlation was previously made in PD, the most frequent neuronal α‐synucleinopathy in which neuroinflammation was mainly observed in gray matter regions, showing pronounced synucleinopathy, such as the substantia nigra 15, 40, 55, 57. Longitudinal PET/MRI studies using the benzodiazepine receptor ligand [11C]‐PK11195 monitored an early and sustained microglia activation in different regions such as the basal ganglia of PD patients 25. In addition, we and others have shown that aggregated α‐syn, myelin debris and neuronal loss trigger immunological responses 4, 12, 20, 37, 66, 83, 93. This suggests a tight spatial relation between myeloid cells and α‐syn loaded neurons in PD. The topographical pattern of a distinct neuroinflammatory response in white matter regions containing high numbers of oligodendroglial α‐syn inclusions together with the lack of myeloid responses in the gray matter leads to the question whether α‐syn bearing oligodendrocytes rather than degenerating neurons induce activation of myeloid cells in MSA. In this respect, we were not able to correlate the number of α‐syn inclusions with CD68+ cells indicating a role of oligodendrogliopathy in activation of the immune response rather than α‐syn itself.
In addition to myeloid cells, astrocytes are known to be activated due to pathological protein aggregation. Interestingly, we detected a complementary image of astrogliosis in comparison to myeloid cell activation in MSA tissue. In line with our observation, previous reports described astrogliosis in striatonigral and olivopontocerebellar regions with severe signs of neurodegeneration 62. Furthermore, analysis of subcortical white matter regions revealed no change in astrocyte numbers and astrocyte‐specific gene expression in MSA patients compared to controls 61. Together, these findings imply an astroglial interaction rather in the microenvironment of degenerating neurons than of oligodendrocytes.
Subsequently to our observation in human MSA post‐mortem tissue, we analyzed MBP29‐hα‐syn mice at two different time points, a pre‐symptomatic (P21) and a symptomatic (P90) disease stage in order to analyze the temporal pattern of neuroinflammation. In contrast to the MSA mouse models expressing human α‐syn under control of the proteolipid protein (PLP‐hα‐syn mice) or the 2′,3′‐cyclic nucleotide 3′ phosphodiesterase (CNP‐hα‐syn mice) promoter, MBP29‐hα‐syn mice show an early oligodendrocyte maturation failure leading to a severe myelin deficit and early onset of neurological symptoms 43, 76, 92. Although PLP‐hα‐syn mice show oligodendroglial α‐syn inclusions, there is no myelin deficit and a later onset of functional deficits in comparison to MBP29‐hα‐syn mice 21, 43. Interestingly, PLP‐hα‐syn mice present an early myeloid immune response mainly in the striatum, the substantia nigra and in white matter tracts without signs of astrogliosis 79, 80. Demyelination, neurodegeneration as well as astrogliosis in the cerebral cortex and the spinal cord were described in CNP‐hα‐syn mice 92. However, a detailed analysis of the inflammatory phenotype is lacking in this mouse model. Taken together, albeit the three transgenic MSA mouse models present some differences, these findings suggest, a tight link between oligodendroglial α‐synucleinopathy and neuroinflammation in MSA transgenic mouse models.
Strikingly, we observed an increased neuroinflammatory response restricted to myeloid cells in areas of more pronounced α‐synucleinopathy already prior to the onset of motor symptoms. In contrast, gray matter regions of MBP29‐hα‐syn mice show only increased astrocytic cell numbers similar to the human post‐mortem examination. This indicates an early inflammatory myeloid response linked to α‐syn accumulation in oligodendrocytes.
An increased number of myeloid cells may be related to either an enhanced proliferation of resident microglia or infiltration of peripheral monocytes and macrophages. Infiltration of peripheral cells in the brain parenchyma was identified in human post‐mortem brain tissue of PD patients as well as its corresponding mouse models 31, 46, 73, 77. Recent studies have identified distinct macrophage subsets located in the choroid plexus, the meninges as well as the perivascular space expressing similar surface markers and contributing to defense mechanisms in neurodegenerative diseases 28, 34. Although these subsets constitute distinct cellular populations, peripheral myeloid cells and resident microglia are still very difficult to distinguish. Furthermore, peripheral monocytes change their expression profile toward a microglia‐like phenotype upon infiltration into the CNS parenchyma 11, 22, 56. In order to more clearly distinguish myeloid cells from peripheral and resident origin different microglia‐specific markers were recently proposed such as Tmem119 7 or P2yr12 23, 35. Moreover, several microglia‐specific gene sets have been suggested; however, the specificity of these markers in the context of synucleinopathies needs to be further explored 13, 14, 23. Very recently, mass cytometry combined with fluorescence‐activated cell sorting was able to distinguish different murine CNS‐associated immune cell subsets including microglia, macrophages and monocytes. Interestingly, the different identified cell populations change in phenotype and frequency during aging as well as in a transgenic mouse model of Alzheimer’s disease and a murine multiple sclerosis model generating a disease‐specific fingerprint 5, 58. In particular, the markers used for analysis of myeloid cells in this study do not allow a clear distinction between peripheral myeloid cells and resident microglia 14, 28.
Our observation suggests a dramatically and early increased proliferation of myeloid cells in a locally restricted pattern in MBP29‐hα‐syn mice. Additionally, in MSA post‐mortem tissue reflecting a very late disease stage we detected an increased, nevertheless not significant (P = 0.056), number of proliferating cells. Under physiological conditions, microglia proliferation peaks within the second postnatal week and declines afterward into a lower turnover rate including proliferation and apoptosis cycles 3, 59. We observed a highly increased proliferative activity of myeloid cells at P21 which remained upregulated in the cc at P90 in MBP29‐hα‐syn mice. Hence, these findings indicate that proliferation is an important mechanism to establish the myeloid cell population in the present MSA mouse model and decreases to a lower but sustained pro‐inflammatory level upon later disease stages. However, the severity of an immune response is not only reflected by the number of myeloid cells but also to a major part by the state of myeloid cell activation. CD68 is linked to lysosomal activity of myeloid cells 91. Enlarged lysosomal structures and elevated gene expression of Trem2, Tlr2 and Tlr4 in white matter regions of MBP29‐hα‐syn mice already at a pre‐symptomatic disease stage demonstrate increased phagocytic activity of myeloid cells in this model. Thus, these data suggest a profound and early proliferation followed by a severe phagocytic activation with enlarged lysosomal structures restricted to white matter regions with high levels of α‐syn inclusions in MBP29‐hα‐syn mice.
Finally, we asked whether α‐syn overexpression in oligodendrocytes triggers this immune response. Primary oligodendrocytes with a constitutive overexpression of α‐syn showed an increased expression of pro‐inflammatory cytokines important for myeloid cell activation and recruitment such as Cxcl10, Ccl2, and Ccl7. These data imply a non‐cell autonomous mode of myeloid cell recruitment and activation induced by α‐syn inclusions in oligodendrocytes. Moreover, we showed that α‐syn inclusions in the putamen negatively correlate with disease progression of MSA‐P patients. Numerous studies indicated that α‐syn aggregates are released from neurons or oligodendrocytes, may spread throughout the CNS, thereby, seeding aggregates in other neuronal or glial cells 16, 27, 44, 47, 51, 68, 72. In this regard, we propose a non‐cell autonomous mechanism of microglia activation via cytokine expression of oligodendroctyes, thereby, playing a detrimental role in disease progression of MSA.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Correlation of α‐syn+ cells with neuroinflammation and disease duration. Α‐syn+ cells per mm2 separated in putaminal white (wm; r sp = −0.80, P = 0.133) and gray matter (gm; r sp = −1.00, P = 0.017) regions of each MSA‐P patient (n = 5) were correlated to the individual disease duration (months) (A) as well as density of CD68+ myeloid cells (cells/per mm2) in these regions (wm: r sp = 0.10, P = 0.950; gm: r sp = −0.20, P = 0.783) (B).
{kind=link}
Figure S2. Expression of human α‐syn in white matter regions of MBP29‐hα‐syn mice. Representative immunofluorescence staining of human α‐syn+ and OLIG2+ cells within the cc of MBP29‐hα‐syn mice (left). Measurement of the α‐syn+ area (%) revealed a 3.0‐fold and 3.8‐fold increase of the cc compared to the ctx and the str in MBP29‐hα‐syn mice (n = 5), respectively (right). Scale bars: 20 µm. Data represent mean ± SEM. Statistical analyses were performed using Friedman one‐way ANOVA. ***: P ≤ 0.001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
{kind=link}
Figure S3. Heat map and gene ontology (GO) analysis of primary oligodendrocytes overexpressing human α‐syn. (A) 81 and 16 genes were up‐ and downregulated, respectively, in α‐syn overexpressing oligodendrocyte (n = 4) compared to GFP controls (n = 3). Heat map presenting z‐scores of the 97 differentially expressed genes illustrates the effect of α‐syn overexpression in oligodendrocytes in vitro. (B) GO analysis of the 81 upregulated genes revealed a functional enrichment of genes associated to pro‐inflammatory processes including cytokine signaling and production.
{kind=link}
Table S1. Overview of demographic and clinical characteristics of human post‐mortem brain tissue of controls and MSA‐P patients. Tissue samples (n = 5 patients per group) were obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam (open access: http://www.brainbank.nl). Control and MSA‐P cohorts do not differ (t test: P > 0.05) in regard of gender, age and post‐mortem delay. Due to their clinical history and neuropathological examination (striatonigral degeneration, demyelination, putaminal α‐syn inclusions, see also Figure 1) MSA patients were diagnosed as parkinsonian subtype (MSA‐P).
Table S2. Primary antibodies used for immunohistochemistry.
Table S3. Secondary antibodies used for immunohistochemistry.
Acknowledgements
This work was supported by the Interdisciplinary Center for Clinical Research (IZKF Erlangen, TP E18 and E24), the Bavarian Research Network for Induced Pluripotent Stem Cells (ForIPS) and the Deutsche Forschungsgemeinschaft (DFG grants SCHL 21021‐1 and INST 410/45‐1 FUGG). AH is a graduate student of the research training group 2162 “Neurodevelopment and Vulnerability of the Central Nervous System” of the Deutsche Forschungsgemeinschaft (DFG GRK2162). SR, MW and JW are members of research training group 2162 of the Deutsche Forschungsgemeinschaft (DFG GRK2162). BE was a graduate student of the IZKF Erlangen and was supported by a research grant of the German Parkinson Association. The authors gratefully acknowledge the Netherlands Brain Bank (NBB) for providing human post‐mortem brain tissue and the excellent support of the Core Unit Genomics at the Institute of Human Genetics of the University Hospital Erlangen. Additionally, the authors thank Beate Winner for scientific discussion and comments on the manuscript as well as Holger Meixner, Ulrike Naumann, Edward Rockenstein, Maria Hirblinger, Claudia Kemming, and Florian Billing for technical assistance.
References
- 1. Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW (2007) Actin‐binding proteins coronin‐1a and IBA‐1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem 55:687–700. [DOI] [PubMed] [Google Scholar]
- 2. Anders S, Pyl PT, Huber W (2015) HTSeq ‐ A Python framework to work with high‐throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Askew K, Li K, Olmos‐Alonso A, Garcia‐Moreno F, Liang Y, Richardson P et al (2017) Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Reports 18:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bauer J, Sminia T, Wouterlood FG, Dijkstra CD (1994) Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res 38:365–375. [DOI] [PubMed] [Google Scholar]
- 5. Becher B, Schlitzer A, Chen J, Mair F, Sumatoh HR, Teng KW et al (2014) High‐dimensional analysis of the murine myeloid cell system. Nat Immunol 15:1181–1189. [DOI] [PubMed] [Google Scholar]
- 6. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300. [Google Scholar]
- 7. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB et al (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci 113:1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Béraud D, Maguire‐Zeiss KA (2012) Misfolded α‐synuclein and toll‐like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat Disord 18:17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolte S, Cordelieres FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. [DOI] [PubMed] [Google Scholar]
- 10. Bottenstein JE, Sato GH (1979) Growth of a rat neuroblastoma cell line in serum‐free supplemented medium. Proc Natl Acad Sci U S A 76:514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brendecke SM, Prinz M (2015) Do not judge a cell by its cover—diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Semin Immunopathol 37:591–605. [DOI] [PubMed] [Google Scholar]
- 12. Brück W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmarch HA, Lassmann H (1995) Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 38:788–796. [DOI] [PubMed] [Google Scholar]
- 13. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G et al (2014) Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP et al (2013) A neurodegeneration‐specific gene‐expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to α‐synuclein deposition. J Neuroinflammation 2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desplats P, Lee H‐J, Bae E‐J, Patrick C, Rockenstein E, Crews L et al (2009) Inclusion formation and neuronal cell death through neuron‐to‐neuron transmission of α‐synuclein. Proc Natl Acad Sci 106:13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dickson DW, Liu WK, Hardy J, Farrer M, Mehta N, Uitti R et al (1999) Widespread alterations of α‐synuclein in multiple system atrophy. Am J Pathol 155:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ettle B, Kerman BE, Valera E, Gillmann C, Schlachetzki JC, Reiprich S et al (2016) α‐Synuclein‐induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol 132:59–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W et al (2013) Toll‐like receptor 4 is required for α‐synuclein dependent activation of microglia and astroglia. Glia 61:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernagut PO, Meissner WG, Biran M, Fantin M, Bassil F, Franconi JM, Tison F (2014) Age‐related motor dysfunction and neuropathology in a transgenic mouse model of multiple system atrophy. Synapse 68:98–106. [DOI] [PubMed] [Google Scholar]
- 22. Flügel A, Bradl M, Kreutzberg GW, Graeber MB (2001) Transformation of donor‐derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. J Neurosci Res 66:74–82. [DOI] [PubMed] [Google Scholar]
- 23. Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA et al (2018) Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of alzheimer’s disease not evident in mouse models. Cell Reports 22:832–847. [DOI] [PubMed] [Google Scholar]
- 24. Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN et al (2003) [11C](R)‐PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61:686–689. [DOI] [PubMed] [Google Scholar]
- 25. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A et al (2006) In vivo imaging of microglial activation with [11C](R)‐PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412. [DOI] [PubMed] [Google Scholar]
- 26. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ et al (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goedert M, Masuda‐Suzukake M, Falcon B (2017) Like prions: the propagation of aggregated tau and α‐synuclein in neurodegeneration. Brain 140:266–278. [DOI] [PubMed] [Google Scholar]
- 28. Goldmann T, Wieghofer P, Jordao MJC, Prutek F, Hagemeyer N, Frenzel K et al (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17:797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gomez‐Nicola D, Fransen NL, Suzzi S, Perry VH (2013) Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33:2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gow A, Friedrich VL, Lazzarini RA (1992) Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol 119:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harms AS, Thome AD, Yan Z, Schonhoff AM, Williams GP, Li X et al (2018) Peripheral monocyte entry is required for α‐Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp Neurol 300:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I (2017) Staining of HLA‐DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol 309:12–22. [DOI] [PubMed] [Google Scholar]
- 33. Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009) The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 26:83–94. [DOI] [PubMed] [Google Scholar]
- 34. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J (2017) Myeloid cells in the central nervous system. Immunity 46:943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L‐c, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397. [DOI] [PubMed] [Google Scholar]
- 37. Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J et al (2016) α‐synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun 479:881–886. [DOI] [PubMed] [Google Scholar]
- 38. Hopperton KE, Mohammad D, Trépanier MO, Giuliano V, Bazinet RP (2018) Markers of microglia in post‐mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry 23:177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huitinga I. (2009) Ethical and legal declaration of the Netherlands Brain Bank. Available at: https://www.brainbank.nl/media/uploads/file/Ethical-declaration.pdf (accessed 10 August 2017).
- 40. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II‐positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 106:518–526. [DOI] [PubMed] [Google Scholar]
- 41. Ishizawa K, Komori T, Arai N, Mizutani T, Hirose T (2008) Glial cytoplasmic inclusions and tissue injury in multiple system atrophy: a quantitative study in white matter (olivopontocerebellar system) and gray matter (nigrostriatal system). Neuropathology 28:249–257. [DOI] [PubMed] [Google Scholar]
- 42. Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T (2004) Microglial activation parallels system degeneration in multiple system atrophy. J Neuropathol Exp Neurol 63:43–52. [DOI] [PubMed] [Google Scholar]
- 43. Kahle PJ, Neumann M, Ozmen L, Müller V, Jacobsen H, Spooren W et al (2002) Hyperphosphorylation and insolubility of α‐synuclein in transgenic mouse oligodendrocytes. EMBO Rep 3:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim C, Lee H‐J, Masliah E, Lee S‐J (2016) Non‐cell‐autonomous neurotoxicity of α‐synuclein through microglial toll‐like receptor 2. Exp Neurobiol 25:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koga S, Dickson DW (2017) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 89:175–184. [DOI] [PubMed] [Google Scholar]
- 46. Kokovay E, Cunningham LA (2005) Bone marrow‐derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson’s disease. Neurobiol Dis 19:471–478. [DOI] [PubMed] [Google Scholar]
- 47. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body–like pathology in long‐term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506. [DOI] [PubMed] [Google Scholar]
- 48. Korzhevskii DE, Kirik OV (2016) Brain microglia and microglial markers. Neurosci Behav Physiol 46:284–290. [Google Scholar]
- 49. Krismer F, Wenning GK (2017) Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol 13:232–243. [DOI] [PubMed] [Google Scholar]
- 50. Lee HJ, Kim C, Lee SJ (2010) α‐synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev 3:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li J‐Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ et al (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host‐to‐graft disease propagation. Nat Med 14:501–503. [DOI] [PubMed] [Google Scholar]
- 52. Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimura J (1998) Myelin degeneration in multiple system atrophy detected by unique antibodies. Am J Pathol 153:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E et al (2014) α‐Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging 35:2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCarthy KD, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McGeer P, Itagaki S, Boyes B, McGeer E (1988) Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1285. [DOI] [PubMed] [Google Scholar]
- 56. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U‐K, Mack M et al (2007) Microglia in the adult brain arise from Ly‐6C hi CCR58+ monocytes only under defined host conditions. Nat Neurosci 10:1544–1553. [DOI] [PubMed] [Google Scholar]
- 57. Mirza B, Hadberg H, Thomsen P, Moos T (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95:425–432. [DOI] [PubMed] [Google Scholar]
- 58. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP et al (2018) High‐dimensional single‐cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48:380–395. [DOI] [PubMed] [Google Scholar]
- 59. Nikodemova M, Kimyon RS, De I, Small AL, Collier LS, Watters JJ (2015) Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J Neuroimmunol 278:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 61. Nykjaer CH, Brudek T, Salvesen L, Pakkenberg B (2017) Changes in the cell population in brain white matter in multiple system atrophy. Mov Disord 32:1074–1082. [DOI] [PubMed] [Google Scholar]
- 62. Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L et al (2004) The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 127:2657–2671. [DOI] [PubMed] [Google Scholar]
- 63. Papp MI, Kahn JE, Lantos PL (1989) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy‐Drager syndrome). J Neurol Sci 94:22. [DOI] [PubMed] [Google Scholar]
- 64. Paxinos G, Franklin K (2013) Paxinos and Franklin’s the mouse brain in stereotaxic coordinates, 4th edn. Cambridge, MA, USA: Academic Press. [Google Scholar]
- 65. Pernot F, Dorandeu F, Beaup C, Peinnequin A (2010) Selection of reference genes for real‐time quantitative reverse transcription‐polymerase chain reaction in hippocampal structure in a murine model of temporal lobe epilepsy with focal seizures. J Neurosci Res 88:1000–1008. [DOI] [PubMed] [Google Scholar]
- 66. Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224. [DOI] [PubMed] [Google Scholar]
- 67. Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14:1227–1235. [DOI] [PubMed] [Google Scholar]
- 68. Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB et al (2015) Evidence for α‐synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci 112:5308–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Quinn N. (1989) Multiple system atrophy–the nature of the beast. J Neurol Neurosurg Psychiatry 52(Suppl):78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Radford R, Rcom‐H’cheo‐Gauthier A, Wong MB, Eaton ED, Quilty M, Blizzard C et al (2015) The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α‐synuclein inclusions. Mol Cell Neurosci 65:68–81. [DOI] [PubMed] [Google Scholar]
- 71. Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19:987–991. [DOI] [PubMed] [Google Scholar]
- 72. Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) α‐synuclein transfers from neurons to oligodendrocytes. Glia 62:387–398. [DOI] [PubMed] [Google Scholar]
- 73. Rodriguez M, Alvarez‐Erviti L, Blesa FJ, Rodríguez‐Oroz MC, Arina A, Melero I et al (2007) Bone‐marrow‐derived cell differentiation into microglia: a study in a progressive mouse model of Parkinson’s disease. Neurobiol Dis 28:316–325. [DOI] [PubMed] [Google Scholar]
- 74. Salvesen L, Ullerup BH, Sunay FB, Brudek T, Lokkegaard A, Agander TK et al (2015) Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy ‐ A stereological study. Neurobiol Dis 74:104–113. [DOI] [PubMed] [Google Scholar]
- 75. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Meth 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G et al (2005) Neurological and neurodegenerative alterations in a transgenic mouse model expressing human α‐synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci 25:10689–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sommer A, Maxreiter F, Krach F, Fadler T, Grosch J, Maroni M et al (2018) Th17 lymphocytes induce neuronal cell death in a human iPSC‐based model of parkinson’s disease. Cell Stem Cell 23:123–131. [DOI] [PubMed] [Google Scholar]
- 78. Song YJ, Halliday GM, Holton JL, Lashley T, O’Sullivan SS, McCann H et al (2009) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68:1073–1083. [DOI] [PubMed] [Google Scholar]
- 79. Stefanova N, Reindl M, Neumann M, Haass C, Poewe W, Kahle PJ, Wenning GK (2005) Oxidative stress in transgenic mice with oligodendroglial α‐Synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am J Pathol 166:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK (2007) Microglial activation mediates neurodegeneration related to oligodendroglial α‐synucleinopathy: implications for multiple system atrophy. Mov Disord 22:2196–2203. [DOI] [PubMed] [Google Scholar]
- 81. Stefanova N, Wenning GK (2015) Animal models of multiple system atrophy. Clin Auton Res 25:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Su X, Federoff HJ, Maguire‐Zeiss KA (2009) Mutant α‐synuclein overexpression mediates early proinflammatory activity. Neurotox Res 16:238–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Su X, Maguire‐Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29:1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Süß P, Kalinichenko L, Baum W, Reichel M, Kornhuber J, Loskarn S et al (2015) Hippocampal structure and function are maintained despite severe innate peripheral inflammation. Brain Behav Immun 49:156–170. [DOI] [PubMed] [Google Scholar]
- 85. Sutthiwarotamakun R (2011) Perpheral Autoimmunity Induces Central Neuro‐Inflammation and Hippocampal Neurogenesis Impairment in a Murine Model of Collagen Induced Rheumatoid Arthritis. Division of Immunology, Infection and Inflammation, Faculty of Medicine. University of Glasgow: Glasgow, pp. 419. [Google Scholar]
- 86. Trias E, Ibarburu S, Barreto‐Nunez R, Babdor J, Maciel TT, Guillo M et al (2016) Post‐paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation 13:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tripathi S, Pohl MO, Zhou Y, Rodriguez‐Frandsen A, Wang G, Stein DA et al (2015) Meta‐and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18:723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ubhi K, Low P, Masliah E (2011) Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci 34:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vieira BD, Radford RA, Chung RS, Guillemin GJ, Pountney DL (2015) Neuroinflammation in multiple system atrophy: response to and cause of α‐synuclein aggregation. Front Cell Neurosci 9:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S (2011) In vivo demonstration that α‐synuclein oligomers are toxic. Proc Natl Acad Sci USA 108(10):4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yamada Y, Doi T, Hamakubo T, Kodama T (1998) Scavenger receptor family proteins: roles for atherosclerosis, host defence and disorders of the central nervous system. Cellular and Molecular. Life Sci CMLS 54:628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K et al (2005) Mouse model of multiple system atrophy α‐synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45:847–859. [DOI] [PubMed] [Google Scholar]
- 93. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML et al (2005) Aggregated α‐synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19:533–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation of α‐syn+ cells with neuroinflammation and disease duration. Α‐syn+ cells per mm2 separated in putaminal white (wm; r sp = −0.80, P = 0.133) and gray matter (gm; r sp = −1.00, P = 0.017) regions of each MSA‐P patient (n = 5) were correlated to the individual disease duration (months) (A) as well as density of CD68+ myeloid cells (cells/per mm2) in these regions (wm: r sp = 0.10, P = 0.950; gm: r sp = −0.20, P = 0.783) (B).
Figure S2. Expression of human α‐syn in white matter regions of MBP29‐hα‐syn mice. Representative immunofluorescence staining of human α‐syn+ and OLIG2+ cells within the cc of MBP29‐hα‐syn mice (left). Measurement of the α‐syn+ area (%) revealed a 3.0‐fold and 3.8‐fold increase of the cc compared to the ctx and the str in MBP29‐hα‐syn mice (n = 5), respectively (right). Scale bars: 20 µm. Data represent mean ± SEM. Statistical analyses were performed using Friedman one‐way ANOVA. ***: P ≤ 0.001. Abbreviation: ctx: cortex, cc: corpus callosum, str: striatum.
Figure S3. Heat map and gene ontology (GO) analysis of primary oligodendrocytes overexpressing human α‐syn. (A) 81 and 16 genes were up‐ and downregulated, respectively, in α‐syn overexpressing oligodendrocyte (n = 4) compared to GFP controls (n = 3). Heat map presenting z‐scores of the 97 differentially expressed genes illustrates the effect of α‐syn overexpression in oligodendrocytes in vitro. (B) GO analysis of the 81 upregulated genes revealed a functional enrichment of genes associated to pro‐inflammatory processes including cytokine signaling and production.
Table S1. Overview of demographic and clinical characteristics of human post‐mortem brain tissue of controls and MSA‐P patients. Tissue samples (n = 5 patients per group) were obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam (open access: http://www.brainbank.nl). Control and MSA‐P cohorts do not differ (t test: P > 0.05) in regard of gender, age and post‐mortem delay. Due to their clinical history and neuropathological examination (striatonigral degeneration, demyelination, putaminal α‐syn inclusions, see also Figure 1) MSA patients were diagnosed as parkinsonian subtype (MSA‐P).
Table S2. Primary antibodies used for immunohistochemistry.
Table S3. Secondary antibodies used for immunohistochemistry.