

ABSTRACT
Human genetic evidence demonstrates that WNT1 mutations cause osteogenesis imperfecta (OI) and early‐onset osteoporosis, implicating WNT1 as a major regulator of bone metabolism. However, its main cellular source and mechanisms of action in bone remain elusive. We generated global and limb bud mesenchymal cell–targeted deletion of Wnt1 in mice. Heterozygous deletion of Wnt1 resulted in mild trabecular osteopenia due to decreased osteoblast function. Targeted deletion of Wnt1 in mesenchymal progenitors led to spontaneous fractures due to impaired osteoblast function and increased bone resorption, mimicking the severe OI phenotype in humans with homozygous WNT1 mutations. Importantly, we showed for the first time that Wnt1 signals strictly in a juxtacrine manner to induce osteoblast differentiation and to suppress osteoclastogenesis, in part via canonical Wnt signaling. In conclusion, mesenchymal cell‐derived Wnt1, acting in short range, is an essential regulator of bone homeostasis and an intriguing target for therapeutic interventions for bone diseases. © 2019 The Authors. Journal of Bone and Mineral Research Published by Wiley Periodicals, Inc.
Keywords: WNT1, OSTEOBLAST, OSTEOCLAST, SPONTANEOUS FRACTURE, RANKL, OPG, JUXTACRINE SIGNALING
Introduction
Despite the recent advances in the treatment of metabolic bone diseases, OI and severe osteoporosis remain therapeutic challenges. We and others recently identified that a heterozygous mutation in WNT1 causes early‐onset osteoporosis whereas homozygous mutations in the gene lead to OI.1, 2, 3, 4 Together with the gain‐of‐function and loss‐of‐function mutations in the WNT signaling receptor low‐density lipoprotein receptor‐related protein 5 and 6 (LRP5/6) and their inhibitor Sclerostin, these data highlight the predominant role of WNT signaling in regulating bone metabolism.5, 6, 7 Not surprisingly, monoclonal anti‐Sclerostin antibody romosozumab, which enhances endogenous Wnt signaling, is currently in late clinical development for the treatment of osteoporosis. Romosozumab promotes bone formation and suppresses bone resorption, leading to uncoupling of these two normally tightly co‐regulated functions, but the mechanisms for this uncoupling remain unclear.8
Wnt ligands are a family of 19 lipid‐modified glycoproteins that play essential roles during development, tissue homeostasis, and cancer. Genetically modified mouse models have suggested that Wnt ligands Wnt3a, Wnt5a, Wnt10b, and Wnt16 regulate bone mass, of which WNT1, WNT3a, and WNT16 have also been linked to osteoporosis in humans.3, 9, 10, 11 Induction of canonical Wnt signaling in osteoblasts promotes osteoblast differentiation and function and suppress osteoclastogenesis by inducing the expression of osteoprotegerin (Opg) in osteoblasts.12 In addition, recent work has demonstrated that Wnt16 and Wnt5a can also directly regulate osteoclast differentiation.13, 14 Recently, osteocyte‐targeted deletion of Wnt1 was shown to lead to spontaneous fractures at early age. Moreover, phenotype of a spontaneous Wnt1 mutant Sway mouse was partially rescued by either treatment with anti‐Sclerostin antibody or by activation of mammalian target of rapamycin (mTOR) signaling.15
Wnt proteins have traditionally been classified as long‐range–acting secreted morphogens. Because of their hydrophobic nature, they unlikely diffuse freely in the extracellular space. Wnt ligands have been proposed to use carrier mechanisms for long‐range signaling such as chaperone proteins, extracellular vesicles, or filopodia‐like cell protrusions to bring Wnt ligands to their target cells.16 Interestingly, recent study showed that short‐range–acting membrane‐tethered Wingless (Wg, Drosophila homolog of Wnt1) was sufficient to rescue Wg loss‐of‐function phenotype in Drosophila.17 Similarly, membrane‐bound Wnt3 signaling to the distance of one to two cells induced intestinal cell differentiation.18 These data suggest that in many biological processes Wnt signaling requires immediate contact between the Wnt‐producing and its target cell.
Like all Wnts (except for Drosophila WntD), Wnt1 secretion depends on O‐acyltransferase porcupine (PORCN) and Wntless (WLS), which catalyze its palmitoylation and chaperone the Wnt ligand to the cell membrane for secretion, respectively.19, 20 While secreted, Wnt1 is bound to cell membrane at specific detergent‐resistant microdomains called lipid rafts as well as to extracellular matrix.21, 22 The best characterized functions of Wnt1 take place during embryonic development, where Wnt1 is expressed in the developing neural tube and is essential for normal brain development.23 In adult tissues, Wnt1 mRNA and protein levels are generally very low or absent, suggesting strictly regulated and context specific pattern of expression. However, Wnt1 was early shown to exhibit oncogenic properties in mouse mammary tumorigenesis,24 and elevated Wnt1 protein level has been shown in breast cancer and gliomas.25, 26
Spontaneous Swaying mutation in mouse Wnt1 gene results in an osteopenic phenotype with frequent spontaneous fractures due to decreased osteoblast activity.27 We previously reported that Wnt1 was expressed in a subset of osteocytes, in hematopoietic progenitor cells and in B cell lineage. Despite the increasing literature connecting the Wnt1 gene to bone‐related defects in humans, the major source of Wnt1 and the mechanisms of Wnt1 action in bone remain elusive. To address these questions, we generated global and limb bud mesenchyme‐targeted Wnt1 knockout mice and analyzed their phenotype. Further, we studied the molecular mechanisms, by which Wnt1 regulates both osteoblast and osteoclast differentiation.
Subjects and Methods
Generation of global Wnt1+/– mice
All mouse studies were approved by The Finnish ethical committee for experimental animals, complying with the international guidelines on the care and use of laboratory animals. Five mice were housed in one cage under standard laboratory conditions (temperature 22°C, light from 8:00 a.m. to 8:00 p.m.) and had free access to tap water and food pellets (R36, 4% fat, 55.7% carbohydrate, 18.5% protein, 3 kcal/g; Labfor, Stockholm, Sweden).
The targeting vector for the Wnt1 gene, PRPGS00167_B‐B10, was obtained from The European Conditional Mouse Mutagenesis Program (EUCOMM) (Supporting Fig. 1), and validated by PCR, restriction enzyme mapping, and sequencing. In the construct, the reporter gene LacZ and selection gene Neo are inserted into intron1 in the Wnt1 gene, leading to generation of a truncated protein or nonsense‐mediated decay. The mouse strain used for the generation of Wnt1LacZ/+ mice was created from G4 embryonic stem (ES) cells derived from mouse 129S6/C57BL/6Ncr using standard methods. The correctly targeted ES cells were then injected into C57BL/6N mouse blastocysts to generate chimeric mice. Germline transmission was achieved by cross‐breeding male chimeras with C57BL/6N females. Genotyping of mice was carried out with DNA extracted from ear marks of 2‐week‐old to 3‐week‐old mice. The following primers were used for genotyping chimeric and WT mice: forward primer (5′‐TTCCACTGGTGCTGCCACGTCA‐3′) and reverse primer (5′‐TGGCAAAAGGGTTCGAGCCGAC‐3′). In order to delete Neo cassette, heterozygous Wnt1LacZ/+ mice were bred with PGK‐Cre mice, which expresses Cre recombinase under the control of phosphoglycerate kinase promoter in all tissues leading to deletion of exons 2 to 4 of the Wnt1 gene and generation of global Wnt1+/– mice (Supporting Fig. 1). The following primers were used for genotyping WT mice: forward primer (5′‐TTCCACTGGTGCTGCCACGTCA‐3′) and reverse primer (5′‐TGGCAAAAGGGTTCGAGCCGAC‐3′), and for Wnt1+/– mice: forward primer (5′‐GCCATATCACATCTGTAGAG‐3′) and reverse primer (5′‐TGGCAAAAGGGTTCGAGCCGAC‐3′). Wnt1+/– and their WT age‐ and gender‐matched littermates were used in all studies.
Generation of conditional Wnt1Prrx1–/– knockout mice
Heterozygous Wnt1LacZ/+ F1 females born from the chimera breeding were subsequently bred with males expressing Flp recombinase in order to delete Neo and LacZ cassettes and to generate floxed Wnt1 allele (Wnt1flox/+). Genotyping of WT and Wnt1flox/+ alleles was performed by PCR using primer pair Wnt15HAse1 (5′‐TGCATTGTGACTTCACATCC‐3′) and Wnt1loxP2R (5′‐TTAAATGGGAATGGTCTCTG‐3′). To generate tissue specific knockout of Wnt1 in early limb bud mesenchyme, Wnt1flox/+ female mice were crossed with Wnt1flox/+ male mice expressing Cre recombinase driven by the prrx1 promoter specifically expressed in early limb mesenchymal cells (Tg(Prrx‐cre)1cjt/J mice) acquired from The Jackson Laboratory (Bar Harbor, ME, USA).28 The presence of the Prrx1‐cre gene was determined using the following primer pair: forward primer (5′‐CCAATTTACTGACCGTACACC‐3′) and reverse primer (5′‐CCCGGCAAAACAGGTAGTTA‐3′).
Micro–computed tomography and whole‐body CT analysis of Wnt1+/– and Wnt1Prrx1 –/– knockout mice
Micro–computed tomography (μCT) analyses were performed on the proximal tibias and lumbar L4 vertebrae using a Skyscan 1070 μCT scanner (Skyscan, Kontich, Belgium) and imaged with an X‐ray tube voltage of 72 kV and current of 138 μA, with a 0.25‐mm aluminum filter. The scanning angular rotation was 182.45 degrees, and the angular increment was 0.45 degrees. Cross‐ sectional images were reconstructed with NRecon 1.4 software (Bruker, Kontich, Belgium). For the analysis of the trabecular bone in tibia, a volumetric region of interest (ROI) excluding the cortical bone was defined at the metaphysis of the tibia starting 50 layers (488 μm) below the lower surface of the growth plate and extending 200 layers distally (1954 μm). For the analysis of cortical bone, a slice at the diaphysis of the tibia starting 5000 μm below the growth plate and extending for 50 layers was defined. For the analysis of the trabecular bone in the fourth lumbar vertebrae, an ROI excluding the cortical bone was defined at the metaphysis of the lumbar L4 vertebrae starting 20 layers (167.4 μm) above the distal growth plate and extending 150 layers proximally (1255.5 μm).
For whole‐body CT scan, isoflurane anesthetized mice were placed onto a dedicated small‐animal CT scanner (Inveon Multimodality; Siemens Medical Solutions, Knoxville, TN, USA) and body temperature was maintained using a heating pad. High‐resolution CT acquisition consisted of 160 projections with the exposure time of 1250 ms, X‐ray voltage of 80 kV, and anode current of 500 µA for a 220‐degree rotation. CT images were reconstructed with a Feldkamp‐based algorithm. Inveon Research Workplace software (Siemens Medical Solutions) was used to generate Supporting Fig. 2C and Supporting Video 1.
Histology and bone histomorphometry
For histological analyses the hindlimb samples were fixed in formalin, decalcified in EDTA, embedded in paraffin, and cut into 5‐μm sections. The sections were deparaffinized, rehydrated, and stained with hematoxylin & eosin.
For dynamic bone histomorphometry, mice were subcutaneously injected with calcein (Sigma‐Aldrich, St. Louis, MO, USA) (20 mg per kg body weight) and demeclocycline (40 mg per kg body weight) at 9 and 2 days for 12‐week‐old mice and 7 and 2 days for 6‐week‐old mice prior to euthanasia, respectively. The right tibias were harvested and fixed in 70% ethanol for 3 days. The fixed bones were dehydrated with acetone and embedded in methyl methacrylate. Undecalcified 4‐μm‐thick sections were cut with a microtome, stained with Von Kossa method for mineralized bone, stained with 2% Toluidine Blue for the analysis of osteoblasts, osteoid, and osteoclasts, or stained with tartrate‐resistant acid phosphatase (TRAP) and counterstained with Toluidine Blue for confirming the analysis of osteoclasts. Consecutive sections were left unstained for the analysis of fluorescence labeling. The bone histomorphometric analysis was performed in the proximal tibia under magnification ×200 in a 0.9‐mm‐high × 1.3‐mm‐wide region 200 μm distal from the growth plate using OsteoMeasure analyzing software (Osteometrics Inc., Decatur, GA, USA). The structure parameters (bone volume [BV/TV], trabecular thickness [Tb.Th], trabecular number [Tb.N], and trabecular separation [Tb.Sp]) were obtained by taking an average of three different measurements from consecutive sections. The structural, dynamic and cellular parameters were calculated and expressed according to the standardized nomenclature.29
Serum analyses
Serum samples were analyzed for P1NP and CTX‐I using Rat/Mouse P1NP and RatLaps assay (IDS) at ValiFinn Ltd. (Oulu, Finland).
Isolation and culture of primary mouse calvarial osteoblasts
Primary calvarial osteoblasts were isolated from newborn mouse using standard techniques. In short, osteoblasts were derived from calvariae with five consecutive 20‐min digestions with 0.1% collagenase and 0.2% dispase in α‐MEM at +37°C. Fractions two through five were pooled. After one passage cells were plated on six‐well plates in α‐MEM supplemented with 100 U/mL penicillin‐streptomycin and 10% fetal bovine serum (FBS). For osteoblast differentiation the cells were stimulated at confluence with 10nM dexamethasone, 50 μg/mL ascorbic acid, and 5mM β‐glycerophosphate.
Differentiation of primary osteoclasts from total bone marrow with PTH
Bone marrow cells were flushed from femur and tibia of 6‐week‐old to 9‐week‐old mice. In cultures using whole bone marrow, 1 × 105 bone marrow cells were seeded in 100 µL α‐MEM supplemented with 10% FBS (Gibco, Grand Island, NY, USA) and 10nM hPTH1‐34 (Millipore, Billerica, MA, USA) in a 96‐well plate. The culture medium was changed every 2 days and RNA was collected and TRAP staining was performed at indicated time points. After TRAP staining, the number of osteoclasts with more than three nuclei was counted.
Quantitative real‐time PCR analysis
Total RNA was prepared using the RNeasy mini Kit (QIAGEN, Valencia, CA, USA) according to the manufacturer's instructions. The cDNA was synthesized with DyNAmo cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). RNA expression of the different genes of interest (Supporting Table 1) was determined using Dynamo Flash SYBR Green qPCR Kit (ThermoFisher Scientific). mRNA levels were normalized to β‐actin expression by using the delta‐delta comparative threshold cycle (2–ΔΔCt) method.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer with a cocktail of protease inhibitors (Complete mini; Thermo Fisher Scientific). The insoluble material was centrifuged (12,000g, 10 min, 4°C), and supernatants were collected. Equivalent amounts of protein were electrophoresed on 10% SDS‐PAGE gels and transferred to nitrocellulose membranes (Maine Manufacturing, LLC, Sanford, ME, USA). Membranes were blocked and incubated with primary antibodies (1:1000 non‐P‐β‐catenin, total β‐catenin; 1:5000, β‐tubulin [Millipore]; and 1:250 Wnt1 [Invitrogen, Carlsbad, CA, USA]) overnight, followed by incubation with horseradish peroxidase (HRP)‐linked secondary antibodies (1:10,000; Cell Signaling Technology, Beverly, MA, USA). The protein bands were visualized with Western Bright Quantum (Advansta Inc., Menlo Park, CA, USA) and LAS‐4000 Luminescent imager (FUJIFILM, Valhalla, NY, USA). Quantification of the density of each band was performed by Image J software (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/).
Viral packaging and infection
Human Wnt1 coding region was cloned to pBabe‐puro vector. Wnt1 and empty viruses were produced in human embryonic kidney (HEK) 293 Phoenix cells according to standard methods. Target cells (C3H10T1/2, MC3T3‐E1) were incubated with viral media containing 4 μg/mL polybrene overnight, followed by selection in puromycin at the concentration of 3 μg/mL for 2 to 3 days, after which the puromycin‐resistant cell pools were expanded and plated for subsequent experiments.
Culture of Wnt1 overexpressing C3H10T1/2 cells
For the RNA and protein analyses, C3H10T1/2‐EV (empty virus) and C3H10T1/2‐Wnt1 cells were seeded in 12‐well plates at the density of 40,000 cells per well. For mixed cultures, 10T1/2‐EV and 10T1/2‐Wnt1 cells were seeded in 12‐well plates at equal amounts of 20,000 and 20,000 cells, respectively. For insert culture, 10T1/2‐EV and 10T1/2‐Wnt1 cells were seeded onto a Millicell cell culture plate insert (Millipore) at a density of 10,000 per well. Millicell inserts were placed in 12‐well plates and 40,000 C3H10T1/2‐EV cells were seeded in the lower chambers. ALP staining, RNA, and protein extraction were performed at 10 days.
Proliferation assay
C3H10T1/2‐EV (empty virus) or C3H10T1/2‐Wnt1 cells were seeded in 96‐well plates at a density of 1500 cells/well. The CellTiter 96® Non‐radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) was used to detect cell viability at indicated time points. Briefly, 20 μL dye solution was added to each well followed by incubation at 37°C for 4 hours. The Solubilization/Stop Solution was then added to the culture wells to solubilize the formazan product, and the absorbance at 570 nm was recorded using a 96‐well plate reader.
Osteoblast and osteoclast co‐cultures
The osteoblastic cells (MC3T3‐E1, C3H10T1/2, primary calvarial osteoblast from WT or Wnt1+/– mice) were plated on 24‐well plates at a density of 10,000 cells/well in α‐MEM with 10% FBS. Next day, nonadherent cells harvested from WT C57Bl/6N mouse bone marrow were plated on top of osteoblastic cells at a density of 1.5 × 106/well (on the top of MC3T3‐E1 and primary cells) or 6.6 × 104 (on top of C3H10T1/2 cells). The cells were stimulated either with 10–8M vitamin D and 10–6M prostaglandin E2 (MC3T3‐E1 cells, primary cells) or with 10 ng/mL M‐CSF and 50 ng/mL hRANKL (C3H10T1/2). The culture medium was changed every 3 days and TRAP staining was performed at indicated time points. For low‐density versus high‐density experiments, 1 × 0.1 and 1 × 104 C3H10T1/2 cells /well were plated on 48‐well plates, respectively. To study osteoblast‐osteoclast interaction mediated by secreted factors, the co‐culture was performed by plating the nonadherent bone marrow cells on the 12‐well plate at a density of 1.32 ×105/well and C3H10T1/2‐EV/Wnt1 cells onto a Millicell cell culture plate insert (Millipore) at a density of 5800 per well.
Co‐culture of Wnt1 overexpressing C3H10T1/2 cells and Raw264.7 cell line
C3H10T1/2‐EV (empty virus) or C3H10T1/2‐Wnt1 cells were seeded in 96‐well plates at the density of 1.68 × 104 cells/well. After attachment to the bottom of the wells, C3H10T1/2 cells were treated with 10 μg/mL mitomycin C (Sigma‐Aldrich) for 2 hours followed by careful washing with PBS. Then Raw264.7 cells were plated on top of C3H10T1/2 cells at a density of 3 × 103 cells/well. The cells were stimulated with 50 ng/mL RANKL to induce osteoclast differentiation. For the experiments with inhibitors, the co‐culture cells were continuously treated with 10 μM XAV939 (Tocris Bioscience, Bristol, UK), 10μM KN93 (Tocris), or 5 μM SP600125 (Tocris). The culture medium was changed every 2 days and TRAP staining was performed at indicated time points.
β‐Catenin immunofluorescence
C3H10T1/2‐Wnt1 cells used for immunofluorescence were seeded on coverslips at the density of 1 × 104/well in 12‐well plates and treated with 10 μg/mL mitomycin C (Sigma‐Aldrich) for 2 hours. C3H10T1/2‐EV cells were seeded on top of C3H10T1/2‐Wnt1 cells at the density of 9 × 104 cells/well and cultured overnight. The cells were then washed in PBS and fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature. After 30 min of blocking in 5% Normal Goat Serum Control (ThermoFisher Scientific) diluted in PBS/3%BSA/0.05%Triton X‐100 (blocking buffer), sections were incubated overnight at 4°C with anti‐Wnt1 rabbit polyclonal antibody (Abcam, Cambridge, MA, USA) and anti‐β‐catenin mouse monoclonal antibody (BD Biosciences, San Jose, CA, USA), diluted 1:200 in blocking buffer. After washing, cells were incubated with Alex Fluor‐594‐conjugated goat anti‐rabbit IgG and Alexa Fluor‐488 conjugated goat anti‐mouse IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) 1:500 for 1 hour. Slides were mounted in mounting medium for fluorescence with DAPI (Vector Laboratories, Burlingame, CA, USA).
Confocal microscopy
Immunofluorescence analysis was performed by confocal laser scanning microscope (LSM780; Zeiss, Jena, Germany) equipped with four laser lines: 458 nm, 488 nm, 543 nm, and 405 nm. The fluorescence intensity was measured by using ImageJ software on the original photographs. In 10T1/2‐EV or 10T1/2‐Wnt1 culture, the mean nuclear and cytoplasmic fluorescence intensity were generated by analyzing 10 cells per field. In the mixed culture, we chose two 10T1/2‐EV cells that were in contact with a 10T1/2‐Wnt1 cell (Fig. 4 A, yellow arrow) and two 10T1/2‐EV cells which were not in contact with a 10T1/2‐Wnt1 cell (Fig. 4 A, white arrow) per field for intensity measurement. In total, nine fields from two duplicate slides were analyzed. For analyzing autocrine effect of Wnt1, the mean nuclear and cytoplasmic fluorescence intensity of 10T1/2‐Wnt1 cell in mixed culture was measured.
Figure 4.
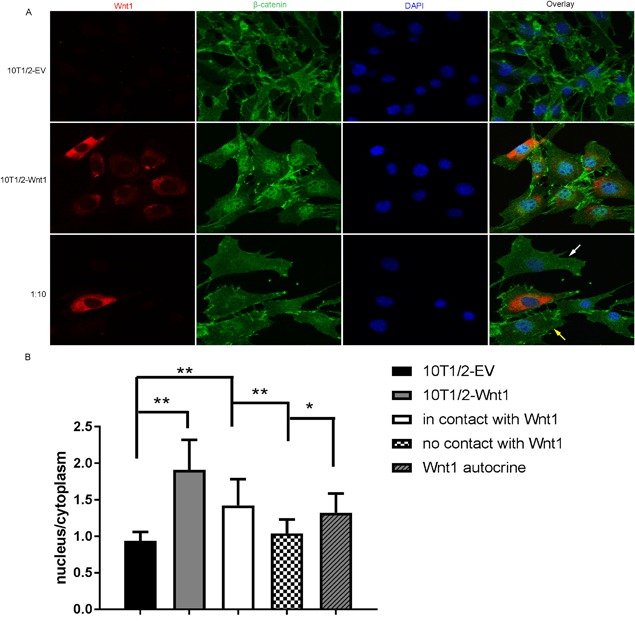
Wnt1 increased the nuclear accumulation of β‐catenin in neighboring cells. (A) Immunofluorescence staining for Wnt1 (red) and β‐catenin (green) in control C3H10T1/2‐EV cells, mitomycin C‐treated Wnt1 overexpressing C3H10T1/2 cells or mixed culture of mitomycin C‐treated Wnt1 overexpressing C3H10T1/2 cells with control cells in 1:10 ratio. (B) Quantification of the β‐catenin fluorescence intensity in the nuclear and the cytoplasmic compartments, presented as a ratio of nuclear intensity to cytoplasmic intensity. Nonparametric values were analyzed with the Steel‐Dwass method. Data are presented as mean ± SD (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001.
Opg ELISA
Conditioned medium from co‐culture of Wnt1 overexpressing C3H10T1/2 cells and Raw264.7 cells was collected and centrifuged at 16128g rpm for 5 min at 4°C. The supernatants were transferred to fresh tubes and stored at –80°C until used. OPG protein level in supernatants was determined using Quantikine osteoprotegerin Immunoassay (MOP00; R&D Systems, Minneapolis, MN, USA).
Histochemical analysis of osteoblast and osteoclast cultures
For ALP and Von Kossa staining, the osteoblast cell culture plates were fixed in 3.7% formaldehyde, rinsed in water, and stained according to standard protocols. Osteoclast cultures were fixed and stained for TRAP according to manufacturer's instructions (Sigma‐Aldrich).
Statistical analysis
Parametric data were compared with a t test. Nonparametric comparisons for all pairs used the Steel‐Dwass method. Values of p < 0.05 were considered significant (*p < 0.05; **p < 0.01; ***p < 0.001). All the data in the graphs are expressed as mean ± SD.
Results
Heterozygous deletion of Wnt1 results in mild osteopenia
Wnt1‐deficient mice had been previously published but were no longer available, which led us to generate novel Wnt1 mutant mice that allowed for producing both global and conditional Wnt1‐deficient mice (Supporting Fig. 1). As reported,23 we did not find any surviving homozygous Wnt1 pups at birth. However, at embryonic day 18.5 (E18.5) Wnt1–/– were found at the expected Mendelian ratio (data not shown). Wnt1 is required for the development of several areas in the central nervous system, which explains the neonatal mortality in absence of Wnt1.23
Because the heterozygous mutation in WNT1 was sufficient to cause an osteoporotic phenotype in humans, we first analyzed the bone phenotype of Wnt1+/– mice. Wnt1+/– mice developed normally and their body weights did not differ from the littermate controls at 6 or 12 weeks of age. X‐ray imaging of control and Wnt1+/– limbs did not detect long‐bone fractures in Wnt1+/– mice or differences in the bone lengths between the groups (Fig. 1 A). Micro–computed tomography (μCT) analysis of Wnt1+/– mice in tibia revealed a mild trabecular osteopenia demonstrated by 23.8% reduction in bone volume/tissue volume (BV/TV, p < 0.01) and 20.5% in trabecular number (Tb.N, p < 0.05) with a subsequent increase in trabecular separation (Tb.Sp, p < 0.05) at 12 weeks of age compared to controls (Fig. 1 B). μCT analysis of fourth lumbar vertebrae (L4) of Wnt1+/– mice showed a mild trabecular osteopenia demonstrated by 16% reduction in BV/TV (p < 0.05) and 17.5% reduction in bone mineral density (BMD, p < 0.05) at 12 weeks of age compared to controls (Fig. 1 C, D). We next performed histomorphometric analysis to understand the mechanisms underlining the osteopenic phenotype. Surprisingly, we did not observe significant differences in any of the histomorphometric parameters analyzed (Supporting Fig. 2A, B). However, there were nonsignificant decreases in mineral apposition and bone formation rates (MAR and BFR/BV, p = 0.14 and p = 0.56, respectively), whereas the number of osteoclasts and eroded surface were numerically increased but the changes were not significant (N.Oc/B.Pm, p = 0.39; ES/BS, p = 0.11) (Supporting Fig. 2B). Taken together, these data suggest that the mild osteopenia in Wnt1+/– mice develops due to modest alterations in bone formation and resorption.
Figure 1.
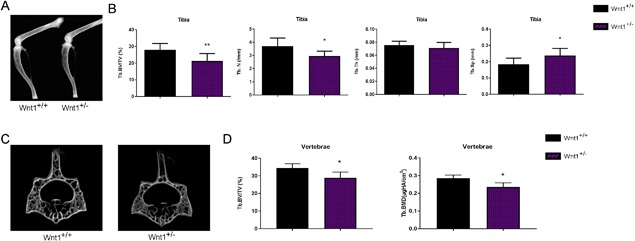
Heterozygous deletion of Wnt1 results in mild osteopenia. (A) Hind limbs of 12‐week‐old Wnt1+/+ and Wnt1+/– mice imaged by X‐ray. (B) μCT analysis showing BV/TV, Tb.N, Tb.Th, and Tb.Sp in Wnt1+/+ and Wnt1+/– mouse tibias at 12 weeks of age. (C) μCT images of vertebral bone of Wnt1+/+ and Wnt1+/– at the age of 12 weeks. (D) μCT analysis showing trabecular BV/TV and trabecular BMD of lumbar vertebrae L4 of Wnt1+/+ and Wnt1+/– mice at 12 weeks of age. Data are presented as mean ± SD (n = 8). In B and D, *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was performed by Student's t test. BV/TV = bone volume per tissue volume; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; BMD = bone mineral density.
Deletion of Wnt1 in long‐bone mesenchymal cells leads to severe osteopenia and spontaneous fractures
We had previously detected Wnt1 expression in vivo in osteocytes, B cells, and in hematopoietic lineage–negative cells,3 part of which are likely of mesenchymal origin. To test which cell lineage is the major source of Wnt1 in bone we next generated conditional knockout mice, in which the Wnt1 gene was deleted in limb bud mesenchymal cells with Prrx1‐Cre (Supporting Fig. 1A).28 Wnt1Prrx1 –/– mice were viable but developed spontaneous long‐bone fractures as early as 4 weeks of age (Fig. 2 A, Supporting Fig. 2, and Supporting Video 1). The penetrance of the fracture phenotype in the tibia was 100% in male Wnt1Prrx1 –/– mice and 80% in females (Fig. 2 B). Fractures were most frequent in mid‐tibia but were also found in other long bones targeted by Prrx1‐Cre. Fractures did heal because callus tissue was found in long‐bone X‐rays and in histological sections of the fracture sites (Fig. 2 A–C). μCT analysis of proximal tibia showed severe trabecular osteopenia and reduced cortical thickness in the diaphysis of Wnt1Prrx1 –/– mice compared to controls at both 6 weeks and 12 weeks of age (Fig. 2 D–G), but the phenotype was more severe at the age of 6 weeks. Wnt1Prrx1 –/– animals indeed presented with an OI phenotype with early spontaneous fractures and low bone mass in both trabecular and cortical compartments, similar to the patients carrying the homozygous WNT1 mutation.
Figure 2.
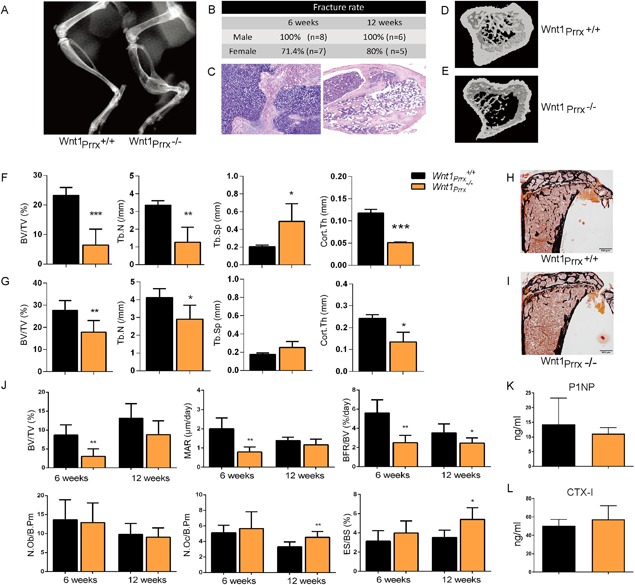
Deletion of Wnt1 in limb mesenchymal cells leads to severe osteopenia and spontaneous fractures. (A) Hind limbs of 6‐week‐old Wnt1Prrx1 +/+ and Wnt1Prrx1 –/– mice imaged by X‐ray. (B) Fracture frequency of the right tibia presented as percentage of Wnt1Prrx1 –/– mice at 6 and 12 weeks of age, evaluated by X‐ray. (C) Representative histological sections of Wnt1Prrx1 –/– fracture sites stained by H&E from early (left) and late (right) stage fractures. (D, E) μCT images of Wnt1Prrx1 +/+ (D) and Wnt1Prrx1 –/– (E) mice proximal tibias at 6 weeks of age. (F, G) μCT analysis showing BV/TV, Tb.N, Tb.Sp, and Cort.Th in Wnt1Prrx1 +/+ and Wnt1Prrx1 –/– mouse tibias at 6 weeks (F) and 12 weeks (G) of age (n = 6). (H, I) Von Kossa staining of tibia sections of Wnt1Prrx1 +/+ (H) and Wnt1Prrx1 –/– (I) mice at 12 weeks of age. (J) Histomorphometry of 6‐week‐old and 12‐week‐old Wnt1Prrx1 +/+ and Wnt1Prrx1 –/– mouse tibias: BV/TV, MAR, BFR/BV, N.Ob/B.Pm, N.Oc/B.Pm, and ES/BS. (n = 6). (K, L) Serum P1NP and CTX‐I levels in 12‐week‐old Wnt1Prrx1 +/+ and Wnt1Prrx1 –/– mice (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001. All data are presented as mean ± SD. BV/TV = bone volume per tissue volume; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; Cort.Th = cortical thickness; MAR = mineral apposition rate; BFR/BV = bone formation rate; N.Ob/B.Pm = number of osteoblasts per bone perimeter; N.Oc/B.Pm = number of osteoclasts per bone perimeter; ES/BS = eroded surface per bone surface.
Histomorphometric analyses confirmed the osteopenic phenotype of Wnt1Prrx1 –/– mice (Fig. 2 H–J, Supporting Fig. 2C). Interestingly, Wnt1 deficiency did not alter the osteoblast number or surface but osteoblast activity was severely impaired in Wnt1Prrx1 –/– mice at 6 weeks of age shown by decreased MAR, mineralized surface (MS/BS), BFR/BV, and osteoid thickness (O.Th) (Fig. 2 J, Supporting Fig. 2C). As in μCT analysis the phenotype was somewhat milder at 12 weeks, but MS/BS and BFR/BV remained significantly lower in Wnt1Prrx1 –/– mice compared to controls, although osteoblast activity is overall lower than at 6 weeks (Fig. 2 J, Supporting Fig. 2C). Surprisingly, bone resorption was enhanced in Wnt1Prrx1 –/– mice at 12 weeks indicated by increased osteoclast number (N.Oc/B.Pm), osteoclast surface (Oc.S/B.Pm), and eroded surface (ES/BS) (Fig. 2 J, Supporting Fig. 2C), further contributing to the osteopenic phenotype. We did not capture the altered bone formation or resorption of Wnt1Prrx1 –/– mice in the measurements of serum markers of bone metabolism (CTX for bone resorption and P1NP for bone formation), likely because Wnt1 was deleted merely in the limbs but not in the axial skeleton (Fig. 2 K, L).
Mesenchymal cell–derived Wnt1 stimulates osteoblast differentiation in a juxtacrine manner
To elucidate the mechanisms by which Wnt1 regulates osteoblast differentiation and activity, we first analyzed the differentiation of Wnt1+/– primary calvarial cells. Wnt1+/– calvarial osteoblast progenitors exhibited impaired osteoblast differentiation and function shown by decreased alkaline phosphatase (ALP) activity and mRNA expression and impaired formation of mineralized bone nodules (Fig. 3 A, B). Wnt1+/– cells also expressed lower levels of Wnt target genes Dkk1 and SOST as well as OPG, an osteoblast‐derived inhibitor of osteoclastogenesis (Fig. 3 B). Conversely, retroviral overexpression of Wnt1 in mesenchymal progenitor cell line C3H10T1/2, which does not express endogenous Wnt1, led to increased osteoblast differentiation shown by increased ALP activity and expression (Fig. 3 C, D). Expectedly, Wnt1 overexpression robustly induced the expression of Wnt target genes (Fig. 3 D).
Figure 3.
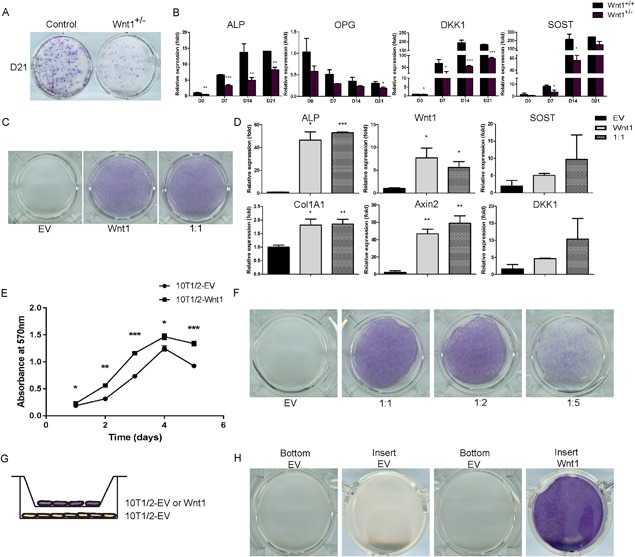
Mesenchymal cell‐derived Wnt1 stimulates osteoblast differentiation in a juxtacrine manner. (A, B) Calvarial osteoblasts from Wnt1+/+ and Wnt1+/– mice were induced to differentiate for indicated times. (A) ALP staining of control and Wnt1+/– calvarial osteoblast cultures. (B) mRNA expression levels of osteoblast‐related genes during calvarial cell differentiation measured by quantitative qRT‐PCR, was normalized to β‐actin expression and presented as a relative fold change to day 0 expression. (C) Mixed culture of Wnt1 overexpressing C3H10T1/2 cells with control EV‐transduced cells were stained for ALP at 7 days posttransduction. (D) mRNA expression of osteoblastic and Wnt target genes quantified by qRT‐PCR in cultures with 1:1 ratio of Wnt1 and control cells (EV). (E) Wnt1‐overexpressing C3H10T1/2 cells or control cells were seeded at density of 1500 cells/well. The CellTiter 96® Non‐radioactive Cell Proliferation Assay was used to detect cell viability at indicated time points. Results were presented as mean absorbance at 570 nm from triplicate assays. (F) Wnt1‐overexpressing C3H10T1/2 cells were growth‐arrested by mitomycin C and C3H10T1/2‐control (EV) cells were plated on top at different ratios keeping the total number constant. ALP staining was performed at D7. (G) Schematic of the method used to study Wnt1 effects mediated by secreted factors using cell culture well inserts. (H) ALP staining of cell culture wells (bottom) and inserts (insert) at D10. All cell culture experiments were repeated at least three independent times with at least three replicate wells. Staining and mRNA data are presented from one representative experiment as mean ± SD (n ≥ 3). B, D, and E: *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was performed by Student's t test. EV = empty virus.
Wnt ligands are secreted proteins that bind to cell membranes, extracellular matrix, and may also be trafficked in lipoprotein‐containing argosomes to induce short‐range and long‐range signaling.30 Despite extensive efforts, we were not able to produce Wnt1 conditioned medium, but instead when mixed with control cells Wnt1 could efficiently induce osteoblastic differentiation, increase the amount of active nonphosphorylated β‐catenin, and induce the expression of Wnt target genes (Fig. 3 D; Supporting Fig. 3).
The Wnt β‐catenin–dependent canonical pathway has been shown to induce proliferation of mesenchymal stem cells31 and indeed Wnt1 overexpression stimulated the proliferation of C3H10T1/2 cells (Fig. 3 E). To study whether Wnt1 could induce osteoblastic differentiation in neighboring control cells and to exclude the effect of enhanced proliferation of Wnt1 overexpressing cells, we cultured growth‐arrested C3H10T1/2 cells stably overexpressing Wnt1 cells with control cells at different ratios. Reduction of the ratio between control and Wnt1 overexpressing cells led to decreased ALP staining, suggesting that Wnt1 would require cell‐cell contact or at least very close proximity to induce osteoblast differentiation in the neighboring cell (Fig. 3 F).
To further investigate this, we used cell culture well inserts that allow for diffusion of humoral factors and small particles such as argosomes between the two compartments (Fig. 3 G). We found that Wnt1‐overexpressing cells in the insert were not able to induce differentiation of the cells at the bottom of the well, indicating that Wnt1 functions in a juxtacrine manner; ie, it induces osteoblastic differentiation only when in immediate proximity to the target cell (Fig. 3 H).
Wnt1 requires cell‐cell contact to induce canonical Wnt signaling
Activation of the canonical Wnt signaling pathway leads to nuclear accumulation of activated β‐catenin. To investigate if Wnt1 indeed requires direct cell‐cell contact to induce Wnt signaling we plated control, Wnt1‐overexpressing, or Wnt1‐overexpressing with control cells in 1:10 ratio on coverslips. Wnt1‐overexpressing cells were treated with mitomycin C to halt their proliferation. The cells were fixed and stained for Wnt1 and β‐catenin with polyclonal and monoclonal antibodies, respectively, and with DAPI to visualize the nucleus. We imaged the samples with confocal microscope and quantified the intensity of nuclear versus cytoplasmic β‐catenin staining. Wnt1 stimulated the nuclear accumulation of β‐catenin most in the Wnt1‐overexpressing cultures, in which it could likely work both in an autocrine and juxtacrine manner (Fig. 4 A, B). In the mixed cultures of Wnt1‐overexpressing and control cells, nuclear β‐catenin was found in Wnt1 cells confirming the autocrine mechanism of action (Fig. 4 A, B; Supporting Fig. 4). In addition to the Wnt1‐overexpressing cells, we found nuclear translocation of β‐catenin control cells only when in direct contact with the Wnt1 cell. We did not observe β‐catenin nuclear translocation in cells without contact with Wnt1‐overexpressing cells, even if they were in very close range. These data show that Wnt1 requires direct cell‐cell contact for its biological functions.
Wnt1 directly suppresses osteoclastogenesis in a juxtacrine manner
Because we did observe increased osteoclast number and eroded surface in Wnt1Prrx1 –/– mice at 12 weeks, we next asked how does Wnt1 regulate osteoclast differentiation? There was no osteoclast phenotype in Wnt1+/– mice in vivo, and although parathyroid hormone (PTH) treatment of total bone marrow from Wnt1+/– mice showed a trend towards increased osteoclastogenesis, co‐culture experiments of control and Wnt1+/– calvarial cells mixed with control and Wnt1+/– hematopoietic cells did not indicate any significant osteoclast phenotype in Wnt1+/– cells in vitro (Supporting Fig. 5A–D). However, overexpression of Wnt1 in MC3T3‐E1 cells that can support osteoclastogenesis, when stimulated with vitamin D3 and prostaglandin E2, resulted in almost complete inhibition of osteoclastogenesis (Supporting Fig. 6A, B). Receptor activator of nuclear factor kappa‐B ligand (RANKL) is a cytokine that induces osteoclastogenesis by binding to its receptor RANK leading to induced expression of downstream target genes such as Nfatc1.32 OPG is a decoy receptor for RANKL that inhibits osteoclastogenesis and is a direct target gene of canonical Wnt signaling.12 We found that Wnt1 overexpression induced OPG production in E1 cells, providing a potential mechanism by which Wnt1 could control osteoclastogenesis indirectly (Supporting Fig. 6C).
To test this in another approach, we overexpressed Wnt1 in C3H10T1/2 cells that do not support osteoclastogenesis, when stimulated with vitamin D3 and prostaglandin E2. We mixed Wnt1 or control C3H10T1/2 cells at different densities with WT hematopoietic bone marrow cells and induced osteoclast differentiation directly by supplementing the culture medium with macrophage colony‐stimulating factor (M‐CSF) and RANKL. Again, Wnt1‐overexpressing cells inhibited osteoclastogenesis, accompanied with decreased expression of downstream target of RANKL signaling Nfatc1 and osteoclast markers Tracp and Ctsk (Fig. 5 A–C). In contrast to MC3T3‐E1 cells, Wnt1 overexpression did not have any significant effect on OPG or RANKL mRNA expression in C3H10T1/2 cells, possibly due to the different cellular origin of the cell lines (mesenchymal progenitor versus preosteoblast) (data not shown). Interestingly, this suppression of osteoclastogenesis was not seen at low cell density of Wnt1‐overexpressing C3H10T1/2 cells, but the formation of osteoclasts was even enhanced. We found that at early stages of these low‐density cultures before addition of RANKL to the medium, OPG expression was actually decreased with highly increased RANKL/OPG ratio. This could at least in part contribute to the enhanced osteoclast formation observed in the low‐density cell cultures. Taken together these data suggest that Wnt1 functions in an autocrine/juxtacrine manner to regulate OPG expression at least in MC3T3‐E1 cells but also raises the question whether Wnt1 could directly suppress osteoclastogenesis.
Figure 5.
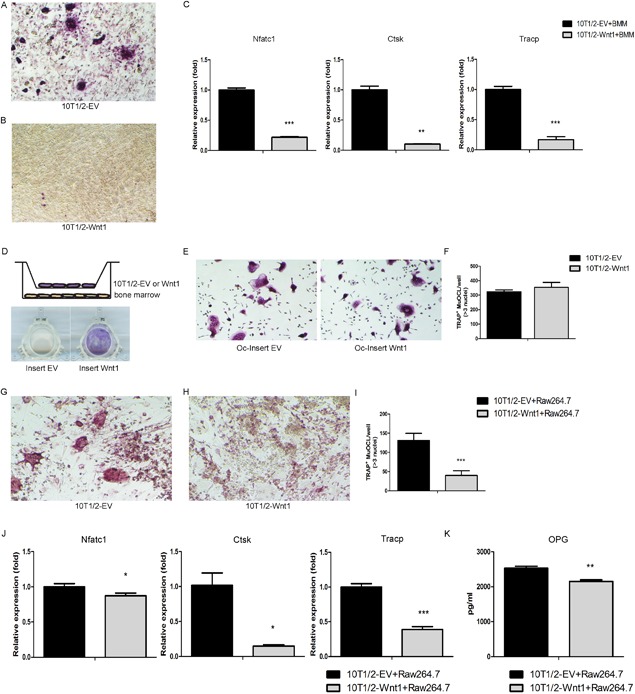
Wnt1 directly suppresses osteoclastogenesis in a juxtacrine manner. (A, B) C3H10T1/2 control (A) or Wnt1‐overexpressing (B) cells were plated on the bottom of cell culture wells and primary bone marrow macrophages (BMM) on top. Osteoclastogenesis was induced by M‐CSF and RANKL. At D5 the cultures were stained for TRAP to demonstrate mature multinucleated osteoclasts. (C) mRNA expression of osteoclast‐specific genes measured by qRT‐PCR in C3H10T1/2 control or Wnt1 overexpressing cells co‐cultured with nonadherent hematopoietic cells at D5. (D) Schematic of the method to study effect of secreted factors from C3H10T1/2‐EV/Wnt1 cells on osteoclastogenesis. Osteoclastogenesis was induced by M‐CSF and RANKL. ALP staining of cell culture inserts at D5 demonstrating the effect of Wnt1 in 10T1/2‐Wnt1 cells. (E, F) TRAP staining of bone marrow cells on cell culture wells after removing the cell culture inserts (E); TRAP‐positive cells were counted from five fields of each three wells per condition (F). (G–I) C3H10T1/2 control (G) or Wnt1‐overexpressing cells (H) were plated on the bottom of cell culture wells and growth‐arrested with mitomycin C and Raw264.7 cells were plated on top. Osteoclastogenesis was induced with RANKL. At D5 the cultures were stained for TRAP to evaluate osteoclastogenesis (I). (J) mRNA expression of osteoclastic genes measured by qRT‐PCR in C3H10T1/2 co‐cultured with Raw264.7 cells at D5. (K) C3H10T1/2 control or Wnt1‐overexpressing cells were co‐cultured with Raw264.7 cells and osteoclastogenesis was induced by RANKL. The supernatants were collected to measure OPG levels by ELISA. All cell culture experiments were repeated three independent times with at least three replicate wells per condition. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was performed by Student's t test. TRAP = tartrate‐resistant acid phosphatase.
To explore this farther, we next tested whether the major effect of Wnt1 would be derived from regulating cytokine expression or by direct juxtacrine mechanism as in mesenchymal cells. In insert cultures of Wnt1‐overexpressing and control C3H10T1/2 (in inserts) and WT hematopoietic cells (in wells), we found no suppression of osteoclastogenesis indicating that putative Wnt1‐induced secretion of OPG is not sufficient to suppress osteoclastogenesis (Fig. 5 D–F). Rather, our data suggests that Wnt1 (and OPG) directly suppresses osteoclast differentiation in a juxtacrine manner.
To exclude the possible effects of enhanced proliferation of Wnt1‐overexpressing cells and the effects of Wnt1 on the heterogenous population of bone marrow hematopoietic cells we co‐cultured growth‐arrested Wnt1 or control C3H10T1/2 cells with murine secondary monocyte/macrophage‐like cell line RAW264.7 cells and induced osteoclast differentiation with RANKL. Wnt1‐overexpressing cells directly inhibited osteoclastogenesis also in this model, accompanied with decreased expression of osteoclast markers Ctsk and Tracp (Fig. 5 G–J). This was not due to increased production of soluble OPG because the OPG protein levels were actually decreased in the medium of Wnt1‐overexpressing cultures mixed with RAW264.7 cells (Fig. 5 K). Based on our data that Wnt1‐overexpressing cells suppress osteoclastogenesis only when at sufficient density and in physical contact with the osteoclast progenitors, we conclude that mesenchymal cell–derived Wnt1 can directly inhibit osteoclastogenesis in a juxtacrine manner.
Wnt1 suppresses osteoclastogenesis through canonical Wnt signaling
Different Wnt ligands and branches of the Wnt signaling have variable effects on osteoclast differentiation. Because of the technical restrictions of the co‐culture system necessary to study Wnt1 effects, we investigated the mechanism of Wnt1 action on osteoclastogenesis in C3H10T1/2‐RAW264.7 co‐cultures using small molecular inhibitors XAV939 (axin‐dependent canonical signaling), KN93 (calcium‐dependent noncanonical signaling), and SP600125 (JNK‐dependent noncanonical signaling).33, 34, 35 Inhibition of calcium signaling with KN93 strongly inhibited osteoclastogenesis in all cultures and Wnt1 did not modify this effect (Fig. 6 A). Inhibition of axin and thus canonical signaling with XAV939 slightly enhanced osteoclastogenesis in control cultures and completely rescued osteoclastogenesis in Wnt1‐overexpressing co‐cultures, while JNK inhibitor partially blocked Wnt1 effect on osteoclast differentiation (Fig. 6 A, B). Accordingly, XAV939 significantly suppressed the expression of Wnt1 target genes Axin2 and TCF1 in the Wnt1‐overexpressing cultures and partially rescued the expression of osteoclast marker Ctsk (Fig. 6 C–E). We also observed that Wnt1 overexpression suppressed OPG expression that was even further inhibited by addition of XAV939, while RANKL expression was unchanged, resulting in increased RANKL/OPG ratio in Wnt1 overexressing cultures (Fig. 6 F–H). These data support our conclusion that Wnt1 directly inhibits osteoclastogenesis independent of OPG under these experimental conditions.
Figure 6.
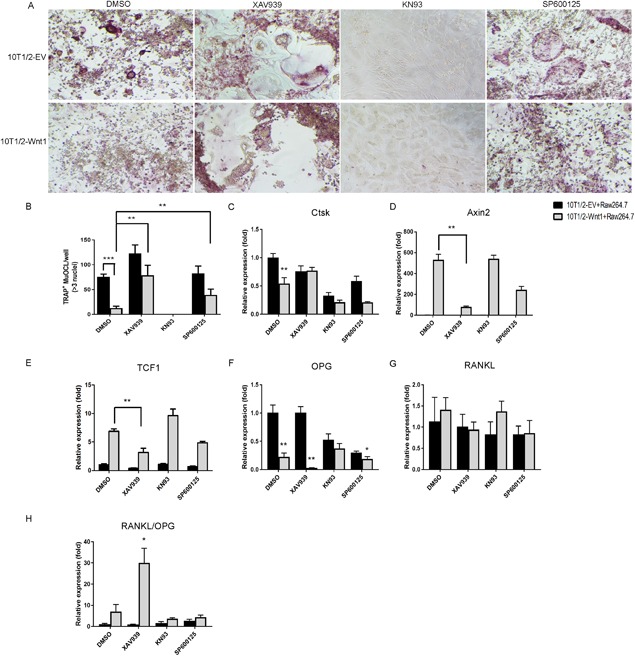
Wnt1 suppresses osteoclastogenesis through canonical Wnt signaling pathway. (A, B) C3H10T1/2 control or Wnt1‐overexpressing cells (A) were plated on the bottom of cell culture wells and growth‐arrested by 2.5 μg/mL mitomycin C treatment for 2 hours. Raw264.7 cells were plated on top of them. Osteoclastogenesis was induced by 50 ng/mL RANKL. Cells were treated with DMSO, 10μM XAV939, 10 μM KN93, or 5μM SP600125. (B) At D5 the cultures were stained for TRAP to demonstrate mature multinucleated osteoclasts. (C–H) mRNA expression of osteoclast‐specific and Wnt target genes was measured by qRT‐PCR. All cell culture experiments were repeated three independent times with at least three replicate wells per condition. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis was performed by Student's t test.
Discussion
We and others have recently identified Wnt1 as a key Wnt ligand regulating bone mass in humans.1, 2, 3, 4 To address the question of the main cellular source of Wnt1 in bone, we generated novel global and conditional mesenchymal progenitor‐targeted Wnt1‐deficient mice. In previous studies, Wnt signaling has been shown to promote mesenchymal cell proliferation, osteoblast differentiation, and osteoblast activity.36 Several Wnt ligands, such as Wnt3a, Wnt5a, Wnt7a, and Wnt10b, have been shown to play a major role in skeletal development and/or regulation of bone formation.36 Although many of these ligands can elicit canonical β‐catenin–dependent Wnt signaling similarly to Wnt1, our data clearly shows that mesenchymal cell–derived Wnt1 has an essential nonredundant function in regulating bone metabolism in vivo.
Global heterozygous deletion of Wnt1 led to mild osteopenic phenotype but no fractures were observed. In humans, however, heterozygous loss of function mutation in WNT1 results in early onset osteoporosis with high fracture incidence already at relatively young age.3 This difference in the observed phenotypes is not surprising, considering species differences including body size, lifespan, and posture. Homozygous deletion of Wnt1 in mesenchymal progenitors led in turn to severe osteoporosis and spontaneous fractures, resembling the form of human OI caused by homozygous Wnt1 mutation. Prrx1‐Cre–driven deletion of Wnt1 led to decreased bone mass in both cortical and trabecular compartments of long bones due to impaired osteoblast function as well as enhanced bone resorption. These data show that the role of Wnt1 in regulating bone metabolism is distinct from many other components of the Wnt signaling pathway, as for example the deletion of Wnt16 leads to decreased cortical bone mass and deletion of Wnt10b to decreased trabecular bone mass, osteoblast‐targeted Wnt5a knockouts exhibit low trabecular bone mass whereas Sfrp4 knockouts have high trabecular bone mass but decreased amount of cortical bone.14, 37 Interestingly these factors also have different effects on bone formation and resorption. Taken together, these findings highlight the complexity of the Wnt signaling pathway but also illustrate how the multiple inputs from different Wnt ligands/modulators and canonical and noncanonical pathways are integrated to control bone homeostasis.
Wnt1 can activate the canonical, β‐catenin–mediated Wnt signaling pathway through Lrp5/6 receptors.3, 38 Extensive evidence supports the positive role of canonical Wnt signaling and β‐catenin in promoting mesenchymal progenitor cell commitment to and progression on the osteoblast lineage.39, 40 However, in mature osteoblasts β‐catenin appears to mainly regulate osteoblast‐dependent osteoclastogenesis via the expression of Opg.12, 41 Deletion of Wnt1 in the early limb bud mesenchyme did not alter the number of osteoblasts on bone surface, but their activity was impaired, shown by decreased MAR and MS/BS, which resulted in overall reduced bone formation. Although β‐catenin does not appear to regulate bone formation activity in mature osteoblasts, gain‐of‐function of LRP5/6 in mature osteoblasts as well as blocking Sclerostin activity does induce high bone mass due to increased bone formation.5, 6, 7 Wnt1 has not been shown to activate noncanonical Wnt receptors/pathways and, although we cannot at present rule out this possibility, it is likely that Wnt1 activates osteoblast function via canonical signaling.
Sclerostin does specifically inhibit Wnt1‐type ligand binding to the first β‐propeller of the LRP5/6 receptor and subsequent activation of the receptor.38 This is clinically very relevant as anti‐Sclerostin antibodies such as romosozumab stimulate bone formation and increase bone mass in humans.8 Romosozumab could thus be an optimal treatment option for patients with Wnt1 mutations. This is supported by the findings of Joeng and colleagues,15 who showed that anti‐Sclerostin antibody treatment partially rescued the bone phenotype of Wnt1 mutation–carrying Swaying mouse model.
In addition to its effects on osteoblasts, we found that deletion of Wnt1 in mesenchymal cells resulted in increased bone resorption. OPG expression was decreased in Wnt+/– calvarial cells but in Wnt1‐overexpressing cell lines OPG mRNA expression and protein secretion were variable depending on the experimental setup and the cell line used. These data suggest that Wnt1 has context dependent effects on OPG expression and that at least part of the Wnt1 effect on osteoclastogenesis is likely mediated via OPG. In an insert culture system using mesenchymal C3H10T1/2 cells, we showed that the suppressive effect of Wnt1 on osteoclastogenesis requires a close proximity between the Wnt1‐producing and the osteoclast progenitor cell. Moreover, our data show that Wnt1 has an OPG‐independent direct juxtacrine effect in suppressing osteoclastogenesis. Interestingly, recent studies indicate that binding of OPG on the osteoblast cell surface via heparin sulfate does greatly enhance the ability of OPG to inhibit RANKL and subsequently osteoclastogenesis.42, 43 Thus, based on our data we conclude that membrane‐bound Wnt1 and OPG regulate osteoclastogenesis in concert in a juxtacrine manner.
Wnt signaling has very complex direct effects on osteoclastogenesis. Wnt3‐induced canonical Wnt signaling via LRP5/6 receptors suppresses early osteoclast differentiation, whereas Wnt16 was recently shown to directly inhibit osteoclastogenesis via a noncanonical pathway.5, 14 Wnt5a, however, does promote osteoclast differentiation via Ror2 receptor.44 As Wnt1 functions as a membrane‐tethered molecule, co‐culture systems with Wnt1‐producing and hematopoietic cells must be used. This has made it difficult to identify the exact signaling pathways mediating the Wnt1 effect in osteoclasts, but using a panel of inhibitors for components of canonical and noncanonical branches we could show that inhibition of axin‐dependent canonical signaling does rescue osteoclastogenesis in Wnt1‐overexpressing co‐cultures. To underline the complexity of Wnt signaling in osteoclasts, blocking JNK activity resulted in partial rescue of osteoclastogenesis. We conclude that the canonical pathway is the main driver of the direct Wnt1 effect on osteoclastogenesis but JNK‐dependent noncanonical signaling may also contribute to the suppression of osteoclast differentiation.
We show here for the first time that a Wnt ligand functions only in short range in regulating bone cells in vitro using multiple approaches. First, we did not detect any Wnt1 in the culture medium by Western blot or IP, nor did conditioned medium from Wnt1‐expressing cells induce differentiation of C3H10T1/2 cells. When Wnt1‐overexpressing C3H10T1/2 cells were mixed with control cells allowing physical contact, reduction in the ratio between Wnt1 and control cells led to decreased ALP activity. Second, in insert cultures, we found that Wnt1‐overexpressing cells in the insert (and not in contact with the target cells) were not able to induce differentiation of mesenchymal progenitor cells or to suppress osteoclastogenesis of hematopoietic progenitors in the bottom of the well. Wnt1‐overexpressing cells did suppress the osteoclastogenesis of both hematopoietic progenitors as well as RAW264.7, when in contact with the target cells. Finally, we show using confocal imaging that Wnt1 expression induced nuclear accumulation of β‐catenin in the overexpressing cell itself but also in the neighboring cells in direct physical contact with the Wnt1‐expressing cell. Nuclear translocation of β‐catenin was not observed in cells that were close to but not in physical contact with the Wnt1 source cell. These data show that Wnt1 functions in a juxtacrine manner requiring cell‐cell contact to its target cells.
Wnt1 mRNA is expressed in bone and in bone cells at very low levels but we have detected Wnt1 expression in some osteocytes, lineage negative pool of hematopoietic and mesenchymal progenitor cells, and in B cell lineage.3 Together with its restricted expression pattern and juxtacrine mode of action it is actually very surprising that Wnt1 deletion in limb mesenchymal cells leads to such a striking phenotype affecting both osteoblasts and osteoclasts. Wnt1 is palmitoylated, making it hydrophobic, which then limits Wnt dispersion and range of biological actions.45 Some membrane‐bound Wnt could be incorporated to secretory vesicles and carried to their target cells.46 Our results showed that separating the Wnt1‐producing cells from the target cells by porous membrane (that would allow exosomes to pass) led to total blockage of Wnt1 effects and that actual physical contact was required between the Wnt1 source and target cells to induce canonical Wnt signaling. These data show that vesicle‐mediated transfer is not responsible for transmission of Wnt1, but osteoblast‐derived Wnt1 is more likely to be tethered on the cell membrane of the secreting cell. Wnt1 secretion to the cell membrane requires the expression of Wls.19 Interestingly, targeted knockout of Wls in mature osteoblasts results in a similar but milder bone phenotype to that of our Wnt1Prrx1 –/–, which could suggest that indeed lack of functional Wnt1 is the major contributor to the WlsOcn –/– phenotype.47
Recently, Joeng and colleagues15 reported that Dmp1‐Cre–driven Wnt1 deletion in osteocytes results in osteopenia and spontaneous fractures similarly to our phenotype of Wnt1Prrx1 –/– mice. Moreover, their data suggested that Wnt1 would in part function through the noncanonical mTOR signaling pathway. Phenotypic comparison of these available targeted Wnt1‐deficient mouse models provide interesting insight into the Wnt1 function in bone. First, deletion of Wnt1 in early mesenchymal progenitors results in a more severe bone phenotype with 100% of the male Wnt1Prrx1 –/– mice having fractures compared to 67% in DMP1‐Cre–driven knockouts. Second, Wnt1Prrx1 –/– exhibited more severe reduction in histomorphometric parameters of bone formation as well as increased number of osteoclasts and eroded surface that were not seen in the DMP1‐Cre model. Taken together these data indicate that Wnt1 derived from mesenchymal cells promotes osteoblast but suppresses osteoclast differentiation and function but Wnt1 produced in mature osteoblasts and osteocytes regulates only bone formation.
To conclude, our data show that mesenchymal cell–derived Wnt1 is a key regulator of bone metabolism affecting both trabecular and cortical compartments. We show for the first time that Wnt1 functions in bone cells in a juxtacrine manner requiring physical contact to its target cells, supporting the recent evidence of short‐range signaling mode of Wnt proteins. Moreover, Wnt1 has a dual function to induce both osteoblast differentiation and activity as well as to suppress osteoclast differentiation. Enhancing Wnt1 signaling through innovative approaches is a very intriguing target for development of future therapies for patients with OI as well as with osteoporosis.
Disclosures
All authors state that they have no conflicts of interest.
Supporting information
Supplementary Figures S1.
Supplementary Movie S1.
Acknowledgments
This study was funded by the Academy of Finland (298625, 268535, and 139165 to RK); Emil Aaltonen Foundation; Sigrid Juselius Foundation; Finnish Cultural Foundation; and the work at TCDM by funding provided by the University of Turku and Biocenter Finland. We are grateful to Merja Lakkisto, Dr. Fuping Zhang, the staff at TCDM, and the staff of Turku Central Animal Laboratory for their excellent technical assistance.
Authors’ roles: RK and OM conceived the idea for the study. RK, FW, and KT designed the experiments. FW, KT, PR, and JL collected mouse samples. FW, KT, PR, VNP, TP, RA, and KN performed the experiments. FW, KT, KN, AR, RB, and RK analyzed the data. RK supervised the work, acquired the funding, and drafted the manuscript. FW, KT, and RK edited the manuscript and all authors accepted the final version of the manuscript.
References
- 1. Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50(5):345–8. [DOI] [PubMed] [Google Scholar]
- 2. Keupp K, Beleggia F, Kayserili H, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laine CM, Joeng KS, Campeau PM, et al. WNT1 mutations in early‐onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368(19):1809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pyott SM, Tran TT, Leistritz DF, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013;92(4):590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL‐receptor‐related protein 5. N Engl J Med. 2002;346(20):1513–21. [DOI] [PubMed] [Google Scholar]
- 6. Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot‐containing protein. Am J Hum Genet. 2001;68(3):577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gong Y, Slee RB, Fukai N, et al. LDL receptor‐related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–23. [DOI] [PubMed] [Google Scholar]
- 8. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532–43. [DOI] [PubMed] [Google Scholar]
- 9. Korvala J, Loija M, Makitie O, et al. Rare variations in WNT3A and DKK1 may predispose carriers to primary osteoporosis. Eur J Med Genet. 2012;55(10):515–9. [DOI] [PubMed] [Google Scholar]
- 10. Monroe DG, McGee‐Lawrence ME, Oursler MJ, Westendorf JJ. Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng HF, Tobias JH, Duncan E, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8(7):e1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Glass DA 2nd, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–64. [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi Y, Thirukonda GJ, Nakamura Y, et al. Wnt16 regulates osteoclast differentiation in conjunction with Wnt5a. Biochem Biophys Res Commun. 2015;463(4):1278–83. [DOI] [PubMed] [Google Scholar]
- 14. Moverare‐Skrtic S, Henning P, Liu X, et al. Osteoblast‐derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. 2014;20(11):1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joeng KS, Lee YC, Lim J, et al. Osteocyte‐specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. 2017;127(7):2678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takada S, Fujimori S, Shinozuka T, Takada R, Mii Y. Differences in the secretion and transport of Wnt proteins. J Biochem. 2017;161(1):1–7. [DOI] [PubMed] [Google Scholar]
- 17. Alexandre C, Baena‐Lopez A, Vincent JP. Patterning and growth control by membrane‐tethered Wingless. Nature. 2014;505(7482):180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farin HF, Jordens I, Mosa MH, et al. Visualization of a short‐range Wnt gradient in the intestinal stem‐cell niche. Nature. 2016;530(7590):340–3. [DOI] [PubMed] [Google Scholar]
- 19. Banziger C, Soldini D, Schutt C, Zipperlen P, Hausmann G, Basler K. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125(3):509–22. [DOI] [PubMed] [Google Scholar]
- 20. Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996;10(24):3116–28. [DOI] [PubMed] [Google Scholar]
- 21. Bradley RS, Brown AM. The proto‐oncogene int‐1 encodes a secreted protein associated with the extracellular matrix. EMBO J. 1990;9(5):1569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhai L, Chaturvedi D, Cumberledge S. Drosophila wnt‐1 undergoes a hydrophobic modification and is targeted to lipid rafts, a process that requires porcupine. J Biol Chem. 2004;279(32):33220–7. [DOI] [PubMed] [Google Scholar]
- 23. McMahon AP, Bradley A. The Wnt‐1 (int‐1) proto‐oncogene is required for development of a large region of the mouse brain. Cell. 1990;62(6):1073–85. [DOI] [PubMed] [Google Scholar]
- 24. Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int‐1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55(4):619–25. [DOI] [PubMed] [Google Scholar]
- 25. Liu C, Tu Y, Sun X, et al. Wnt/beta‐Catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med. 2011;11(2):105–12. [DOI] [PubMed] [Google Scholar]
- 26. Wong SC, Lo SF, Lee KC, Yam JW, Chan JK, Wendy Hsiao WL. Expression of frizzled‐related protein and Wnt‐signalling molecules in invasive human breast tumours. J Pathol. 2002;196(2):145–53. [DOI] [PubMed] [Google Scholar]
- 27. Joeng KS, Lee YC, Jiang MM, et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet. 2014;23(15):4035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. [DOI] [PubMed] [Google Scholar]
- 29. Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mikels AJ, Nusse R. Wnts as ligands: processing, secretion and reception. Oncogene. 2006;25(57):7461–8. [DOI] [PubMed] [Google Scholar]
- 31. Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Deliv Rev. 2010;62(12):1149–55. [DOI] [PubMed] [Google Scholar]
- 32. Negishi‐Koga T, Takayanagi H. Ca2+‐NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol Rev. 2009;231(1):241–56. [DOI] [PubMed] [Google Scholar]
- 33. Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N‐terminal kinase. Proc Natl Acad Sci U S.A. 2001;98(24):13681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hegyi B, Chen‐Izu Y, Jian Z, Shimkunas R, Izu LT, Banyasz T. KN‐93 inhibits IKr in mammalian cardiomyocytes. J Mol Cell Cardiol. 2015; 89(Pt B):173–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H, Hao J, Hong CC. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/beta‐catenin signaling. ACS Chem Biol. 2011;6(2):192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–92. [DOI] [PubMed] [Google Scholar]
- 37. Kiper POS, Saito H, Gori F, et al. Cortical‐bone fragility—insights from sFRP4 deficiency in Pyle's disease. N Engl J Med. 2016;374(26):2553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ettenberg SA, Charlat O, Daley MP, et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand‐binding regions by LRP6 antibodies. Proc Natl Acad Sci U S.A. 2010;107(35):15473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Day TF, Guo X, Garrett‐Beal L, Yang Y. Wnt/beta‐catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50. [DOI] [PubMed] [Google Scholar]
- 40. Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta‐catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38. [DOI] [PubMed] [Google Scholar]
- 41. Kramer I, Halleux C, Keller H, et al. Osteocyte Wnt/beta‐catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30(12):3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li M, Yang S, Xu D. Heparan sulfate regulates the structure and function of osteoprotegerin in osteoclastogenesis. J Biol Chem. 2016;291(46):24160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nozawa S, Inubushi T, Irie F, et al. Osteoblastic heparan sulfate regulates osteoprotegerin function and bone mass. JCI Insight. 2018;3(3):e89624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maeda K, Kobayashi Y, Udagawa N, et al. Wnt5a‐Ror2 signaling between osteoblast‐lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18(3):405–12. [DOI] [PubMed] [Google Scholar]
- 45. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. 2014;346(6205):1248012. [DOI] [PubMed] [Google Scholar]
- 46. Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14(10):1036–45. [DOI] [PubMed] [Google Scholar]
- 47. Zhong Z, Zylstra‐Diegel CR, Schumacher CA, et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci U S.A. 2012;109(33):E2197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1.
Supplementary Movie S1.