

Abstract
Microtubule-associated protein tau associates with Src family tyrosine kinase Fyn and is tyrosine phosphorylated by Fyn. The presence of tyrosine phosphorylated tau in AD and the involvement of Fyn in AD has drawn attention to the tau-Fyn complex. In this study, a tau-Fyn double knockout (DKO) mouse was generated to investigate the role of the complex. DKO mice resembled Fyn KO in novel object recognition and contextual fear conditioning tasks and resembled tau KO mice in the pole test and protection from pentylenetetrazole-induced seizures. In glutamate-induced Ca2+ response, Fyn KO was decreased relative to WT and DKO had a greater reduction relative to Fyn KO, suggesting that tau may have a Fyn-independent role. Since tau KO resembled WT in its Ca2+ response, we investigated whether MAP2 served to compensate for tau, since the MAP2 level was increased in tau KO but decreased in DKO mice. We found that like tau, MAP2 increased Fyn activity. Moreover, tau KO neurons had increased density of dendritic MAP2-Fyn complexes relative to WT neurons. Therefore, we hypothesize that in the tau KO, the absence of tau would be compensated by MAP2, especially in the dendrites, where tau-Fyn complexes are of critical importance. In the DKO, decreased levels of MAP2 made compensation more difficult, thus revealing the effect of tau in the Ca2+ response.
Keywords: tau, Fyn, MAP2, calcium, proximity ligation assay
Introduction
Tau was initially discovered as a microtubule-associated protein enriched in axons (Binder et al., 1986; Weingarten et al., 1975). Interest in tau dramatically increased after hyperphosphorylated tau was found in neurofibrillary tangles (NFT), a major hallmark of Alzheimer’s disease (AD) (Grundke-Iqbal et al., 1986; Kosik et al., 1986; Nukina and Ihara, 1986; Wood et al., 1986), and after mutations in the tau gene were found to cause frontotemporal dementia with Parkinsonism linked to chromosome 17 (Clark et al., 1998; Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998). Subsequent studies have revealed new functions for tau, such as the regulation of motor-driven axonal transport (Dixit et al., 2008; Trinczek et al., 1999), formation and trafficking of stress granules (Vanderweyde et al., 2016), and postsynaptic scaffolding (Ittner et al., 2010).
However, despite the multiple roles played by tau, initial studies of five lines of independently generated tau KO mice did not suggest any gross deficits or loss of viability (Dawson et al., 2001; Fujio et al., 2007; Harada et al., 1994; Tan et al., 2018; Tucker et al., 2001). Further testing of tau KO mice has yielded controversial results, as some studies reported minor memory deficits (Ahmed et al., 2014; Ikegami et al., 2000; Lei et al., 2014; Ma et al., 2014) while a larger number of studies reported no cognitive deficits (Dawson et al., 2010; Dawson et al., 2001; Ittner et al., 2010; Kimura et al., 2014; Lei et al., 2012; Li et al., 2014; Morris et al., 2013; Regan et al., 2015; Roberson et al., 2007; Tan et al., 2018; van Hummel et al., 2016). Morphological studies of tau KO mice reported alterations in small caliber axons as the only abnormality (Harada et al., 1994) while low density neuronal cultures from another line of tau KO mouse indicated slowed axonal development, although brain development appeared normal (Dawson et al., 2001). The lack of more pronounced behavioral and morphological phenotypes has largely been attributed to functional compensation by other microtubule-binding proteins such as microtubule associated protein 1A/1B (MAP1A/1B) and MAP2 (Harada et al., 1994; Ma et al., 2014). While it is thought that compensating for a loss of microtubule stabilizing activity is important, given the additional functions for tau, other compensatory mechanisms may be needed if these functions are critical for mouse viability and depend on tau.
Following our finding that tau could associate with the cytoplasmic face of the plasma membrane (Brandt et al., 1995), we found that the proline-rich domain of tau interacts with the SH3 domain of Src family non-receptor tyrosine kinases (SFK) such as Fyn and Src (Lee et al., 1998) and that Fyn phosphorylated tau on Tyr18 (Lee et al., 2004). In addition, we showed that tau increased the auto-phosphorylation of Fyn as well as the enzymatic activity of Fyn (Sharma et al., 2007). Also, tau was critical for nerve growth factor (NGF)-induced mitogen activated protein kinase (MAPK) activation, suggesting a role for tau in signal transduction (Leugers et al., 2013; Leugers and Lee, 2010). Furthermore, ERK activity, contributed by a Fyn/ERK/S6 signaling pathway, can also mediate tau expression in the somatodendritic domain (Li and Gotz, 2017).
Interest in the tau-Fyn interaction has been increased by the presence of phospho-tyr18-tau in NFTs (Bhaskar et al., 2010; Lee et al., 2004) and reported alterations of Fyn expression in AD brain (Ho et al., 2005; Shirazi and Wood, 1993). Additionally, Fyn has also been implicated in AD models, where depletion of Fyn provided neuroprotective effects in mice expressing amyloid precursor protein, and in primary cultured neurons treated with beta-amyloid peptide(Chin et al., 2004; Kaufman et al., 2015; Lambert et al., 1998). Furthermore, Fyn is highly expressed in the central nervous system (Umemori et al., 1992) and phosphorylates the N-methyl-D-aspartate receptor (NR) subunit 2B (NR2B) at Y1472 residue (Nakazawa et al., 2001; Rong et al., 2001), which facilitates the interaction between the NR and the postsynaptic density 95 protein (PSD-95) (Tezuka et al., 1999) and regulates long-term potentiation (LTP) and memory formation (Kojima et al., 1997; Stein et al., 1992). Tau has been implicated in targeting Fyn to the postsynaptic space, as tau KO mice showed decreased levels of Fyn and pY1472 NR2B in synaptosome preparations (Ittner et al., 2010). As a result, the PSD-NR interaction was disrupted, leading to protection from both pentylenetetrazole (PTZ) induced seizures and amyloid-β induced excitotoxicity (Ittner et al., 2010; Roberson et al., 2007). However, electrophysiological studies of tau KO resembled WT (Ittner et al., 2010; Kimura et al., 2014; Roberson et al., 2011), further underlying the need to elucidate the function of the tau-Fyn interaction and its significance in neuronal cell function.
To further understand the impact of the combined action of tau and Fyn, we generated a tau-Fyn double knockout (tau−/−/Fyn−/−) mouse, characterized its behavioral, biochemical, and neurophysiological properties, and found a role for tau in the neuron’s calcium response that is independent of Fyn. Additionally, we identified the location of tau-Fyn complexes in neurons and determined that in dendrites, the loss of such complexes led to compensation mediated by MAP2.
Methods and Materials
Mice
Tau KO (C57BL/6) mice (Dawson et al., 2001) and Fyn KO (C57BL/6/S129) mice (Stein et al., 1992) were obtained from Jackson Laboratories and crossed to generate heterozygotes. The heterozygotes were then bred to generate tau−/−/Fyn−/− and tau+/+/Fyn+/+ mice (Fig. S1A). Mice were genotyped as described by Jackson Laboratories. All mice were housed in non-barrier facilities with maximum of 5 mice to each cage. Mice had access to water and food ad libitum. After the creation of DKO and WT mice, each colony was maintained separately. All procedures and animal care were approved by the University of Iowa Institutional Animal Care and Use Committee and in compliance with the NIH Guide for the Care and Use of Laboratory Mice. The mice were fed NIH31 irradiated modified open formula mouse/rat diet. The pathogens for which the mice were tested are listed in Fig. S1C.
When the homozygous Fyn KO trait was bred into a C57BL/6 background, the mice exhibited a 50% chance of developing non-obstructive hydrocephalus postpartum (Goto et al., 2008). The DKO mice also developed hydrocephalus. Magnetic resonance imaging (MRI) was used to differentiate normal from hydrocephalic mice. For behavioral and biochemical assays, mice with lateral ventricle volume ≤0.2 units, as assessed by MRI, were included while those with lateral ventricle volume >0.2 units were excluded. The experimenter was blinded to mice genotype and the mice were randomly assigned to different testing chambers during the behavioral assays. In general, the same group of animals underwent the following tasks, ordered from least traumatizing to most traumatizing: Open field was performed on 8 weeks old mice, novel object recognition on 9 weeks old mice, contextual fear conditioning on 10 weeks old mice, and PTZ induced seizures on 12 weeks old mice (Fig S1B). Pole test was done on a different cohort of mice aged at 12 months.
Magnetic resonance imaging
Varian Unity/Inova 4.7 T small-bore MRI system (Varian, Inc., Palo Alto, CA; Small Animal Imaging Facility, University of Iowa) with an in-plane resolution of 0.13 x 0.25 mm2 and 0.6 mm slice thickness was used. Coronal images were collected and lateral ventricle volume sizes were analyzed with ImageJ (Carter et al., 2012). A lateral ventricle volume ≤0.2 units was normal, ≥0.2 and ≤ 2 units was moderate, and ≥ 2 units was severe hydrocephalic.
Open field test
The open field test was done at 8 weeks as described (Coryell et al., 2007) with a dimension of 40.6 × 40.6 × 36.8 cm open-field chamber (San Diego Instruments, San Diego, CA), 55 lux, for 20 min. Total activity was measured by total beam breaks and central activity was measured by beam breaks in center (15.2 × 15.2 cm). 14 WT (14 male), 20 Tau KO (9 male, 11 female), 18 Fyn KO (15 male, 3 female), 13 (10 male, 3 female) DKO mice were tested. Since there were no statistically significant differences between males and females, their results were combined.
Pole test
The pole test was done at 12 months of age as described (Lei et al., 2014). Trials were excluded if the mouse jumped or slid down the pole. Between tests, the pole was cleaned using 70% ethanol. 9 WT, 13 tau KO, 11 Fyn KO, 18 DKO male mice were tested.
Novel object recognition
The novel object recognition test was done at 10 weeks as described (Tang et al., 2001). The time each animal spent exploring objects (defined as nose within an inch of the object) was manually determined. Discrimination index [DI = (X1-X2) / (X1+X2)] was calculated. 17 WT (12 male, 5 female), 17 tau KO (12 male, 5 female), 28 Fyn KO (16 male, 12 female), 18 DKO (14 male, 4 female) mice were used. Since there were no statistically significant differences between males and females, their results were combined.
Contextual fear conditioning
Contextual fear conditioning was done at 11 weeks as described (Sowers et al., 2013), using a near-infrared video-equipped fear conditioning chamber (Med Associates, Inc., St. Albums, VT). Freezing was scored with VideoFreeze software (Med Associates, Inc.). 32 WT (27 male, 5 female), 29 tau KO (21 male, 8 female), 21 Fyn KO (14 male, 7 female), 20 DKO (14 male, 6 female) mice were tested. Since there were no statistically significant differences between males and females, their results were combined.
Pentylenetetrazole-induced seizures
Pentylenetetrazole (PTZ)-induced seizure was done at 12 weeks as described-(Roberson et al., 2007). A modified Racine seizure scale was used: 0: normal behavior; 1: immobility; 2: tail extension; 3: forelimb clonus; 4: generalized clonic activity; 5: bouncing; 6: tonic extension; 7: death (Loscher et al., 1991; Racine, 1972). 25 WT (14 male, 11 female), 21 tau KO (9 male, 12 female), 36 (25 male, 11 female) Fyn KO, and 26 DKO (19 male, 7 female) mice were used. Since there were no statistically significant differences between males and females, their results were combined.
Tissue preparation and subcellular fractionation
PSD preps were isolated from hippocampi of 9–12 months old mice (Ittner et al., 2010; Lopes et al., 2016b; Milnerwood et al., 2010). As described, crude synaptosomal membranes were resuspended with 1% Triton X-100 containing buffer and after centrifugation; the supernatant containing non-PSD membranes was retained as the “soluble non-PSD” fraction. The pellet was resuspended in 1% Triton X-100, 1% deoxycholic acid, 1% SDS containing buffer and after centrifugation, the supernatant was retained as the triton “insoluble PSD” fraction.
Primary hippocampal neuron culture
Primary hippocampal neuronal culture was performed as described (Beaudoin et al., 2012). Hippocampi from P0 pups were isolated and digested in 1 mg/ml trypsin (Sigma-T7409) and sequentially triturated with Pasteur pipettes. Cells were plated with MEM media (Gibco) containing 10% horse serum and 1µM insulin (Sigma-I5500) and maintained with Neurobasal-A Plus and 2% B27 Plus (Gibco) at 37 °C and 5% CO2 until harvest. 50% of media was changed every 3 days.
In situ proximity ligation assay (PLA) and immunofluorescence
PLA components were purchased from Sigma (Duolink® in situ) and the assay was performed per manufacturer’s instructions. Cells were imaged using confocal microscopy (Leica SP8 STED) and exported to ImageJ for analysis.
Primary antibodies used were DA9, Fyn3, MAP2, Src, and phospho-Src-Tyr418 and tubulin, as described in Table 1. Secondary antibodies used were anti-mouse Alexa 488 (1:250, Molecular Probes), anti-chicken Alexa 633 (1:1000, Thermo Fisher), and anti-rat Alexa 647 (1:250 Jackson Immunoresearch).
To quantify the density of PLA puncta, ten random areas on each coverslip were photographed. Axons were identified by either tau or tubulin positive staining and MAP2 negative staining; dendrites were identified by MAP2 positive staining. The lengths of all visible processes were measured using ImageJ and all visible PLA puncta were manually counted. For each of the 10 areas, the densities of dendritic or axonal PLA puncta were calculated by dividing the number of PLA puncta located on dendrites or axons by the total dendrite or axon length, respectively. The experiment was performed 3 times. In Fig. 4, the density of PLA in dendrites was expressed relative to the density in axons, where the average value was set as “1”. In Fig. 8, the density of PLA in tau KO mice was expressed relative to the density in WT mice, where the average value was set as “1”. In Fig. S2, for each neuron, the proximal axon length was defined as the average length of the dendrites from that neuron.
Ca2+ imaging
Ca2+ imaging was performed on primary hippocampal neurons (DIV 11–12) as described (Kim et al., 2009). Hippocampal neurons were loaded with 2μM Fura-2/AM (Invitrogen) and then mounted on an inverted microscope (Olympus IX-71). The cells were perfused with HEPES buffered Hank’s salt solution (HH buffer) to establish baseline and then stimulated with 100 µM Glutamate, 10 µM Glycine, and 0.2 µM Tetrodotoxin (TTX) in HH buffer. [Ca2+]i changes were continuously recorded by alternately exciting fluorescence at 340 nm (12-nm bandpass) and 380 nm (12-nm bandpass) using a Polychrome V monochromator (TILL Photonics, Munich, Germany). Fluorescence was recorded at 530 nm (50-nm bandpass) using an IMAGO charge-coupled device camera (640 × 480 pixels; TILL Photonics). A 20x objective (numerical aperture = 0.75, Olympus, Japan) and a 2×2 binning set at room temperature was used. [Ca2+]i was derived using the formula [Ca2+]i = Kdβ(R − Rmin)/(Rmax − R), with Kd=275nM (Shuttleworth and Thompson, 1991), R =F340nm/F380nm, Rmin= 0.21, Rmax= 3.45, and β= 6.97. Data were analyzed using TILLvisION version 4.0.12 software (TILL Photonics). 3 independent experiments were conducted, each using hippocampal neurons derived from a single pup from each of the four different genotypes.
Immunoprecipitation
3T3 (NIH) cells were grown in α-MEM supplemented with 10% fetal bovine serum (Hyclone) and transfected with Fyn alone or co-transfected with Fyn and with MAP2c. Cells were harvested with RIPA lysis buffer. Fyn was IPed with 1µg Fyn antibody (mouse monoclonal antibody [mAb], sc-434, Santa Cruz Biotechnology) and immunoprecipitated with 10 uL protein G plus-agarose (CalBiochem). After washing, protein G bead pellets were resuspended in 2X laemmli buffer and subjected to Western blot analysis. The mouse monoclonal antibody against Fyn was directed against a Fyn specific region and does not cross-react with Src. Therefore, when the Fyn IP was probed with anti-pSFK, the signal was specifically activated Fyn. 3T3 cells were obtained in 1990 from Dr. Frank McKeon (University of Houston). 3T3 cells have not been reported as contaminated or misidentified (International Cell Line Authentication Committee, October 14, 2018).
Plasmids
pRc/CMV expressing the N-terminal fragment of human MAP-2c was synthesized by site-directed mutagenesis using the pRc/CMV MAP2c plasmid (Albala et al., 1993) as template. Residue 320 on MAP-2c was mutated to a stop codon. The construct was sequenced to verify the MAP2c sequence and the mutation site.
In vitro phosphorylation of tubulin
In vitro phosphorylation of tubulin was performed as described (Sharma et al., 2007). Fyn or truncated MAP2c (human N-MAP2c residue 1–319) were expressed in 3T3 cells and IPed using 1µg anti-Fyn (mAb, sc-434, Santa Cruz Biotechnology) or 4 µg anti-MAP2 (HM-2, mAb, Sigma, Cat # M4403) and Protein G beads (CalBiochem). The beads containing Fyn were resuspended in 100 µL buffer (0.1% Triton, 50mM Tris pH7.5, 150 mM NaCl, 1 mM EDTA, 1mM AEBSF) and equally divided into two tubes. N-MAP2c beads were added to one of these tubes and nutated at 4°C for 1 hour, centrifuged, and supernatant discarded. Control tube was similarly treated, where Protein G beads were added to beads containing Fyn. 10 µg taxol-stabilized tubulin and kinase reaction buffer was added to both tubes and incubated for 5 min at 37 °C. A third tube containing only tubulin, kinase reaction buffer, and 1µg mouse non-specific IgG was used as control. Samples were subjected to western blot analysis.
Western blot analysis
Western blotting was performed as described (Lee et al., 1998). Primary antibodies used were described in Table 1. Antibody characterization was performed by confirming that WT mice yielded the reactivity as expected for the antigen on western blots. Quantification was done by densitometry using ImageJ. Protein levels from the PSD fraction were normalized to β-actin (Fig. 5) and protein levels from crude lysates were normalized to GAPDH (Fig. 7A), with values subsequently normalized to WT levels, which were represented as “1”. In Fig. 7C, pSFK was normalized to total Fyn and values from Fyn+MAP2c were normalized to Fyn alone condition with the latter represented as “1”. In Fig. 7D, 4G10 was normalized to both tubulin and Fyn and the Fyn+N-MAP2c condition was normalized to the Fyn alone condition with the latter represented as “1”.
Table 1.
Antibodies
| Antibody name | Structure of Immunogen | Manufacturer, catalog number | Species Raised in | Polyclonal/monoclonal; concentration used |
|---|---|---|---|---|
| DA9 (tau) | Purified paired helical filaments from human AD brain | Peter Davies RRID: AB_2716723 | Mouse | Monoclonal; 1:50 for PLA |
| Tau5 (tau) | Purified bovine microtubule-associated proteins | Late Lester I. Binder RRID: AB_2721194 | Mouse | Monoclonal; 1:10,000 |
| Fyn 3 | Synthetic peptide corresponding to residues 29–48 of human Fyn | Santa Cruz Biotechnology sc-16 RRID: AB_631528 | Rabbit | Polyclonal; 1:250 for PLA 1:1000 for western |
| Fyn 15 | Residues 85–206 of human Fyn | Santa Cruz Biotechnology sc-434 RRID: AB_627642 | Mouse | Monoclonal |
| MAP2 | Recombinant protein corresponding to residues 377–1505 of human MAP2 | Invitrogen/ThermoFisher #PA1–10005 RRID: AB_1076848 | Chicken | Polyclonal; 1:1000 |
| HM-2 (MAP2) | Rat brain microtubule-associated proteins | Sigma #M4403 RRID: AB_477193 | Mouse | Monoclonal; 1:250 for PLA 1:1000 for western |
| 36D10 (Src) | Synthetic peptide corresponding to residues near the carboxy terminus of human Src | Cell Signaling Technology #2109 RRID: AB_2106059 | Rabbit | Polyclonal; 1:250 for PLA |
| GD-11 (Src) | Recombinant chicken pp60src purified from extracts of E. coli containing plasmid plac-Src | Millipore #05–184 RRID: AB_2302631 | Mouse | Monoclonal; 1:1,000 |
| Phospho-Src (Tyr419) | Synthetic phospho-peptide derived from the region of human Src that contains tyrosine 419 | Invitrogen/ThermoFisher #44–660G RRID: AB_2533714 | Rabbit | Polyclonal; 1:250 for PLA |
| Phospho-Src (Tyr416) | Synthetic phospho-peptide corresponding to residues surrounding Tyr419 of human Src | Cell Signaling Technology #2101 RRID: AB_331697 | Rabbit | Polyclonal; 1:1,000 |
| YL1/2 (α tubulin) | Yeast tubulin | Accurate Chemical #YSRTMCA77P RRID: AB_2687885 | Rat | Monoclonal; 1:50 for IF 1:1000 for western |
| SP15 (Synaptophysin) | Human brain proteins immunoprecipitated with anti-synaptophysin | Millipore #MAB329-C RRID: AB_94786 | Mouse | Monoclonal; 1:1,000 |
| NR2B | Fusion protein with amino acids 20–271 of rat NR2B | One World Labs/StressMarq #SMC-333D RRID: AB_10599356 | Mouse | Monoclonal: 1:1,000 |
| Phospho-NR2B (Tyr1472) | Synthetic phospho-peptide corresponding to residues surrounding the phospho-tyr1472 of NMDA NR2B | PhosphoSolutions #p1516–1472 RRID: AB_2492182 | Rabbit | Polyclonal; 1:1,000 |
| PSD95 (EP2652Y) | Synthetic peptide corresponding to residues in the N-terminus of human PSD95 | Millipore #04–1066 RRID: AB_1977415 | Rabbit | Monoclonal, 1:1,000 |
| 4G10 (Phospho-tyrosine) | Phosphotyramine-KLH | Millipore #05–321 RRID: AB_309678 | Mouse | Monoclonal; 1:1,000 |
| GAPDH | GAPDH from rabbit muscle | Chemicon/Millipore #MAB374 RRID: AB_2107445 | Mouse | Monoclonal; 1:25,000 |
| JLA20 (Actin) | Cytoskeletal proteins from chicken gizzards | University of Iowa Developmental Studies Hybridoma Bank RRID: AB_528068 | Mouse | Monoclonal; 1:100 |
Statistical Analysis
Statistical analysis using two-way ANOVA was used for Fig. 1D and E;. The analysis was carried out with GraphPad Prism 7.0. For Fig. 3B and 3E, generalized linear mixed model with a Bonferroni correction for multiple comparisons was carried out with SAS version 9.4;. Otherwise, statistical analysis was carried out with GraphPad Prism 7.0 using ordinary one-way ANOVA with Tukey’s post hoc multiple comparison or unpaired parametric t-test;. p≤0.05 was considered as statistically significant, with p≤0.05 denoted as *, p≤0.01 denoted as **, p≤0.001 denoted as ***, and p<0.0001 denoted as ****. Degrees of freedom was abbreviated as “DF”.
Results
Generation of tau-Fyn DKO mice
In order to investigate the downstream effects of the tau-Fyn interaction, we generated tau−/−/Fyn−/−double knockout mice (DKO) by breeding tau KO (C57BL/6) mice(Dawson et al., 2001) with Fyn KO (C57BL/6-S129) mice(Stein et al., 1992) (Fig. S1). Polymerase chain reaction (Fig. 1A) was used for genotyping and western blotting (Fig. 1B) was used to confirm the loss of tau and Fyn in DKO mice. WT mice, confirmed by the presence of tau and Fyn, were also generated. Tau KO and Fyn KO mice were assayed as controls.
During breeding, DKO animals exhibited a propensity to develop non-obstructive hydrocephalus postpartum, consistent with the observation that having the homozygous Fyn KO trait on a C57BL/6 background increased the occurrence of hydrocephalus (Goto et al., 2008). While mice with severe hydrocephalus could be identified within 6 weeks of age by their domed heads and hunched backs, less severe levels of hydrocephalus escaped visual detection. Therefore, to identify mice with moderate hydrocephalus, MRI was used to image the lateral ventricle. Quantification of the images allowed us to classify mice as being normal or having moderate or severe hydrocephalus (Fig. 1C). Behavioral tests showed that both Fyn KO and DKO mice with mild and severe hydrocephalus had increased total movements in the open field test (Fig. 1D) and decreased total freezing in contextual fear conditioning (Fig. 1E) when compared to normal mice of the same genotype. While there were no interactions between genotype and hydrocephalus status in either test (Table 2A and Table 2B), the data suggested that hydrocephalus caused the animal to be hyperactive. Thus, subsequent behavioral and biochemical tests were performed using only non-hydrocephalic mice with normal brain structure that had been screened by MRI. Beginning at eight weeks of age, the four genotypes of mice underwent a series of motor and behavioral tests.
Fig. 1: Confirmed by MRI, tau−/−/Fyn−/− double knockout (DKO) mice with hydrocephalus exhibited behavioral abnormalities.

A) Polymerase chain reaction (PCR) products from heterozygous (Het), WT, and DKO mice are shown.
B) Western blot of crude brain lysate showed that DKO mice did not express tau or Fyn.
C) Examples of T2 weighted MRI of coronal Fyn KO mouse brains with normal, moderate, or severe hydrocephalus are shown. DKO mouse brains had similar structural abnormalities and are not shown.
D) In the open field test, Fyn KO and DKO mice with hydrocephalus (moderate or severe) had increased total movements relative to mice with no hydrocephalus. n=18 Fyn KO, 13 DKO, 24 Fyn KO Mod Hydro, 17 Fyn KO Severe Hydro, 19 DKO hydro.
E) In contextual fear conditioning, Fyn KO and DKO mice with moderate or severe hydrocephalus had decreased freezing relative to mice with no hydrocephalus. n=21 Fyn KO, 20 DKO, 33 Fyn KO hydro, 23 DKO hydro.
Mean ± SD are shown. Two-way ANOVA with Tukey’s post-hoc multiple comparison was used for both panels D and E. Statistical results are shown in Tables 2A and 2B.
Table 2A.
Statistical results for Two-way analysis for Fig. 1 D
| Source of Variation | % of total variation | P value |
|---|---|---|
| Interaction | 1.216 | 0.2452 |
| Hydrocephalus status | 61.71 | <0.0001 |
| Genotype | 4.5 | 0.0017 |
| ANOVA table | F (DFn, DFd) | P value | DF |
|---|---|---|---|
| Interaction | F (2, 84) = 1.429 | P=0.2452 | 2 |
| Hydrocephalus status | F (2, 84) = 72.5 | P<0.0001 | 2 |
| Genotype | F (1, 84) = 10.58 | P=0.0017 | 1 |
| Residual | 84 |
| Tukey’s multiple comparison test | Mean Diff. | 95.00% CI of diff. | Summary | Adjusted P Value |
|---|---|---|---|---|
| Normal:Fyn KO vs. Normal:DKO | −122.6 | −388.4 to 143.2 | ns | 0.759 |
| Normal:Fyn KO vs. Mod Hydro:Fyn KO | −456.2 | −688.8 to −223.6 | **** | <0.0001 |
| Normal:Fyn KO vs. Mod Hydro:DKO | −565.9 | −870.4 to −261.4 | **** | <0.0001 |
| Normal:Fyn KO vs. Severe Hydro:Fyn KO | −744.4 | −996.7 to −492.2 | **** | <0.0001 |
| Normal:Fyn KO vs. Severe Hydro:DKO | −1078 | −1395 to −761.5 | **** | <0.0001 |
| Normal:DKO vs. Mod Hydro:Fyn KO | −333.6 | −584.4 to −82.72 | ** | 0.0028 |
| Normal:DKO vs. Mod Hydro:DKO | −443.3 | −762 to −124.6 | ** | 0.0015 |
| Normal:DKO vs. Severe Hydro:Fyn KO | −621.8 | −891 to −352.6 | **** | <0.0001 |
| Normal:DKO vs. Severe Hydro:DKO | −955.8 | −1286 to −625.2 | **** | <0.0001 |
| Mod Hydro:Fyn KO vs. Mod Hydro:DKO | −109.7 | −401.3 to 181.8 | ns | 0.881 |
| Mod Hydro:Fyn KO vs. Severe Hydro:Fyn KO | −288.3 | −524.7 to −51.8 | ** | 0.0079 |
| Mod Hydro:Fyn KO vs. Severe Hydro:DKO | −622.3 | −926.8 to −317.7 | **** | <0.0001 |
| Mod Hydro:DKO vs. Severe Hydro:Fyn KO | −178.5 | −486 to 129 | ns | 0.5399 |
| Mod Hydro:DKO vs. Severe Hydro:DKO | −512.5 | −875 to −150.1 | ** | 0.0012 |
| Severe Hydro:Fyn KO vs. Severe Hydro:DKO | −334 | −653.8 to −14.17 | * | 0.0354 |
Table 2B.
Statistical results for Two-way analysis for Fig. 1 E
| Source of Variation | % of total variation | P value |
|---|---|---|
| Interaction | 0.6272 | 0.6502 |
| Hydrocephalus status | 32.13 | <0.0001 |
| Genotype | 0.06779 | 0.7605 |
| ANOVA table | F (DFn, DFd) | P value | DF |
|---|---|---|---|
| Interaction | F (2, 91) = 0.4325 | P=0.6502 | 2 |
| Hydrocephalus status | F (2, 91) = 22.15 | P<0.0001 | 2 |
| Genotype | F (1, 91) = 0.0935 | P=0.7605 | 1 |
| Residual | 91 |
| Tukey’s multiple comparison test | Mean Diff. | 95.00% CI of diff. | Summary | Adjusted P Value |
|---|---|---|---|---|
| Normal:Fyn KO vs. Normal:DKO | 16.74 | −32.89 to 66.37 | ns | 0.9225 |
| Normal:Fyn KO vs. Mod Hydro:Fyn KO | 78.99 | 28.7 to 129.3 | *** | 0.0002 |
| Normal:Fyn KO vs. Mod Hydro:DKO | 72.84 | 19.14 to 126.5 | ** | 0.0021 |
| Normal:Fyn KO vs. Severe Hydro:Fyn KO | 93.36 | 38.55 to 148.2 | **** | <0.0001 |
| Normal:Fyn KO vs. Severe Hydro:DKO | 93.5 | 27.5 to 159.5 | ** | 0.0011 |
| Normal:DKO vs. Mod Hydro:Fyn KO | 62.25 | 11.36 to 113.1 | ** | 0.0075 |
| Normal:DKO vs. Mod Hydro:DKO | 56.1 | 1.838 to 110.4 | * | 0.0384 |
| Normal:DKO vs. Severe Hydro:Fyn KO | 76.62 | 21.26 to 132 | ** | 0.0016 |
| Normal:DKO vs. Severe Hydro:DKO | 76.76 | 10.31 to 143.2 | * | 0.014 |
| Mod Hydro:Fyn KO vs. Mod Hydro:DKO | −6.154 | −61.02 to 48.71 | ns | 0.9995 |
| Mod Hydro:Fyn KO vs. Severe Hydro:Fyn KO | 14.37 | −41.58 to 70.32 | ns | 0.9753 |
| Mod Hydro:Fyn KO vs. Severe Hydro:DKO | 14.51 | −52.44 to 81.46 | ns | 0.9884 |
| Mod Hydro:DKO vs. Severe Hydro:Fyn KO | 20.52 | −38.51 to 79.55 | ns | 0.9128 |
| Mod Hydro:DKO vs. Severe Hydro:DKO | 20.66 | −48.88 to 90.2 | ns | 0.9538 |
| Severe Hydro:Fyn KO vs. Severe Hydro:DKO | 0.1387 | −70.26 to 70.54 | ns | >0.9999 |
DKO mice recapitulate specific behaviors of Fyn KO and tau KO mice
In the open field apparatus, mice of the four genotypes had similar total movements and percent movement in the center (Fig. 2A, 2B). This indicated an absence of motor deficits or anxiety phenotype at young age. However, it is known that tau KO mice exhibited motor deficits when aged to 12 months and older (Lei et al., 2014; Lopes et al., 2016a; Ma et al., 2014; Morris et al., 2013). Therefore, to determine the effect of tau/Fyn depletion on motor function, we performed the pole test on the mice at 12 months of age. We found that both tau KO and DKO mice required more time to descend the pole relative to WT and Fyn KO mice (Fig. 2C). Statistical results are shown in Table 3.
Fig. 2: Young mice from all genotypes had no motor deficits while older tau KO and DKO mice developed motor deficits.

A) 2 months old mice from the four genotypes, with no hydrocephalus, were tested in the open field apparatus. All mice had the same number of total movements, indicating no gross motor deficit. (One-way ANOVA: F(3, 61)=0.4983, p=0.6848, DF:3). n= 14 WT, 20 tau KO, 18 Fyn KO, 13 DKO. (8 wks).
B) The same cohorts of mice from A) also showed no difference in the % movement in the center, indicating no anxiety phenotype.
C) Motor function was assessed in 12 months old mice using the pole test. Tau KO and DKO mice took more time to descend the pole relative to WT and Fyn KO mice, indicating that tau was involved in the motor pathway. WT vs tau KO: p=0.0006, WT vs Fyn KO: p=0.8402, WT vs DKO: p=0.0201, tau KO vs Fyn KO: p<0.0001, tau KO vs DKO: p=0.3529, Fyn KO vs DKO: p<0.0007. n= 9 WT, 13 tau KO, 11 Fyn KO, 18 DKO (12 mo).
Mean ± SD are shown. Ordinary one-way ANOVA with Tukey’s multiple comparison was used. Statistical results are shown in Table 3.
Table 3.
Statistical results for One-way ANOVA and t-test analyses
| Figure | Test | F (DFn, DFd) | t | P value | DF |
|---|---|---|---|---|---|
| 2A | One-way ANOVA | F(3, 61)=0.4983 | 0.6848 | 3 | |
| 2B | One-way ANOVA | F(3, 61)=2.323 | 0.0839 | 3 | |
| 2C | One-way ANOVA | F(3, 47)=12.8 | <0.0001 | 3 | |
| 3A, left | One-way ANOVA | F=(3, 76) 1.182 | 0.3221 | 3 | |
| 3A, right | One-way ANOVA | F(3, 76)=19.87 | <0.0001 | 3 | |
| 3C | One-way ANOVA | F(3, 98)=12.61 | <0.0001 | 3 | |
| 3D | One-way ANOVA | F(3, 98) =2.683 | 0.0509 | 3 | |
| 3F | One-way ANOVA | F (3, 104) =28.7 | <0.0001 | 3 | |
| 4E | t-test | 6.647 | <0.0001 | 55 | |
| 4J | t-test | 6.505 | <0.0001 | 74 | |
| 4O | t-test | 7.984 | <0.0001 | 50 | |
| 5D, pY1472 | One-way ANOVA | F(3, 32) =7.079 | 0.009 | 3 | |
| 5D, pSFK | One-way ANOVA | F(3, 41)=23.71 | <0.0001 | 3 | |
| 5D, Fyn | t-test | 0.8365 | 0.416 | 15 | |
| 5D, tau | One-way ANOVA | F(3, 32)=241 | <0.0001 | 3 | |
| 5D, PSD95 | One-way ANOVA | F(3, 41)=0.5573 | 0.6462 | 3 | |
| 5D, NR2B | One-way ANOVA | F(3, 31)=1.152 | 0.3439 | 3 | |
| 5D, Src | One-way ANOVA | F(3, 48)=0.3771 | 0.7699 | 3 | |
| 6B | One-way ANOVA | F(3, 508)=1.279 | 0.2808 | 3 | |
| 6C | One-way ANOVA | F(3, 512) =41.7 | <0.0001 | 3 | |
| 7A | One-way ANOVA | F(3,27)=11.49 | <0.0001 | 3 | |
| 7C | t-test | 2.958 | 0.0254 | 6 | |
| 7D | t-test | 3.640 | 0.0108 | 6 | |
| 8G | t-test | 6.268 | <0.0001 | 166 | |
| 8H | t-test | 3.063 | 0.0033 | 58 | |
| 8I | t-test | 5.629 | <0.0001 | 56 | |
| S2A | t-test | 0.08309 | 0.9346 | 20 | |
| S3, tau | t-test | 9.2 | 0.0027 | 3 | |
| S3, MAP2c | t-test | 6.946 | 0.0061 | 3 | |
| S4 | One-way ANOVA | F(3, 30)=36.88 | <0.0001 | 3 | |
| S5 | One-way ANOVA | F(2, 52)= 3.978 | 0.0247 | 2 |
In novel object recognition, mice from the four genotypes displayed equal preferences for two identical objects on training day (Fig. 3A, left). However, on testing day, both Fyn KO and DKO mice performed significantly worse than WT and tau KO mice in interacting with the new object (Fig. 3A, right). There were no differences between WT and tau KO mice (p=0.9012), and between Fyn KO and DKO mice (p=0.1301). The absence of anxiety as monitored by the open field test suggested that Fyn KO and DKO had a cognitive deficit.
Fig. 3: In memory tasks, DKO recapitulated the behavior of Fyn KO mice whereas in PTZ induced seizures, DKO mimicked the tau KO mice.

A) In novel object recognition, there was no difference in object preference on training day (day 4, left panel) while on testing day (day 5, right panel), Fyn KO and DKO mice had significantly reduced interaction with the novel object ***p=0.0004, ****p≤ 0.0001. n= 17 WT, 17 tau KO, 28 Fyn KO, 18 DKO. (9 wks).
B) In contextual fear conditioning, using minute by minute measurements, WT and tau KO mice spent more time freezing compared to Fyn KO and DKO mice on training day at 8th minute (left). On testing day, minute by minute freezing for mice from four genotypes is also displayed (right). n= 32 WT, 29 tau KO, 21 Fyn KO, 20 DKO (10 wks).
C) Fyn KO and DKO mice spent less time freezing than WT (p=0.0002, p<0.0001, respectively) and tau KO mice (p=0.0075, p=0.0004, respectively). **p= 0.0075, ***p= 0.0002 or 0.0004, ****p≤ 0.0001.
D) Ratio of time freezing in first minute of day 2 to time freezing in last minute of day 1 show an average of 1 for all genotypes, indicating a learning rather than memory deficit for Fyn KO and DKO mice.
E) In PTZ induced seizures, tau KO and DKO mice were equally protected while Fyn KO mice were only moderately protected as measured in latency to reach each seizure stages 3, 4, and 5. n= 25 WT, 21 tau KO, 36 Fyn KO, and 26 DKO (12 wks).
F) For maximum seizure stage reached, tau KO and DKO mice were similarly protected relative to WT and Fyn KO mice; Fyn KO mice were only slightly protected relative to WT mice. **p= 0.0011, ***p= 0.0003, ****p< 0.0001. n= 25 WT, 21 tau KO, 36 Fyn KO, and 26 DKO.
Mean ± SD are shown. Ordinary one-way ANOVA with Tukey’s post-hoc multiple comparison was used for A, C, D, and F; statistical results are shown in Table 3. Generalized linear mixed model with a Bonferroni correction for multiple comparisons was done for Fig. 3B and 3E; statistical results are shown in Tables 4A, 4B, and 4C.
In contextual fear conditioning, Fyn KO and DKO mice spent significantly less time freezing as compared to WT and tau KO mice on training day in the 8th minute (Fig. 3B, left). On testing day, Fyn KO and DKO mice also had less total time freezing compared to WT and tau KO mice (Fig. 3C). There was no statistically significant difference between Fyn KO and DKO mice (p=0.8442) and between WT and tau KO mice (p=0.7076) (Fig. 3C). To assess learning versus memory, the amount of freezing during the first minute of the testing day was divided by the amount of freezing during the last minute of the training day. There was no significant difference between the ratios of the four genotypes (Fig. 3D), indicating that the mice did not differ with respect to memory and that the deficits exhibited by Fyn KO and DKO mice on day 2 were due to a learning deficit (Statistical results are shown in Table 3, Table 4A, Table 4B, and Table 4C.).
Table 4A.
Mixed model analysis for Fig. 3B Left panel with Bonferroni correction for multiple comparisons
| TIME | COMPARISON | ESTIMATE | p | SIG |
|---|---|---|---|---|
| 1 | DKO vs. FynKO | 0.6291 | 0.8474 | N |
| 1 | DKO vs. WT | 0.6401 | 0.8311 | N |
| 1 | DKO vs. tauKO | 1.9273 | 0.5262 | N |
| 1 | FynKO vs. WT | 0.0110 | 0.9970 | N |
| 1 | FynKO vs. tauKO | 1.2982 | 0.6650 | N |
| 1 | WT vs. tauKO | 1.2872 | 0.6339 | N |
| 2 | DKO vs. FynKO | 1.7170 | 0.5994 | N |
| 2 | DKO vs. WT | 1.0905 | 0.7163 | N |
| 2 | DKO vs. tauKO | 2.3444 | 0.4408 | N |
| 2 | FynKO vs. WT | −0.6265 | 0.8322 | N |
| 2 | FynKO vs. tauKO | 0.6274 | 0.8342 | N |
| 2 | WT vs. tauKO | 1.2539 | 0.6427 | N |
| 3 | DKO vs. FynKO | 3.0319 | 0.3538 | N |
| 3 | DKO vs. WT | 1.8500 | 0.5376 | N |
| 3 | DKO vs. tauKO | 2.7264 | 0.3701 | N |
| 3 | FynKO vs. WT | −1.1820 | 0.6893 | N |
| 3 | FynKO vs. tauKO | −0.3055 | 0.9188 | N |
| 3 | WT vs. tauKO | 0.8764 | 0.7457 | N |
| 4 | DKO vs. FynKO | 1.6329 | 0.6174 | N |
| 4 | DKO vs. WT | 1.2023 | 0.6887 | N |
| 4 | DKO vs. tauKO | 5.6073 | 0.0655 | N |
| 4 | FynKO vs. WT | −0.4307 | 0.8842 | N |
| 4 | FynKO vs. tauKO | 3.9743 | 0.1852 | N |
| 4 | WT vs. tauKO | 4.4050 | 0.1035 | N |
| 5 | DKO vs. FynKO | −8.7601 | 0.0075 | N |
| 5 | DKO vs. WT | −7.9998 | 0.0078 | N |
| 5 | DKO vs. tauKO | −5.4129 | 0.0754 | N |
| 5 | FynKO vs. WT | 0.7603 | 0.7971 | N |
| 5 | FynKO vs. tauKO | 3.3472 | 0.2644 | N |
| 5 | WT vs. tauKO | 2.5869 | 0.3386 | N |
| 6 | DKO vs. FynKO | −2.1675 | 0.5073 | N |
| 6 | DKO vs. WT | −12.3745 | 0.0000 | Y |
| 6 | DKO vs. tauKO | −7.2455 | 0.0174 | N |
| 6 | FynKO vs. WT | −10.2070 | 0.0006 | Y |
| 6 | FynKO vs. tauKO | −5.0780 | 0.0906 | N |
| 6 | WT vs. tauKO | 5.1291 | 0.0580 | N |
| 7 | DKO vs. FynKO | −4.2064 | 0.1984 | N |
| 7 | DKO vs. WT | −16.6781 | <0.0001 | Y |
| 7 | DKO vs. tauKO | −11.1661 | 0.0003 | Y |
| 7 | FynKO vs. WT | −12.4716 | <0.0001 | Y |
| 7 | FynKO vs. tauKO | −6.9597 | 0.0205 | N |
| 7 | WT vs. tauKO | 5.5120 | 0.0417 | N |
| 8 | DKO vs. FynKO | −1.6272 | 0.6186 | N |
| 8 | DKO vs. WT | −13.9440 | <0.0001 | Y |
| 8 | DKO vs. tauKO | −11.9485 | 0.0001 | Y |
| 8 | FynKO vs. WT | −12.3168 | <0.0001 | Y |
| 8 | FynKO vs. tauKO | −10.3214 | 0.0006 | Y |
| 8 | WT vs. tauKO | 1.9954 | 0.4604 | N |
Bonferroni alpha: 0.0010
Table 4B.
Mixed model analysis for Fig. 3B Right panel with Bonferroni correction for multiple comparisons
| TIME | COMPARISON | ESTIMATE | p | SIG |
|---|---|---|---|---|
| 1 | DKO vs. FynKO | 2.2744 | 0.6234 | N |
| 1 | DKO vs. WT | −7.1572 | 0.0907 | N |
| 1 | DKO vs. tauKO | −4.6809 | 0.2775 | N |
| 1 | FynKO vs. WT | −9.4316 | 0.0238 | N |
| 1 | FynKO vs. tauKO | −6.9553 | 0.1019 | N |
| 1 | WT vs. tauKO | 2.4763 | 0.5147 | N |
| 2 | DKO vs. FynKO | 6.0068 | 0.1950 | N |
| 2 | DKO vs. WT | −10.7224 | 0.0114 | N |
| 2 | DKO vs. tauKO | −7.7736 | 0.0716 | N |
| 2 | FynKO vs. WT | −16.7292 | 0.0001 | Y |
| 2 | FynKO vs. tauKO | −13.7804 | 0.0012 | Y |
| 2 | WT vs. tauKO | 2.9488 | 0.4379 | N |
| 3 | DKO vs. FynKO | −1.7001 | 0.7136 | N |
| 3 | DKO vs. WT | −15.0940 | 0.0004 | Y |
| 3 | DKO vs. tauKO | −12.8988 | 0.0029 | N |
| 3 | FynKO vs. WT | −13.3939 | 0.0014 | Y |
| 3 | FynKO vs. tauKO | −11.1987 | 0.0086 | N |
| 3 | WT vs. tauKO | 2.1952 | 0.5636 | N |
| 4 | DKO vs. FynKO | 1.3731 | 0.7669 | N |
| 4 | DKO vs. WT | −14.5618 | 0.0006 | Y |
| 4 | DKO vs. tauKO | −8.5779 | 0.0469 | N |
| 4 | FynKO vs. WT | −15.9349 | 0.0001 | Y |
| 4 | FynKO vs. tauKO | −9.9510 | 0.0195 | N |
| 4 | WT vs. tauKO | 5.9839 | 0.1158 | N |
| 5 | DKO vs. FynKO | 2.8370 | 0.5402 | N |
| 5 | DKO vs. WT | −15.3582 | 0.0003 | Y |
| 5 | DKO vs. tauKO | −7.8090 | 0.0704 | N |
| 5 | FynKO vs. WT | −18.1952 | <0.0001 | Y |
| 5 | FynKO vs. tauKO | −10.6460 | 0.0125 | N |
| 5 | WT vs. tauKO | 7.5492 | 0.0474 | N |
| 6 | DKO vs. FynKO | 2.2504 | 0.6270 | N |
| 6 | DKO vs. WT | −11.8780 | 0.0051 | N |
| 6 | DKO vs. tauKO | −7.8613 | 0.0685 | N |
| 6 | FynKO vs. WT | −14.1284 | 0.0007 | Y |
| 6 | FynKO vs. tauKO | −10.1118 | 0.0176 | N |
| 6 | WT vs. tauKO | 4.0166 | 0.2908 | N |
Bonferroni alpha: 0.0014
Table 4C.
Mixed model analysis for Fig. 3E with Bonferroni correction for multiple comparisons
| STAGE | COMPARISON | ESTIMATE | p | SIG |
|---|---|---|---|---|
| 0 | DKO vs. FynKO | 0.0143 | 0.9998 | N |
| 0 | DKO vs. WT | 0.0141 | 0.9999 | N |
| 0 | DKO vs. tauKO | 0.0994 | 0.9990 | N |
| 0 | FynKO vs. WT | −0.0003 | 1.0000 | N |
| 0 | FynKO vs. tauKO | 0.0851 | 0.9991 | N |
| 0 | WT vs. tauKO | 0.0853 | 0.9992 | N |
| 1 | DKO vs. FynKO | −3.2785 | 0.9635 | N |
| 1 | DKO vs. WT | 29.4387 | 0.7045 | N |
| 1 | DKO vs. tauKO | −34.5160 | 0.6712 | N |
| 1 | FynKO vs. WT | 32.7172 | 0.6517 | N |
| 1 | FynKO vs. tauKO | −31.2374 | 0.6826 | N |
| 1 | WT vs. tauKO | −63.9547 | 0.4357 | N |
| 2 | DKO vs. FynKO | 165.3434 | 0.0207 | N |
| 2 | DKO vs. WT | 238.8587 | 0.0022 | N |
| 2 | DKO vs. tauKO | −227.2064 | 0.0053 | N |
| 2 | FynKO vs. WT | 73.5153 | 0.3084 | N |
| 2 | FynKO vs. tauKO | −392.5498 | <0.0001 | Y |
| 2 | WT vs. tauKO | −466.0651 | <0.0001 | Y |
| 3 | DKO vs. FynKO | 359.3370 | <0.0001 | Y |
| 3 | DKO vs. WT | 655.9156 | <0.0001 | Y |
| 3 | DKO vs. tauKO | 35.3229 | 0.6640 | N |
| 3 | FynKO vs. WT | 296.5786 | <0.0001 | Y |
| 3 | FynKO vs. tauKO | −324.0141 | <0.0001 | Y |
| 3 | WT vs. tauKO | −620.5928 | <0.0001 | Y |
| 4 | DKO vs. FynKO | 321.9984 | <0.0001 | Y |
| 4 | DKO vs. WT | 695.7664 | <0.0001 | Y |
| 4 | DKO vs. tauKO | −33.8987 | 0.6768 | N |
| 4 | FynKO vs. WT | 373.7679 | <0.0001 | Y |
| 4 | FynKO vs. tauKO | −355.8972 | <0.0001 | Y |
| 4 | WT vs. tauKO | −729.6651 | <0.0001 | Y |
| 5 | DKO vs. FynKO | 325.3455 | <0.0001 | Y |
| 5 | DKO vs. WT | 649.5228 | <0.0001 | Y |
| 5 | DKO vs. tauKO | 60.3394 | 0.4581 | N |
| 5 | FynKO vs. WT | 324.1773 | <0.0001 | Y |
| 5 | FynKO vs. tauKO | −265.0062 | 0.0005 | Y |
| 5 | WT vs. tauKO | −589.1835 | <0.0001 | Y |
| 6 | DKO vs. FynKO | 270.9310 | 0.0002 | Y |
| 6 | DKO vs. WT | 552.2541 | <0.0001 | Y |
| 6 | DKO vs. tauKO | 39.9090 | 0.6236 | N |
| 6 | FynKO vs. WT | 281.3231 | 0.0001 | Y |
| 6 | FynKO vs. tauKO | −231.0221 | 0.0025 | N |
| 6 | WT vs. tauKO | −512.3451 | <0.0001 | Y |
| 7 | DKO vs. FynKO | 117.4032 | 0.1001 | N |
| 7 | DKO vs. WT | 425.3341 | <0.0001 | Y |
| 7 | DKO vs. tauKO | 1.3589 | 0.9867 | N |
| 7 | FynKO vs. WT | 307.9309 | <0.0001 | Y |
| 7 | FynKO vs. tauKO | −116.0443 | 0.1276 | N |
| 7 | WT vs. tauKO | −423.9752 | <0.0001 | Y |
Bonferroni alpha: 0.0010
Previous reports have also shown that tau ablation protected against pentylenetetrazole (PTZ) induced seizures (Roberson et al., 2007). This property has been attributed to tau depletion reducing levels of Fyn at the post-synaptic region leading to disrupted NR stability within the post-synaptic density(Ittner et al., 2010). In terms of both latency to develop seizures, DKO and tau KO mice were equally and dramatically protected against PTZ whereas Fyn KO mice were only moderately protected relative to WT mice as measured by latency to reach each seizure stages 3, 4, and 5 (Fig. 3E). In terms of maximum seizure stage reached, tau KO and DKO were also significantly protected relative to WT mice whereas Fyn KO mice were only moderately protective relative to WT (p<0.0001, p<0.0001, p=0.0011 respectively, Fig. 3F). There was no significant difference between tau KO and DKO mice (p=0.9752; Fig. 3F). To summarize, in motor tasks and PTZ induced seizures, DKO mice resembled tau KO mice while in cognitive and memory tasks, DKO mice resembled Fyn KO mice.
Localization of Tau- SFK complexes in WT primary hippocampal culture
To further investigate the function of the tau/Fyn complex, the location of such complexes needs to be elucidated. Therefore, we have used proximity ligation assays (PLA) to localize tau-Fyn complexes in WT primary hippocampal cultures (Fig. 4A). PLAs are able to detect endogenous protein-protein interactions in situ; signals are recovered when the two PLA probes lie within 40 nm of each other, using rolling circle amplification to yield a marker for the interaction (Gullberg et al., 2004) (Soderberg et al., 2006). As a control, tau KO cultures were similarly probed. Tau-Fyn complexes were found in all cellular compartments of WT hippocampal neurons (axons (Fig. 4B), dendrites (Fig. 4C), and cell bodies (Fig. 4D)). For quantitation, PLA dots were counted in both dendrites and axons. Dot intensity was not used because the intensity of a PLA signal reflected the rolling circle step, making the intensity level an artificial characteristic. Upon quantitation, the density of tau-Fyn complexes in dendrites (number of PLA puncta/process length) was 2.54 fold higher than that in axons (Fig. 4E). As expected, there were no tau-Fyn complexes in tau KO neurons (Fig. S2B). Since we have previously shown that tau increased Fyn auto-phosphorylation (Sharma et al., 2007), complexes between tau and activated SFK (pSFK) were also examined (Fig. 4F–J). Similar to the tau-Fyn complexes, the density of tau-pSFK complexes in dendrites was 2.04 fold higher than that in axons (Fig. 4J). However, since the pSFK antibody cannot distinguish between activated Fyn and activated Src, tau-Src complexes were also examined as a control. The density of tau-Src complexes in dendrites was increased by 2.78 fold relative to that in the axon (Fig. 4O), indicating that tau-Src complexes may also contribute to the tau-pSFK signal in both dendrites and axons. The presence of tau-pSFK in axons did not agree with the observation that pSFKs were found only in the somatodendritic compartment (Li and Gotz, 2017). However, the PLA assay is capable of eliciting signal from low levels of antigen, as amplification is used. Therefore, a low level of pSFK must exist in the axon.
Fig. 4: Tau-Fyn, tau-pSFK, and tau-Src complexes are present in neurons.

A,F,K) Using WT hippocampal neurons, tau-Fyn (A), tau-pSFK (F), and tau-Src (K) complexes were identified using proximity ligation assay as described in Materials and Methods. Scale bar: 25µm.
B-D, G-I, L-N) Tau-Fyn, tau-pSFK, and tau-Src complexes were found in axons (B, G, L), dendrites (C, H, M), and cell bodies (D, I, N), respectively. Scale bar: 10µm (B, C, G, H, L, M); 20 µm (D, I, N).
E, J, O) PLA complex density in WT neurons was quantitated as described in Materials and Methods.
At least 3 experiments were analyzed. ****p<0.0001. Mean ± SD values are shown. Statistical results are shown in Table 3.
Since tau is known to be enriched in axons, we found it surprising that the density of tau-SFK complexes was higher in the dendrites relative to axons. However, we noted that axons were significantly longer and more numerous than dendrites and that the density of PLA puncta was relatively high in the proximal axon but dramatically decreased in the distal axon whereas the density of PLA puncta in the shorter dendrites was uniformly high. In fact, if only the proximal axon was used to calculate density, the density of tau-Fyn complexes in axons would match that of the dendrites (Fig. S2A). Therefore, when the total axon length was used, the calculated density in axons was lower than that in dendrites.
Fyn KO and DKO hippocampal insoluble PSD fractions have decreased phospho-SFK and phospho-Y1472 NR2B levels
Because the density of tau-Fyn complexes in dendrites exceeded that in axons and because tau has been shown to target Fyn to the post-synaptic density to affect NR2B phosphorylation (Ittner et al., 2010), we wanted to determine if NR2B phosphorylation was altered in the DKO mice. Crude hippocampal synaptosomes were fractionated into “soluble non-PSD” and “insoluble PSD” fractions using Triton X-100 (Fig. 5A) (Ittner et al., 2010; Lopes et al., 2016b; Milnerwood et al., 2010). The presence of synaptophysin in the “soluble non-PSD” and the presence of PSD-95 in the “insoluble PSD” fractions (Fig. 5B) demonstrated the separation of PSD and non-PSD membranes from crude WT synaptosomes; tau and Fyn were present in both fractions (Fig. 5B). In examining the “insoluble PSD” fraction from 9–12 month old hippocampus, PSD-95 and total NR2B levels were not significantly different between the four genotypes (One-way ANOVA p=0.6462, p=0.3439, respectively; Fig. 5C, 5D). However, relative to WT, pY1472-NR2B levels were decreased by 54.6 % in Fyn KO and 64.0 % in DKO insoluble PSD fractions, with Fyn KO and DKO not being significantly different from each other (p=0.8871; Fig. 5C, 5D). WT and tau KO were also not significantly different from each other (p=0.8284; Fig. 5C, 5D). Lastly, tau level was decreased by 13.4% in Fyn KO PSD fraction, relative to WT (p<0.0414, Fig. 5D). While this finding was consistent with the reduction of total tau found in Fyn KO primary cultured cortical neurons (Li and Gotz, 2017), the extent of tau reduction was less in the PSD fraction of mouse hippocampal lysates. The difference may be attributed to the different cellular fractions examined, with Li et al examining total tau expression from primary cortical neurons (Li and Gotz, 2017) while we examined tau expression from the PSD 95 fraction of mouse hippocampal lysate. Once tau was associated with the PSD95 protein complex, its expression may possibly have been less affected by Fyn depletion.
We then examined whether Src might be compensating for Fyn. In the PSD fraction, although the Src level was unchanged in the four genotypes (p=0.7699; Fig. 5C, 5D), the activated SFK (pSFK) level in Fyn KO and DKO was decreased by 73.2% and 84.0% with respect to that of WT. There was no significant difference between tau KO and WT (p=0.9946) and between Fyn KO and DKO (p=0.8512; Fig. 5C, 5D). While the pSFK antibody cannot distinguish between activated Fyn and activated Src, all four genotypes had similar Src levels in the PSD but only Fyn KO and DKO had decreased pSFK levels. Since the absence of Fyn did not alter Src levels but reduced pSFK levels, we concluded that most of the activated tyrosine kinase in WT and tau KO PSD fractions was Fyn that was responsible for phosphorylating Y1472-NR2B.
Fig. 5: Fyn KO and DKO hippocampal PSD fraction had decreased phospho-SFK and phospho-Y1472 NR2B levels.

A) Schematic for separating insoluble PSD and soluble non-PSD fractions from crude synaptosomes.
B) Soluble non-PSD fraction isolated from crude hippocampal synaptosomes of WT mice showed synaptophysin, a pre-synaptic marker, while insoluble PSD fraction showed PSD95, a post-synaptic marker. Fyn and tau were found in both fractions.
C) Western blotting of hippocampal insoluble PSD fractions prepared from WT, tau KO, Fyn KO, and DKO.
D) Blots were quantitated by densitometry and normalized. At least 3 replicate experiments were performed, examining a total of at least 8 mice per genotype. For pY1472-NR2B, relative to WT, levels were decreased in Fyn KO and in DKO (*p=0.045, *p=0.0381, respectively). Relative to tau KO, levels were also significantly decreased in Fyn KO and in DKO (**p=0.0058, **p=0.0048, respectively). For pSFK, relative to WT, levels were decreased in Fyn KO and in DKO (****p<0.0001, ****p<0.0001, respectively); relative to tau KO, levels were also decreased in Fyn KO and in DKO (****p<0.0001, ****p<0.0001, respectively). For Tau: WT vs Fyn KO * p=0.0414.
Unpaired two-tailed parametric t-test was used for Fyn and ordinary one-way ANOVA with Tukey’s post-hoc multiple comparison was used for others. Statistical results are shown in Table 3.
Decreased glutamate-induced Ca2+ response in Fyn KO and DKO primary hippocampal neurons
Since phosphorylation of NR2B has been shown to be involved in facilitating Ca2+ response upon NR activation (Nakazawa et al., 2001; Rong et al., 2001; Tezuka et al., 1999), we were interested in determining the glutamate-induced Ca2+ influx of DKO neurons. In addition, depleting tau with shRNA in WT neurons had already been shown to affect glutamate-induced, NMDA receptor-dependent Ca2+ influx (Miyamoto et al., 2017), so we were also interested in the response of tau KO neurons. Sample tracings of [Ca2+]i from five individual cells of each genotype are shown in Fig. 6A. At baseline, unstimulated intracellular [Ca2+]i from the four genotypes were similar (p=0.2808; Fig. 6B). Upon stimulation, Fyn KO neurons had a 29.9% reduction and DKO neurons had a 53.1% reduction relative to WT neurons; tau KO neurons were not significantly different from WT neurons (p=0.7264). However, Ca2+ response in DKO neurons was significantly different from Fyn KO neurons (Fig. 6C).
Fig. 6: Glutamate induced Ca2+ response was impaired in Fyn KO and DKO hippocampal neurons.

A) Ca2+ response traces from five representative neurons from each genotype are shown; traces were obtained in response to glutamate and glycine as described in Materials and Methods. n=3 experiments.
B) No difference in baseline intracellular [Ca2+]i was found among the hippocampal neurons from the different genotypes.
C) Upon stimulation, Fyn KO neurons had a 29.9% reduction (****p<0.0001) and DKO neurons had a 53.1% reduction (****p<0.0001) in Ca2+ response relative to WT neurons. Compared to tau KO neurons, calcium influx for Fyn KO and DKO neurons were also significantly reduced (***p=0.004, ****p<0.0001 respectively). The reduction of the DKO neurons was significantly different from that of Fyn KO neurons (***p=0.0001).
Ordinary one-way ANOVA with Tukey’s post-hoc multiple comparison was used. Statistical results are shown in Table 3.
The fact that DKO neurons had an even greater reduction relative to Fyn KO neurons suggests that tau had a Fyn-independent role in regulating Ca2+ response that was unmasked in the absence of Fyn. In addition, the absence of any changes in the Ca2+ response of tau KO neurons suggested that another protein could compensate for tau in the tau KO. However, this compensatory effect was apparently lost in the DKO, leading to an exacerbated decrease in Ca2+ influx.
Microtubule-associated protein 2 (MAP2) was altered in KOs and like tau, could increase Fyn activity
Our finding that tau KO had no memory deficits, no changes in pY1472-NR2B, and no changes in glutamate-induced Ca2+ response suggested that either Fyn did not require tau to mediate these functions or that a different protein was compensating for the loss of tau with respect to its Fyn interaction. Interestingly, another microtubule-associated protein, MAP2, has been found to be upregulated in tau KO mice (Ma et al., 2014) and in agreement with the previous data, we also found MAP2 to be increased by 48.9% in the tau KO mice (p=0.021, Fig. 7A). However, while it has been shown that MAP2 compensated for the loss of tau by maintaining microtubule stability, as measured by tubulin acetylation (Ma et al., 2014), the possibility of other compensating mechanisms remained unexplored.
Fig. 7: Microtubule-associated protein 2 (MAP2) is increased in tau KO and its association with Fyn increases Fyn activity.

A) Crude mouse brain lysates from four genotypes were probed for MAP2, with GAPDH as control. Relative to WT mice, MAP2 was increased in tau KO mice (*p=0.021), decreased in Fyn KO mice (*p=0.044) as well as in DKO mice (**p=0.0045). Relative to tau KO mice, MAP2 was decreased in Fyn KO mice (**p=0.0021) and DKO mice (***p=0.0006). 7 WT, 8 tau KO, 8 Fyn KO, and 8 DKO mice were used.
B) Lysates from 3T3 cells confirmed transfection with Fyn alone or Fyn plus MAP2c.
C) Fyn IP from cells co-transfected with Fyn plus MAP2c showed increased pSFK levels relative to Fyn IP from cells transfected with Fyn alone. n=4 independent experiments were conducted. (p=0.0254)
D) Tubulin was incubated either alone, with Fyn, or with Fyn plus N-MAP2c, then probed with anti-phospho-tyrosine (4G10). n=4 independent experiments. (p=0.0108)
Mean ± SD values are shown. One-way ANOVA or unpaired two-tailed parametric t-test was used. Statistical results are shown in Table 3.
Similar to tau, MAP2 has a PXXP domain that interacts with the SH3 domain of Fyn (Sontag et al., 2012; Zamora-Leon et al., 2001). While we have shown that for tau, this interaction potentiated Fyn and Src activities (Sharma et al., 2007), the ability of MAP2 to exhibit a similar property has not been demonstrated. To investigate, MAP2c, the shortest isoform in the MAP2 family was tested for its ability to increase auto-phosphorylation of Fyn and to increase the enzymatic activity of Fyn. To examine auto-phosphorylation of Fyn, as detected by antibodies against activated SFK, 3T3 cells were transfected with Fyn alone or with MAP2c (Fig. 7B). After Fyn was immunoprecipitated (IPed) and probed for activated SFK, we found that the phosphorylation of Fyn expressed with MAP2c was increased by 61.9% relative to Fyn expressed alone (Fig. 7C). This indicated that the presence of MAP2c increased the auto-phosphorylation of Fyn. To investigate the ability of MAP2c to increase the enzymatic activity of Fyn, in vitro kinase assays were conducted using tubulin, a known substrate for Fyn (Marie-Cardine et al., 1995). Because MAP2c can bind tubulin, an N-terminal fragment of MAP2c (N-MAP2c) containing the Fyn binding motif but lacking the microtubule binding domain was used. Fyn and N-MAP2c were separately expressed in 3T3 cells and IP’ed for use in the kinase reaction. Tubulin was incubated either alone, with Fyn, or with pre-incubated Fyn and N-MAP2c. The presence of N-MAP2c increased tyrosine phosphorylation of tubulin by 59.4% relative to tubulin incubated with Fyn alone (p=0.0108; Fig. 7D). Our data provide evidence that similar to tau, MAP2 can increase Fyn activity. As an additional indication that MAP2 resembled tau with regards to potentiating SFK activity, we examined the ability of MAP2c to sustain SFK activation in stimulated 3T3 cells. In this system, SFK activation was indirectly assessed and we had previously shown that tau was able to prolong SFK activation following stimulation (Sharma et al., 2007). When MAP2 was compared to tau in this assay, we found that MAP2 resembled tau, prolonging SFK activation in cells (Fig. S3).
Interestingly, when MAP2 levels were assessed in Fyn KO and DKO mice, MAP2 was decreased by 23.6% and 33.2 %, respectively, relative to WT mice (p=0.044 and p=0.0045; Fig. 7A), with no significant difference between the two (p=0.438; Fig. 7A). The labeling of MAP2 in 12-month-old hippocampal sections from the four genotypes confirmed the biochemical result (Fig. S4). Compared to WT, hippocampal sections of Fyn KO and DKO mice appeared to display shorter MAP2 fibers and weaker MAP2 intensity while tau KO mice appeared to show stronger MAP2 intensity (Fig. S4). Therefore, in regulating MAP2 expression levels, Fyn appeared to act downstream of tau, since the DKO resembled the Fyn KO. In the DKO, the decrease in MAP2 most likely eliminated the ability of MAP2 to compensate for the loss of tau. Thus, when DKO neurons experienced a further decrease in calcium influx, relative to the Fyn KO, we were able to identify a Fyn-independent effect that tau had on glutamate-induced calcium influx. In the tau KO, it was likely that this effect was not detected because of the increased MAP2 level.
Tau KO neurons had more dendritic MAP2-Fyn complexes than WT neurons
To determine whether MAP2-Fyn complexes might compensate for the loss of tau-Fyn complexes in the tau KO, MAP2-Fyn and MAP2-pSFK complexes were visualized in WT and tau KO primary hippocampal cultures using PLA (Fig. 8). Relative to WT dendrites, tau KO dendrites had a 32.5% increase in MAP2-Fyn density (Fig. 8A, 8B, 8G) and a 39.9% increase in MAP2-pSFK density (Fig. 8C, 8D, 8H). As expected, we did not find any MAP2-containing complexes in axons, identified as MAP2 negative and tubulin positive processes (Fig. 8, open arrowheads). In tau KO neurons, MAP2-Src complex density was increased 36.8% relative to WT neurons (Fig. 8E, 8F, 8I). Together, these data support the notion that MAP2 compensates for tau in terms of interacting with SFKs in dendrites.
Fig. 8: Tau KO neurons have more MAP2-Fyn, MAP2-pSFK, and MAP2-Src complexes relative to WT neurons.
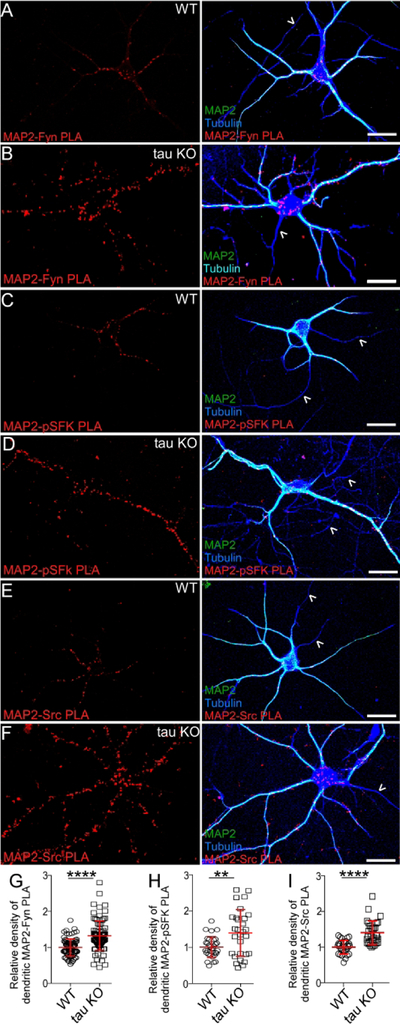
MAP2-Fyn (A, B), MAP2-pSFK (C, D), and MAP2-Src complexes (E, F) were detected in WT neurons (A, C, E) or tau KO neurons (B, D, F) using proximity ligation assay. No MAP2-SFK complexes were visualized in axons (open arrow heads). Scale bar: 25µm. MAP2-Fyn (G), MAP2-pSFK (H), and MAP2-Src (I) complex densities were quantitated in both WT and tau KO neurons. At least 3 experiments were analyzed. Mean ± SD values are shown. ****p<0.0001, **p=0.0033. Statistical results are shown in Table 3.
Discussion
In the present study, we generated tau−/−/Fyn−/− DKO mice to investigate the combined effect of tau and Fyn and found that they resembled tau KO mice in motor tasks and protection from PTZ while resembling Fyn KO mice in cognitive tasks. DKO and Fyn KO mice also had decreased Y1472-NR2B phosphorylation levels in hippocampal insoluble PSD fractions. These changes were accompanied by a reduction in glutamate-induced Ca2+ response in Fyn KO and DKO primary hippocampal neurons, with DKO neurons having a more severe reduction than Fyn KO neurons, indicating that tau may have a Fyn-independent effect on regulating Ca2+ influx. Together with our findings that MAP2 potentiated Fyn activity and that dendritic MAP2-Fyn complexes were increased in tau KO neurons relative to WT neurons, our data suggest that MAP2 may compensate for the loss of tau by increasing the levels of MAP2-Fyn and MAP2-pSFK complexes in dendrites.
In motor tasks, our aged tau KO and DKO mice had deficits in the pole test, which was consistent with results obtained by other laboratories (Ikegami et al., 2000; Lei et al., 2014; Lopes et al., 2016a; Morris et al., 2013). Since aged Fyn KO mice did not exhibit such deficits, we concluded that tau, not Fyn, was involved in the motor pathway. Another known property for tau KO mice is their protection from PTZ-induced seizures and Aβ induced excitotoxicity (Roberson et al., 2007), traits attributed to reduced Fyn levels at the post-synaptic region and disrupted NR stability within the post-synaptic density(Ittner et al., 2010). However, in PTZ-induced seizures, our finding that tau KO mice had greater protection than Fyn KO mice suggested that tau may participate in Fyn-independent pathways to regulate seizure susceptibility. If tau acted only with Fyn, Fyn KO mice would have shown protection against PTZ equal to that of the tau KO. The fact that in response to PTZ, DKO and tau KO mice had similar seizure thresholds argued that Fyn’s protective effect against seizure was encompassed by tau’s protective effect and did not represent a separate pathway since the DKO mice did not exhibit an additional level of protection relative to tau KO mice. Besides interacting with Fyn and affecting NR2B-PSD95 association, tau has been reported to affect two other pathways that regulate seizure susceptibility. One pathway involves SynGAP1, a site-specific inhibitor of Ras, where it has been found that reducing SynGAP1 in tau KO mice increased susceptibility to PTZ-induced seizure (Bi et al., 2017). A second pathway has been reported where the loss of tau prevented the depletion of Kv4.2, a dendritic potassium channel, and mitigated hyperexcitability in an AD mouse model (Hall et al., 2015). These data indicate that tau is involved in other mechanisms that modulate seizure susceptibility and excitotoxicity in dendrites. We speculate that one or both of these mechanisms will be Fyn-independent.
Our data also provide evidence that Fyn plays a role in regulating PTZ seizure severity, as our non-hydrocephalic Fyn KO mice had decreased PTZ-induced seizure susceptibility. Our results disagreed with previous reports that Fyn KO mice had either increased seizure susceptibility (Miyakawa et al., 1996) or similar susceptibility (Kojima et al., 1998) relative to WT mice. However, these previous studies likely included mice with undetected mild/moderate hydrocephalus. By using MRI to identify Fyn KO mice with mild/moderate hydrocephalus, we had found that mice with hydrocephalus had increased seizure susceptibility compared to WT (Fig. S5). In addition, when a cohort of superficially normal Fyn KO mice, with and without hydrocephalus, was analyzed as a group, the seizure threshold resembled that of WT (Fig. S5). Therefore, because prior studies were not aware that the Fyn KO mice might have had mild/moderate hydrocephalus (Kojima et al., 1998; Miyakawa et al., 1996), their data did not reflect the sole contribution of the Fyn KO trait. Our data suggest that the Fyn KO trait conferred mild protection against seizures.
In agreement with prior studies where mice lacking Fyn, but not other members of Src family non-receptor tyrosine kinases, had deficits in LTP and performance in the Morris water maze (Grant et al., 1992), we found Fyn, not tau, to be critically involved in cognitive tasks (Dawson et al., 2010; Dawson et al., 2001; Ittner et al., 2010; Kimura et al., 2014; Lei et al., 2012; Li et al., 2014; Morris et al., 2013; Roberson et al., 2007; van Hummel et al., 2016). In contextual fear conditioning, both Fyn KO and DKO mice already showed deficits on training day, where the mice differed from WT at the 5th (DKO) or 6th (Fyn KO) minute (Fig. 3B, left). Thus, it is possible that Fyn depletion could have changed the way animals perceived pain, leading to a decrease in the saliency of the shock and weakening the association between shock and the chamber, with learning and memory being normal. Another possibility is that the expression of freezing behavior itself was impaired. Future studies using cue induced freezing or predator odor induced freezing would determine whether Fyn and/or tau depletion affected freezing behavior. In Fyn KO and DKO mice, the cognitive deficits were accompanied by decreases in Y1472-NR2B phosphorylation levels in the hippocampal insoluble PSD fractions while WT and tau KO mice had no memory deficits and no decreases in pY1472-NR2B. The similarity in the levels of pY1472-NR2B in our WT and tau KO mice agree with other findings using the same tau KO line used here (Lopes et al., 2016b). However, another report using a different tau KO line found reduced levels of pY1472-NR2B (Ittner et al., 2010). The fact that different lines of tau KO mice exhibited different phenotypes underlines the complexity of the analysis of tau KO mice (Ke et al., 2012; Lei et al., 2014).
In the brain, tyrosine phosphorylation of NR2B is important in regulating receptor trafficking, gating kinetics, and Ca2+ permeability (reviewed by (Groveman et al., 2012; Kalia et al., 2004; Trepanier et al., 2012)). Since Fyn KO and DKO mice had reduced levels of pY1472-NR2B, we examined calcium influx using hippocampal neurons of each genotype. As expected, Fyn KO neurons had reduced glutamate-induced Ca2+ response relative to WT neurons. Interestingly, DKO neurons had an even greater reduction relative to Fyn KO neurons while tau KO neurons had a response similar to WT neurons, suggesting that tau had a Fyn-independent role in regulating Ca2+ response that was unmasked in the absence of Fyn. Possible mechanisms include interactions of tau with other Ca2+ permeable channels such as voltage gated Ca2+ channels, regulators of NMDARs, or other components of the PSD, with tau modulating signaling pathways that increase intracellular Ca2+ levels. Additional studies are needed to explore these nonexclusive possibilities. Our Ca2+ response data from the tau KO neurons agree with a report showing that brain slices from tau KO mice had no change in glutamate-induced Ca2+ response profiles (Bi et al., 2017) and is consistent with the overall picture of the tau KO having no changes in excitatory post-synaptic currents (EPSCs), AMPA/NMDA current ratios, and LTP (Ittner et al., 2010; Kimura et al., 2014; Roberson et al., 2011). However, when tau was acutely depleted by shRNA in WT neurons, a decrease in Ca2+ response after glutamate stimulation was revealed (Miyamoto et al., 2017). This suggested that extended tau depletion, achieved through genetic ablation in a tau KO mouse, may be fundamentally different from acute tau depletion achieved through the use of tau shRNA. Indeed, although no permanent deficits in axonal development were reported in neurons from tau KO mice (Dawson et al., 2001; Harada et al., 1994), WT neurons treated with tau antisense oligonucleotides or shRNA exhibited dramatic deficits in axon formation defective migration properties, abnormalities in mitochondria transport, and developmental deficits (Caceres and Kosik, 1990); (Sapir et al., 2012). These findings indicate that comparing neurons from a knockout animal to those created by treating WT neurons with shRNA may be less straightforward than expected. Successful creation of a KO animal may be required to select genetic alterations that allow pups to be born and animals to be bred. Moreover, the extent of compensation in the tau KO mouse has been analyzed by microarray analysis and it was found that in the brain, several signal transduction genes such as fosB and c-fos were up-regulated 5–24 fold (Oyama et al., 2004). Based on these findings, we demonstrated that tau was required for NGF-induced AP-1 activation (Leugers and Lee, 2010). The fact that the loss of tau required up-regulation of both fosB and c-fos for compensation suggested that AP-1 activation was a critical tau function required by a mouse to survive.
Our finding that tau KO had no memory deficits, no changes in pY1472-NR2B, and no changes in glutamate induced Ca2+ response suggested that either Fyn did not require tau to mediate these functions or that another protein was compensating for the loss of tau with respect to its Fyn interaction. Interestingly, other microtubule-associated proteins (MAPs), MAP1A/1B and MAP2, are upregulated in tau KO mice (Harada et al., 1994; Ma et al., 2014) and in agreement with previous data reported for our tau KO mouse line (Ma et al., 2014), we found MAP2 to be increased in our tau KO mice (Fig. 7A). In contrast, MAP2 was significantly decreased in Fyn KO mice, correlating with the reduction and shortening of MAP2 positive dendrites described previously (Kojima et al., 1997). In the DKO mice, MAP2 was decreased, indicating that the Fyn effect superseded the tau effect on MAP2 levels. The mechanisms by which Fyn depletion down-regulated or tau depletion up-regulated MAP2 are not known. However, since Fyn has been shown to affect protein translation (White et al., 2008) and tau, fos activation (Leugers and Lee, 2010), the effects of Fyn may be downstream of those of tau. Therefore, in the DKO, the Fyn KO trait resulted in the loss of MAP2-mediated compensation for the loss of tau whereas in the tau KO, up-regulated MAP2 levels would be capable of compensating for the loss of tau. In examining the glutamate induced Ca2+ response, the response in tau KO resembled those in WT due to compensation by MAP2. In contrast, because of the depletion of Fyn, DKO neurons had decreased MAP2 expression and therefore lacked both tau and MAP2. Given that DKO neurons had a larger decrease in glutamate induced Ca2+ response compared to Fyn KO neurons, which only had depleted MAP2, tau depletion must be responsible for the additional decrease in the calcium response of DKO neurons. In addition, since the decrease was revealed in the absence of Fyn, we propose that tau can affect calcium in a Fyn-independent manner and that MAP2 might also perform this function (Fig. 6C).
In the tau KO mice, if the tau-Fyn interaction was of critical importance, one would predict that compensation for the loss of the tau-Fyn interaction would occur. Our finding that tau KO neurons had a significantly higher density of MAP2-Fyn complexes in the dendrites relative to WT neurons suggested that MAP2 would have an increased role in interacting with Fyn in the absence of dendritic tau. We also showed that similar to tau, MAP2 also potentiated Fyn activity (Fig. 7C, 7D), leading us to hypothesize that MAP2 would have an increased role in interacting with Fyn, affecting NR2B phosphorylation and NR function in the absence of dendritic tau. Future studies using gain-of-function and loss-of-function experiments would further test this hypothesis.
In summary, we generated a tau−/−/Fyn−/− DKO mouse and found that tau was capable of regulating Ca2+ influx in a Fyn-independent manner, a property that we propose to be mimicked by MAP2 in the absence of tau. As dysregulation of Ca2+ influx is a known mechanism leading to excitotoxity in AD, our findings identify tau as a new therapeutic target in the regulation of neuronal Ca2+. Most importantly, increased levels of dendritic MAP2-Fyn complexes in the tau KO highlight the importance of the tau-Fyn interaction and indicate the need to compensate if tau is lost. Our findings suggest that the association and activation of Fyn by tau in dendrites are critical neuronal functions.
Supplementary Material
Significance Statement.
The downstream effect of the interaction between microtubule-associated protein tau and Src family non-receptor tyrosine kinase Fyn was investigated with a tau/Fyn double KO mouse. We demonstrate that tau has a Fyn-independent role in glutamate-induced calcium response and that MAP2 can compensate for tau in interacting with Fyn in dendrites. Our findings underline the importance of the tau-Fyn interaction in developing neurons.
Acknowledgements
We thank Drs. Peter Davies for generously providing the DA9 antibody, Stefan Strack for valuable suggestions, Bridget Shafit-Zagardo for MAP2c plasmid, Dan Thedens for help with MRI, Patrick Ten Eyck for help with statistical analysis, Skye Souter for technical assistance, and Craig Morita for help with figures. We acknowledge the University of Iowa Medical Scientist Training Program for support to Guanghao Liu and the Holden Comprehensive Cancer Center (NIH/NCI P30CA086862) for support at the University of Iowa Central Microscopy Research Facility and Small Animal Imaging Facility.
Funding
G Lee received support from NIH/NIA R01 AG017753 and Alzheimer’s Association IIRG-12-241042. G Liu is supported by NIH/NIA F30 AG054134 and by Iowa Neuroscience Institute Kwak-Ferguson Fellowship. YMU is supported by NIH/NINDS R01 NS096246.
Footnotes
Conflict of Interest Disclosure
No competing interests declared.
Data Accessibility
Data available on request from the authors.
References
- Ahmed T, Van der Jeugd A, Blum D, Galas MC, D’Hooge R, Buee L, and Balschun D. 2014. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging 35:2474–2478. [DOI] [PubMed] [Google Scholar]
- Albala JS, Kalcheva N, and Shafit-Zagardo B. 1993. Characterization of the transcripts encoding two isoforms of human microtubule-associated protein-2 (MAP-2). Gene 136:377–378. [DOI] [PubMed] [Google Scholar]
- Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, and Arikkath J. 2012. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7:1741–1754. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Hobbs GA, Yen SH, and Lee G. 2010. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol 36:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Muller J, Parmar J, Jonquieres GV, Stefen H, Guccione E, Fath T, Housley GD, Klugmann M, Ke YD, and Ittner LM. 2017. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun 8:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, and Rebhun LI. 1986. Differential localization of MAP-2 and tau in mammalian neurons in situ. Ann N Y Acad Sci 466:145–166. [DOI] [PubMed] [Google Scholar]
- Brandt R, Leger J, and Lee G. 1995. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol 131:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, and Kosik KS. 1990. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343:461–463. [DOI] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, Nishimura DY, Searby CC, Bugge K, and Sheffield VC. 2012. Abnormal development of NG2+PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat Med 18:1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, and Mucke L. 2004. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci 24:4692–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, and Wilhelmsen KC. 1998. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A 95:13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha XM, Price M, Schnizler MK, and Wemmie JA. 2007. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry 62:1140–1148. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, and Laskowitz DT. 2010. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience 169:516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, and Vitek MP. 2001. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 114:1179–1187. [DOI] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, and Holzbaur EL. 2008. Differential regulation of dynein and kinesin motor proteins by tau. Science 319:1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio K, Sato M, Uemura T, Sato T, Sato-Harada R, and Harada A. 2007. 14–3-3 proteins and protein phosphatases are not reduced in tau-deficient mice. Neuroreport 18:1049–1052. [DOI] [PubMed] [Google Scholar]
- Goto J, Tezuka T, Nakazawa T, Sagara H, and Yamamoto T. 2008. Loss of Fyn tyrosine kinase on the C57BL/6 genetic background causes hydrocephalus with defects in oligodendrocyte development. Mol Cell Neurosci 38:203–212. [DOI] [PubMed] [Google Scholar]
- Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, and Kandel ER. 1992. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258:1903–1910. [DOI] [PubMed] [Google Scholar]
- Groveman BR, Feng S, Fang XQ, Pflueger M, Lin SX, Bienkiewicz EA, and Yu X. 2012. The regulation of N-methyl-D-aspartate receptors by Src kinase. FEBS J 279:20–28. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, and Binder LI. 1986. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullberg M, Gustafsdottir SM, Schallmeiner E, Jarvius J, Bjarnegard M, Betsholtz C, Landegren U, and Fredriksson S. 2004. Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci U S A 101:8420–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, and Strittmatter SM. 2016. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 139:526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, Hoffman DA, and Roberson ED. 2015. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci 35:6221–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, and Hirokawa N. 1994. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369:488–491. [DOI] [PubMed] [Google Scholar]
- Ho GJ, Hashimoto M, Adame A, Izu M, Alford MF, Thal LJ, Hansen LA, and Masliah E. 2005. Altered p59Fyn kinase expression accompanies disease progression in Alzheimer’s disease: implications for its functional role. Neurobiol Aging 26:625–635. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, and Heutink P. 1998. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, and Hirokawa N. 2000. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett 279:129–132. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, and Gotz J. 2010. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142:387–397. [DOI] [PubMed] [Google Scholar]
- Kalia LV, Gingrich JR, and Salter MW. 2004. Src in synaptic transmission and plasticity. Oncogene 23:8007–8016. [DOI] [PubMed] [Google Scholar]
- Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, and Strittmatter SM. 2015. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 77:953–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, Ittner A, and Ittner LM. 2012. Lessons from tau-deficient mice. International journal of Alzheimer’s disease 2012:873270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kang S, Lee JY, and Rhim H. 2009. High throughput screening assay of alpha(1G) T-type Ca2+ channels and comparison with patch-clamp studies. Comb Chem High Throughput Screen 12:296–302. [DOI] [PubMed] [Google Scholar]
- Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, Brown C, Hashikawa T, Murayama M, Seok H, Sotiropoulos I, Kim E, Collingridge GL, Takashima A, and Cho K. 2014. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci 369:20130144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Ishibashi H, Obata K, and Kandel ER. 1998. Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-methyl-D-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem 5:429–445. [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, and Kandel ER. 1997. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci U S A 94:4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, and Selkoe DJ. 1986. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83:4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, and Klein WL. 1998. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, and Panchamoorthy G. 1998. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci 111 ( Pt 21):3167–3177. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, and Ksiezak-Reding H. 2004. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci 24:2304–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, and Bush AI. 2012. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18:291–295. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, and Bush AI. 2014. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leugers CJ, Koh JY, Hong W, and Lee G. 2013. Tau in MAPK activation. Front Neurol 4:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leugers CJ, and Lee G. 2010. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J Biol Chem 285:19125–19134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, and Gotz J. 2017. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J 36:3120–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hall AM, Kelinske M, and Roberson ED. 2014. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol Aging 35:2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes S, Lopes A, Pinto V, Guimaraes MR, Sardinha VM, Duarte-Silva S, Pinheiro S, Pizarro J, Oliveira JF, Sousa N, Leite-Almeida H, and Sotiropoulos I. 2016a. Absence of Tau triggers age-dependent sciatic nerve morphofunctional deficits and motor impairment. Aging Cell 15:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes S, Vaz-Silva J, Pinto V, Dalla C, Kokras N, Bedenk B, Mack N, Czisch M, Almeida OF, Sousa N, and Sotiropoulos I. 2016b. Tau protein is essential for stress-induced brain pathology. Proc Natl Acad Sci U S A 113:E3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Honack D, Fassbender CP, and Nolting B. 1991. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res 8:171–189. [DOI] [PubMed] [Google Scholar]
- Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Kiosea NC, Nazari S, Chen PP, Nothias F, Chan P, Teng E, Frautschy SA, and Cole GM. 2014. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. J Neurosci 34:7124–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie-Cardine A, Kirchgessner H, Eckerskorn C, Meuer SC, and Schraven B. 1995. Human T lymphocyte activation induces tyrosine phosphorylation of alpha-tubulin and its association with the SH2 domain of the p59fyn protein tyrosine kinase. Eur J Immunol 25:3290–3297. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, and Raymond LA. 2010. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 65:178–190. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Tateishi K, and Niki H. 1996. Susceptibility to drug-induced seizures of Fyn tyrosine kinase-deficient mice. Neuroreport 7:2723–2726. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Stein L, Thomas R, Djukic B, Taneja P, Knox J, Vossel K, and Mucke L. 2017. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener 12:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Hamto P, Adame A, Devidze N, Masliah E, and Mucke L. 2013. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol Aging 34:1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, and Yamamoto T. 2001. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem 276:693–699. [DOI] [PubMed] [Google Scholar]
- Nukina N, and Ihara Y. 1986. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem 99:1541–1544. [DOI] [PubMed] [Google Scholar]
- Oyama F, Kotliarova S, Harada A, Ito M, Miyazaki H, Ueyama Y, Hirokawa N, Nukina N, and Ihara Y. 2004. Gem GTPase and tau: morphological changes induced by gem GTPase in cho cells are antagonized by tau. J Biol Chem 279:27272–27277. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, and Schellenberg GD. 1998. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. [DOI] [PubMed] [Google Scholar]
- Racine RJ 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. [DOI] [PubMed] [Google Scholar]
- Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, Ryu JH, Whitcomb DJ, and Cho K. 2015. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci 35:4804–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, and Mucke L. 2011. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31:700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, and Mucke L. 2007. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316:750–754. [DOI] [PubMed] [Google Scholar]
- Rong Y, Lu X, Bernard A, Khrestchatisky M, and Baudry M. 2001. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J Neurochem 79:382–390. [DOI] [PubMed] [Google Scholar]
- Sapir T, Frotscher M, Levy T, Mandelkow EM, and Reiner O. 2012. Tau’s role in the developing brain: implications for intellectual disability. Hum Mol Genet 21:1681–1692. [DOI] [PubMed] [Google Scholar]
- Sharma VM, Litersky JM, Bhaskar K, and Lee G. 2007. Tau impacts on growth-factor-stimulated actin remodeling. J Cell Sci 120:748–757. [DOI] [PubMed] [Google Scholar]
- Shirazi SK, and Wood JG. 1993. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport 4:435–437. [DOI] [PubMed] [Google Scholar]
- Shuttleworth TJ, and Thompson JL. 1991. Effect of temperature on receptor-activated changes in [Ca2+]i and their determination using fluorescent probes. J Biol Chem 266:1410–1414. [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, and Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3:995–1000. [DOI] [PubMed] [Google Scholar]
- Sontag JM, Nunbhakdi-Craig V, White CL 3rd, Halpain S, and Sontag E. 2012. The protein phosphatase PP2A/Balpha binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: implications for tauopathies. J Biol Chem 287:14984–14993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers LP, Loo L, Wu Y, Campbell E, Ulrich JD, Wu S, Paemka L, Wassink T, Meyer K, Bing X, El-Shanti H, Usachev YM, Ueno N, Manak JR, Shepherd AJ, Ferguson PJ, Darbro BW, Richerson GB, Mohapatra DP, Wemmie JA, and Bassuk AG. 2013. Disruption of the non-canonical Wnt gene PRICKLE2 leads to autism-like behaviors with evidence for hippocampal synaptic dysfunction. Mol Psychiatry 18:1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, and Ghetti B. 1998. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PL, Lee HM, Rich S, and Soriano P. 1992. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell 70:741–750. [DOI] [PubMed] [Google Scholar]
- Tan DCS, Yao S, Ittner A, Bertz J, Ke YD, Ittner LM, and Delerue F. 2018. Generation of a New Tau Knockout (tauDeltaex1) Line Using CRISPR/Cas9 Genome Editing in Mice. J Alzheimers Dis 62:571–578. [DOI] [PubMed] [Google Scholar]
- Tang YP, Wang H, Feng R, Kyin M, and Tsien JZ. 2001. Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology 41:779–790. [DOI] [PubMed] [Google Scholar]
- Tezuka T, Umemori H, Akiyama T, Nakanishi S, and Yamamoto T. 1999. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc Natl Acad Sci U S A 96:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepanier CH, Jackson MF, and MacDonald JF. 2012. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J 279:12–19. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, and Mandelkow E. 1999. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci 112 (Pt 14):2355–2367. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, and Barde YA. 2001. Neurotrophins are required for nerve growth during development. Nat Neurosci 4:29–37. [DOI] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ, Gunther EC, Nygaard HB, and Strittmatter SM. 2013. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79:887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, and Strittmatter SM. 2012. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, and Strittmatter SM. 2013. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 7:37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Wanaka A, Kato H, Takeuchi M, Tohyama M, and Yamamoto T. 1992. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res Mol Brain Res 16:303–310. [DOI] [PubMed] [Google Scholar]
- van Hummel A, Bi M, Ippati S, van der Hoven J, Volkerling A, Lee WS, Tan DC, Bongers A, Ittner A, Ke YD, and Ittner LM. 2016. No Overt Deficits in Aged Tau-Deficient C57Bl/6.Mapttm1(EGFP)Kit GFP Knockin Mice. PLoS One 11:e0163236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, and Wolozin B. 2016. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep 15:1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, and Kirschner MW. 1975. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R, Gonsior C, Kramer-Albers EM, Stohr N, Huttelmaier S, and Trotter J. 2008. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J Cell Biol 181:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, and Binder LI. 1986. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc Natl Acad Sci U S A 83:4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora-Leon SP, Lee G, Davies P, and Shafit-Zagardo B. 2001. Binding of Fyn to MAP-2c through an SH3 binding domain. Regulation of the interaction by ERK2. J Biol Chem 276:39950–39958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.