

Abstract
Introduction
Turoctocog alfa pegol (N8‐GP) is a site‐specific, 40 kDa glycoPEGylated recombinant factor VIII (FVIII) product with an extended half‐life. The comprehensive main phase of the pivotal pathfinder 2 trial showed N8‐GP dosed every 4 days (Q4D) provided favourable safety and efficacy for preventing bleeds in 175 patients with haemophilia A.
Aim and methods
We investigated the safety and efficacy of N8‐GP prophylaxis when administered weekly (Q7D) for 24 weeks to patients with low bleeding rates in the pathfinder 2 extension trial. Patients (≥12 years) with ≤2 bleeds during the preceding 6 months of the pathfinder 2 main phase were eligible for randomization to receive N8‐GP 50 IU/kg Q4D or 75 IU/kg Q7D. Safety and efficacy endpoints were incidence of FVIII inhibitors and annualized bleeding rate (ABR), respectively.
Results
Fifty‐five of 143 (38.5%) patients on prophylaxis who continued into the extension phase were randomized to receive 50 IU/kg Q4D (n = 17) or 75 IU/kg Q7D (n = 38). Nine patients in the Q7D cohort reverted to 50 IU/kg Q4D. No inhibitors were detected. In both cohorts, >50% of patients experienced no bleeds. Median ABR for overall, joint, spontaneous, traumatic and muscle was 0.00 for both cohorts. Overall estimated success rate for treating bleeding episodes was 87.5%; 94.7% of bleeds were controlled with ≤2 injections.
Conclusions
Weekly N8‐GP was well tolerated and efficacious and may benefit selected “low bleeder” patients with haemophilia A.
Keywords: efficacy, FVIII, haemophilia A, N8‐GP, once‐weekly prophylaxis, safety
1. INTRODUCTION
Although prophylaxis with factor VIII (FVIII) is widely considered to be the standard of care for patients with severe haemophilia A,1, 2, 3, 4, 5, 6 barriers to its widespread implementation remain.2, 6 A key barrier is the need for frequent injections (3‐4 times/wk, translating to ~182 injections/y) due to the relatively short plasma half‐life of standard FVIII products.1, 3, 7, 8, 9, 10 Many patients struggle to integrate prophylactic schedules into their daily lives, leading to missed injections8 and increased bleeding episodes.2, 5 Therefore, prophylactic strategies offering effective haemostatic coverage with more convenient administration schedules are needed.
FVIII replacement therapy used to treat acute breakthrough bleeding episodes can also be problematic as multiple injections may be required over several days.3, 11
Limitations of standard recombinant FVIII (rFVIII) products have driven improvements in both prophylactic and on‐demand treatment for patients with severe haemophilia A.4, 7, 12 Prolonging the FVIII circulation time has been a primary objective,13 and several strategies have been developed.9, 14, 15, 16, 17
N8‐GP (turoctocog alfa pegol; Novo Nordisk, Bagsværd, Denmark) is a novel, extended half‐life (EHL) rFVIII product produced by a site‐specific 40 kDa glycoPEGylation of turoctocog alfa, a B‐domain‐truncated rFVIII.9, 14 N8‐GP has a half‐life of 19 hours, corresponding to a 1.6‐fold prolongation compared to patients’ previous plasma‐derived or standard rFVIII products.9 In the main phase of the comprehensive, phase III pathfinder 2 trial,14 N8‐GP provided effective prophylaxis and maintained a low observed median annualized bleeding rate (ABR) of 1.18 when dosed every 4 days (Q4D) in 175 patients over 299 days (mean duration). N8‐GP was also effective for treating bleeding episodes (83.6% were resolved with a single injection and 95.5% with up to two injections).14
In pathfinder 2, the main phase was followed by a two‐part extension phase. The main objective of pathfinder 2 extension phase part 1 was to investigate the safety and efficacy of N8‐GP for prophylaxis and treatment of bleeds when administered Q4D vs weekly (Q7D) to a randomized subset of patients who had low bleeding rates in the trial's main phase (≤2 bleeds in the last 6 months of the main phase). The extension phase provided an opportunity to explore whether selected patients could be managed with a once‐weekly prophylactic schedule, while retaining the favourable safety and efficacy profile of N8‐GP and reducing the treatment burden to 52 injections/y. Here, we report the findings from the pathfinder 2 extension phase part 1.
2. TRIAL DESIGN, PATIENTS, OBJECTIVES AND ENDPOINTS
Details of pathfinder 2 have been previously published,14 but are described briefly here.
2.1. Trial design
The trial was conducted in accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice guidelines,18 the Declaration of Helsinki19 and all applicable regulatory requirements. Trial approval was obtained from independent ethics committees or institutional review boards of participating sites. All patients provided written informed consent.
pathfinder 2 is a multicentre, multinational, open‐label, phase III trial evaluating the safety, pharmacokinetics and efficacy of N8‐GP when used for the prophylaxis and treatment of bleeding episodes. It comprised a non‐randomized main phase (completed and reported14) and an ongoing two‐part extension phase with both randomized and non‐randomized patient cohorts (Figure 1).
Figure 1.
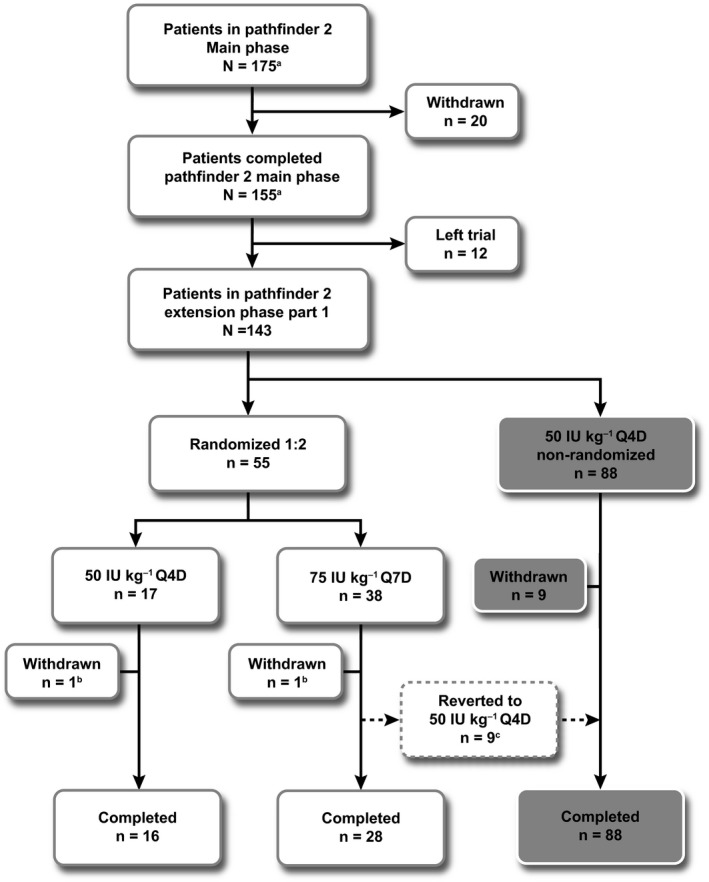
Patient flow through pathfinder 2 extension phase for patients who received N8‐GP prophylaxis. aPatients who participated in the pathfinder 2 main phase and received N8‐GP prophylaxis. The last patient last visit of pathfinder 2 extension phase part 1 was 3 March 2015. bWithdrawn due to SAEs considered unlikely to be N8‐GP related. cReverted due to investigator's discretion (X‐ray changes showing worsened pre‐existent arthropathy [n = 1]) or bleeding episodes (n = 8). The patients withdrawn from randomization due to bleeding episodes were equally distributed over time; within first month (n = 2), second month (n = 2), third month (n = 2) and after 5 months (n = 2). n, number of patients; Q4D, every 4 d; Q7D, every 7 d; SAE, serious adverse event. Trial registered at http://www.clinicaltrials.gov (NCT01480180)
In the non‐randomized main phase, patients received N8‐GP prophylaxis as a single bolus dose of 50 IU/kg Q4D (twice‐weekly dosing was permitted at the investigator's discretion). After the main phase, all patients were given the option of continuing treatment in the extension phase (Figure 1).
Using the interactive voice/web‐response system in extension phase part 1, a subset of eligible patients was randomized to receive 50 IU/kg Q4D (50Q4D) or 75 IU/kg Q7D (75Q7D) N8‐GP prophylaxis for 24 weeks. Patients who experienced ≥3 bleeds during the preceding 6 months, and those unwilling to be randomized, continued to receive N8‐GP prophylaxis at 50Q4D (non‐randomized patients).
Randomized patients allocated to 75Q7D could revert to 50Q4D at any time at the investigator's discretion. In addition, any patient receiving N8‐GP prophylaxis Q7D was required to revert to Q4D if ≥2 spontaneous or one severe bleeding episode requiring hospitalization occurred over an 8‐week period.
Any bleeding episode was treated with N8‐GP 20‐75 IU/kg, depending on the severity and location of the bleed.
2.2. Patients
Male patients aged ≥12 years with severe haemophilia A (FVIII <1%), a history of ≥150 exposure days to FVIII and without a history of FVIII inhibitors (≥0.6 Bethesda Units [BU]), were eligible for inclusion in pathfinder 2. For randomization eligibility in the extension phase, patients must have experienced ≤2 bleeds (spontaneous or traumatic) during the preceding 6 months of the main trial phase and be willing to undergo randomization.
2.3. Objectives and endpoints
The coprimary objectives were to evaluate the immunogenicity of N8‐GP and assess its clinical efficacy when used for once‐weekly prophylaxis. These primary endpoints were, respectively, incidence of FVIII inhibitors (≥0.6 BU) and ABR.
Secondary safety endpoints comprised adverse events (AEs), serious AEs (SAEs) and changes in vital signs. The secondary efficacy objective was to investigate the clinical efficacy of N8‐GP when used to treat bleeding episodes, as measured using a four‐point haemostatic response scale (excellent, good, moderate, none); “excellent” and “good” were considered treatment successes; “moderate”, “none” and missing evaluations were considered failures. The bleeding episodes were categorized as mild/moderate and severe.
2.4. Analytical methods and statistical analysis
See Appendix S1.
3. RESULTS
3.1. Patients
Of 175 patients initially enrolled into pathfinder 2 and allocated to receive prophylaxis with N8‐GP 50Q4D,14 143 continued into the extension phase (Figure 1); 120 met the randomization inclusion criteria, of which 55 received N8‐GP prophylaxis at 50Q4D (n = 17) or 75Q7D (n = 38). The remaining 88 continued to receive 50Q4D in the non‐randomized treatment cohort (Figure 1). Of the 120 patients who fulfilled the randomization criteria, 65 (54%) preferred to stay on their treatment regimen. Randomized patient demographics and baseline characteristics are shown in Table 1.
Table 1.
Demographics and baseline characteristics of randomized patients
|
50 IU/kg Q4D |
75 IU/kg Q7D |
Total | |
|---|---|---|---|
| Number of patients | 17 | 38 | 55 |
| Age in years, mean (SD) | 26.4 (11.0) | 30.9 (15.0) | 29.5 (13.9) |
| Race, n (%) | |||
| Caucasian | 14 (82.4) | 34 (89.5) | 48 (87.3) |
| Black/African American | – | 1 (2.6) | 1 (1.8) |
| Asian | 3 (17.6) | 3 (7.9) | 6 (10.9) |
| Weight in kg, mean (SD) | 77.2 (16.8) | 78.6 (15.2) | 78.2 (15.6) |
| BMI in kg/m2, mean (SD) | 24.6 (4.1) | 25.5 (4.2) | 25.2 (4.2) |
BMI, body mass index; n, number of patients; Q4D, every 4 d; Q7D, every 7 d; SD, standard deviation.
One patient from each of the randomized prophylaxis cohorts was withdrawn from the trial at the investigator's discretion, due to SAEs considered unlikely to be related to N8‐GP (Figure 1): one hepatocellular carcinoma (50Q4D) and one ankle fracture (75Q7D). There were nine other withdrawals from the 75Q7D cohort (23.7%) due to bleeding episodes (n = 8) or for safety reasons (n = 1; X‐ray changes showing worsened pre‐existent arthropathy). These nine reverted to the 50Q4D regimen (non‐randomized treatment) due to the predefined safety criteria in the trial protocol. Patients withdrawn from randomization due to bleeding episodes were equally distributed over time, within first (n = 2), second (n = 2) and third month (n = 2) and after 5 months (n = 2). The patients who switched from the randomized 75Q7D to the non‐randomized 50Q4D cohort (n = 8) remained “low bleeders” throughout the remaining treatment period.
In total, 16 and 28 patients in the 50Q4D and 75Q7D cohorts, respectively, completed the randomized treatment period of the extension phase (Figure 1). Overall, there were 1539 exposure days to N8‐GP in the randomized treatment period (Table 2).
Table 2.
Exposure to N8‐GP in the randomized treatment cohorts
| Mean (SD) |
50 IU/kg Q4D |
75 IU/kg Q7D |
Total |
|---|---|---|---|
| Number of patients | 17 | 38 | 55 |
| Total days in randomizationa | 167.8 (10.1) | 145.5 (48.3) | – |
| EDs | 43.0 (2.6) | 21.3 (6.4) | – |
| Number of doses | 43.1 (2.7) | 21.3 (6.4) | – |
| Number of PPX dosesa | 41.5 (2.6) | 20.4 (7.0) | – |
| Number of doses used for treatment of bleeding episodes | 2.6 (2.0) | 2.1 (1.0) | 2.2 (1.4) |
| Overall EDs in randomizationa | 731 | 808 | 1539 |
EDs, exposure days; PPX, prophylaxis; Q4D, every 4 d; Q7D, every 7 d; SD, standard deviation.
Total number across all patients.
3.2. Safety
No randomized patients developed FVIII inhibitors during the extension phase. No anti‐N8‐GP antibodies (neutralizing or non‐neutralizing) were detected. Overall, 108 AEs were reported in 36 (65.5%) patients (50Q4D: 37 in 10 patients; 75Q7D: 71 in 26 patients). The vast majority of AEs (105) were mild or moderate in severity. Four SAEs in four patients were reported (anal abscess, right hip prosthesis dislocation, hepatocellular carcinoma [all 50Q4D] and ankle fracture [75Q7D]); all were judged unlikely to be related to N8‐GP treatment. All recovered apart from the patient with hepatocellular carcinoma. Five AEs in five patients (rash, thrombocytopenia, increase in aspartate aminotransferase and headache [75Q7D]; purpura [50Q4D]) were considered to be related to N8‐GP treatment; most of which resolved (except thrombocytopenia). The patient with a skin rash had two events as follows: the first was a moderate rash occurring 90 minutes after commencing N8‐GP treatment and appeared several times over 3 weeks, after which the patient recovered. The second event was a mild rash occurring 1 week after the patient was randomized to Q7D; the patient gradually developed an itching rash covering arms, back, stomach and scrotum. No treatment was given and the rash cleared; the patient remained in the trial. The patient with thrombocytopenia was negative for hepatitis B and C, and human immunodeficiency virus and entered with a platelet count of 165 000/µL, falling to 130 000/µL at the time of the last visit of the extension phase part 1; the patient remained in the trial. Laboratory safety parameters, vital signs and physical examinations did not reveal any clinically relevant changes following N8‐GP treatment. No thromboembolic events were reported during the extension phase.
One patient entered the extension phase with anti‐polyethylene glycol (PEG) antibodies. Three patients had low‐titre (≤2) anti‐PEG antibodies at the end of the extension phase part 1 (50Q4D: n = 1; 75Q7D: n = 2); one from each cohort remained positive at the last reported visit. A further three (50Q4D: n = 1; 75Q7D: n = 2) had one or two positive samples for low‐titre anti‐PEG antibodies during the extension phase. In all, no patients with anti‐PEG antibodies reported AEs such as itching and rash. One patient reported eczema that was considered as a medical event of special interest not related to N8‐GP. FVIII activity did not differ between patients with or without anti‐PEG antibodies.
3.3. Prophylaxis
The proportion of patients who experienced no bleeding episodes while receiving N8‐GP prophylaxis was 52.9% (9/17) in the 50Q4D cohort and 57.9% (22/38) in the 75Q7D cohort. Observed median ABR (interquartile range) was 0.00 (0.00‐2.23) and 0.00 (0.00‐2.36) for patients randomized to 50Q4D and 75Q7D, respectively. Observed mean ABR (95% confidence interval [CI]) was 1.66 (0.69‐4.04) and 1.65 (0.87‐3.13) for patients on 50Q4D and 75Q7D regimens, respectively (Table 3). Imputed median and mean ABR data representing the predefined, confirmatory test are presented in Table S1.
Table 3.
Observed ABRs for patients randomized to receive N8‐GP prophylaxis
|
50 IU/kg Q4D |
75 IU/kg Q7D |
|
|---|---|---|
| Number of patients | 17 | 38 |
| Withdrawn from randomization | 1 | 10c |
| Patients with bleeding episodesa, n (%) | 8 (47.1) | 16 (42.1) |
| No. of bleeding episodesa | 13 | 25 |
| Mean observed treatment period in yearsb | 0.46 | 0.40 |
| Observed median ABR (IQR) | 0.00 (0.00‐2.23) | 0.00 (0.00‐2.36) |
| Observed mean ABR (95% CI) | 1.66 (0.69‐4.04) | 1.65 (0.87‐3.13) |
ABR, annualized bleeding rate (total number of bleeds/total exposure time); CI, confidence interval; IQR, interquartile range; n, number of patients; Q4D, every 4 d; Q7D, every 7 d.
Bleeding episodes treated with N8‐GP.
Observed time for completers and planned time for withdrawn patients.
Of the 10 patients who withdrew from the 75 IU/kg cohort, nine reverted to the 50 IU/kg non‐randomized cohort.
The relationship between overall observed/spontaneous ABR and predose FVIII activity levels for each patient is shown in Figures S1A,B, respectively. No significant correlations between predose FVIII activity and ABR were observed.
In all, 13 bleeding episodes occurred in eight patients randomized to 50Q4D; 25 occurred in 16 patients in the 75Q7D cohort (Table 3). In both cohorts, all bleeding episodes were mild or moderate, and as in the main phase,14 most were spontaneous (50Q4D: 53.8% [7/13 bleeds]; 75Q7D: 64.0% [16/25 bleeds]) (Figure 2). For the 50Q4D cohort, all bleeds were distributed throughout the 4‐day dosing interval, with most occurring between days 2 and 4 (8/13 bleeds: 61.5%); while for the 75Q7D cohort, most bleeds occurred on days 6 and 7, since the last N8‐GP dose (20/25 bleeds: 80.0%) (Figure 3). Joints were the most common bleed location for patients on the 50Q4D regimen (53.8% [7/13 bleeds; estimated mean ABR: 0.89; median ABR: 0.00]) (Figure 2) and those on 75Q7D (68.0% [17/25 bleeds; estimated mean ABR: 2.78; median ABR: 0.00]). Other bleed locations in the 50Q4D cohort were muscle or subcutaneous (4/13 bleeds, 30.8%) and mucosal or “other” (2/13 bleeds, 15.4%). In the 75Q7D cohort, all other bleeding episodes were muscular (5/25 bleeds, 20.0%) or subcutaneous (3/25 bleeds, 12.0%) (see Table S1).
Figure 2.
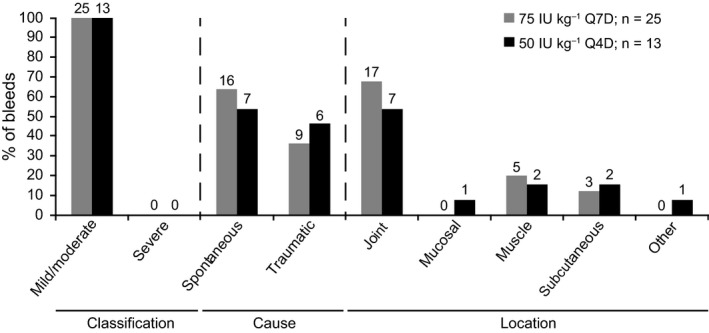
Bleeding details for patients randomized to receive N8‐GP 50 IU/kg Q4D or 75 IU/kg Q7D prophylaxis. The number of bleeds is reported above each bar. The total exposure time for the 50 IU/kg Q4D cohort was eight patient‐years and 15 patient‐years for the 75 IU/kg cohort. n, number of bleeds; Q4D, every 4 d; Q7D, every 7 d
Figure 3.
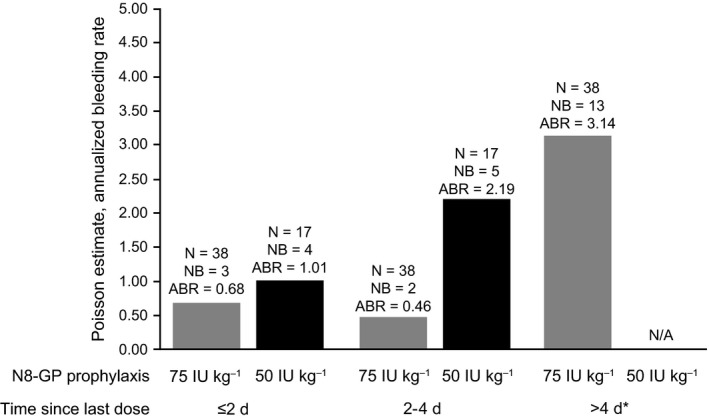
Observed mean ABR for patients randomized to receive N8‐GP 50 IU/kg Q4D or 75 IU/kg Q7D prophylaxis by time since last N8‐GP dose. If the exact time of a bleed was not known, the bleed was grouped into a time interval. If a bleed could be grouped to more than one interval, it was grouped to the one closest to the last N8‐GP dose. *The N, NB and ABR for >4 d since N8‐GP last dose were not presented for 50 IU/kg Q4D patients due to the limited amount of the data observed for Q4D patients with more than 4 d since N8‐GP last dose. ABR, annualized bleeding rate (total number of bleeding episodes/total exposure time); n, number of patients; N/A, not applicable; NB, number of patients with bleeds; Q4D, every 4 d; Q7D, every 7 d
3.4. Treatment of bleeding episodes
All treatment of bleeding episodes in the 50Q4D cohort (100%) and 80% in the 75Q7D cohort were considered a success (Table 4), where the overall estimated success rate for treating bleeding episodes was 87.5%. Due to the low patient numbers in both cohorts, a statistical comparison of effectiveness of the treatment of bleeds could not be conducted. Overall, 94.7% of bleeding episodes were controlled with ≤2 injections: 92.3% of bleeds (12/13 bleeds) and 96.0% of bleeds (24/25 bleeds) in the 50Q4D and 75Q7D cohorts, respectively. All remaining bleeds were controlled with seven (50Q4D, a spontaneous wrist bleed) and three (75Q7D, a traumatic ankle bleed) injections, respectively. The mean number of injections to control a bleed in both cohorts was similar (1.5 in 50Q4D; 1.3 in 75Q7D).
Table 4.
Haemostatic response to treatment of bleeding episodes with N8‐GP in patients randomized to receive N8‐GP prophylaxis
|
50 IU/kg Q4D |
75 IU/kg Q7D |
Total | |
|---|---|---|---|
| Number of bleeding episodes, na | 13 | 25 | 38 |
| Success, n (%) | |||
| Excellent | 8 (61.5) | 9 (36.0) | 17 (44.7) |
| Good | 5 (38.5) | 11 (44.0) | 16 (42.1) |
| Failure, n (%) | |||
| Moderate | 0 (0.0) | 3 (12.0) | 3 (7.9) |
| None | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Missing | 0 (0.0) | 2 (8.0) | 2 (5.3) |
| Success, n (%) | 13 (100.0) | 20 (80.0) | 33 (86.8) |
| Estimated success rateb | 87.5 | ||
| 95% CI | 71.7‐95.1 | ||
CI, confidence interval; n, number of bleeding episodes; Q4D, every 4 d; Q7D, every 7 d.
Bleeding episodes treated with N8‐GP.
The estimated success rate that is rated “excellent” or “good”; the planned analysis was based on cumulative. Analysed using logistic regression accounting for repeated measures within patient assuming compound symmetry working correlation. Missing responses are counted as failure.
3.5. N8‐GP consumption
In the 50Q4D and 75Q7D cohorts, respective median (range) N8‐GP dose from bleed start to stop was 52.3 (32.0‐298.0 IU/kg) and 76.7 (22.0‐157.0 IU/kg), while the median (range) time to control a bleed was 12.8 (0‐358 hours) and 18.5 (0‐469 hours), respectively. The quantity of N8‐GP used to treat a bleed depended on the severity of the bleed and when it occurred in relation to the dosing interval.
For patients in the 75Q7D cohort, the median prophylaxis dose used was higher than the 50Q4D cohort (77.2 vs 52.3 IU/kg for 50Q4D), but the median annualized consumption for prophylaxis (4005 vs 4752 IU/kg annually for 50Q4D) and total overall annualized consumption (4046 vs 4907 IU/kg annually for 50Q4D) was lower than the 50Q4D cohort. Patients in the 75Q7D cohort received fewer annual doses (650 IU/kg/y less, corresponding to 39 doses less/y) than the 50Q4D cohort. Once‐weekly prophylaxis with N8‐GP resulted in up to 43% fewer injections/y (from 91.25 to 52) in patients previously dosed with N8‐GP Q4D. Similarly, Q4D prophylaxis with N8‐GP during the main phase of pathfinder 2 resulted in up to 42% (from 156 to 91) and 50% (from 182 to 91) fewer injections/y in patients dosed with their previous FVIII product three times weekly and every other day, respectively.
3.6. FVIII activity
FVIII plasma activity was measured using a chromogenic assay (see Appendix S1). Mean peak FVIII activity levels (assessed 30 minutes postdosing) were similar before (1.33 IU/mL) and after (1.36 IU/mL) randomization for the 50Q4D cohort, while mean peak levels for the 75Q7D cohort increased from 1.40 IU/mL before randomization to 1.98 IU/mL afterwards, due to the higher dose. Predose FVIII activity levels (trough) before randomization were the same for both randomized cohorts (0.04 IU/mL). The nine patients who reverted to their previous Q4D regimen had similar mean (95% CI) predose FVIII activity levels (0.03 [0.02‐0.06] IU/mL). During the extension phase, mean (95% CI) predose FVIII levels were 0.03 (0.02‐0.05 IU/mL) for the 50Q4D cohort and 0.012 (0.01‐0.02 IU/mL) for the 75Q7D cohort.
4. CONCLUSIONS
The main finding of this extension trial is that effective prophylaxis was demonstrated at 75Q7D in patients with a low bleeding phenotype (≤2 bleeds during the preceding 6 months of the trial's main phase with a dosing regimen of 50Q4D). Importantly, 58% of patients on the 75Q7D regimen experienced no bleeding episodes during the 24‐week randomization period, which was a similar percentage to patients in the 50Q4D (53%) dosing regimen.
Observed median ABR for overall, joint, spontaneous, traumatic and muscle was 0.00 for both cohorts. No correlation between predose FVIII activity and ABR was seen; the aetiology of bleeding in patients with haemophilia is multifactorial, and additional observations will be required to understand fully the ideal dosing regimen for most patients. These observations suggest that N8‐GP at 75Q7D provides effective prophylaxis for a subset of selected “low bleeder” patients.
The subset of patients who participated in the randomized part of the extension trial had an observed mean ABR of 1.16 when treated with 50Q4D during the trial's main phase, consistent with the inclusion criteria for randomization (low bleeders). Those who continued to receive 50Q4D maintained a low observed mean ABR upon completion of the extension phase (1.66). For those randomized to 75Q7D, the observed mean ABR before entering randomization was 1.11. In the extension trial, these patients had an observed mean ABR of 1.65, whereas the observed mean ABR for all non‐randomized patients who received 50Q4D in the main phase of the pathfinder 2 trial was 3.04.14
The extension phase confirmed that N8‐GP treatment is well tolerated: no inhibitors were detected and there were no unexpected safety concerns. The haemostatic effect of N8‐GP when used to treat bleeding episodes was also confirmed in the extension phase: overall, estimated success rate for treating bleeding episodes was 87.5%, similar to the haemostatic success rate reported for the trial's main phase (84.2%).14 The proportion of bleeding episodes successfully treated with ≤2 injections was similar between the main (95.5%)14 and extension (94.7%) phases. As expected, overall N8‐GP consumption was lower in 75Q7D (up to 650 IU/kg/y less) than the 50Q4D cohort, thereby reducing the treatment burden for patients with severe haemophilia A. Additionally, patients in the 75Q7D cohort received fewer annual injections (52.0 injections/y) to maintain adequate plasma FVIII activity levels when compared to patients on standard FVIII products (~182 injections/y),1, 7, 8 which is mainly attributed to the EHL property of N8‐GP.
Promising results have also been achieved in recent studies of other EHL products.15, 16, 17, 20 To date, Q7D dosing with an EHL product has been assessed in two other phase III trials.15, 20 A partially randomized trial of rFVIII Fc fusion protein (rFVIIIFc) randomized 47 adult and adolescent patients, who had received only on‐demand treatment prior to trial entry, to receive either on‐demand rFVIIIFc treatment or weekly rFVIIIFc prophylaxis (65 IU/kg Q7D).15 Observed median ABR in the 23 patients who received weekly rFVIIIFc prophylaxis was 3.6. The proportion who experienced no bleeding episodes while on rFVIIIFc prophylaxis Q7D was 17.4%; three (13%) experienced >5 bleeding episodes.15 Another partially randomized trial included adults and adolescents with severe haemophilia A who received B‐domain‐deleted rFVIII conjugated with 60‐kDa PEG (BAY 94‐9027) for 36 weeks prophylactically at intervals determined following a 10‐week run‐in period on 25 IU/kg twice weekly.20 Patients with >1 bleed during the run‐in period received 30‐40 IU/kg twice weekly; patients with ≤1 bleed were randomized to Q5D (45‐60 IU/kg) or Q7D (60 IU/kg) prophylaxis (1:1) for 26 additional weeks. Observed median ABR in the 32 low‐bleeding patients randomized to receive BAY 94‐9027 Q7D was 0.96, and 50% of patients experienced no bleeding episodes.20
Limitations of the extension phase include the small patient populations in both treatment cohorts (50Q4D [n = 17] and 75Q7D [n = 38]) and the limited trial length of 24 weeks. Randomized patients were all low bleeders too, so should not be considered representative of the entire population. Randomization involved more frequent clinic visits, and patients may have perceived that reduced dosing frequency may increase bleeding, possibly influencing the remaining 65 patients eligible for randomization but who chose to remain on Q4D dosing. Nevertheless, the extension phase identified a proportion of patients randomized to receive 75Q7D who did not bleed during the 24‐week trial period, informing providers of a patient subset who may benefit from Q7D prophylaxis in the real‐life treatment setting.
Despite numerous treatment advances, recent retrospective21 and longitudinal, prospective22 data collected across Europe and the United States still show unacceptably high bleeding rates in patients with severe haemophilia A. In Europe, most patients with severe haemophilia A on prophylaxis were treated ≥3 times per week with standard FVIII products; their median ABR ranged from 1.0 to 4.0.21 Further research is warranted to explore potential benefits of prophylactic schedules incorporating more prolonged dosing intervals offered by products with a longer half‐life. Nevertheless, results achieved to date with N8‐GP in the pathfinder 2 trial, together with those from recent studies investigating other FVIII products with an EHL,15, 16, 17 suggest that reducing the treatment burden for patients with severe haemophilia A is a realistic and achievable goal for selected cohorts.4, 7
In conclusion, data from the randomized part of the extension phase of the pivotal pathfinder 2 trial demonstrate an efficacious prophylactic effect of N8‐GP when dosed at 75 IU/kg Q7D; 58% of patients receiving this regimen experienced no bleeding episodes during the 24‐week extension period. These findings suggest that weekly N8‐GP may provide effective prophylaxis with a reduced treatment burden for a selected subset of low‐bleeding patients with severe haemophilia A.
DISCLOSURES
NC has received support to attend conferences from Bayer, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Shire and SOBI, and advisory boards from LFB, Shire and SOBI. She has served as a paid consultant for Bayer and was the Investigator who led research for CSL Behring. CA has received research support from Bayer, Octapharma, Pfizer and Shire, and has served as a paid consultant for Bayer, Biogen Idec, CSL Behring, Novo Nordisk, Pfizer, Shire and SOBI. ME has received research support from Pfizer and has served as a paid consultant for Bayer, CSL Behring, Genentech, Hemabiologics, Kedrion, Novo Nordisk, Octapharma, Pfizer and Shire. PAH received research support from Bayer, Octapharma, Pfizer and Shire, and has served as a paid consultant for Bayer, Biogen Idec, CSL Behring, Novo Nordisk, Pfizer, Shire and SOBI. SK received a grant from Bayer, Bioverativ, Daiichi Sankyo, Grifols and Novo Nordisk, and attended advisory boards for Bayer, Bioverativ and Novo Nordisk. RK received research support from Baxalta (Shire), Bayer, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Shire and SOBI, and has served as a paid consultant for Baxalta (Shire), Bayer, CSL Behring, Novo Nordisk, Octapharma, Pfizer, Shire and SOBI. MM has nothing to disclose. CN received research support and/or was a Principal Investigator for Alnylam, Baxalta (Shire), Bayer, Biogen Idec/SOBI, CSL Behring, Novo Nordisk, Octapharma and Pfizer. He attended scientific advisory boards and received honoraria and/or travel support from Baxalta (Shire), Bayer, Biogen Idec/SOBI, CSL Behring, LFB, Novo Nordisk, Octapharma and Pfizer. AW participated in advisory boards for Baxalta (Shire), Novo Nordisk, Octapharma and Bayer. ES attended and received funds for advisory boards from Bayer, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Shire and SOBI. She attended Speaker Bureaus for and received funds from Bayer, Bioverativ, CSL Behring, Grifols, Kedrion, Novo Nordisk, Octapharma, Pfizer, Roche, Shire and SOBI. MS has received grant support from Bayer, Bioverativ, Chugai, CSL Behring, Novo Nordisk, Pfizer, Roche and Shire; personal fees from Bayer, Bioverativ, Chugai, Novo Nordisk, Sysmex and Shire. AL and SMT are employees of Novo Nordisk A/S. SRL has received grant support and personal fees from Novo Nordisk A/S and has served as a paid consultant for Novo Nordisk A/S.
Supporting information
ACKNOWLEDGEMENTS
This trial was sponsored by Novo Nordisk A/S (Bagsværd, Denmark). The authors thank the patients and their families/caregivers and the investigators, pharmacists, nurses and trial staff at each centre for participating in the trial. The authors directed the data analysis and the development of the manuscript. The sponsor was responsible for trial operations, including data analysis. The Principal Investigator (Dr Nicola Curry) assumes full responsibility for the accuracy and completeness of the reported data and for the conformity of the manuscript to the trial protocol and the final trial report. The authors would also like to thank Frank Driessler (Novo Nordisk Health Care AG) for his scientific advice. Medical writing support was provided by Jo Fetterman, PhD (PAREXEL, UK). This work was funded by Novo Nordisk A/S (Bagsværd, Denmark). Novo Nordisk's policy on data sharing may be found at https://www.novonordisk-trials.com/how-access-clinical-trial-datasets.
Curry N, Albayrak C, Escobar M, et al. Once‐weekly prophylaxis with glycoPEGylated recombinant factor VIII (N8‐GP) in severe haemophilia A: Safety and efficacy results from pathfinder 2 (randomized phase III trial). Haemophilia. 2019;25:373–381. 10.1111/hae.13712
Trial registration: Trial registered at http://www.clinicaltrials.gov (NCT01480180)
REFERENCES
- 1. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535‐544. [DOI] [PubMed] [Google Scholar]
- 2. Zappa S, McDaniel M, Marandola J, Allen G. Treatment trends for haemophilia A and haemophilia B in the United States: results from the 2010 practice patterns survey. Haemophilia. 2012;18:e140‐e153. [DOI] [PubMed] [Google Scholar]
- 3. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐47. [DOI] [PubMed] [Google Scholar]
- 4. Jimenez‐Yuste V, Auerswald G, Benson G, et al. Achieving and maintaining an optimal trough level for prophylaxis in haemophilia: the past, the present and the future. Blood Transfus. 2014;12:314‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on‐demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richards M, Altisent C, Batorova A, et al. Should prophylaxis be used in adolescent and adult patients with severe haemophilia? An European survey of practice and outcome data. Haemophilia. 2007;13:473‐479. [DOI] [PubMed] [Google Scholar]
- 7. Fogarty PF. Biological rationale for new drugs in the bleeding disorders pipeline. Hematology Am Soc Hematol Educ Program. 2011;2011:397‐404. [DOI] [PubMed] [Google Scholar]
- 8. Hacker MR, Geraghty S, Manco‐Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7:392‐396. [DOI] [PubMed] [Google Scholar]
- 9. Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first‐in‐human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11:670‐678. [DOI] [PubMed] [Google Scholar]
- 10. World Federation of Hemophilia (WFH) . Guidelines for the Management of Hemophilia. https://www1.wfh.org/publication/files/pdf-1472.pdf. Accessed 31 January 2017.
- 11. Rickard KA. Guidelines for therapy and optimal dosages of coagulation factors for treatment of bleeding and surgery in haemophilia. Haemophilia. 1995;1(Suppl 1):8‐13. [DOI] [PubMed] [Google Scholar]
- 12. Franchini M, Mannucci PM. The history of hemophilia. Semin Thromb Hemost. 2014;40:571‐576. [DOI] [PubMed] [Google Scholar]
- 13. Lillicrap D. Improvements in factor concentrates. Curr Opin Hematol. 2010;17:393‐397. [DOI] [PubMed] [Google Scholar]
- 14. Giangrande P, Andreeva T, Chowdary P, et al. Clinical evaluation of glycoPEGylated recombinant FVIII: Efficacy and safety in severe haemophilia A. Thromb Haemost. 2017;117:252‐261. [DOI] [PubMed] [Google Scholar]
- 15. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Young G, Mahlangu J, Kulkarni R, et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J Thromb Haemost. 2015;13:967‐977. [DOI] [PubMed] [Google Scholar]
- 17. Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full‐length, recombinant factor VIII for prophylactic and on‐demand treatment of severe hemophilia A. Blood. 2015;126:1078‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. ICH . Tripartite harmonised guideline: Good Clinical Practice: Consolidated Guideline (E6). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed 31 January 2017.
- 19. World Medical Association . Declaration of Helsinki ‐ Ethical Principles for Medical Research Involving Human Subjects. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ Accessed 31 January 2017.
- 20. Reding MT, Ng HJ, Poulsen LH, et al. Safety and efficacy of BAY 94‐9027, a prolonged‐half‐life factor VIII. J Thromb Haemost. 2017;15:411‐419. [DOI] [PubMed] [Google Scholar]
- 21. Berntorp E, Dolan G, Hay C, et al. European retrospective study of real‐life haemophilia treatment. Haemophilia. 2017;23:105‐114. [DOI] [PubMed] [Google Scholar]
- 22. Mazepa MA, Monahan PE, Baker JR, Riske BK, Soucie JM. Men with severe hemophilia in the United States: birth cohort analysis of a large national database. Blood. 2016;127:3073‐3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials