

Abstract
Nucleotide coenzymes dot the map of metabolic pathways providing energy to drive the reactions of the pathway and play an important role in regulating and controlling energy metabolism through their shared potential energy, which is widely unobserved due to the paradox that the energy in the coenzyme pools cannot be determined from the concentration of the coenzyme couples. The potential energy of the nucleotide couples in the mitochondria or the cytoplasm is expressed in the enzyme reactions in which they take part. The energy in these couples, [NAD+]/[NADH], [NADP+]/[NADPH], [acetyl CoA]/[CoA], and [ATP]/[ADP]x[Pi], regulates energy metabolism. The energy contained in the couples can be altered by suppling energy equivalents in the form of ketones, such as, D‐β‐hydroxybutyrate to overcome insulin resistance, to restore antioxidants capacity, to form potential treatments for Alzheimer's and Parkinson's diseases, to enhance life span, and to increase physiological performance. © 2019 IUBMB Life, 71(5):565–579, 2019
Keywords: free NADPH, free NADH, ATP, nucleotide coenzyme, β hydroxybutyrate, acetylCoA, Parkinson's disease
Abbreviations
- 3PG
3‐phosphoglycerate
- Acetyl CoA
acetyl coenzyme A
- CD38
cluster of differentiation 38, cyclic ADP ribose hydrolase
- DHAP
dihydroxyacetone phosphate
- FADH2
flavin adenine dinucleotide (reduced)
- FDP
fructose 1,6‐diphosphate
- FMN
flavin mononucleotide
- FOXO
forkhead box O transcription factors
- GAP
glyceraldehyde 3‐phosphate
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GSH
glutathione (reduced)
- GSSG
glutathione (oxidized)
- Keq
K equilibrium
- Kj
kilojoules
- LDH
Lactate dehydrogenase
- NAMPT
nicotinamide phosphoribosyltransferase
- NAD(H)
nicotinamide dinucleotide(reduced)
- NADP[H]
phosphorylated nicotinamide dinucleotide(reduced)
- NMN
nicotinamide mononucleotide
- PARP
poly(ADP‐ribose) polymerase
- Pi
inorganic phosphate
- Q, [QH2], CoQ
Coenzyme Q
INTRODUCTION
The four “great” nucleotide coenzyme couples [NAD+]/[NADH], [NADP+]/[NADPH], [acetyl CoA]/[CoA] and [ATP]/[ADP]x[Pi] all contain AMP. These coenzymes may be relics of an earlier stage of evolution when metabolism was mediated by relatively inefficient polynucleotide catalysis prior to the development of protein enzymes 1. The couples ultimately obtain their energy from the transfer of electrons from the oxidation of nutrients, making water and CO2. These couples can both donate or accept energy. The coenzymes are reactants that distribute energy throughout the metabolic pathways. The ratio of the free coenzyme couples determines the direction and extent of the enzymatic reactions and concentrations of the substrates and products within the compartment where the coenzyme couple is localized (Fig. 1).
Figure 1.
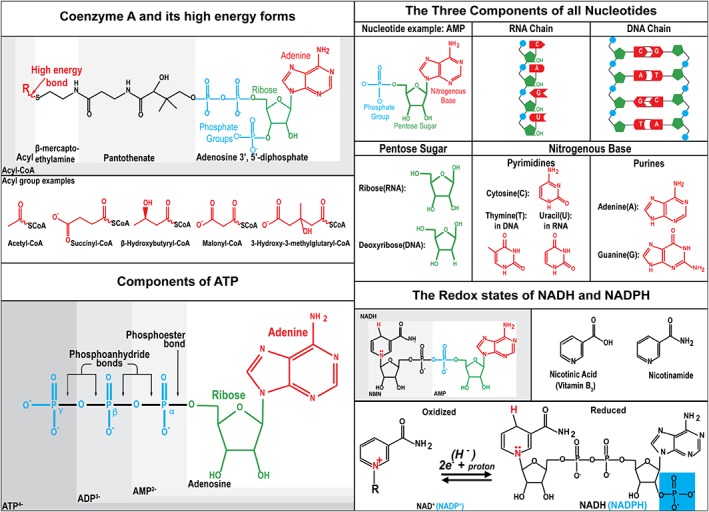
Structure and derivation of the controlling coenzyme couples. There are four “great” nucleotide coenzyme couples [ATP]/[ADP] [Pi], [Acetyl CoA]/[CoASH], [NAD+]/[NADH] and [NADP+]/[NADPH]. Three components: The nucleotide coenzymes are modified ribonucleotides. All nucleotides are composed of three parts, a nitrogenous base, a pentose sugar, and a phosphate group. Their free diffusion within the mitochondria or cytoplasm and the large number of reactions in which they participate has allowed them to evolve to become major regulators of metabolism. ATP can be reversibly converted from high‐energy form ATP to low‐energy form, ADP, to an even lower energy form, AMP, by forming or breaking one or two of the phosphoanhydride bonds. Coenzyme A is a nucleotide coenzyme containing pantothenic acid, vitamin B5 and a sulfhydryl on β mercaptoethylamine. The sulfhydryl group can form thioesters to make a high‐energy form of acyl CoA. Transfer of the acyl group leaves behind the low‐energy CoASH. There are many potential acyl groups that can form the high energy thioester bond. Nicotinamide adenine dinucleotide is composed of the vitamin B3 nicotinic acid. It is a dinucleotide. The first nucleotide AMP is used by enzymes for recognition and can be phosphorylated at the 2′ position in ribose to make NADP+, which can be recognized by a set of enzymes. The second nucleotide, nicotinamide mononucleotide (NMN) contains the base nicotinamide which is derived from nicotinic acid (vitamin B3, niacin). The NAD(P)H coenzymes act as electron donors and receptors. Interestingly, they accept pairs of electrons and not single electrons. The net reaction is equivalent to the transfer of a hydride ion which is two electrons and a proton. It is interesting that NADH can only transfer two electrons at a time while O2 is reactive with other free radicals. This keeps the main reducer in the cell from forming a substrate cycle with O2.
The map of metabolic pathways is covered thoroughly by the great controlling nucleotide coenzymes. By taking part in many reactions, the coenzymes assume the energy of the collection of reactions in which they take part. All the reactions are in turn regulated; meaning that the extent and direction of reactions are determined by the potential energy of the coenzyme couple (Fig. 2).
Figure 2.
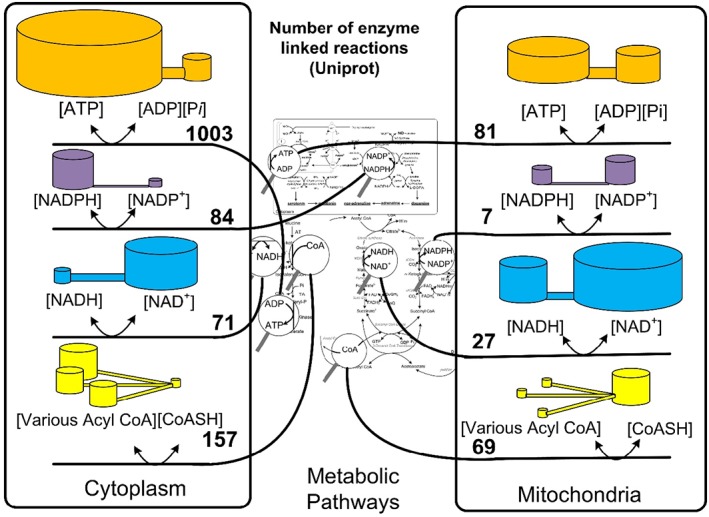
The relative numbers of enzyme‐linked reactions in which the controlling nucleotide couples are reactants and products. The magnifying glasses in the center show nucleotide coenzymes. The lines from the magnifying glasses sort the reactions to either the cytoplasm or the mitochondria. Numbers of enzymes were taken from Uniprot July, 2018 using search parameters human, location (cytoplasm or mitochondrion matrix) and the names ATP, NADH, NADPH, and CoA. The volume of the cylinders is proportional to the concentrations of thermodynamically activated and inactivated free nucleotides. The ratios and scales do not represent a particular cell or mitochondria. The illustration is meant to display graphically that the potential energy of these couples can vary and that all the numbers of enzymes listed are regulated and controlled by the same chemical potential or redox potential of the nucleotide coenzymes.
The distinction is drawn between cofactors that are permanently attached to the enzymes and coenzymes that are released from the enzyme. The free diffusion of the coenzymes allows them to coordinate and influence all the reactions that share the coenzymes and are within diffusion limits. Coenzymes act as regulators of metabolism only when they are free to diffuse. Because cofactors, such as flavin mononucleotide (FMN) and flavin adenine dinucleotide (FADH2), do not readily dissociate from the enzyme they bind, they control only the enzyme to which they are bound.
THE MEASUREMENT OF COENZYME COUPLES
Paradoxically, the potential energy of the coenzyme couples cannot be determined in tissue lysates by measuring the concentrations of each component of the couple directly. The method of measuring the energy in the coenzyme couples indirectly has gone largely unappreciated. However, modern analytical techniques such as the use of stable isotope dilution mass spectrometry render values for metabolites in accord with previous analytical methods 2, 3. Understanding how the nucleotide coenzyme's energy can be measured and how they regulate metabolism and can be manipulated to control metabolism is the subject of this review.
Table 1 shows the measure of total cellular contents of pyridine nucleotides in rat liver in micromole per gram wet weight 4. The ratio of the free [NAD+]/[NADH] ratios is calculated from the following equation 5, 6, 7, 8:
Table 1.
Total and free NAD(P) nucleotide levels in rat live
| NADH | |||||
|---|---|---|---|---|---|
| Dietary state | Total NAD μg/g | Total NADH μg/g | Total [NAD]/[NADH] | Free cytoplasmic [NAD+]/[NADH] from lactate dehydrogenase | Free Mitochondrial [NAD+]/[NADH] from β‐hydroxybutyrate dehydrogenase |
| Fed | 0.76 | 0.14 | 5.4 | 1,164 | 7.74 |
| Fasted | 0.82 | 0.16 | 5.1 | 564 | 5.84 |
| NADPH | |||||
| Total NADP μg/g | Total NADPH μg/g | Total [NADP]/[NADPH] | Free cytoplasmic [NADP+]/[NADPH] from isocitrate‐dehydrogenase | ||
| Fed | 0.067 | 0.31 | 0.22 | 0.0101 | |
| Fasted | 0.080 | 0.36 | 0.22 | 0.00442 | |
Variations in Total Cellular NAD Content
Measurement of total NAD is widely used. It is a composite of free and bound as well as mitochondrial and cytoplasmic NAD. Total NAD cannot be used in the mass action ratio upon which activation energy, K equilibrium (K eq), Gibbs free energy, ΔE, and rate equations are dependent. Rat liver total NAD content is around 0.8 μmol/g and NADH about 0.15 μmol/g, whereas total NADP is about 0.07 and total NADPH about 0.35 mmol/g 9. Total tissue content of NAD in mouse tissues varies only twofold from a low of 0.24 nmol/mg protein in heart to a high of 0.60 nmol/mg protein in spleen 10. Total NAD content declines from about 3 to 2 ng/mg protein in human skin from age 40 to 80 11.
NAD can be made de novo from tryptophan by way of formyl kynurenine and quinolinic acid. It can be made from nicotinic acid (Preiss‐Handler pathway). It can also be made by the so‐called “salvage pathway” where nicotinamide made from NAD degradation by poly(ADP‐ribose) polymerase (PARP), sirtuins, or cluster of differentiation 38 (CD38) is transformed by nicotinamide phosphoribosyltransferase (NAMPT) to NMN and back to NAD 11. NADP is synthesized by NAD kinase.
Oral administration of nicotinamide mononucleotide, a direct precursor of NAD can reverse the age‐induced decreases in tissue NAD content as well as some of the age‐induced abnormalities in glucose metabolism 12, 13. Age‐induced decreases in tissue content of NAD can also be reversed by administration of nicotinamide riboside 14.
Feeding a ketogenic diet can increase total NAD in hippocampus of the rat within 2 days 15, 16 as well as in humans 17. Oxidative stress inducing poly ADP ribosylation is a leading cause of NAD loss 18. Oxidative stress can also be combated by NADPH produced by the mitochondrial isocitrate dehydrogenase 2, 19. Ketosis also increases forkhead box transcription factors (FOXO), which increase antioxidant enzymes and NAMPT expression 20. Increases in the tissue content of d‐β‐hydroxybutyrate extend the life span of nematodes 21 and possibly humans as well 22.
Free Cytoplasmic and Mitochondrial NADH and NADPH
Veech and Krebs found that the ratios obtained for the total levels of the coenzyme couples were very different from the ratios for the free coenzyme couples calculated from the ratios of enzyme substrates to products. The free cytoplasmic NAD couple was calculated from the levels of the reactants of the lactate dehydrogenase (LDH) reaction 6, 23. The free cytoplasmic NADP couple was calculated from the levels of the reactants of the isocitrate dehydrogenase reaction 8. The free mitochondrial NAD couple was calculated from the levels of the reactants of the d‐β‐hydroxybutyrate dehydrogenase reaction 7. The ratio of the free NAD couple in the cytoplasm was found to be 215 times that of the measured ratio using total levels. The ratio for the free cytoplasmic NADP couple was found to be 1/22nd of the measure found using total levels 8. The free cytoplasmic NADP couple was found to be more than 100,000 times lower than the free NAD couple in the same compartment. The redox potential of the free cytoplasmic NAD couple was calculated to be −0.19 V. The redox potential of the cytosolic NADP couple was calculated to be −0.42 V, the lowest of any cellular redox couple 9.
The ratios of the free coenzyme couples cannot be measured directly in tissue extracts, and so they were calculated from measurements of the substrates and K eq of the reactions after correction for tissue pH and tissue free [Mg2+]. The agreement between the calculated free coenzyme couple ratios using two different enzymatic reactions in which the couple participates increased confidence in the accuracy of several of the calculated ratios. Table 2 lists the enzymes and reactions in the form of mass action ratios that were used historically to measure coenzyme couples.
Table 2.
The reaction and K eq of high‐activity enzymes used to calculate the ratios of the free coenzyme couples
| Enzymes | Reactions | K eq | References | |
|---|---|---|---|---|
| High‐activity cytoplasmic NAD‐linked dehydrogenases | ||||
| Lactate dehydrogenase EC 1.1.1.27 |
|
=1.11 × 10−11 M | 7 | |
| Malate dehydrogenase NAD (cytoplasmic) EC 1.1.1.37 |
|
=2.86 × 10−12 M | 24 | |
| Glycerol‐3‐phosphate dehydrogenase EC 1.1.1.8 |
|
=1.35 × 10−11 M | 25 | |
| Glyceraldehyde 3P dehydrogenase EC 1.2.1.12 |
|
=0.51 × 10−7M | 26 | |
| High‐activity mitochondrial NAD‐linked dehydrogenases | ||||
| β‐Hydroxybutyrate dehydrogenase EC 1.1.1.30 |
|
= 4.93 × 10−9 M | 7 | |
| Glutamate Dehydrogenase EC 1.4.1.2 |
|
=3.87 ×10−13 M2 | 27 | |
| High‐activity cytoplasmic NADP‐linked dehydrogenases | ||||
| Isocitrate dehydrogenase EC 1.1.1.41 |
|
=1.17 M | 28 | |
| Glutathione reductase EC 1.6.4.2 |
|
=100 × 10−7 | 29 | |
| 6‐P Gluconate dehydrogenase EC 1.1.1.44 |
|
=1.72 × 10−1 M | 30 | |
| Malic enzyme EC 1.1.1.39 |
|
=3.44 × 10−2 M | 31 | |
| High‐activity kinases at 1 mM free [Mg 2+ ] | ||||
| Creatine phosphokinase EC 2.7.3.2 |
|
=1.66 × 10−9 M−1 | 32 | |
| Phosphoglycerate kinase EC 2.7.2.3 |
|
=3,600 | 26 | |
| Combined KGAPDH + K3PGK |
|
=1830 × 10−10 | 26 | |
| Adenylate kinase EC 2.7.4.3 |
|
=1.05 | 32 | |
| Acetyl CoA enzymes cytoplasm | ||||
| Citrate cleavage enzyme cytoplasm EC 4.1.3.8 |
|
=0.985 M | 33 | |
| Acetyl CoA enzymes mitochondria | ||||
| Citrate synthase EC 2.3.3.1 |
|
=2.24 × 106 | 24 | |
| Isomerases | ||||
| Triose phosphate isomerase EC 5.3.1.1 |
|
=22 | 34 | |
| Aldolase EC 4.1.2.13 |
|
=0.99 × 10−4 M | 34 | |
| Transaminases | ||||
| Aspartate amino transferase EC 2.6.1.1 |
|
=6.61 | 8 | |
| Alanine amino transferase EC 2.6.1.2 |
|
=1.52 | 8 | |
The concentrations of substrates and products and the K eq of these reactions plug into the mass action equation allowing one to solve for the coenzyme couples: [NAD+]/[NADH], [NADP+]/[NADPH], [acetyl CoA]/[CoA] and [ATP]/[ADP]x[Pi]. The K eq constants all assume a pH of zero, a convention used in calculating K eq, which must be adjusted to account for physiological pH. Abbreviations: DHAP: dihydroxyacetone phosphate; FDP: fructose 1,6‐diphosphate; GAP: glyceraldehyde 3‐phosphate; GAPDH: glyceraldehyde 3‐phosphate dehydrogenase; 3PG: 3‐ phosphoglycerate; 3PGK: 3‐ phosphoglycerate kinase.
The free intracellular [Mg2+] was estimated from the measured [citrate]/[isocitrate] ratio 35 and the equation:
where K a is the acid dissociation constants, K b is the Mg2+ binding constants, and Ki Acon is the equilibrium constant of the aconitase reaction. The K eq were measured at ionic strength = 0.25 M and at 38 °C with substrates at near physiological concentrations. ΔG is calculated from these K eq constants according to the following equation
Unlike enzymes for the other coenzyme couples, there was found to be only one high activity enzyme reacting with the free [acetyl CoA]/[CoA] coenzyme couple in each compartment. Citrate synthase occurs in the mitochondria and citrate cleavage enzyme (ATP‐citrate lyase) in the cytoplasm. No value for the free NADP couple in mitochondria is given, because it is difficult to measure. Two of the strongest attempts yielded results similar, but just slightly more reduced than the cytoplasmic NADP couple 36, 37.
In a reaction at true equilibrium, there is no net flux in either direction. These reactions which have high enzyme capacity, such as, in glycolysis where the enzyme capacity exceeds the flux through the pathway by a factor of 100‐fold, are said to be in “near equilibrium” 38. It has been estimated the maximum deviation between the redox potential of the free cytoplasmic [NAD+]/ [NADH] and that calculated from the measured [pyruvate]/[l‐lactate] ratio would be no more than 2 mV out of 190 mV. 39.
Because of the high activity of the glyceraldehyde 3‐phosphate dehydrogenase, (GAPDH), (EC 1.1.1.29)—3‐phosphoglycerate (3PG) kinase (EC 2.7.3.2)—triose phosphate isomerase (EC 5.3.1.1) system located in cytoplasm, and because the energy of the [ATP]/[ADP] × [Pi] ratio is algebraically related to the redox state of the NAD redox couple 40. This system was first used to determine the phosphorylation potential of the [ATP]/[ADP] × [Pi] couple. More about this will be said when talking about four critical reactions of ATP.
The results of measuring the coenzyme couples in their compartmental context give some valuable insights. The measurement of total amounts of nucleotide coenzymes directly has different goals but does not give an accurate picture of the chemical potential in the compartment of interest and includes both bound and free nucleotides from both compartments.
THE MEASURED COENZYME COUPLES
The coenzyme couple ratio can be used to calculate ΔG for specific reactions of ATP or CoA. For reactions of NADP and NAD, the redox potential E of the couple can be derived from the Nernst equation to provide a voltage relative to the hydrogen electrode. Based on the measured values, the selective rational for each coenzyme couple begins to emerge. Compartmentalization avoids substrate cycling, separating the activation of the nucleotide coenzymes from their consumption. ATP is made primarily in the mitochondria and consumed primarily in the cytoplasm. Cytoplasmic NAD receives electrons in the cytoplasm from glycolysis and shuttles them to the mitochondria. NADP(H) is compartmentalized by the 2’phosphate and the set of enzymes that distinguish NADP(H) from NAD(H). Although cytoplasmic NAD is a collector of electrons, NADPH is a donor of electrons.
Cytoplasmic [NAD+]/[NADH]
The four cytoplasmic dehydrogenases shown in Table 2 have similar redox potentials and are in near‐equilibrium with the same free [NAD+]/[NADH] ratio. In cytoplasm, the redox potential of the free cytoplasmic [NAD+]/[NADH] is normally about −0.17 to −0.19 V. This NAD‐couple is therefore poised to accept reducing equivalents produced by glycolysis.
Mitochondrial [NAD+]/[NADH]
The free mitochondrial [NAD+]/[NADH] calculated from the above equations are between 5 and 10 41, with a redox potential of about −0.28 V. This lower redox potential of the mitochondrial NAD couple compared to the free cytoplasm NAD couple makes more energy available to the electron transport system for the synthesis of three ATP, ADP, and inorganic phosphate (Pi).
The method of using the law of mass action and equilibrium to measure the NADH couple relies on the assumption that the coenzyme couple is near equilibrium. In the case of conditions of hypoxia and also in cancer cells where the Warburg effect shifts the utilization of glucose to primarily anaerobic glycolysis the levels of total, [NAD+]/[NADH] were shown to be unstable and varied considerably when looking at the [lactate]/[pyruvate] couple alone 42. This points to the importance of using more than one of the sets of metabolites to measure the free NADH couple. The analysis was also done without freeze clamping. Freeze clamping is simply crushing the tissue with a tool that brings two blocks of aluminum chilled in liquid nitrogen together to rapidly cease all reactions. The procedure is followed by perchloric acid treatment of the tissue. The fact that it is difficult to measure does not mean that the effect of the enzyme couple goes away, it just means that in some circumstances, it cannot be easily measured. It also points out that the high activity enzymes are not only measures of the free cytoplasmic NADH couple, but they also play a significant role in determining the NADH couple.
Cytoplasmic [NADP+]/[NADPH]
The K eq constant of the NADP‐linked reactions range from about 0.1 to 1 M. The K eq of the cytoplasmic NAD‐linked dehydrogenases are of the order of 10−4 at pH 7.0. The segregation of the dehydrogenases in their compartments fits with their intrinsic equilibrium constants. The redox ratio of the free cytoplasmic [NADP+]/NADPH] commonly range from 0.01 to 0.002 and a redox potential −0.42 V, the most negative in the cell. This NADP system is thus poised to provide reducing equivalents to reactions such as lipid synthesis, the reduction in other reducing agents such as dihydrobiopterin to tetrahydrobiopterin, oxidized glutathione (GSSG) to two molecules of glutathione (GSH) and oxidized to reduced thioredoxin. Through these reductions, the free cytoplasmic [NADP+]/NADPH] couple determines the redox potential on pathways related to inflammation, redox signaling, hypoxia response, antioxidant response, apoptosis, and mitophagy and should provide many answers if applied in these fields. The NADP couple also provides the low reduction potential necessary for the destruction of oxygen free radicals 2.
D‐β‐HYDROXYBUTYRATE REDUCES CYTOPLASMIC NADP+ TO NADPH
The very first measurement of the cytoplasmic NADP couple in the liver of rats that were either fed normally or fasted for 48 h showed that the d‐beta‐hydroxybutyrate produced as a result of fasting‐induced ketosis led to a reduction in the cytoplasmic NADP couple. Feeding exogenous ketone bodies led to a similar finding. The mechanism by which d‐β‐hydroxybutyrate ultimately transfers electrons to cytoplasmic NADP+ is shown in Fig. 3.
Figure 3.
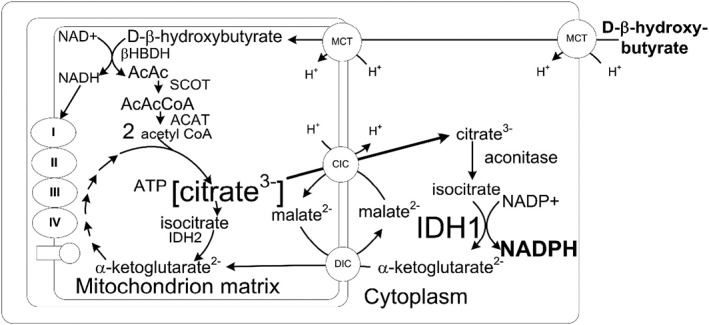
The metabolism of d‐β‐hydroxybutyrate produces cytoplasmic NADPH. The metabolism of d‐β‐hydroxybutyrate in heart or brain leads to the formation of mitochondrial acetyl CoA and citrate 3, 43. The mitochondrial citrate is transported to cytoplasm by the citrate–isocitrate carrier where it is converted to NADPH by the NADP‐linked isocitrate dehydrogenase. It is generally thought that NADPH is made in the hexose monophosphate shunt. However, this occurs when the cell is replete with glucose. In the absence of glucose metabolism d‐β‐hydroxybutyrate can produce NADPH 44. During situations where intracellular glucose is low, the NADPH appears to be made from intramitochondrial citrate, which is exported to cytoplasm where it is converted to isocitrate which produces NADPH during its conversion to α ketoglutarate via the cytoplasmic isocitrate dehydrogenase (IDH1) 2.
THE ΔG O OF ACETYL COA HYDROLYSIS
Using the following equation:
the standard free energy for the hydrolysis of acetyl‐CoA as shown for the reaction below at pH 7.0 is −35.75 kJ/mol 24.
The standard free energy of ATP hydrolysis in the absence of Mg2+ was a very similar −35.69 kJ/mol. This value changed slightly to −31.8 kJ/mol when free [Mg2+] was present at 1 mM 45. Therefore, altered Mg2+ levels may slightly alter the standard free energy of these two “great” free nucleotide couples[ATP]/[ADP] and [acetyl‐CoA]/[CoA].
ΔG′ OF CYTOPLASMIC ATP HYDROLYSIS
The energy of ATP hydrolysis is the same whether it is produced by glycolysis in the red blood cell lacking mitochondria or by oxidative phosphorylation in mitochondrial containing tissue, two very different mechanisms 40. The ΔG′ of ATP in the cell is the “still point in the turning world” 46. It is maintained in a very narrow band between −56 and −59 kJ/mol. Although redox states for NAD(P) can vary appreciably, the ΔG′ of ATP remains within narrow limits due to the essential functions it controls. There are four important reactions in which the ΔG′ of ATP takes part. The first two allowed the accurate measurement of ATP phosphorylation potential, which allows one to derive the relation of ATP in the other two processes.
The ΔG′ of ATP in Glycolysis
The glycolytic pathway, at the GAPDH—3PG kinase reaction, provides the intersection of two of the “great” free nucleotide couples: the free [ATP]/[ADP] × [Pi] with the free cytoplasmic [NAD+]/[NADH] 40 where the value of the combined constant at free [Mg2+] was 1.65 × 10–7 M−1.
It is from these reactions and their similarity to the metabolites of the creatine kinase reaction (EC 2.7.3.2) that the ΔG′ of ATP can be determined 40.
The ΔG′ of ATP in the Span of the Electron Transport Chain
The energy within the span of the electron transport system between mitochondrial NADH dehydrogenase and Coenzyme Q 43 also reflects the energy of the free [ATP]/[ADP] × [Pi] couple. The free energy equation for the reaction is shown as follows:
where z is the number of electrons transferred and F is the Faraday constant, 8.3145 kJ/mol/oK. The redox energy contained between mitochondrial NADH and mitochondrial Coenzyme Q can be determined by metabolite measurements using the same principle illustrated earlier. The redox state of the mitochondrial NAD‐couple can be determined by measuring the metabolites of the β‐hydroxybutyrate dehydrogenase (EC 1.1.1.30) or the glutamate dehydrogenase (EC 1.4.1.2) system 7. The redox state of the mitochondrial CoQ system can be determined by measuring the metabolites of the redox state of the succinate dehydrogenase system (EC 1.3.5.1) using the following equation:
The ΔG′ of ATP and the Membrane Potential, EM/C, Across the Inner Mitochondrial Membrane
The energy of the proton gradient between the mitochondrial matrix and intramembrane space has been determined to be between −140 and −120 mV 43. As there are four protons ejected from the matrix per site, but not two as suggested by Mitchell 48 then:
The energy of the mitochondrial proton gradient also reflects the ΔG′ of ATP.
The ΔG′ of ATP and The Sodium Potassium Atpase
The ΔG′ of ATP hydrolysis is also expressed in the gradient of inorganic ions across the plasma membrane. This is seen in perfused working heart where the gradient of the Na + gradient generated by the 3Na+/2 K+ ATPase (EC 3.6.1.37) in hearts with a resting membrane potential of about −83 mV and an open K+ channel is given by 49.
Although K+ is essentially permeant between intra and extracellular phases such as nerve or muscle, Cl− is the permeant ion in other lower voltage tissues. The resting membrane potentials in liver cells (hepatocytes) was shown by direct measurement with intracellular electrodes to be in the range between −50 and −20 mV 50, 51, 52. The resting membrane potential is widely thought to be a “diffusion potential” resulting from the higher intracellular K+ concentration relative to the extracellular concentration. Liver, like neuronal or muscle tissues, also has a high intracellular K+ concentration but has a resting membrane potential equivalent to the [Cl−]out/in potential showing that the resting membrane potential is not a diffusion potential, but rather is a Gibbs–Donnan near equilibrium potential where the resting membrane potential assumes the voltage of the most permeant ion, K+ in the case of high voltage tissues and Cl− in the case of lower voltage tissues 52, 53
A system of ion transport systems are linked to the 3Na+/2 K+ ATPase and to one another to bring the energy gradients of the nine inorganic ions into near equilibrium with the ΔG′ of ATP hydrolysis indicated by the following equation 54:
This equation indicates that the gradients of the inorganic ions between the cytoplasm and extracellular space are related to one another and to the energy of ATP hydrolysis.
REGULATION AND CONTROL
Researchers who study enzyme kinetics have yet to sufficiently explain the global mechanisms that regulate the concentrations of metabolites and flux rates for metabolic pathways in vivo. The current state of knowledge cannot account for the level of regulation observed in organisms 55. Biochemical simulations limiting metabolic regulation to effects on enzyme kinetics do not provide enough elasticity for the changes observed. The kinetics of the [ATP]/[ADP] × [Pi] couple functioning simultaneously on hundreds of enzymes can be used to demonstrate how the interaction of coenzyme couples with multiple enzymes can greatly influence the regulation of the rates of reactions and the concentrations of metabolites in these pathways.
A practical way to observe the stability of the [ATP]/[ADP] × [Pi] ratio is to consider a gymnast on uneven parallel bars. She flies up to the high bar and wraps her fingers. Her muscles go from being relaxed to tetanic contraction in approximately 30 ms, fast enough to assure that her hands can grip the bar before her momentum would rip the bar out of her grasp. Her rate of ATP utilization goes from about 0.01 μmol ATP/g/s to approximately 5 μmol ATP/g/s. At this high rate of energy consumption, the available ATP (approximately 7 μmol ATP/g) would last about 1 s 56. Fortunately, there are more than a thousand enzymes connected to the cytoplasmic ATP pool. They respond immediately to the altered flux to regulate the [ATP]/[ADP] × [Pi]. Among those enzymes that respond are creatine kinase, which taps into the reservoir of creatine phosphate to offer another 3–5 s of sustained ATP concentration.
ADP accumulation is limited by adenylate kinase:
AMP accumulation is limited by AMP deaminase:
The depolymerization of glucose from glycogen followed by glucose oxidation to lactate has an overall reaction of
and provides another 30 s of sustained [ATP]/[ADP] × [Pi] ratio.
It is interesting to note that all this regulation of ATP by the [ATP]/[ADP] × [Pi] goes on inside the cytoplasm before any nutrients or oxygen is transported to the tissue. The coenzyme couples work locally and affect only the cell. However, that does not mean there are not important implications outside the cell and in relation to other tissues. The muscle's consumption of ATP puts a demand on glucose, which is needed in the brain. Humans have evolved to compensate for this competition by producing pyruvate dehydrogenase kinase 4 which limits glucose's entry into oxidative phosphorylation. This has the effect that muscles consume fatty acids for energy in addition to glucose, preserving some glucose for the brain 57. The brain requires glucose because the blood–brain barrier blocks the majority of fatty acids from entering the brain. The problem of longer periods of exertion is solved by the production of D‐β‐hydroxybutyrate in these situations. D‐β‐hydroxybutyrate functions as an alternative fuel for the brain and the muscles. The implication of using D‐β‐hydroxybutyrate to fuel the brain makes D‐β‐hydroxybutyrate a singularly unique metabolite with potential to treat disease.
ALTERING THE FREE LEVELS OF CONTROLLING COENZYMES FOR DISEASE TREATMENT
We have made use of the ability of the four nucleotide coenzymes to govern metabolism in studies of the liver 1, perfused rat heart 4, and more recently the brain 5. In these studies, one metabolite stood out for its ability to control the ratios of the “great” controlling nucleotides. On their own, the nucleotide coenzyme couples regulate all of metabolism, keeping the concentration of metabolites within certain limits by the shared pools and the law of mass action. When the ratios of the nucleotide coenzyme couples can be altered for therapeutic use they become the great controlling nucleotide coenzymes. A metabolite that can significantly change the ratios of the controlling nucleotide coenzymes is D‐β‐hydroxybutyrate 43, 44 and therefore this metabolite shows great promise for the therapeutic treatment of diseases, which are characterized by altered energy metabolism.
Two diseases will be used to illustrate how increasing the level of the d‐β‐hydroxybutyrate, can control the potential energy of the “great” regulating coenzymes to some day delay the pathology of the diseases 58. The understanding that changes in the energy of the coenzyme couples may potentially delay disease onset or change the rate of progression provides novel insights that can lead to new treatments of diverse diseases, some of which currently have no effective therapy. As D‐β‐hydroxybutyrate can overcome insulin resistance ketosis can be an effective treatment for Alzheimer's disease.
ALZHEIMER'S DISEASE
We demonstrated in the model of a working heart that perfusing the rat heart with glucose alone was unable to supply large amounts of acetyl‐CoA for energy production to the Krebs cycle. However, when insulin, which stimulates the activity of pyruvate dehydrogenase, was added to the perfusate acetyl‐CoA levels were increased by an amazing eightfold 59, 60. Importantly, supplementation of the perfusion media with ketone bodies increased the acetyl‐CoA even more, to about 15‐fold 43. A major effect of insulin is the stimulation of mitochondrial pyruvate dehydrogenase 59, 60. It was clear that the increased energy metabolism stimulated by insulin addition compensated for the insulin resistance by activating pyruvate dehydrogenase to increase acetyl‐CoA levels for the Krebs cycle. These data show that the metabolism of ketone bodies can overcome limitations in energy metabolism induced by insulin resistance.
Dementia afflicts about 48% of the US population after 85 years of life 61. There is currently no efficacious treatment for the pathology that drives Alzheimer's disease. Alzheimer's disease patients exhibit decreased glucose utilization and oxidative capacity in the brain, especially in the hippocampus 62. Systemic insulin resistance or type II diabetes has been found to be associated with an increased incidence of Alzheimer's disease 63. Insulin resistance was also demonstrated in brain slices from Alzheimer's patients 64. Hippocampal insulin resistance, as evidenced by decreased fluorodeoxyglucose uptake, precedes clinical cognitive impairment by many years 62, 63, 64. Patients with mild cognitive impairment at risk for the development of Alzheimer's disease were found to have a decrease in FDG uptake, but no defect in 11C acetoacetate uptake indicating no decrease in neuronal number, but a specific decrease in brain glucose metabolism 65, 66. The data also suggest that the primary energy impairment in Alzheimer's disease brain is the PDH reaction 67.
The hypothesis that Alzheimer's disease results from insulin resistance in the brain led to a trial of intranasal insulin administration as a possible treatment. In the pilot study, memory improvement was demonstrated after 2–4 months of insulin treatment 68. The therapeutic effectiveness of intranasal insulin, while demonstrating the pathophysiological principle of improving energy metabolism to treat Alzheimer's disease, has practical difficulties as a therapy, while ketosis through the use of exogenous ketone bodies to correct the energy deficits does not 69.
D‐β‐Hydroxybutyrate Can Replace Glucose and Insulin for the Synthesis Of Mitochondrial Acetyl‐Coa and Nadh
The increase in the ΔG′ of ATP observed in the working perfused heart 43 led directly to the increase in physiological performance in animals 70 and in endurance athletes 71. The same increase in acetyl coenzyme A (acetyl CoA) induced by the metabolism of d‐β‐hydroxybutyrate in heart can also be potentially used as a therapy overcoming the impairment of brain PDH activity in Alzheimer's disease 72.
Administration of ketone bodies has been shown to decrease the severity of pathological changes in both Alzheimer's and Parkinson's disease tissue culture models 58. Triple transgenic mouse models of Alzheimer's disease fed a ketone ester diet for 4–7 months showed decreased anxiety and improved memory function in behavioral tests 73. At autopsy of the ketone fed mice showed decreased brain β‐amyloid and phosphorylated tau levels 73. These same ketone fed mice showed an increased mitochondrial free [NAD+]/[NADH] ratio and marked increased hippocampal acetyl‐CoA and citrate levels showing that ketone feeding was able to restore energy metabolism 3, and this likely occurs by the same mechanism through which ketone bodies increase energy metabolism in perfused, insulin‐deficient rat hearts 43.These findings in Alzheimer's model mice strongly suggest that feeding ketone esters can improve most of the pathological findings in Alzheimer's disease 67. Indeed, a single subject fed ketone esters for 7 years showed significant clinical improvement when on this treatment 74. A recent more detailed review discusses the role of cerebral insulin deficiency and its impairment of pyruvate dehydrogenase activity and how this defect is at the center of the etiology of Alzheimer's disease and can be corrected by feeding ketone body esters 67.
The β‐oxidation pathway generates both NADH and FADH2, which results in the feeding of electrons to both electron transport chain complex I and complex II, resulting in reduction of both the NADH‐binding site and the coenzyme Q couple. This yields no increase in the redox span and hence no increase in the ΔG′ of ATP.
GIVING EXOGENOUS KETONES
During chronic fasting a human puts out 150 g of ketone bodies/24 h 75. This quantity of ketone bodies is roughly 1.5 mol per day resulting in a blood level of β‐hydroxybutyrate of 7 m molar 76, 77. To differentiate the metabolic differences caused by ketone body from those caused by the elevation of free fatty acids, it was desirable to give ketone bodies themselves.
Giving the ketone body as the acid would result in administration of a mole of H+ per day which was unacceptable. Giving the ketone body as a salt would result in administration of 1.5 mol/day of cation, either Na+ of a mixture, which would exceed permissible Na+ limits. This difficulty could be overcome by synthesizing an ester of the d‐β‐hydroxybutyric acid. The usual ester component would be glycerol, but this was undesirable because of the large glucose precursor load giving a mole of glycerol would present.
Several acetoacetate esters, some with racemic 1,3 butanediol are available. These are unsuitable for the purposes intended. Acetoacetate oxidizes the mitochondrial NAD couple thus decreasing the ΔG′ of ATP. Racemic 1,3 butanediol reduces the mitochondrial Q couple, which also decreases the ΔG′ of ATP hydrolysis.
An optimal alternative for use in a ketone ester would be d‐β‐hydroxybutyrate—R‐1,3 butanediol monoester, which had been shown to be metabolizable without complication, in humans 78. Importantly R‐1,3 butanediol is metabolized rapidly to d‐β‐hydroxybutyrate 79 therefore giving an ester made up of d‐β‐hydroxybutyrate and R‐1,3 butanediol, results in the administration only of ketone bodies without added substrate burden. The d‐β‐hydroxybutyrate—R 1,3 butanediol has proven nontoxic in human tests 80 and has a declared Generally recognized as safe (GRAS) status.
In view of the observations by a trained physician on a single patient over 7 years, a larger double‐blind study using ketone ester treatment is warranted.
PARKINSON'S DISEASE
Parkinson's disease is characterized by decreased dopamine production by dopaminergic neurons in the substantia nigra of the brain. Parkinsonism can be induced in animals and humans by administration of the oxidant and heroin analogue MPP+ (1‐methyl‐4‐phenylpyridinium), which is taken up by the dopamine transporter in dopaminergic neurons resulting in cell death. The toxicity of this compound can be overcome in dopaminergic cell cultures by the addition of 4 mM d‐β‐hydroxybutyrate 58 (Fig. 4).
Figure 4.
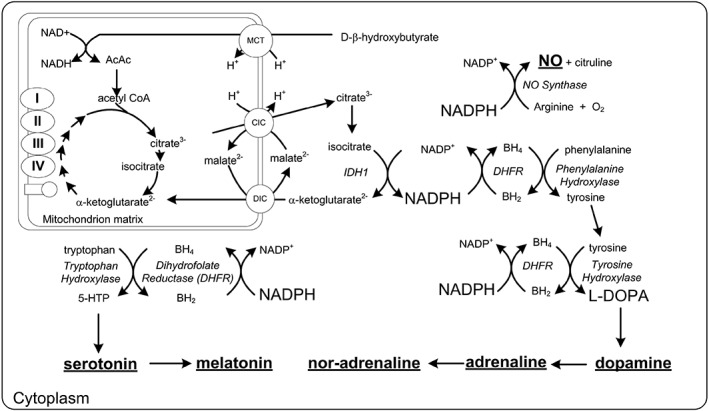
Neurotransmitter synthesis and its relation to the BH4/BH2 ratio and the controlling free [NADP+]/[NADPH] couple. Reducing the NADP couple potentially increases the synthesis of dopamine and other neurotransmitters. Abbreviations: AcAc: acetoacetate; BH2: dihydrobiopterin; BH4: tetrahydrobiopterin; CIC: citrate isocitrate carrier; DIC: dicarboxylate carrier; 5‐HTP 5‐hydroxytryptophan; IDH1: isocitrate dehydrogenase 1; L‐DOPA: L‐3,4‐dihydroxyphenylalanine; MCT: monocarboxylate transporter
In a pilot study, feeding a ketogenic diet lead to an average improvement of 43% on Universal Parkinson's Disease Rating Scale in a small group of Parkinsonian patients 81. The limited clinical trial showed ketogenic diet was capable of improving symptoms responsive to administration of dopamine precursor 82. The metabolism of ketone bodies causes reduction in the cytoplasmic free [NADP+]/[NADPH] ratio. The enzyme dihydrofolate reductase (EC 1.5.1.3) catalyzes the reduction of dihydrobiopterin (BH2) to tetrahydrobiopterin (BH4) in the reaction:
BH4 is a required coenzyme for the synthesis of dopamine as shown in Figure 5 and in this way increased ketone body metabolism and NADPH levels may increase the synthesis of dopamine. Just as in measuring the NADPH couple, Crabtree demonstrated it is not the total BH4 that is critical for the impact of BH4 on NOS, but rather the ratio of [BH4]/[BH2] which is determined by the [NADP+]/[NADPH] couple.
Figure 5.
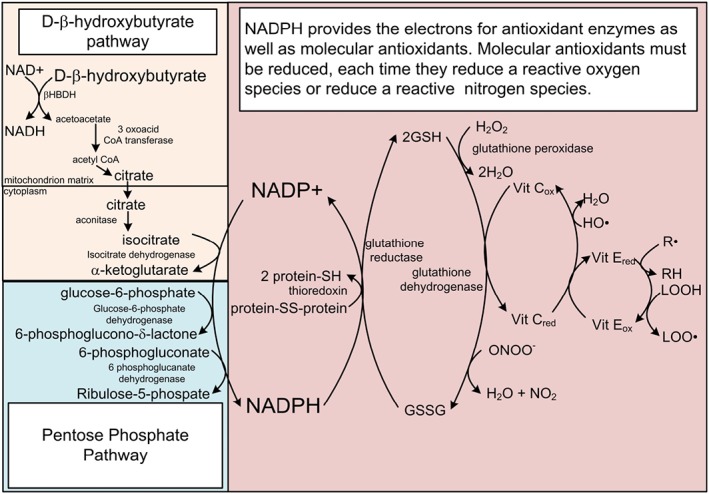
The free [NADP+]/[NADPH] couple controls the redox state of secondary redox couples linked to ROS and RNS detoxification. NADPH provides the reducing potential to counteract ROS and RNS through the reduction of antioxidants that are oxidized each time they quench a ROS or RNS. Abbreviations: GSH: reduced glutathione; GSSG: oxidized glutathione; R: free radical on a carbon atom; LOOH: lipid peroxide; LOO: lipid peroxide radical.
L‐DOPA is the standard of treatment for Parkinson's and that might lead one to assume that dopamine and dopaminergic neurons are the only type of neurons involved in the progression of Parkinson's. Necropsies by Braak et al. 83 correlating Lewy neurites and Lewy bodies to progression of symptoms showed six stages of progression. The dopaminergic neurons of the substantia nigra pars compacta did not show Lewy body staining until the third stage. In stages one and two, the Raphe nucleus that produces serotonin, and locus coeruleus the major producer of noradrenalin were affected. The dorsal motor nucleus of vagus is also affected before the substantia nigra. In stage 4, the Amygdala, nucleus of Meynert and hippocampus are affected. Finally, in stages 5 and 6, the cingulate, temporal, frontal, parietal, and occipital cortex are affected. The ability of the NADPH couple to restore the production of several neurotransmitters other than dopamine may allow therapies that replace other neurotransmitters lost in Parkinson's disease that go untreated by dopamine therapy.
The redox sensitive chaperone protein DJ‐1 84 that has a variant that is associated with PARK7 inherited risk factor for Parkinson's disease is also regulated by the NADPH couple. DJ‐1 has been associated with protection from alpha synuclein aggregation and also the regulation of Nrf2. Without the understanding that the NADPH couple exists and drives the functional status of DJ‐1, it would be difficult to untangle the role played by DJ‐1 in Parkinson's. The ability to measure the NADPH couple should contribute to future research regarding DJ‐1.
The ability of low levels of ketone bodies to correct the tremor and inability to concentrate in patients with long‐standing Parkinson's disease who have become refractory to the effects of dopa therapy is a major advance in the treatment of Parkinson's disease. The ability of ketone body metabolism to correct the symptoms of tremor and impaired cognition suggests that the production of dopamine is increased and further that the dopaminergic neurons are simply functionally impaired but not destroyed. This implies that the administration of ketones during dopa therapy might extend the length of the therapeutic window of dopa therapy beyond the usual 5–7 years.
KETOSIS AND EXTENSION OF LIFE SPAN
Since 1935, it has been known that a 30% decrease in calorie intake resulted in a 50% increase in life span in rats 85. This general observation has been extended across species 86. Calorie restriction, like fasting, increases the levels of ketone bodies in the circulation. In 1956, Harman proposed that the mechanism of aging, like that of radiation, results from damage to tissue from reactive oxygen species (ROS) 87. It has been shown in Caenorhabdtitis elegans, that d‐, but not l‐beta‐hydroxybutyrate, extended life span 21. It has more recently been shown that a ketogenic diet increases the mean life span (health span) of mice 88 suggesting a link between the longevity benefits of calorie restriction and ketone body metabolism. A number of factors have been implicated in the extension of life span including: an increased [NAD+]/[NADH] increasing NAD+‐linked sirtuin histone deacetylase activity, decreased mTOR kinase activity and decreased signaling through insulin‐like growth factor receptors 89. The proteins involved influence the activity of many transcription factors making a complex picture. However, no simple pathway involving small molecules has evolved to date.
We have also shown that ketone bodies decrease mortality in mice exposed to γ‐radiation 2. The metabolism of ketone bodies increases the redox potential of the free cytoplasmic [NADP+]/[NADPH] system 44 overcoming energy deficits associated with insulin resistance by increasing the ΔG′ of ATP hydrolysis 43. d‐β‐hydroxybutyrate inhibits class I histone deacetylases 90, which promotes the transcription of antioxidant enzymes. Feeding ketone esters or a ketogenic diet decreases glucose load and hence decreases insulin signaling through the insulin/IGF‐1/FOXO pathway. Mutations in this pathway in C. elegans first demonstrated that loss of function in single genes resulted in greatly extended life span 91. Feeding ketone esters or a ketogenic diet also has been shown to ameliorate age‐associated neurodegenerative conditions such as Parkinson's 81 or Alzheimer's disease 67. The ability of the metabolism of ketone bodies to delay aging and associated phenotypes such as ROS production, insulin resistance, and neurodegeneration suggest that ketones are promising candidates to promote a healthy life span.
In contrast to the life span extending effects of ketosis, which we have attributed largely to its effects of reducing the cytoplasmic free [NADP+]/[NADPH] and oxidizing the cytoplasmic free [NAD+]/[NADH], administration of antioxidants, such as vitamin C and E, in large multicenter trials have resulted in an increase in all‐cause mortality 92. Therefore, administration of these so‐called antioxidants, without proper understanding of cellular redox state appears to be without benefit and could even be associated with negative health outcomes.
SUMMARY AND OUTLOOK
Understanding the control of metabolic pathways by the “great” controlling nucleotide coenzyme couples and changes in their chemical energies by the simple metabolite d‐β‐hydroxybutyrate offer new biochemical and therapeutic opportunities. These opportunities include increasing the efficiency of aerobic exercise, treating diseases of insulin resistance, combating reactive oxygen toxicity and extending lifespan. The understanding that the NADPH couple can change and influence other couples such as thioredoxin, tetrahydrobiopterin, glutathione, or the redox controlled protein modifications by hydrogen peroxide and other oxidants should provide new understanding in inflammation, hypoxia, mitophagy, autophagy, apoptosis that could not be discovered without an understanding of the great controlling nucleotide coenzymes and their couples and how to measure those couples.
AUTHOR CONTRIBUTIONS
R.L.V. wrote the manuscript; M.T.K. edited and contributed to all the sections; R.P. edited and contributed to sections; Y.K. edited and contributed to sections; P.C.B. edited and contributed to all the sections; W.C. edited and contributed to all the sections.
CONFLICT OF INTERESTS
No competing interests are reported.
ACKNOWLEDGEMENTS
Sam Lally and William Curtis contributed to the graphics.
REFERENCES
- 1. White, H. B. (1982) Evolution of Coenzymes and the Origin of Pyridine Nucleotides. In The Pyridine Nucleotide Coenzymes. (Everse, Johannes editor) Academic Press, New York, NY. [Google Scholar]
- 2. Curtis, W. , Kemper, M. L. , Miller, A. L. , Pawlosky, R. , King, M. T. , et al. (2017) Mitigation of damage from reactive oxygen species and ionizing radiation by ketone body esters In Ketogenic Diet and Metabolic Therapy. (Masino S. A., editor.); p. 254–270 Oxford University Press, Oxford. [Google Scholar]
- 3. Pawlosky, R. J. , Kemper, M. F. , Kashiwaya, Y. , King, M. T. , Mattson, M. P. , et al. (2017) Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer's disease. J. Neurochem. 141, 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krebs, H. A. , and Veech, R. L. (1969) Interrelations between diphospho‐ and triphospho‐pyridine nucleotides In Mitochondria: Structure and Function. (Ernster L., and Drohota Z., editors.); p. 101–110 Academic Press, New York, NY. [Google Scholar]
- 5. Holzer, H. , Schultz, G. , and Lynen, F. (1956) Bestimmung des quotienten DPNH/DPN in lebenden hefezellen durch analyse stationarer alkohol und acetaldehyde konzentrationen. Biochem Z 332, 252–263. [PubMed] [Google Scholar]
- 6. Bucher, T. , and Klingenberg, M. (1958) Wege des Wasserstoffs in der lebendigen Organisation. Angew. Chem. 70, 552–570. [Google Scholar]
- 7. Williamson, D. H. , Lund, P. , and Krebs, H. A. (1967) The redox state of free nicotinamide‐adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Veech, R. L. , Eggleston, L. V. , and Krebs, H. A. (1969) The redox state of free nicotinamide‐adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J 115, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krebs, H. A. , and Veech, R. L. (1969) Pyridine nucleotide interrelations In The Energy Level and Metabolic Conntrol in Mitochondria. (Papa S., Tager J. M., Quagliariello E., and Slater E. C., editors.); p. 329–384 Adriatica Editrice, Bari, Italy. [Google Scholar]
- 10. Aksoy, P. , White, T. A. , Thompson, M. , and Chini, E. N. (2006) Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 345, 1386–1392. [DOI] [PubMed] [Google Scholar]
- 11. Massudi, H. , Grant, R. , Braidy, N. , Guest, J. , Farnsworth, B. , et al. (2012) Age‐associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7, e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshino, J. , Mills, K. F. , Yoon, M. J. , and Imai, S. (2011) Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab. 14, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mills, K. F. , Yoshida, S. , Stein, L. R. , Grozio, A. , Kubota, S. , et al. (2016) Long‐term Administration of Nicotinamide Mononucleotide Mitigates age‐Associated Physiological Decline in mice. Cell Metab. 24, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshino, J. , Baur, J. A. , and Imai, S. I. (2018) NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 27, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elamin, M. , Ruskin, D. N. , Masino, S. A. , and Sacchetti, P. (2017) Ketone‐based metabolic therapy: is increased NAD(+) a primary mechanism? Front. Mol. Neurosci. 10, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elamin, M. , Ruskin, D. N. , Masino, S. A. , and Sacchetti, P. (2018) Ketogenic diet modulates NAD(+)‐dependent enzymes and reduces DNA damage in hippocampus. Front. Cell. Neurosci. 12, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xin, L. , Ipek, O. , Beaumont, M. , Shevlyakova, M. , Christinat, N. , et al. (2018) Nutritional ketosis increases NAD(+)/NADH ratio in healthy human brain: an in vivo study by (31)P‐MRS. Front. Nutr. 5, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Du, L. , Zhang, X. , Han, Y. Y. , Burke, N. A. , Kochanek, P. M. , et al. (2003) Intra‐mitochondrial poly(ADP‐ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 278, 18426–18433. [DOI] [PubMed] [Google Scholar]
- 19. Jo, S. H. , Son, M. K. , Koh, H. J. , Lee, S. M. , Song, I. H. , et al. (2001) Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+−dependent isocitrate dehydrogenase. J. Biol. Chem. 276, 16168–16176. [DOI] [PubMed] [Google Scholar]
- 20. Khaidizar, F. D. , Nakahata, Y. , Kume, A. , Sumizawa, K. , Kohno, K. , et al. (2017) Nicotinamide phosphoribosyltransferase delays cellular senescence by upregulating SIRT1 activity and antioxidant gene expression in mouse cells. Genes Cells 22, 982–992. [DOI] [PubMed] [Google Scholar]
- 21. Edwards, C. , Canfield, J. , Copes, N. , Rehan, M. , Lipps, D. , et al. (2014) D‐beta‐hydroxybutyrate extends lifespan in C. elegans . Aging 6, 621–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veech, R. L. , Bradshaw, P. C. , Clarke, K. , Curtis, W. , Pawlosky, R. , et al. (2017) Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 69, 305–314. [DOI] [PubMed] [Google Scholar]
- 23. Holzer, H. (1956) Bestimmung des Quotienten DPNH/DPN in lebenden Hefezellen durch Analyse stationarer Alkohol‐und Acetaldehyde‐Konzentrationen. Biochem. Z. 328, 252–263. [PubMed] [Google Scholar]
- 24. Guynn, R. W. , Gelberg, H. J. , and Veech, R. L. (1973) Equilibrium constants of the malate dehydrogenase, citrate synthase, citrate lyase, and acetyl coenzyme a hydrolysis reactions under physiological conditions. J. Biol. Chem. 248, 6957–6965. [PubMed] [Google Scholar]
- 25. Russman W. (1967) Ph.D Dissertation, Dissertation Munchen.
- 26. Cornell, N. W. , Leadbetter, M. , and Veech, R. L. (1979) Effects of free magnesium concentration and ionic strength on equilibrium constants for the glyceraldehyde phosphate dehydrogenase and phosphoglycerate kinase reactions. J. Biol. Chem. 254, 6522–6527. [PubMed] [Google Scholar]
- 27. Engle, P. C. , and Dalziel, K. (1967) The equilibrium constants of the glutamate dehydrogenase systems. Biochem. J 105, 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Londesborough, J. C. , and Dalziel, K. (1968) The equilibrium constant of the isocitrate dehydrogenase reaction. Biochem. J 110, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scott, E. M. , Duncan, I. W. , and Ekstrand, V. (1963) Purification and properties of glutathione reducatase of human erythrocytes. J. Biol. Chem. 238, 3928–3933. [PubMed] [Google Scholar]
- 30. Villet, R. H. , and Dalziel, K. (1969) The nature of the carbon dioxide substrate and equilibrium constant of the 6‐phosphogluconate dehydrogenase reaction. Biochem. J. 115, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Veech R.L. (1968) Measurment of respiratory metabolites in animal tissues. DPhil in Biochemistry, Oxford.
- 32. Lawson, J. W. R. , and Veech, R. L. (1979) Effects of pH and free Mg 2+ on the Keq of the creatine kinase reaction and other phosphate hydrolyses and phosphate transfer reactions. J. Biol. Chem. 254, 6528–6537. [PubMed] [Google Scholar]
- 33. Guynn, R. W. , Webster, L. T., Jr. , and Veech, R. L. (1974) Equilibrium constants of the reactions of acetyl coenzyme a synthetase and the hydrolysis of adenosine triphosphate to adenosine monophosphate and inorganic pyrophosphate. J. Biol. Chem. 249, 3248–3254. [PubMed] [Google Scholar]
- 34. Veech, R. L. , Raijman, L. , Dalziel, K. , and Krebs, H. A. (1969) Disequilibrium in the triose phosphate isomerase system in rat liver. Biochem. J. 115, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Veloso, D. , Guynn, R. W. , Oskarsson, M. , and Veech, R. L. (1973) The concentrations of free and bound magnesium in rat tissues. Relative constancy of free Mg 2+ concentrations. J. Biol. Chem. 248, 4811–4819. [PubMed] [Google Scholar]
- 36.Tischler, M. E., Friedrichs, D., Coll, K., and Williamson, J. R. (1977) Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 184, 222–236. [DOI] [PubMed] [Google Scholar]
- 37.Sallin, O., Reymond, L., Gondrand, C., Raith, F., Koch, B., and Johnsson K. (2018) Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. eLife 7, e32638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kashiwaya, Y. , Sato, K. , Tsuchiya, N. , Thomas, S. , Fell, D. A. , et al. (1994) Control of glucose utilization in working perfused rat heart. J. Biol. Chem. 269, 25502–25514. [PubMed] [Google Scholar]
- 39. Bucher, T. , and Russmann, W. (1964) Equilibrium and nonequilibrium in the glycolytic system. Angew. Chem. Int. Ed. Engl. 3, 426–439. [Google Scholar]
- 40. Veech, R. L. , Lawson, J. W. R. , Cornell, N. W. , and Krebs, H. A. (1979) Cytosolic phosphorylation potential. J. Biol. Chem. 254, 6538–6547. [PubMed] [Google Scholar]
- 41. Veech, R. L. (1978) Regulation of coenzyme potential by near equilibrium reactions In Microenvironmentsandmetaboliccompartmentation. (Srere P. A., and Estabrook R. W., editors.) ; p. 17–64 Acedemic Press, New York, NY. [Google Scholar]
- 42. Sun, F. , Dai, C. , Xie, J. , and Hu, X. (2012) Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLos one 7, e34525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sato, K. , Kashiwaya, Y. , Keon, C. A. , Tsuchiya, N. , King, M. T. , et al. (1995) Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 9, 651–658. [DOI] [PubMed] [Google Scholar]
- 44. Kashiwaya, Y. , King, M. T. , and Veech, R. L. (1997) Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. Am. J. Cardiol. 80, 50A–64A. [DOI] [PubMed] [Google Scholar]
- 45. Guynn, R. W. , and Veech, R. L. (1973) The equilibrium constants of the adenosine triphosphate hydrolysis and the adenosine triphosphate‐citrate lyase reactions. J. Biol. Chem. 248, 6966–6972. [PubMed] [Google Scholar]
- 46. Eliot, T. S. (1936)Burnt Norton. Faber, London. [Google Scholar]
- 47. Bergman, C. , Kashiwaya, Y. , and Veech, R. L. (2010) The effect of pH and free Mg2+ on ATP linked enzymes and the calculation of Gibbs free energy of ATP hydrolysis. J. Phys. Chem. B 114, 16137–16146. [DOI] [PubMed] [Google Scholar]
- 48. Mitchell, P. (1966) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 41, 445–502. [DOI] [PubMed] [Google Scholar]
- 49. Masuda, T. , Dobson, G. P. , and Veech, R. L. (1990) The Gibbs‐Donnan near‐equilibrium system of heart. J. Biol. Chem. 265, 20321–20334. [PubMed] [Google Scholar]
- 50. Wondergem, R. , and Harder, D. R. (1980) Membrane potential measurements during rat liver regeneration. J. Cell Physiol. 102, 193–197. [DOI] [PubMed] [Google Scholar]
- 51. Wondergem, R. , and Castillo, L. B. (1986) Effect of temperature on transmembrane potential of mouse liver cells. Am. J. Physiol. 251, C603–C613. [DOI] [PubMed] [Google Scholar]
- 52. Veech, R. L. , Gates, D. N. , Crutchfield, C. W. , Gitomer, W. L. , Kashiwaya, Y. , et al. (1994) Metabolic hyperpolarization of liver by ethanol: the importance of mg 2+ and H + in determining impermeant intracellular anionic charge and energy of metabolic reactions. Alcohol. Clin. Exp. Res. 18, 1040–1056. [DOI] [PubMed] [Google Scholar]
- 53. Veech, R. L. , Kashiwaya, Y. , and King, M. T. (1995) The resting potential of cells are measures of electrical work not of ionic currents. Intergr. Physiol. Behav. Sci. 30, 283–306. [DOI] [PubMed] [Google Scholar]
- 54. Veech, R. L. , Kashiwaya, Y. , Gates, D. N. , King, M. T. , and Clarke, K. (2002) The energetics of ion distribution: the origin of the resting electric potential of cells. IUBMB Life 54, 241–252. [DOI] [PubMed] [Google Scholar]
- 55. Cornish‐Bowden, A. (2012)Fundamentals of Enzyme Kinetics. Wiley‐Blackwell, Weinheim, Germany. [Google Scholar]
- 56. Meyer, R. A. , and Wiseman, R. W. (2011) The metabolic systems: control of ATP synthesis in skeletal muscle In ACSM's Advanced Exercise Physiology, 2nd edn (Tipton C. M., Sawka M. N., Tate C. A., and Terjung R. L., editors.) Lippincott Williams & Wilkins, Baltimore, MD. [Google Scholar]
- 57. Neufer, P. D. , and Booth, F. (2005) Exercise Controls Gene Expression. Am. Sci. 93, 28. [Google Scholar]
- 58. Kashiwaya, Y. , Takeshima, T. , Mori, N. , Nakashima, K. , Clarke, K. , et al. (2000) D‐beta ‐Hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc. Natl. Acad. Sci. USA 97, 5440–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mukherjee, C. , and Jungas, R. L. (1975) Activation of pyruvate dehydrogenase in adipose tissue by insulin. Evidence for an effect of insulin on pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 148, 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taylor, S. I. , Mukherjee, C. , and Jungas, R. L. (1975) Regulation of pyruvate dehydrogenase in isolated rat liver mitochondria. Effects of octanoate, oxidation‐reduction state, and adenosine triphosphate to adenosine diphosphate ratio. J. Biol. Chem. 250, 2028–2035. [PubMed] [Google Scholar]
- 61. Evans, D. A. , Funkenstein, H. H. , Albert, M. S. , Scherr, P. A. , Cook, N. R. , et al. (1989) Prevalence of Alzheimer's disease in a community population of older persons. Higher than previously reported [see comments]. JAMA 262, 2551–2556. [PubMed] [Google Scholar]
- 62. Hoyer, S. (1996) Oxidative metabolism deficiencies in brains of patients with Alzheimer's disease. Acta Neurol. Scand. Suppl. 165, 18–24. [DOI] [PubMed] [Google Scholar]
- 63. Kuusisto, J. , Koivisto, K. , Mykkanen, L. , Helkala, E. L. , Vanhanen, M. , et al. (1997) Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ (Clinical research ed) 315, 1045–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Talbot, K. , Wang, H. Y. , Kazi, H. , Han, L. Y. , Bakshi, K. P. , et al. (2012) Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF‐1 resistance, IRS‐1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Castellano, C. A. , Nugent, S. , Paquet, N. , Tremblay, S. , Bocti, C. , et al. (2015) Lower brain 18F‐fluorodeoxyglucose uptake but normal 11C‐acetoacetate metabolism in mild Alzheimer's disease dementia. J. Alzheimers Dis. 43, 1343–1353. [DOI] [PubMed] [Google Scholar]
- 66. Willette, A. A. , Bendlin, B. B. , Starks, E. J. , Birdsill, A. C. , Johnson, S. C. , et al. (2015) Association of Insulin Resistance with Cerebral Glucose Uptake in late middle‐aged adults at risk for Alzheimer disease. JAMA Neurol. 72, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Veech, R. L. , and King, M. T. (2017) Alzheimer's disease: causes and treatment In Ketogenic Diet and Metabolic Therapy. (Masino S. A., editor.) ; p. 241–253 Oxford University Press, Oxford. [Google Scholar]
- 68. Craft, S. , Claxton, A. , Baker, L. D. , Hanson, A. J. , Cholerton, B. , et al. (2017) Effects of regular and long‐acting insulin on cognition and Alzheimer's disease biomarkers: a pilot clinical trial. J. Alzheimers Dis. 57, 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Taylor, M. K. , Sullivan, D. K. , Mahnken, J. D. , Burns, J. M. , and Swerdlow, R. H. (2018) Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement (N Y) 4, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Murray, A. J. , Knight, N. S. , Cole, M. A. , Cochlin, L. E. , Carter, E. , et al. (2016) Novel ketone diet enhances physical and cognitive performance. FASEB J 30, 4021–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cox, P. J. , Kirk, T. , Ashmore, T. , Willerton, K. , Evans, R. , et al. (2016) Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 24, 256–268. [DOI] [PubMed] [Google Scholar]
- 72. Klyuyeva, A. , Tuganova, A. , Kedishvili, N. Y. , and Popov, K. M. (2018) Tissue‐specific kinase expression and activity regulates flux through the pyruvate dehydrogenase complex. J. Biol. Chem. 10.1074/jbc.RA118.006433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kashiwaya, Y. , Bergman, C. , Lee, J. H. , Wan, R. , Todd King, M. , et al. (2013) A ketone ester diet exhibits anxiolytic and cognition‐sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiol. Aging 34, 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Newport, M. T. , Vanitallie, T. B. , Kashiwaya, Y. , King, M. T. , and Veech, R. L. (2015) A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer's disease. Alzheimers Dement. 11, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reichard, G. A. , Owen, O. E. , Haff, A. C. , Paul, P. , and Bortz, W. M. (1974) Ketone‐body production and oxidation in fasting obese humans. J. Clin. Invest 53, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Owen, O. E. , Morgan, A. P. , Kemp, H. G. , Sullivan, J. M. , Herrera, M. G. , et al. (1967) Brain metabolism during fasting. J. Clin. Invest. 46, 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cahill, G. F., Jr. (1970) Starvation in man. N. Engl. J. Med. 282, 668–675. [DOI] [PubMed] [Google Scholar]
- 78. Kies, C. , Tobin, R. B. , Fox, H. M. , and Mehlman, M. A. (1973) Utilization of 1,3‐butanediol and nonspecific nitrogen in human adults. J. Nutr. 103, 1155–1163. [DOI] [PubMed] [Google Scholar]
- 79. Mehlman, M. A. , and Veech, R. L. (1972) Redox and phosphorylation states and metabolite concentrations in frozen clamped livers of rats fed diets containing 1,3‐butanediol and DL‐carnitine. J. Nutr. 102, 45–51. [DOI] [PubMed] [Google Scholar]
- 80. Clarke, K. , Tchabanenko, K. , Pawlosky, R. , Carter, E. , Knight, N. S. , et al. (2012) Oral 28‐day and developmental toxicity studies of (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate. Regul. Toxicol. Pharmacol. 63, 196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vanitallie, T. B. , Nonas, C. , Di Rocco, A. , Boyar, K. , Hyams, K. , et al. (2005) Treatment of Parkinson disease with diet‐induced hyperketonemia: a feasibility study. Neurology 64, 728–730. [DOI] [PubMed] [Google Scholar]
- 82. Cotzias, G. C. , Van Woert, M. H. , and Schiffer, L. M. (1967) Aromatic amino acids and modification of parkinsonism. N. Engl. J. Med. 276, 374–379. [DOI] [PubMed] [Google Scholar]
- 83. Braak, H. , Del Tredici, K. , Rüb, U. , de Vos, R. A. I. , Jansen Steur, E. N. H. , et al. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- 84. Bjorkblom, B. , Adilbayeva, A. , Maple‐Grodem, J. , Piston, D. , Okvist, M. , et al. (2013) Parkinson disease protein DJ‐1 binds metals and protects against metal‐induced cytotoxicity. J. Biol. Chem. 288, 22809–22820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McCay, C. M. , Crowell, M. F. , and Maynard, L. A. (1935) The effect of retarded growth upon the lenght of life span and upon the ultimate body size. J. Nutr. 10, 63–79. [PubMed] [Google Scholar]
- 86. Fontana, L. , Partridge, L. , and Longo, V. D. (2010) Extending healthy life span‐‐from yeast to humans. Science 328, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Harman, D. (1956) Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. [DOI] [PubMed] [Google Scholar]
- 88. Roberts, M. N. , Wallace, M. A. , Tomilov, A. A. , Zhou, Z. , Marcotte, G. R. , et al. (2017) A Ketogenic diet extends longevity and Healthspan in adult mice. Cell Metab. e535, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pan, H. , and Finkel, T. (2017) Key proteins and pathways that regulate lifespan. J. Biol. Chem. 292, 6452–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shimazu, T. , Hirschey, M. D. , Newman, J. , He, W. , Shirakawa, K. , et al. (2013) Suppression of oxidative stress by beta‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kenyon, C. (2011) The first long‐lived mutants: discovery of the insulin/IGF‐1 pathway for ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bjelakovic, G. , Nikolova, D. , and Gluud, C. (2013) Antioxidant supplements to prevent mortality. JAMA 310, 1178–1179. [DOI] [PubMed] [Google Scholar]