

SUMMARY
Human BRCA1 mutation carriers and BRCA1-deficient mouse mammary glands contain an abnormal population of mammary luminal progenitors that can form 3D colonies in a hormone-independent manner. The intrinsic cellular regulatory defect in these presumptive breast cancer precursors is not known. We have discovered that nuclear factor kappaB (NF-κB) (p52/RelB) is persistently activated in a subset of BRCA1-deficient mammary luminal progenitors. Hormone-independent luminal progenitor colony formation required NF-κB, ataxia telangiectasia-mutated (ATM), and the inhibitor of kappaB kinase, IKKα. Progesterone (P4)-stimulated proliferation resulted in a marked enhancement of DNA damage foci in Brca1−/− mouse mammary. In vivo, NF-κB inhibition prevented recovery of Brca1−/− hormone-independent colony-forming cells. The majority of human BRCA1mut/+ mammary glands showed marked lobular expression of nuclear NF-κB. We conclude that the aberrant proliferative capacity of Brca1−/− luminal progenitor cells is linked to the replication-associated DNA damage response, where proliferation of mammary progenitors is perpetuated by damage-induced, autologous NF-κB signaling.
Graphical Abstract
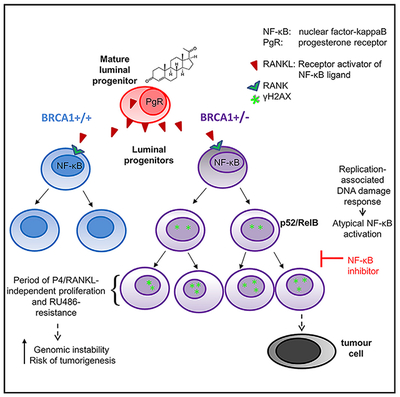
In Brief
BRCA1 mutations increase the risk for breast cancer and have been linked with hormone-independent abnormal proliferation of mammary luminal progenitors. In this study, Sau et al. find that DNA-damage-induced NF-κB activation in BRCA1-deficient luminal progenitors is a cellular mechanism required for hormone-uncoupled proliferation, increasing the risk of genomic instability and transformation.
INTRODUCTION
The hereditary breast and ovarian cancer gene-derived protein, BRCA1, participates in homology-directed repair (HDR) of double-stranded DNA (dsDNA) breaks (Jasin, 2002) and is a component of several multi-protein complexes that mediate chromosomal maintenance and integrity during the cell cycle (Huen et al., 2010). In the absence of BRCA proteins, replication fork stalling is increased and forks are rendered unstable, leading to fork collapse and dsDNA breaks (Schlacher et al., 2012), which can activate the ataxia telangiectasia-mutated (ATM) checkpoint kinase (Lee and Paull, 2005; Willis et al., 2014). The BRCA1 gene is haploinsufficient for stalled fork protection, but not HDR, under conditions mimicking replication stress (Pathania et al., 2014). Therefore replication stress is predicted to generate dsDNA breaks and a DNA damage response (DDR) in HDR-competent BRCA1 heterozygous mammary cells. BRCA1 also has a role in telomere maintenance. Studies of BRCA1-mutated cells and knockdown mammary cells demonstrated perturbation of telomere lengths, shortening of the single stranded 3’ telomere overhang, and other abnormalities (Cabuy et al., 2008; Rosen, 2013).
The cellular architecture of the mammary gland consists of two epithelial lineages: smooth muscle actin-positive myoepithelium surrounding an inner layer of luminal epithelial cells. These differentiated cells are products of a hierarchy beginning with a stem cell capable of self-renewal and repopulating all lineages following transplantation into a cleared mammary fat pad (Visvader and Stingl, 2014). Transit amplification results in several progenitor subpopulations that include luminal progenitor cells (LPs) and basal progenitors (Stingl et al., 2006; Shackleton et al., 2006; Asselin-Labat et al., 2007). Fluorescence-activated cell sorting (FACS)-enriched LPs termed colony-forming cells (CFCs) form acinar colonies in 3-dimensional cultures (Shehata et al., 2012).
The progesterone receptor (PR) antagonist RU486 (mifepristone) has been shown to prevent mammary cancer in Brca1/Tp53-deficient mice (Poole et al., 2006), while BRCA1 mutation carriers who have undergone oophorectomy have reduced breast cancer risk (Eisen et al., 2005; Kramer et al., 2005). Paradoxically, breast cancers arising in this population typically belong to the estrogen receptor/progesterone receptor (ER/PR) and HER2 triple-negative basal subtype (Molyneux and Smalley, 2011). Human and mouse BRCA1-deficient LP cells accumulate abnormal numbers of LPs that are able to form colonies in the absence of B27, which contains progesterone (P4) (Lim et al., 2009). Taken together, these characteristics indicate the presence of an intrinsic defect in BRCA1-deficient progenitor cells that renders their proliferation hormone sensitive but independent in vitro.
Nuclear factor kappaB (NF-κB) is a transcription factor comprised of dimeric members of the Rel family whose activity is regulated by the inhibitor of κBα (IκBα) kinases (IKKs); IKKα, β, and γ (NEMO). There are two main NF-κB pathways (Hayden and Ghosh, 2008; Vallabhapurapu and Karin, 2009). The canonical pathway consists primarily of nuclear p65/p50 and is activated following phosphorylation and subsequent degradation of IκBα by IKKα/β. p65 phosphorylation at serine 536 also induces canonical activity (Sakurai et al., 1999). The alternative NF-κB pathway (p100/p52) is regulated by IKKα homodimers that phosphorylate p100 to signal its partial proteolytic processing to mature p52. p52 then translocates to the nucleus with RelB to mediate transcriptional regulation (Dejardin, 2006; Vallabhapurapu and Karin, 2009). Importantly, NF-κB can also be activated in an atypical manner by DNA damage (Hadian and Krappmann, 2011; Miyamoto, 2011). This pathway involves formation of a complex between ATM, NEMO and IKKs resulting in canonical NF-κB activation. Alternative NF-κB activity in response to DNA damage has also previously been described (Josson et al., 2006).
NF-κB is required for normal proliferation and branching in the mouse mammary epithelium (Brantley et al., 2001). Knockin mice lacking IKKα catalytic function fail to undergo lobuloalveloar expansion during pregnancy (Cao et al., 2001). Thus, IKKα and alternative NF-κB activity are transiently required for amplification of ductal and alveolar cells. Signal activation of NF-κB is mediated by members of the tumor necrosis factor alpha (TNF-α) receptor family including the receptor activator of NF-κB (RANK). P4 induces RANK ligand (RANKL) expression in PR-positive luminal cells (Brisken et al., 1998), thought to result in proliferation of stem and progenitor cells that mature under the control of lactogenic hormones during pregnancy (Asselin-Labat et al., 2010; Joshi et al., 2010).
NF-κB also contributes critical signaling in cancer cells and is often altered in both solid and hematopoietic human malignancies. Through transcriptional regulation of a wide spectrum of genes, NF-κB can promote proliferation, angiogenesis, metastasis, tumor promotion, inflammation, and cell survival (Baud and Karin, 2009). Importantly, genetic inhibition of NF-κB can prevent or attenuate mammary cancers in mice (Cao et al., 2007; Pratt et al., 2009).
In this study, we have sought to determine the underlying defect(s) and account for hormone-mediated signaling pathways that promote accumulation of B27 factor-independent progenitor cells in BRCA1-deficient mammary glands. We have identified a unifying mechanism that integrates genomic instability-induced DNA damage with proliferative signaling in BRCA1-deficient mammary epithelial cells (MECs) involving ATM and NF-κB activation.
RESULTS
NF-κB Is Activated in BRCA1-Deficient Luminal Progenitors
Cells deficient in BRCA1 function are distinctly susceptible to replication stress (Schlacher et al., 2012) as well as telomere dysfunction (Cabuy et al., 2008; Sedic et al., 2015), both of which can activate a DDR. Since genotoxic stress resulting in the DDR can activate NF-κB through ATM:NEMO (Hadian and Krappmann, 2011; Miyamoto, 2011), we hypothesized that NF-κB might be activated in BRCA1-deficient mammary progenitor cells as a consequence. Absence of BRCA1 protein and genomic PCR confirmed recombination in mammary progenitors from 10-week-old Brca1f/f ;MMTV-cre+(Brca1−/−), but not Brca1f/f;cre− (control/Brca1+/+), mice (Figures S1A and S1B). We initially performed an immunoblot of protein extracts from 3-day cultures of primary mouse lineage-negative cells (Lin−; hematopoietic and endothelial cells removed) from Brca1−/− and control mice and detected a 6-fold increase in p52, the transcriptionally active form of p100/p52 (Figure 1A). MECs from these mice and Brca1f/+;MMTV-cre (Brca1+/−) were enriched by FACS then cytospun for immunofluorescence (IF) analysis. Control and Brca1−/− FACS-sorted CD24+/CD29lo MEC subpopulations were further enriched based on CD61 expression to separate mature LPs (ML) (CD61−) and the balance of LP (CD61+) populations (Asselin-Labat et al., 2007). Representative dot plots are shown in Figure 1B. Sorted cells were subjected to IF for p100/p52. Nuclear p52 was increased in ~50% of Brca1−/− LPs and <20% of MLs compared with <5% in LPs and MLs in Brca1+/+ (Figures 1C–1F). Approximately 30% of Brca1+/− mouse LP cells, which approximate Brca1mut/+ carriers, also contained nuclear p52 (Figures 1G and 1H). Figure S1C shows higher-magnification images of p100/p52 in the CD24+/CD49+ population, which contains both LPs and MLs. Nuclear RelB was similarly increased in ~35% of CD24+/CD49f+ nuclei from Brca1−/− mice (Figures S1D and S1E). The same population of BRCA1−/− MECs did not show an increase in overall numbers of nuclei containing p65 by IF, but the average intensity of nuclear p65 was increased by ~2-fold (Figures S1F–S1H).
Figure 1. The Alternative NF-κB Pathway Is Constitutively Active in BRCA1-Deficient Mammary Progenitor Cells.
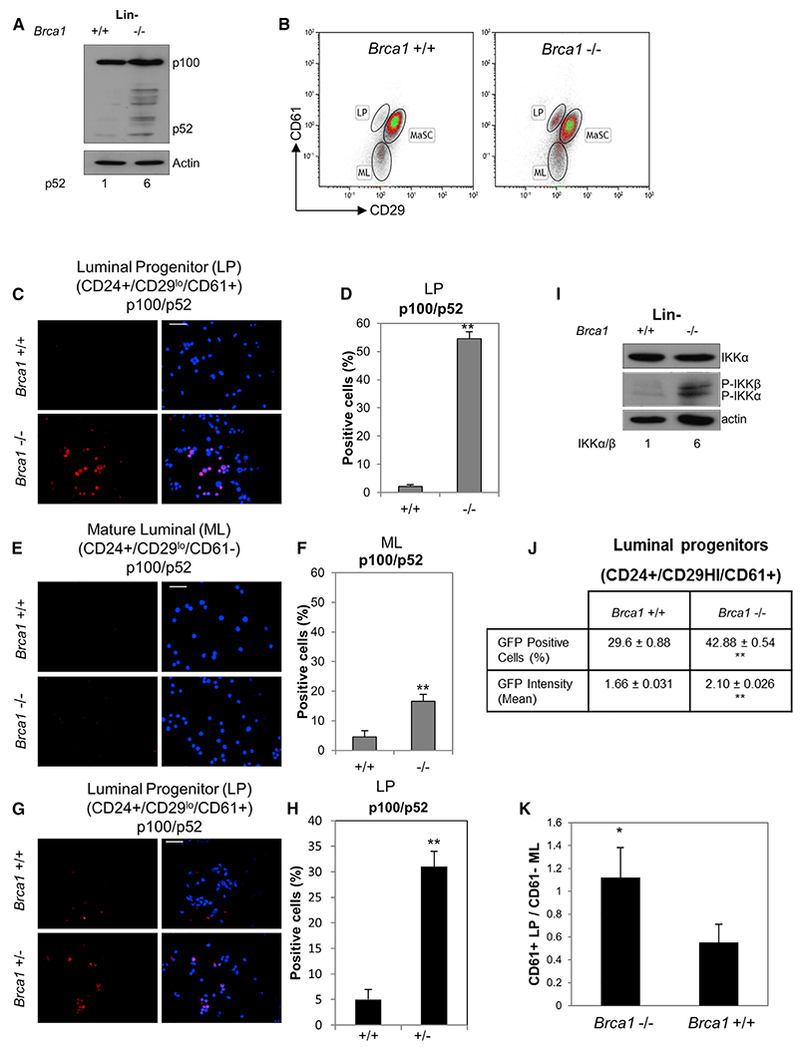
(A) Lin− Brca1+/+ and Brca1−/− cells were expanded in monolayer culture for 3 days, and protein extracts were subjected to immunoblot with anti-p100/p52. Numbers below the blots represent relative density normalized to actin.
(B) Representative FACS images of primary MECs from Brca1+/+ and Brca1−/− mice enriched based on CD24/CD29/CD61 immunophenotype. Mammary stem cell (MaSC) (CD24+/CD29hi/CD61+), luminal progenitor (LP) (CD24+/CD29lo/CD61+), and mature luminal (ML) (CD24+/CD29lo/CD61−) cell populations are shown.
(C and E) Brca1−/− and Brca1+/+ LP and ML cell fractions were FACS sorted, cytospun, and immunostained. Representative images are shown for anti-p100/p52. Scale bar, 20 μm.
(D and F) Percentage of p52-positive cells in LP- and ML-enriched populations, respectively. Bars on graphs are ±SEM (**p < 0.01, unpaired t test).
(G) CD24hi/CD29lo/CD61+ primary cells from Brca1+/− mice were FACS sorted, cytospun, and immunostained with anti-p100/p52. Representative image is shown (n = 15). Scale bar, 20 μm.
(H) Graph showing percentage of Brca1+/− p100/p52-positive cells ± SEM values derived from 15 fields. **p < 0.01, unpaired t test.
(I) Lin− cells as in (A) were immunoblotted with anti-IKKα and P-IKKα/β antibodies. Actin reactivity was used as a protein loading control. Numbers below represent relative density of P-IKKα normalized to actin.
(J) FACS-sorted CD24+/CD29hi/CD61+ cells were infected for 72 hr with lenti-NF-κB reporter and GFP expression analyzed by FACS. **p < 0.01, unpaired t test.
(K) Ratio of LP/ML cell populations obtained from FACS analyses of n = 5 Brca1+/+ and five Brca1−/− mice (*p < 0.05, unpaired t test). Bars on graphs are ±SEM.
See also Figure S1.
Increased IKKα and IKKβ phosphorylation was detected in Brca1−/− Lin− cell extracts in the absence of a change in total IKK protein expression (Figure 1I). P-IKKα/β was localized to nuclei and perinuclear regions of CD24+/CD49+ Brca1−/− cells, but not Brca1+/+ cells (Figure S1I). To assess NF-κB-mediated transcription, FACS-sorted LPs were transduced with a lentiviral NF-κB-GFP reporter gene. Brca1−/− cells showed differential GFP expression (Figure 1J). Moreover the increased expression of two NF-κB-responsive genes relative to controls was reduced by the IKK inhibitor BMS-345541 (BMS) (Figure S1J).
The accumulation of Brca1−/− mammary progenitor cells may be a reflection of both proliferation and delayed differentiation. Consistent with this, the ratio of LPs to MLs from 8- to 10-week-old mice was significantly higher in Brca1−/− mice than in control mice (Figure 1K).
BRCA1 Knockdown Induces Phosphorylation of p65 on Ser536 and p100/p52 Processing in Human Mammary Cell Lines
Genotoxic stress-mediated IKKα/β activation is followed by induction of canonical NF-κB (Miyamoto, 2011). To further investigate the canonical NF-κB response to BRCA1 deficiency, we knocked down BRCA1 in rapidly proliferating MECs. Transient expression of the stable S32/36A-IκBα mutant of IκBα (IκBαSR) did not inhibit p52 protein expression in Brca1-depleted HC11 cells (Figure 2A). Phosphorylation of p65 ser536 results in nuclear translocation and heterodimeric DNA binding with p105/p50 (Sakurai et al., 1999). Remarkably, P-Ser536-p65 was strongly induced in mouse HC11 cells independent of IκBαSR expression (Figure 2B). BRCA1 knockdown (KD) in MCF-10A cells, representing a basaloid progenitor (Neve et al., 2006), similarly induced P-ser536-p65 (Figure 2C). Thus activation of NF-κB by depletion of BRCA1 is not restricted to LP cells. It is notable that in addition to LPs, the basal cell fraction of human mammary Brca1mut/+ mammary tissue is also aberrantly expanded (Lim et al., 2009). BRCA1 KD-induced p52 was prevented by a secondary KD of p105/p50 to block the canonical pathway (Figure 2C), indicating that canonical signaling is essential to sustain the alternative pathway. An increase in P-ser536 p65 was also detected in Lin− MECs from Brca1−/− mice (Figure S2A).
Figure 2. BRCA1 Depletion Induces ser536-p65 Phosphorylation and Alternative NF-κB in Human Mammary Epithelial and Breast Cancer Cells.
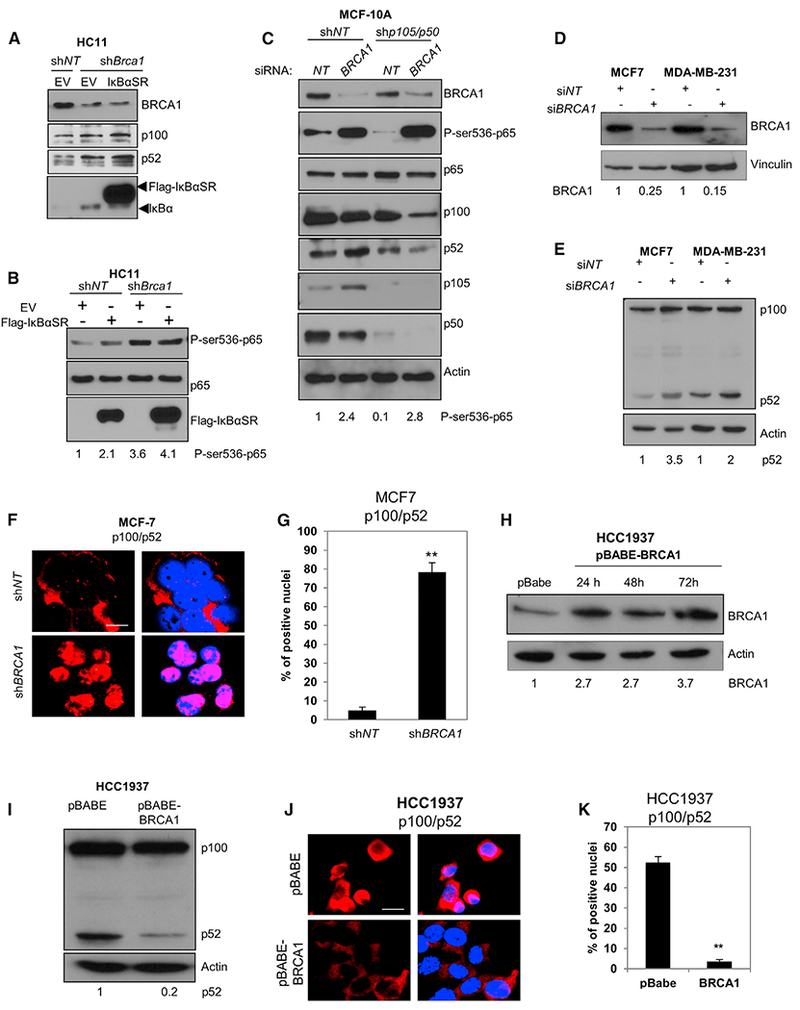
(A) HC11 mouse MECs stably expressing shNT or shBRCA1 were transfected with empty vector (EV) or CMV4-FLAG-IκBαSR and harvested after 72 hr. Immunoblots were reacted with anti-BRCA1, p100/p52, or IκBα.
(B) HC11 cells described in (A) were immunoblotted with P-ser536-p65, p65, and IκBα antibodies.
(C) MCF-10A immortalized human MECs were infected with lenti-shNT or shp105/p50 and then transfected 72 hr later with BRCA1 or NT siRNA. After an additional 72 hr, cells were harvested and immunoblotted with the indicated antibodies. Actin was used as a loading control.
(D and E) MCF-7 and MDA-MB-231 cells were transfected with BRCA1 siRNA (siBRCA1) and non-targeting siRNA (siNT), and cell lysates were immunoblotted 72 hr later with anti-BRCA1 (D) or anti-p100/p52 (E). Numbers below the blots represent relative density normalized to vinculin or actin.
(F) MCF-7 cells were infected with BRCA1 lenti-shRNA (shBRCA1) and non-targeting lenti-shRNA (shNT), cultured for 72 hr, and then immunostained with anti-p100/p52 and RelB antibodies, respectively. Scale bar, 10 μm.
(G) Nuclear fluorescence intensities for p100/p52 and RelB, respectively, were determined using ImageJ and represented as mean intensities normalized to the control (shNT). Bars on graphs are ±SEM (**p < 0.01 unpaired t test).
(H) HCC1937 BRCA1mut/− cells were infected with retrovirus pBABE-BRCA1 or empty vector (pBABE). Cell lysates were collected at indicated times and immunoblotted with anti-BRCA1 antibody. Numbers below the blots represent relative density normalized to actin.
(I) Immunoblot with anti-p100/p52 antibody in extracts from pBABE-BRCA1 and pBABE-infected HCC1937 cells. Numbers below represent relative densities for the p52 band normalized to actin.
(J) Representative images of HCC1937 Brca1mut/− breast cancer cells 72 hr following infection with retrovirus pBABE-BRCA1 or empty vector. Cells were immunostained with anti-p100/p52 and RelB antibodies. Scale bar, 10 μm.
(K) Mean nuclear fluorescence intensity for p100/p52 and RelB in HCC1937 cells normalized to the control (pBABE) (**p < 0.01, unpaired t test). Bars on graphs are ±SEM.
See also Figure S2.
MDA-MB-231 basal breast cancer cells express high endogenous p52 compared to MCF-7 cells. Immunoblot analysis showed a modest increase in mature p52 72hrs after BRCA1 KD while a greater increase was obtained in MCF-7 cells (Figures 2D and 2E). BRCA1 KD also resulted in accumulation of both nuclear p52 (Figures 2F and 2G) and RelB (Figures S2B and S2C). Consistent with the ability of NF-κB to promote cell growth, the increased nuclear p52/RelB resulted in enhanced proliferation in MCF-7 cells, but not MDA-MB-231 cells (Figures S2D and S2E).
To directly address the regulation of alternative NF-κB by BRCA1, we infected HCC1937 BRCA1-mutated human breast cancer cells with retrovirus expressing WT BRCA1 (Figure 2H). p52 protein and both nuclear p52 and RelB were strongly repressed by expression of BRCA1 (Figures 2I–2K).
p100/p52 Is Essential for NF-κB Brca1-Deficient Hormone-Independent CFC Growth
Three dimensional acinar colony formation is a functional assay for LPs where colony numbers represent the frequency of CFCs and colony diameter is indicative of proliferative capacity (Williams et al., 2008). Since P4 can induce RANKL expression in MECs (Brisken et al., 1998), thereby stimulating NF-κB in RANK-expressing MECs (Cao et al., 2001), we speculated that the endogenous level of NF-κB might supplant the requirement for P4 in vitro in Brca1-deficient LPs. Both Brca1−/− and Brca1+/+ mouse LPs formed colonies when cultured in media with B27 containing P4, while only Brca1−/− cells formed colonies in the absence of B27 (Lim et al., 2009). The role of p100/p52 in B27-independent CFCs was directly tested by lentiviral small hairpin RNA (shRNA)-mediated KD of p100/p52 (Figure 3A). p100/p52 KD in Brca1+/+ Lin− cells had a small but significant effect on acinar size and, as expected, these cells did not form colonies in the absence of B27. Similarly, p100/p52 KD in Brca1−/− cells only modestly reduced colony growth in medium with B27. However, the reduction of p100/p52 by different p100 shRNAs in Brca1−/− Lin− cells strongly diminished B27-independent colony sizes and numbers (Figures 3B, 3C, and S3A–S3C). Thus, CFCs do not require p100/p52 for proliferation in the presence of B27, but BRCA1-deficient CFCs need alternative >NF-κB for B27-independent growth. Inhibition of IKKα/β with BMS inhibited primary and secondary mammosphere formation in both Brca1+/+ and Brca1−/− LPs (Figure S3D), indicating that NF-κB activity is necessary for expansion of cell numbers regardless of Brca1 status. Importantly, Brca1+/− mouse mammary cells also formed colonies in the absence of B27 (Figures 3D, 3E, and S3E), confirming that a single wild-type copy in mice is insufficient to prevent hormone-independent growth.
Figure 3. Alternative NF-κB Is Essential for BRCA1-Deficient CFCs in the Absence of B27.
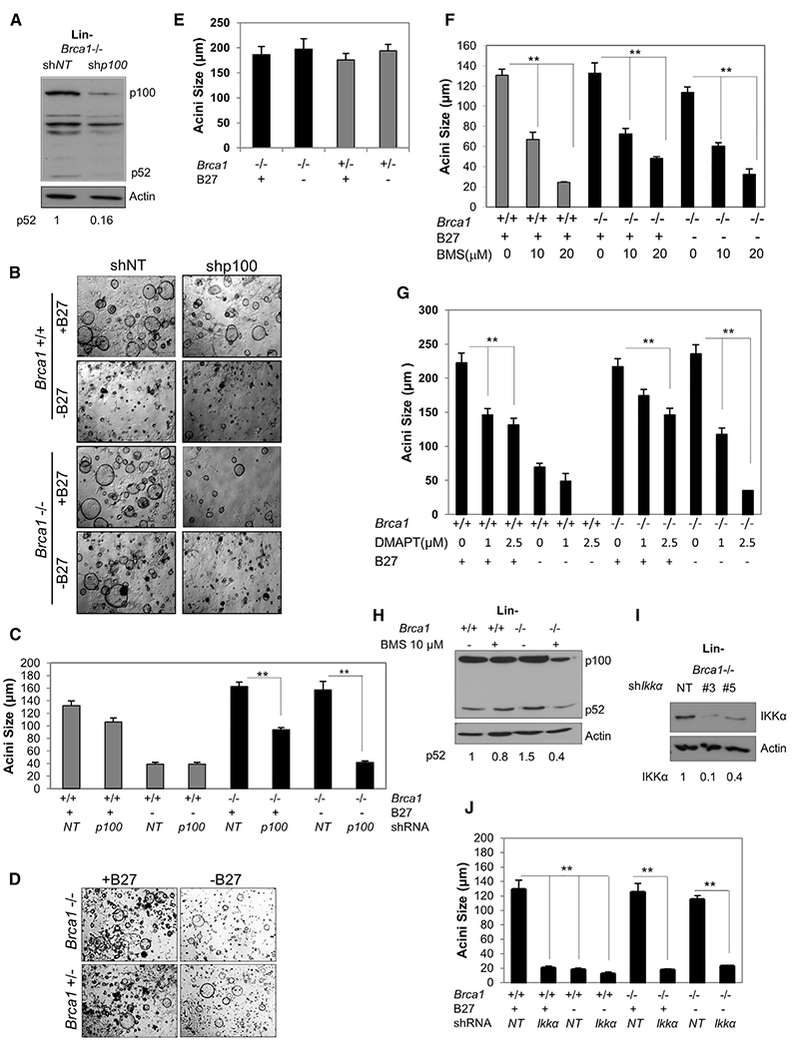
(A) Immunoblot of Brca1−/− Lin− cell extracts collected 72 hr after infection with p100/p52 lenti-shRNA (shp100) or non-targeting lenti-shRNA (shNT). Relative densities normalized to actin are shown below.
(B) Representative images of CFC assays of Lin- cells in the presence or absence of the B27 media supplement. Cells were infected just prior to plating with lenti-shp100 or shNT. Scale bar, 200 μm.
(C) Acini diameters from Lin− cells infected with shp100 or shNT and plated on Matrigel. Bars on graphs are ±SEM (**p < 0.01, two-way ANOVA followed by Tukey test).
(D) Representative images of CFC assays in the presence or absence of B27 supplement using Lin− cells from Brca1−/− and Brca1+/− mice.
(E) Diameters of acini formed by Lin- Brca1−/− and Brca1+/− MECs in the presence or absence of B27.
(F) Diameters of acini formed by CFCs from Lin− cells in the presence of vehicle (0) or 10 μM or 20 μM BMS.
(G) Diameters of acini formed by CFCs from Lin− cells in the presence of vehicle (0) or 1 or 2.5μM DMAPT.
(H) Cell lysates were collected from Lin− cells after 72 hr of 10 μM BMS treatment and immunoblotted with anti-p100/p52. Relative densities normalized to actin are shown below.
(I) Cell lysates were collected from Brca1−/− Lin− cells infected with two different IKKα lenti-shRNAs or non-targeting lenti-shRNA and immunoblotted for IKKα. Relative densities normalized to actin are shown below.
(J) Acini diameters from CFCs derived from Lin− cells infected with IKKα lenti-shRNAs or non-targeting lenti-shRNA and plated on Matrigel. Bars on graphs are ±SEM (**p < 0.01, two-way ANOVA followed by Tukey test). See also Figure S3.
To determine if IKK signaling has a role in the B27-independent progenitor proliferation, CFC assays were performed in the presence or absence of B27 and/or BMS. BMS and a second IKK inhibitor, dimethylamino-parthenolide (DMAPT) (Neelakantan et al., 2009), reduced both control and Brca1−/− colony sizes in all culture conditions (Figures 3F, 3G, and S3F–S3H). The reduction in p52 in 3-day monolayer cultures of Lin− cells in the presence of BMS (Figure 3H) or DMAPT (Figure S3I) was confirmed by immunoblot.
IKKα specifically activates alternative NF-κB and promotes canonical signaling through IKKα/β dimers. Therefore, similar to the IKK inhibitors, KD of IKKα with two different shRNAs completely prevented colony formation independent of Brca1status or the presence of B27 (Figures 3I and 3J).
Atm Is Required for B27-Independent Mammary Progenitor Cell Proliferation
DDR-induced NF-κB involves the ATM-mediated activation of IKKs (Miyamoto, 2011). Activated ATM (P-ser1981 ATM) was detected by IF in the CD24+/CD49f+ progenitor cells from Brca1−/−, but not Brca1+/+, mammary glands (Figures 4A and 4B). Similar results were obtained in MCF-7 (shBRCA1) cells, but not MCF-7(shNT) cells (Figure S4A).
Figure 4. ATM Is Activated, Interacts with NEMO, and Is Required for Brca1-Deficient B27-Independent Colony Formation.
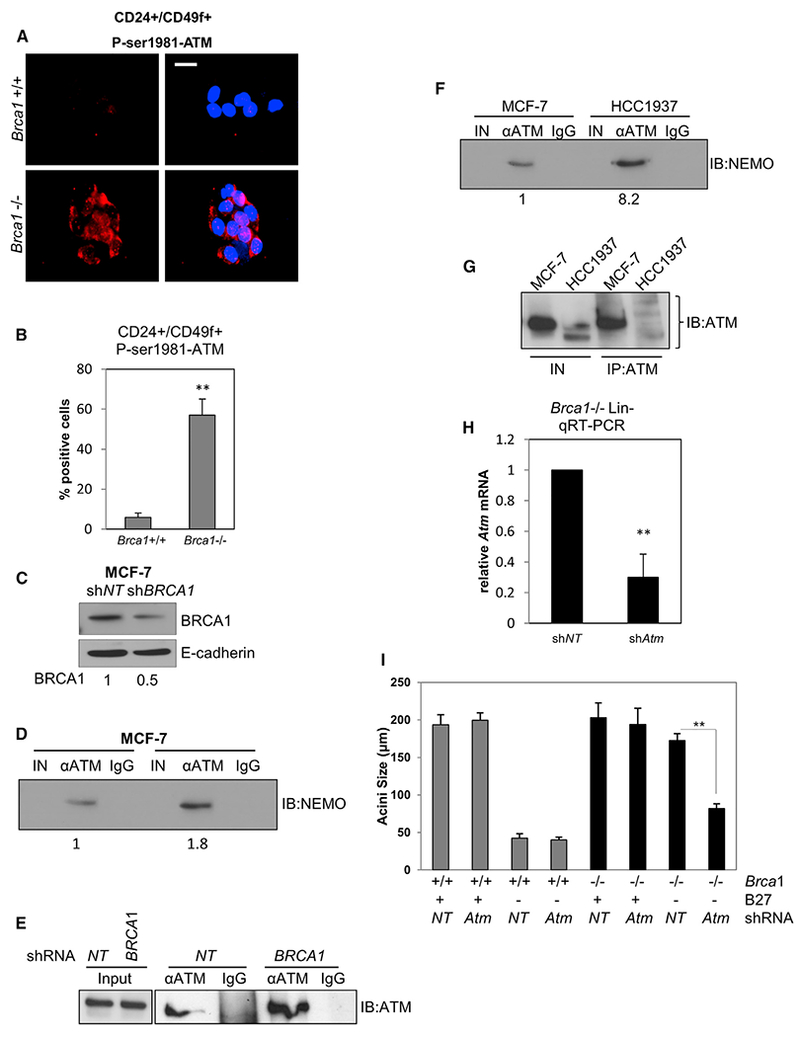
(A) CD24+/CD49f+ FACS-sorted cells from Brca1+/+ and Brca1−/− mice were cultured for 72 hr and then immunostained with anti-phospho ATM. Scale bar, 10 μm.
(B) Nuclear fluorescence intensity for P-ser1981-ATM was determined using ImageJ and represented as mean intensities normalized to the control (Brca1+/+). **p < 0.01, unpaired t test.
(C) MCF-7 cells were infected with BRCA1 lenti shRNA (shBRCA1) and non-targeting shRNA (shNT) for 72 hr. Cell lysates were collected and immunoblotted with anti-BRCA1 normalized to E-cadherin.
(D) 20 μg input lysate (IN), α-ATM immunoprecipitate (IP), or non-immune IgG IP derived from MCF-7 cells infected with shBRCA1 shNT was immunoblotted (IB) to detect NEMO. Relative densities normalized to IB:ATM in (E) are shown below.
(E and G) Duplicate IN and ATM IPs from cells in (D) and (F), respectively, were immunoblotted on a separate 6% polyacrylamide gel to facilitate immunoblot of high-molecular-weight ATM.
(F) Twenty μg of input lysate (IN), α-ATM IP or non-immune IgG IP derived from MCF-7 cells and HCC1937 were subjected to immunoblot (IB) to detect NEMO. Results are typical of two separate experiments.
(H) qRT-PCR for ATM mRNA in Lin- cells infected with lenti-shRNA to ATM (shATM) or non-targeting lenti-shRNA (shNT) normalized to the control (shNT). Bars on graphs are ±SEM (**p < 0.01, unpaired t test).
(I) Acini diameters from CFC assays of Lin- cells infected with shATM or shNT. Bars on graphs are ±SEM (**p < 0.01, two-way ANOVA followed by Tukey test).
See also Figure S4.
ATM and NEMO interact in a complex following DNA damage resulting in downstream NF-κB activation (McCool and Miyamoto, 2012). If this pathway is engaged in cells deficient for BRCA1, we expected that this association would be enhanced in BRCA1-deficient cells. Immunoprecipitation (IP) of ATM followed by immunoblot for NEMO revealed an increase in NEMO:ATM complexes in MCF-7 (shBRCA1) compared with MCF-7 (NT shRNA) cells (Figures 4C–4E). Moreover, more NEMO was detected in complex with ATM in HCC1937 cells compared with MCF-7 cells (Figure 4F), despite lower levels of ATM in the HCC1937 IP (Figure 4G). We next performed CFC assays using Lin- cells depleted of Atm after infection with Atm lenti-shRNA (shAtm) or non-targeting lenti-shRNA (shNT) (Figure 4H) in the presence or absence of B27. While ATM KD did not affect colony sizes in either the Brca1−/− or control cells in the presence of B27, loss of ATM drastically reduced the size of colonies obtained from Brca1−/− cells assayed in the absence of B27 (Figures 4I, S4B, and S4C). Similar results were obtained when CFC assays were performed in the presence or absence of the ATM kinase inhibitor KU-55933 (Figures S4D and S4E). Together, these results indicate a critical requirement for DDR signaling in B27-independent growth.
The NF-κB Inhibitor DMAPT Mediates Prolonged Suppression of the B27-Independent Progenitor Population In Vivo
Although in vitro assays demonstrated the effects of NF-κB inhibition on BRCA1-deficient colony formation, other factors could mediate the growth of aberrant progenitor cells within the intact mammary gland. To assess the effect of NF-κB inhibition in vivo on the B27-independent population of mammary progenitors, control and Brca1−/− mice were administered 40 mg/kg DMAPT intraperitoneally (i.p.) and Lin− mammary cells were collected and subjected to acini assays. Lin− cells from Brca1−/− mice collected on day 1 after final treatment showed strong inhibition of B27-independent colony formation (Figure 5A). Remarkably, only partial recovery from inhibition occurred in vivo after 15 days post-treatment representing approximately six mouse estrus cycles (Figure 5B). Immunoblot assay of cell extracts from 3-day cultures of Lin− MECs from each treatment group for p100/p52 confirmed p52 reduction by DMAPT in cells derived from control and Brca1−/− mice (Figure 5C). These results are consistent with a requirement for NF-κB in the generation of genetically unstable aberrant mammary progenitor cells in vivo.
Figure 5. DMAPT Eliminates B27-Independent Progenitor Cell Proliferation In Vivo.
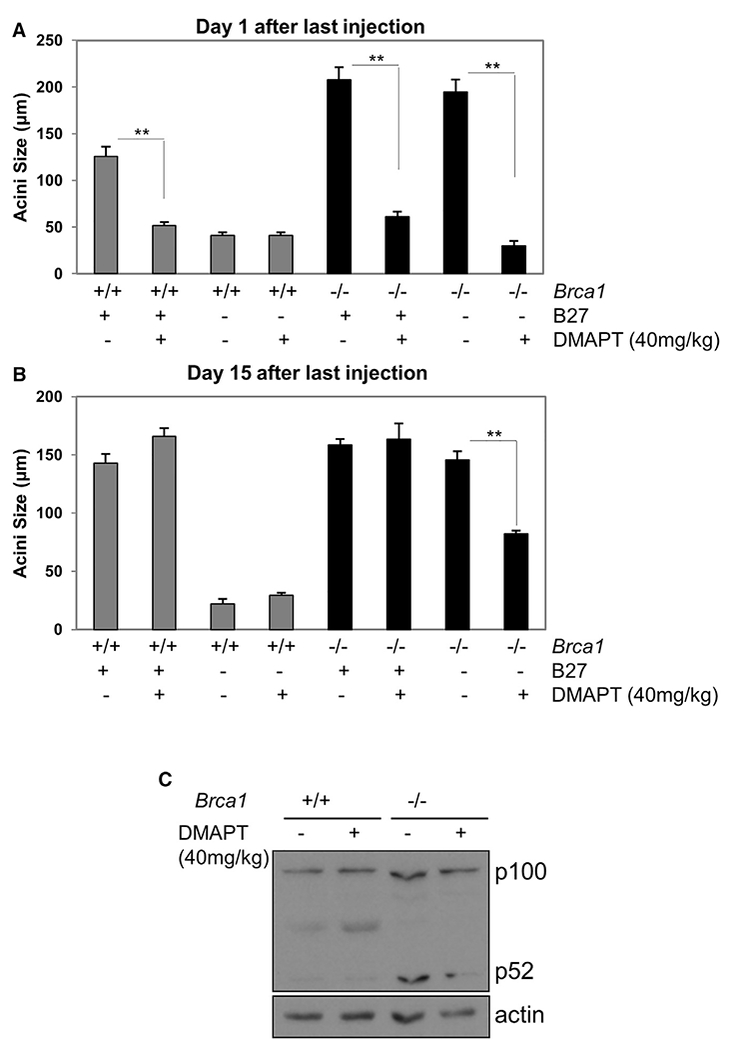
(A and B) Brca1+/+ and Brca1−/− mice were injected i.p. with 40 mg/kg DMAPT or vehicle every 2 days for 2 weeks. Lin− cells were isolated 1 day (A) or 15 days (B) after the last injection and subjected to CFC assay in the presence or absence of B27. Bars on graphs are ±SEM (**p < 0.01, two-way ANOVA followed by Tukey test).
(C) Lin− cells isolated as in (A) were cultured for 2 days in monolayer and lysates subjected to immunoblot to assess p100/p52.
Progesterone Induces DNA Damage in Brca1−/− Mammary Glands
P4 induces proliferation in mammary glands through the RANKL/RANK axis (Cao et al., 2001; Gonzalez-Suarez et al., 2007; Schramek et al., 2010), and the PR antagonist mifepristone inhibits tumor formation in Brca1−/−;Tp53+/− mammary glands (Poole et al., 2006). We speculated that the role of P4 in tumor formation in BRCA1-deficient mammary glands might be through promotion of replication stress. We first assessed DDR foci in FACS-sorted LPs from each mouse group. Strikingly, Brca1−/− LPs contain numerous cells with gH2AX foci that resemble foci in Adriamycin-treated cultures of progenitors compared with few BRCA1+/+ LPs (Figure 6A). Next, we injected Brca1−/− and Brca1+/+ mice daily for 3 days with vehicle or P4 and then isolated mammary glands 24 hr after the final injection. Paraffin sections were immunostained with anti-γH2AX to detect DDR foci. γH2AX foci were sometimes detected in vehicle-treated Brca1−/− ductal, but not control, glands. After P4 injections, weakly stained foci were clearly evident in treated control ducts, although intense γH2AX foci were present in Brca1−/− ducts, consistent with a P4-induced DDR (Figure 6B). These effects were also clearly evident after immunoblot for γH2AX in Lin− cells from saline or P4-treated mice (Figure 6C).
Figure 6. DMAPT Treatment Reduces DNA Damage in BRCA1-Deficient Cells.
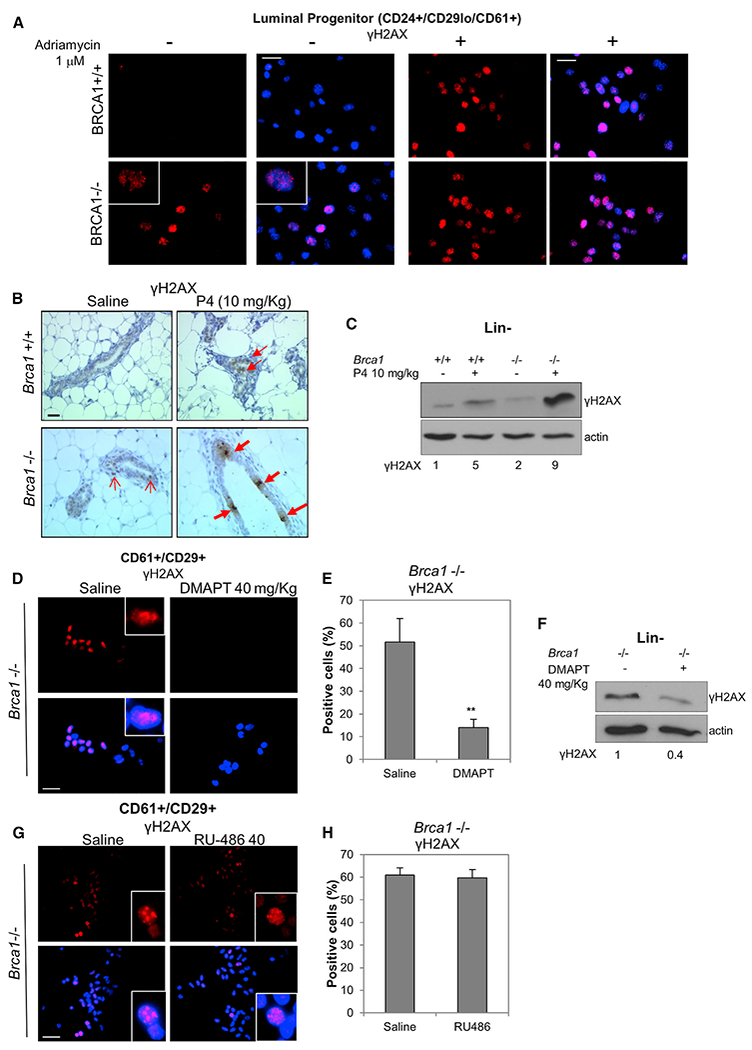
(A) CD24+/CD29lo/CD61+ cells from Brca1+/+ and Brca1−/− mice were FACS-sorted and plated for 24 hr. γH2AX levels were assessed by IF. Adriamycin was added at 1 μM and used as positive control for γH2AX foci. Scale bar, 20 μm.
(B) Brca1+/+ and Brca1−/− mice were injected daily with progesterone at 10 mg/kg or saline for 3 days. Mammary glands were collected the day after the last injection, paraffin embedded, and subjected to IHC with anti-γH2AX antibody. Open and closed arrows indicate light and intense γH2AX foci, respectively. Scale bar, 30 μm.
(C) Same as in (B), but the day after the last injection, Lin− cells were isolated from mouse mammary glands and plated for 72 hr. Cell lysates were then collected and immunoblotted with anti-γH2AX. Relative densities normalized to actin are shown below.
(D) Brca1−/− mice were injected every 2 days over a period of 14 days with 40 mg/kg DMAPT or saline. The day after the last injection, CD29+/CD61+ cells were FACS sorted, cytospun onto slides, and subjected to IF with anti-γH2AX. Scale bar, 20 μm.
(E) The graph indicates the percentage of γH2AX-positive cells from (D). Bars on graphs are ±SEM (**p < 0.01, unpaired t test).
(F) Lin− cells were isolated from Brca1−/− mice injected with DMAPT or saline as in (D) and plated for 72 hr. Cell lysates were immunoblotted with anti-γH2AX. Relative densities normalized to actin are shown below.
(G) BRCA1−/− mice were injected for 3 days with 40 mg/kg RU-486 or saline. The day after the last injection, CD29+/CD61+ cells were FACS sorted, cytospun onto slides, and subjected to IF with anti-γH2AX. Scale bar, 20 μm.
(H) The graph indicates the percentage of γH2AX-positive cells from (G). Bars on graphs are ±SEM.
See also Figure S5.
To investigate the effect of in vivo treatment with an NF-κB inhibitor, CD24+/CD29+/CD61+ LPs from Brca1−/− mice that had been injected with saline or DMAPT were sorted and cytospun and then subjected to IF for γH2AX. DMAPT profoundly reduced the detection of γH2AX foci (Figures 6D and 6E), which was confirmed by immunoblot of Lin− cells from vehicle and DMAPT-treated mice (Figure 6F). Notably, we did not detect increased apoptosis in Brca1−/− mammary ducts following DMAPT treatment (Figure S5). If DDR-induced NF-κB supplants the requirement for the P4 stimulus, allowing LPs to proliferate in a hormone-independent manner, continued replication-associated DNA damage should be resistant to short-term exposure to a PR antagonist. To test this, we treated Brca1−/− mice daily for 5 days with 40 mg/kg of RU486. In contrast to DMAPT, RU486 had no effect on γH2AX foci-containing progenitor cells (Figures 6G and 6H). These data show that P4-induced proliferation induces DNA damage resulting in a DDR in Brca1−/− MECs in vivo. Moreover, DNA damage associated with proliferation persists for an extended period in Brca1−/− MECs in the absence of P4.
p100/p52 Expression in BRCA1-Mutated Human Mammary Glands
If human BRCA1mut/+ mammary glands differentially sustain enhanced DNA damage and induction of the DDR, nuclear NF-κB proteins may also be evident in intact mammary tissue. The presence and localization of p100/p52 in mammary gland sections from eight BRCA1 mutation carriers and ten age-matched normal controls was analyzed by immunohistochemistry (IHC). Almost two-thirds (6/8) of the BRCA1mut/+ cases expressed high-intensity anti-p100/p52 immunostaining in approximately half of the whole lobular units present in the section. Other BRCA1mut/+ lobules (from the same sections) and most control lobules showed less intense staining. Representative images of p100/p52 IF are presented in Figure 7A and show high and low levels of anti-p100/p52 immunostainingin normal and BRCA1mut/+ lobular units. IHC demonstrated that relatively uniform, mostly cytoplasmic expression of p100/p52 was detected in the normal samples. In contrast, many nuclei within BRCA1mut/+ lobules were positive for p52. Examples of IHC in Figure 7B represent sections with a low and high percentage of p52-positivenuclei. Because p52 IF varied across BRCA1mut/+ lobules, we determined the mean percentage of p52-positive nuclei across six lobules with the highest percentages of nuclear p52-positive cells. The results in Figure 7C show that BRCA1mut/+ lobules contained a significantly higher percentage of nuclear p52-positive cells compared to normal lobular sections.
Figure 7. Nuclear p52 Is Differentially Expressed in Luminal Cells of Lobules within BRCA1mut/+ Human Breast Tissue.
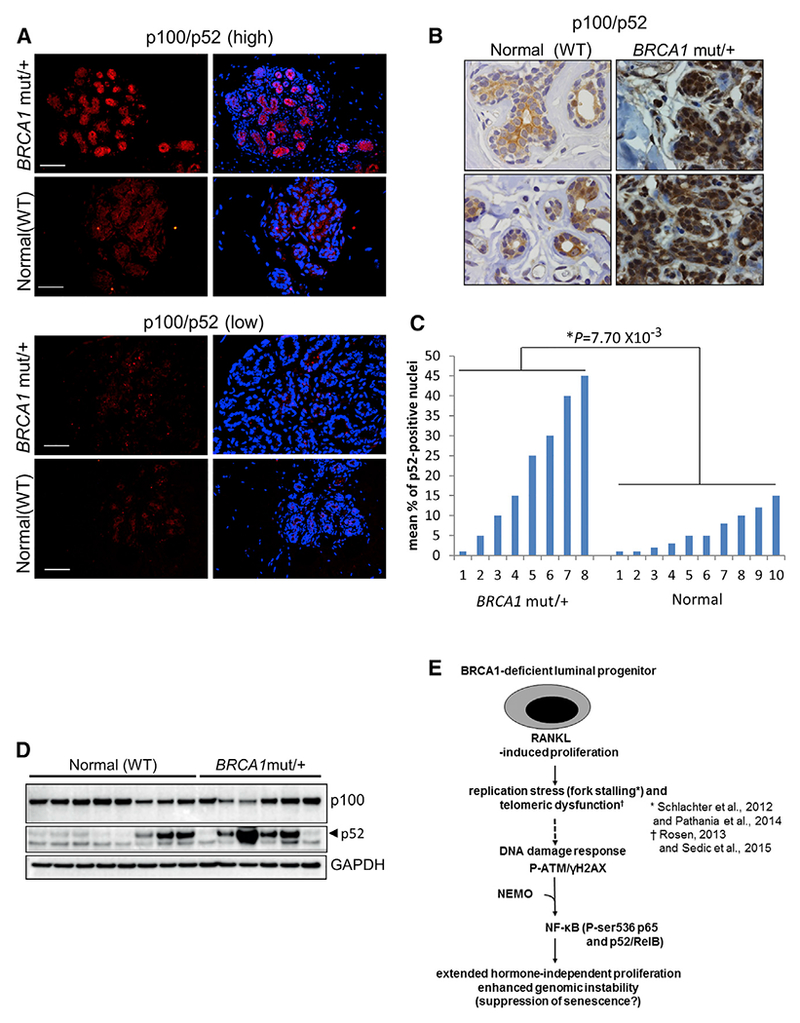
(A) Representative anti-p100/p52 IF on lobular units in deparaffinized sections from BRCA1mut/+ breast tissue or normal breast tissue from reduction mammoplasty. Scale bar, 30 μm.
(B) Examples of anti-p100/p52 IHC on sections of human mammary tissue. The WT and BRCA1mut/+ samples show a low and high percentage of nuclear p52, respectively (63× objective).
(C) Mean percentages of p52-positive nuclei from BRCA1mut/+ and WT patient mammary sections. Values were determined by IHC analysis of 200–300 cells in the six lobules with the highest percentages of nuclear p52 (p = 0.0077, unpaired t test).
(D) Western blot for p100/p52 showing sorted epithelial cells from frozen organoids. Lysates from ~105 cells were loaded in each lane. Samples denoted as “normal” are from reduction mammoplasties of women with verified wild-type BRCA1. BRCA1mut/+ samples are normal breast tissue from patients with no cancer who underwent a prophylactic mastectomy.
(E) Hypothetical model of DDR signaling leading to activation of NF-κB in BRCA1-deficient progenitor cells. p52/RelB promotes proliferation to expand the genomically unstable LP population in the absence of P4. See text for details.
In samples derived from organoid cultures of human MECs from six BRCA1 mutation carriers and eight age matched normal mammoplasty controls, an intense 52-kDa band was detected in four out of six (66%) BRCA1mut/+ lysates compared to three out of eight (37.5%) normal MEC samples (Figure 7D). Although the number of samples is not sufficiently high to obtain significance, p52 expression in BRCA1 mutation carriers appears more consistently elevated compared to normal controls.
DISCUSSION
BRCA1 is a multifunctional protein with roles in cell-cycle control, ubiquitination, and transcriptional regulation (Buckley and Mullan, 2012). In the mammary gland BRCA1 also participates in the regulation of genes that are critical for normal luminal cell differentiation (Gorski et al., 2010; Mullan et al., 2006). Although these functions are likely to be integral to the contribution of BRCA1 in mammary gland function, the role of BRCA1 in DNA damage repair, chromosome duplication, and segregation is thought to be the cornerstone of its tumor suppressor activity (Venkitaraman, 2014).
Signal-induced NF-κB is critically required for expansion of mammary progenitor cells during pregnancy. NF-κB is normally activated by P4-induced RANKL secretion, which stimulates progenitor cell proliferation and mammary gland expansion (Asselin-Labat et al., 2010; Joshi et al., 2010) in an IKKα-dependent manner (Cao et al., 2001). Rapid proliferation induced by hormones or oncogenes can result in replication stress through depletion of the nucleotide pool (Bester et al., 2011). BRCA1 is required for stalled replication fork protection and, critically, BRCA1 deficiency renders cells prone both to replication stress (Pathania et al., 2014; Schlacher et al., 2012) and, in nullizygous cells, defective in HDR. In addition to replication stress, normal telomeric erosion in the transit amplifying luminal cells, particularly in humans, is another potential trigger for the DDR (Soler et al., 2005; Kannan et al., 2013). Since BRCA1 plays a role in telomere maintenance (Rosen, 2013), the DDR is likely to be exacerbated in BRCA1mut/+ cells. While telomere erosion may be an important activator of the DDR in human BRCA1-deficient cells, this may be less likely in mouse LPs due to the presence of very long telomeres (Kipling and Cooke, 1990). Nevertheless, both human and mouse BRCA1-deficient LPs demonstrate aberrant accumulation and B27-independent growth (Lim et al., 2009), suggesting that replication stress plays a significant role in activating the DDR. We reasoned that DNA damage occurring in proliferating cells might be sufficient to activate NF-κB, possibly through the previously described ATM:NEMO-dependent pathway (Miyamoto, 2011). Indeed, nuclear p65 was increased in Brca1−/− LPs and P-ser536-p65 was detected in Lin− cells and markedly after acute BRCA1 KD in MECs. Most prominent was the presence of nuclear p52/RelB and processed p100 (p52) shown in both Brca1−/− and Brca1−/+ LPs. Sequential activation of the canonical and alternative pathways has been postulated to occur through various mechanisms (Müller and Siebenlist, 2003; Hayden and Ghosh, 2008; Shih et al., 2011). Alternative NF-κB then persists largely unaffected by IκBα-mediated inhibition. This is an important aspect of p52/RelB signaling in BRCA1-deficient LPs, as this longer-term proliferative stimulus would be expected to lead to extend beyond RANKL exposure and further replication would lead to subsequent DDR events. The DDR-induced IKK activity in RANK-expressing LPs could also amplify the response to cyclical stimulation by RANKL.
Although KD of BRCA1 in cell lines acutely activated the phosphorylation of p65, we did not detect P-ser536-p65 in immunoblots of patient organoid lysates (not shown). This may a function of the transient nature of p65 signaling, which is subject to rapid inhibition (Hayden and Ghosh, 2008; Chew et al., 2009). It is also likely that rapidly proliferating cell lines rendered deficient for BRCA1 experience exacerbated genotoxic stress and a high-frequency DDR compared to primary cells. Surprisingly, BMS-345541 did not prevent BRCA1 KD-associated P-ser536-p65 (not shown), suggesting that alternative kinase(s) mediate p65 phosphorylation as previously described (Sakurai et al., 1999).
Interestingly, p100/p52 KD primarily affected only Brca1−/− B27-independent CFCs, while IKKα/β inhibitors blocked colony formation regardless of genotype. These results suggest that the canonical pathway is primarily required for the expansion of normal LP cells while the persistent alternative pathway is responsible for driving proliferation of aberrant Brca1−/− LPs. Inhibition of ATM had a similar effect, consistent with a role for the DDR in promoting and maintaining the canonical/alternative NF-κB pathway circuitry.
Although BRCA1 mutations predominantly give rise to triple negative breast cancer, oophorectomy reduces breast cancer risk in BRCA1 mutation carriers (Eisen et al., 2005; Kramer et al., 2005) while long term RU486 treatment potently inhibits mammary cancers in the Brca1;Tp53-deficient mouse model (Poole et al., 2006). The role of P4 in the etiology of BRCA1mut-associated breast cancer is incompletely understood. Despite the activation of NF-κB, over time, unless cells are immortalized or transformed, the P4/RANKL axis would have a role in cyclical re-initiation of proliferation/DDR signaling in BRCA1-deficient progenitors. Therefore, long-term P4 deprivation in vivo would prevent RANKL-relayed replicative/DNA damage signals and consequently DDR-activated NF-κB activity, which can ultimately lead to abnormal mammary epithelial growth and carcinoma in situ (Barham et al., 2015).
We found that lobular cells lining the lumen of acini in BRCA1mut/+, but not normal, breast human tissue express enhanced levels of nuclear p52. Interestingly, luminal cells within lobules of normal and BRCA1mut/+ patients were shown to exhibit markedly reduced average telomere lengths relative to epithelial cells from ducts and non-luminal breast epithelium in lobules (Sedic et al., 2015). Chromosomal damage coupled to replicative stress in luminal cells that have previously undergone numerous divisions could contribute to the activation of NF-κB in lobular units. Transcriptional targets of NF-κB could then promote long-term survival and reduce telomere erosion-associated senescence as described previously (Li and Tergaonkar, 2014).
Both BMS and DMAPT inhibit IKK/NF-κB and had similar inhibitory effects on B27-independent luminal cell acini formation and expression of p52. While DMAPT has been shown to have both NF-κB-dependent and -independent activities in different cell systems (Nakshatri et al., 2015), it has potential therapeutic value as an NF-κB inhibitor (Neelakantan et al., 2009). In vivo treatment of Brca1−/− mice with DMAPT had profound inhibitory effects on the B27-independent progenitor cell population that only gradually recovered over several murine estrus cycles. Since we did not observe an increase in apoptosis in DMAPT-treated Brca1−/− mammary glands, it is likely that DMAPT induced cell-cycle arrest.
Taken together, our data support a model whereby P4/RANKL induced replication of LPs results in a DDR that activates and perpetuates intrinsic NF-κB activity (Figure 7E). The outcome is an expanded cell population capable of extended proliferation in the absence of a hormonal driver. While long-term mifepristone usage is limited by toxicity, longer-term prevention of the “initializing” proliferative signal through inhibition of RANKL signaling or cyclic administration of an IKK inhibitor may be candidate approaches to forestalling prophylactic mastectomy in young women with the BRCA1 mutation.
EXPERIMENTAL PROCEDURES
Cell Lines
MCF-10A, MCF-7, MDA-MB-231, HCC1937, HC11,293T, and PT67 were purchased from the American Type Culture Collection (ATCC) and cultured according to ATCC instructions.
Transfection
Small interfering RNA (siRNA) transfections were performed for as previously described (Lau et al., 2012). siRNAs for BRCA1 (SMARTpool #M-003461–02–0005) and non-targeting siRNA #1 (siGENOME #D-001210–01–05) were purchased from Dharmacon. DNA transfections were performed using Viafect (Promega) according to the manufacturer’s instructions.
Immunoblots and Antibodies
Whole-cell extracts were prepared in RIPA buffer, and immunoblotting was performed as previously described (Lau et al., 2012). Quantification of immune-reactive bands on immunoblots was performed using ImageJ software (www.rsb.info.nih.gov/ij/). The area under curve (AUC) of the specific signal was corrected for the AUC of the loading control.
Mice
Brca1loxP/loxP [FVB;129-Brca1tm2Brn] mice, bearing loxP sites in introns 4 and 13 of the Brca1 gene (obtained from the NCI-Frederick National Laboratories Mouse Repository), were bred to MMTV-cre mice (kind gift of Dr. W.J. Muller, McGill University) (Andrechek et al., 2000). 8- to 12-week-old BRCA1f/f;cre and BRCA1f/f littermates were used for FACS-isolation of cells using CD24/CD49f antibodies. Mouse genotypes were determined by PCR. Animal husbandry was conducted in accordance with Policy 31 of the University of Ottawa and the Canadian Council on Animal Care guidelines.
Mouse Stem and Progenitor Cell Isolation and Assays
Isolation of mammary epithelial subpopulations was performed as described previously (Schramek et al., 2010). For CFC assays, single-cell suspensions of EasySep-derived (STEMCELL Technologies) Lin− cells were seeded in 20 μl Matrigel (BD Biosciences), and acini formation was scored after 15 days. The diameters of at least 30 of the largest acini were measured using Northern Eclipse software and mean and SE calculated. For mammosphere assays, Lin− cells were plated in 96-well low-attachment plates (Corning), and mammosphere formation was scored after 7 days.
Lentivirus Isolation
Lentiviruses were prepared in 293T cells transfected using polyethyleneimine and packaging plasmids psPax2 and pMD2G.
Full details of experimental procedures and reagents with associated references are available as supplemental files.
Supplementary Material
Highlights.
NF-κB is autonomously activated in BRCA1-deficient mammary progenitors
NF-κB and ATM are required for aberrant proliferation of BRCA1-deficient progenitors
BRCA1 prevents a replication-induced DNA damage response and resulting NF-κB activation
ACKNOWLEDGMENTS
These studies were funded by a grant from the Canadian Breast Cancer Foundation (Ontario) to M.A.C.P. We thank V. Tang and G. Bugnot for technical expertise. We thank the Victorian Cancer Biobank (supported by the Victorian Government) and kConFab for coded breast tissue and data. This work was also supported by grants from the Australian National Health and Medical Research Council (NHMRC 1016701) and NHMRC IRIISS, The Victorian State Government OIS, and the National Breast Cancer Foundation (NC-13-32) to J.E.V. and the NIH National Cancer Institute (R01 CA158275) and NIH National Institute of General Medical Sciences (P20GM109005) to P.A.C. E.N. was supported by a Cancer Council Victoria Scholarship and a Cancer Therapeutics CRC Top-Up Scholarship and J.E.V. by a NHMRC Australia Fellowship (1037230). We also thank Dr. B. Sleckman (Washington University) for providing the pLFRU lenti-mouse shATM and shNT plasmids, Dr. L. Gan (U.C.S.F.) for the lenti-kB-dEGFP reporter, and Dr. R. Piva (University of Torino) for the shRNA plasmids targeting NFKB1 (p105/p50).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2016.05.003.
CONFLICTS OF INTEREST
The University of Kentucky holds a patent on DMAPT. A potential royalty to P.A.C. may occur consistent with University of Kentucky policy. P.A.C. is a founder of Leuchemix, Inc.
REFERENCES
- Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, and Muller WJ (2000). Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc. Natl. Acad. Sci. U S A 97, 3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al. (2007). Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol 9, 201–209. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, Yasuda H, Smyth GK, Martin TJ, Lindeman GJ, and Visvader JE (2010). Control of mammary stem cell function by steroid hormone signalling. Nature 465, 798–802. [DOI] [PubMed] [Google Scholar]
- Barham W, Chen L, Tikhomirov O, Onishko H, Gleaves L, Stricker TP, Blackwell TS, and Yull FE (2015). Aberrant activation of NF-κB signaling in mammary epithelium leads to abnormal growth and ductal carcinoma in situ. BMC Cancer 15, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V,and Karin M(2009). Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov 8, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, and Kerem B (2011). Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, Medina D, Matrisian LM, Kerr LD, and Yull FE (2001). Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol. Biol. Cell 12, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisken C, Park S, Vass T, Lydon JP, O’Malley BW, and Weinberg RA (1998). A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc. Natl. Acad. Sci. USA 95, 5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE, and Mullan PB (2012). BRCA1: conductor of the breast stem cell orchestra: the role of BRCA1 in mammary gland development and identification of cell of origin of BRCA1 mutant breast cancer. Stem Cell Rev. 8, 982–993. [DOI] [PubMed] [Google Scholar]
- Cabuy E, Newton C, and Slijepcevic P (2008). BRCA1 knock-down causes telomere dysfunction in mammary epithelial cells. Cytogenet. Genome Res 122, 336–342. [DOI] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, and Karin M (2001). IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107, 763–775. [DOI] [PubMed] [Google Scholar]
- Cao Y, Luo JL, and Karin M (2007). IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 104, 15852–15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, Teo H, Hazra A, Fang CC, Lopez-Collazo E, et al. (2009). WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat. Cell Biol 11, 659–666. [DOI] [PubMed] [Google Scholar]
- Dejardin E (2006). The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem. Pharmacol 72, 1161–1179. [DOI] [PubMed] [Google Scholar]
- Eisen A, Lubinski J, Klijn J, Moller P, Lynch HT, Offit K, Weber B, Rebbeck T, Neuhausen SL, Ghadirian P, et al. (2005). Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. J. Clin. Oncol 23, 7491–7496. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Branstetter D, Armstrong A, Dinh H, Blumberg H, and Dougall WC (2007). RANK overexpression in transgenic mice with mouse mammary tumor virus promoter-controlled RANK increases proliferation and impairs alveolar differentiation in the mammary epithelia and disrupts lumen formation in cultured epithelial acini. Mol. Cell. Biol 27, 1442–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JJ, James CR, Quinn JE, Stewart GE, Staunton KC, Buckley NE, McDyer FA, Kennedy RD, Wilson RH, Mullan PB, and Harkin DP (2010). BRCA1 transcriptionally regulates genes associated with the basal-like phenotype in breast cancer. Breast Cancer Res. Treat 722, 721–731. [DOI] [PubMed] [Google Scholar]
- Hadian K, and Krappmann D (2011). Signals from the nucleus: activation of NF-kappaB by cytosolic ATM in the DNA damage response. Sci. Signal 4, pe2. [DOI] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2008). Shared principles in NF-kappaB signaling. Cell 132, 344–362. [DOI] [PubMed] [Google Scholar]
- Huen MSY, Sy SMH, and Chen J (2010). BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol 11, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M (2002). Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene 21, 8981–8993. [DOI] [PubMed] [Google Scholar]
- Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, Stingl J, Waterhouse PD, and Khokha R (2010). Progesterone induces adult mammary stem cell expansion. Nature 465, 803–807. [DOI] [PubMed] [Google Scholar]
- Josson S, Xu Y, Fang F, Dhar SK, St Clair DK, and St Clair WH (2006). RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 25, 1554–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Huda N, Tu L, Droumeva R, Aubert G, Chavez E, Brinkman RR, Lansdorp P, Emerman J, Abe S, et al. (2013). The luminal progenitor compartment of the normal human mammary gland constitutes a unique site of telomere dysfunction. Stem Cell Reports 1, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipling D, and Cooke HJ (1990). Hypervariable ultra-long telomeres in mice. Nature 347, 400–402. [DOI] [PubMed] [Google Scholar]
- Kramer JL, Velazquez IA, Chen BE, Rosenberg PS, Struewing JP, and Greene MH (2005). Prophylactic oophorectomy reduces breast cancer penetrance during prospective, long-term follow-up of BRCA1 mutation carriers. J. Clin. Oncol 23, 8629–8635. [DOI] [PubMed] [Google Scholar]
- Lau R, Niu MY, and Pratt MAC (2012). cIAP2 represses IKKα/β-mediated activation of MDM2 to prevent p53 degradation. Cell Cycle 11, 4009–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, and Paull TT (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. [DOI] [PubMed] [Google Scholar]
- Li Y, and Tergaonkar V (2014). Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Res. 74, 1639–1644. [DOI] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat M-L, Gyorki DE, Ward T, Partanen A, et al. ; kConFab (2009). Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med 15, 907–913. [DOI] [PubMed] [Google Scholar]
- McCool KW, and Miyamoto S (2012). DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev 246, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S (2011). Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 21, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneux G, and Smalley MJ (2011). The cell of origin of BRCA1 mutation-associated breast cancer: a cautionary tale of gene expression profiling. J. Mammary Gland Biol. Neoplasia 16, 51–55. [DOI] [PubMed] [Google Scholar]
- Mullan PB, Quinn JE, and Harkin DP (2006). The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 25, 5854–5863. [DOI] [PubMed] [Google Scholar]
- Müller JR, and Siebenlist U (2003). Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J. Biol. Chem 278, 12006–12012. [DOI] [PubMed] [Google Scholar]
- Nakshatri H, Appaiah HN, Anjanappa M, Gilley D, Tanaka H, Badve S, Crooks PA, Mathews W, Sweeney C, and Bhat-Nakshatri P (2015). NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 6, e1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakantan S, Nasim S, Guzman ML, Jordan CT, and Crooks PA (2009). Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT(LC-1). Bioorg. Med. Chem. Lett 19, 4346–4349. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. (2014). BRCA1 haploin-sufficiency for replication stress suppression in primary cells. Nat. Commun 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AJ, Li Y, Kim Y, Lin S-CJ, Lee W-H, and Lee EY-HP (2006). Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science 314, 1467–1470. [DOI] [PubMed] [Google Scholar]
- Pratt MAC, Tibbo E, Robertson SJ, Jansson D, Hurst K, Perez-Iratxeta C, Lau R, and Niu MY (2009). The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene 28, 2710–2722. [DOI] [PubMed] [Google Scholar]
- Rosen EM (2013). BRCA1 in the DNA damage response and at telomeres. Front. Genet 4, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, and Toriumi W (1999). IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem 274, 30353–30356. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, et al. (2010). Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P, Keller PJ, Glover E, Richardson AL, Cowan J, et al. (2015). Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun 6, 7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, and Visvader JE (2006). Generation of a functional mammary gland from a single stem cell. Nature 439, 84–88. [DOI] [PubMed] [Google Scholar]
- Shehata M, Teschendorff A, Sharp G, Novcic N, Russell IA, Avril S, Prater M, Eirew P, Caldas C, Watson CJ, and Stingl J (2012). Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 14, R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih VF, Tsui R, Caldwell A, and Hoffmann A (2011). A single NFκB system for both canonical and non-canonical signaling. Cell Res. 21, 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler D, Genescà A, Arnedo G, Egozcue J, and Tusell L (2005). Telomere dysfunction drives chromosomal instability in human mammary epithelial cells. Genes Chromosomes Cancer 44, 339–350. [DOI] [PubMed] [Google Scholar]
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, and Eaves CJ (2006). Purification and unique properties of mammary epithelial stem cells. Nature 439, 993–997. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, and Karin M (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol 27, 693–733. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2014). Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343, 1470–1475. [DOI] [PubMed] [Google Scholar]
- Visvader JE, and Stingl J (2014). Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 28, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CM, Engler AJ, Slone RD, Galante LL, and Schwarzbauer JE (2008). Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 68, 3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.