

Abstract
Background
RYR2, encoding cardiac ryanodine receptor, is the major responsible gene for catecholaminergic polymorphic ventricular tachycardia (CPVT). Meanwhile, KCNJ2, encoding inward‐rectifier potassium channel (IK1), can be the responsible gene for atypical CPVT. We recently encountered a family with CPVT and sought to identify a responsible gene variant.
Methods
A targeted panel sequencing (TPS) was employed in the proband. Copy number variation (CNV) in RYR2 was identified by focusing on read numbers in the TPS and long‐range PCR. Cascade screening was conducted by a Sanger method and long‐range PCR. KCNJ2 wild‐type (WT) or an identified variant was expressed in COS‐1 cells, and whole‐cell currents (IK1) were recorded using patch‐clamp techniques.
Results
A 40‐year‐old female experienced cardiopulmonary arrest while cycling. Her ECG showed sinus bradycardia with prominent U‐waves (≥0.2 mV). She had left ventricular hypertrabeculation at apex. Exercise induced frequent polymorphic ventricular arrhythmias. Her sister died suddenly at age 35 while bouldering. Her father and paternal aunt, with prominent U‐waves, received permanent pacemaker due to sinus node dysfunction. The initial TPS and cascade screening identified a KCNJ2 E118D variant in all three symptomatic patients. However, after focusing on read numbers, we identified a novel exon3 deletion of RYR2 (RYR2‐exon3 deletion) in all of them. Functional analysis revealed that KCNJ2 E118D generated IK1 indistinguishable from KCNJ2 WT, even in the presence of catecholaminergic stimulation.
Conclusions
Focusing on the read numbers in the TPS enabled us to identify a novel CNV, RYR2‐exon3 deletion, which was associated with phenotypic features of this family.
Keywords: catecholaminergic polymorphic ventricular tachycardia, copy number variation, KCNJ2, RYR2, targeted panel sequencing
1. INTRODUCTION
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is characterized by exercise or emotion‐induced polymorphic or bidirectional ventricular arrhythmias (VAs) leading to syncope or sudden cardiac death in the absence of structural heart disease and is often accompanied by sinus node dysfunction (SND) or sinus bradycardia. (Leenhardt et al., 1995; Postma et al., 2005) (Miyata, Ohno, Itoh, & Horie, 2018) Several responsible genes for CPVT have been identified so far, and RYR2 which encodes the cardiac ryanodine receptor (RyR2) accounts for approximately 60% of CPVT cases. (Laitinen et al., 2001; Priori et al., 2001).
The RyR2 is a calcium (Ca2+) release channel on the sarcoplasmic reticulum (SR) and plays a major role in the regulation of intracellular Ca2+ homeostasis. (Kushnir & Marks, 2010) In patients with RYR2‐related CPVT, intracellular Ca2+ overload due to an increased diastolic Ca2+ leakage from SR in the presence of high β‐adrenergic tone has been thought to be the cause of enhanced U‐waves on electrocardiogram (ECG) and VAs which may be coincident with delayed afterdepolarization (DAD)‐induced triggered activity. (Katra & Laurita, 2005; Katra, Oya, Hoeker, & Laurita, 2007; Lieve, van der Werf, & Wilde, 2016; Paavola et al., 2007; Viitasalo et al., 2008).
On the other hand, KCNJ2, encoding inward‐rectifier potassium current (IK1), has also been reported to be responsible for an atypical type of CPVT,(Tester et al., 2006) although KCNJ2 is typically responsible for Andersen–Tawil syndrome (ATS) characterized by (a) VAs with prominent U‐waves (≥0.2 mV) on baseline ECG, (b) periodic paralysis, and (c) dysmorphic features. (Nguyen, Pieper, & Wilders, 2013; Plaster et al., 2001) However, approximately 30% of the disease‐causing KCNJ2 variant carriers show only cardiac phenotypes. (Kimura et al., 2012).
We recently encountered a family with CPVT. Genetic screening of the proband by a targeted panel sequencing using next‐generation sequencer (NGS) identified only a rare KCNJ2 E118D variant. However, after focusing on the read numbers of the NGS, we could identify a novel exon3 deletion of RYR2 (RYR2‐exon3 deletion) without performing multiplex ligation‐dependent probe amplification (MLPA) method. To elucidate the causative roles of these variants, we investigated family members and functional role of the KCNJ2 E118D variant.
2. METHODS
2.1. Clinical evaluation
The proband (Ⅲ‐3) who had CPVT phenotype and her father (Ⅱ‐2), mother (Ⅱ‐3), paternal aunt (Ⅱ‐4), and three children (Ⅳ‐1, Ⅳ‐2, and Ⅳ‐3) were enrolled in this study (Figure 1). Baseline ECGs were available from all the subjects. Prominent U‐waves were defined as having U‐wave amplitude ≥0.2 mV on baseline ECG. Transthoracic echocardiograms (TTEs) were available from the proband (Ⅲ‐3) and her daughter (Ⅳ‐3). Hypertrabeculation or left ventricular non‐compaction (LVNC) localized at left ventricular apex (LVA) was defined as non‐compacted to compacted ratio of ≥2.0 when LVA was observed by short‐axis view of TTE as reported by Jenni et al.(Jenni, Oechslin, Schneider, Attenhofer Jost, & Kaufmann, 2001) The proband (Ⅲ‐3) and her daughter (Ⅳ‐3) underwent exercise stress test using ergometer (30‐watt ramp protocol). The proband (Ⅲ‐3) underwent isoproterenol infusion test (0.015 μg kg–1 min–1). The proband (Ⅲ‐3) also underwent left heart catheterization and subsequent acetylcholine provocation to coronary arteries (up to 100 μg), and programmed electrical stimulation up to three extra stimuli from both right ventricular apex and right ventricular outflow tract.
Figure 1.
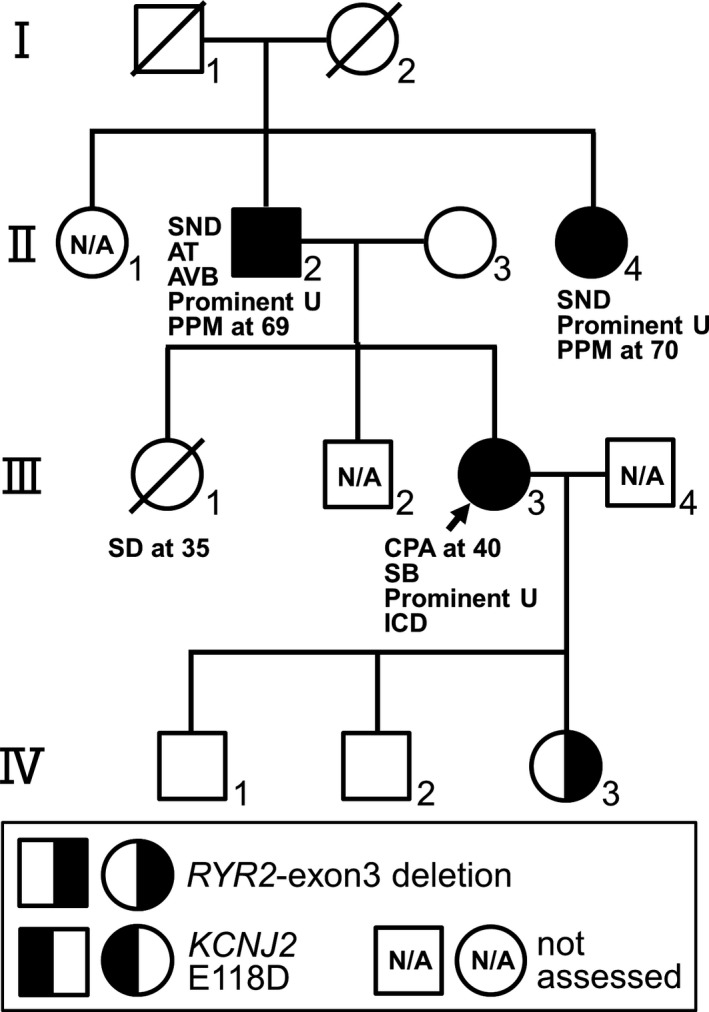
A pedigree and clinical characteristics of the family. AT, atrial tachycardia; AVB, atrioventricular block; CPA, cardiopulmonary arrest; ICD, implantable cardioverter defibrillator; PPM, permanent pacemaker; Prominent U, Prominent U‐waves at rest; SB, sinus bradycardia; SD, sudden death. Carriers of an exon3 deletion of RYR2 (RYR2‐exon3 deletion) and a KCNJ2 E118D variant are shown as the graphic symbols in the bottom
2.2. Genetic analysis
After obtaining appropriate approval from the institutional review board and written informed consent for the genetic analysis from all the subjects of this study, we performed genetic analysis.
Genomic DNA was extracted from peripheral blood lymphocytes as described previously.(Imai et al., 2014) We then performed a targeted panel sequencing of 55 genes of the proband (Ⅲ‐3) using NGS (MiSeq, Illumina, San Diego, CA, USA) (Table 1). Nucleotides substitution was confirmed by a Sanger method. To detect the copy number variations (CNVs), the depths (read numbers) of the NGS reads obtained by the targeted panel sequencing were compared between control and the proband by using the SureCall software (Agilent Technologies, Santa Clara, CA, USA). To confirm the CNV in RYR2, we performed long‐range PCR using KOD‐Plus‐Neo (Toyobo, Osaka, Japan). Forward primer (5′‐CACAGAACAGGACCAAGTTAGAGGC‐3′) and reverse primer (5′‐CATTACCTTCCTGACACACTTCATCCTAG‐3′) were designed to amplify a region that includes the deleted site of RYR2. The 5’ position of the forward primer and the reverse primer was 287,712 and 291,655 in the NCBI Reference Sequence NC_000001.10 (GRCh37), respectively. The PCR products were loaded into 0.8% agarose gels with Tris‐Acetate‐EDTA buffer and electrophoresed. Then, the bands were extracted and purified using the QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany), which were directly sequenced to confirm the precise location of the deletion in RYR2. Cascade screening of family members, her father (Ⅱ‐2), mother (Ⅱ‐3), paternal aunt (Ⅱ‐4), and three children (Ⅳ‐1, Ⅳ‐2 and Ⅳ‐3), was conducted by a Sanger method and a long‐range PCR.
Table 1.
Fifty‐five genes analyzed by the targeted panel sequencing
| AKAP9 | ANKB | CACNA1C | CACNA2D1 | CACNB2 | CALM1 |
| CALM2 | CALM3 | CAMK2D | CASQ2 | CAV3 | CTNNA3 |
| DES | DPP6 | DSC2 | DSG2 | DSP | GJA1 |
| GJA5 | GPD1L | HCN4 | JUP | KCNA5 | KCND3 |
| KCNE1 | KCNE2 | KCNE3 | KCNE4 | KCNE5 | KCNH2 |
| KCNIP2 | KCNJ2 | KCNJ3 | KCNJ5 | KCNJ8 | KCNN2 |
| KCNQ1 | LMNA | MOG1 | NCS1 | PKP2 | RANGRF |
| RYR2 | SCN10A | SCN1B | SCN2B | SCN3B | SCN4B |
| SCN5A | SLC8A1 | SNTA1 | TCAP | TMEM4 | TRDN |
| TRPM4 |
2.3. Mutagenesis and heterologous expression
Human KCNJ2 wild‐type (WT) cDNA (NM_000891.1) subcloned into pCMS‐EGFP vector (KCNJ2 WT) was used to engineer a KCNJ2 mutant.(Kimura et al., 2012) Site‐directed mutagenesis (KCNJ2 E118D) was performed by using the QuikChangeⅡSite‐Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions. The presence of the mutation was confirmed by direct sequencing.
Using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), 1.0 μg of KCNJ2 WT cDNA or 1.0 μg of KCNJ2 E118D cDNA were transiently transfected into COS‐1 cells, and maintained in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin‐streptomycin in a 5% CO2 incubator at 37°C for 24–36 hr before current recordings. Cells exhibiting green fluorescence were chosen for the current recordings.
2.4. Electrophysiology
Membrane currents (IK1) were recorded using whole‐cell patch‐clamp techniques at room temperature (23–25℃). The bath solution for recording IK1 contained (in mmol/L) 140 NaCl, 5.4 KCl, 0.5 MgCl2, 1.8 CaCl2, 0.33 NaH2PO4, 5.5 glucose, 5 HEPES (pH 7.4 with NaOH), and the pipette solution contained (in mmol/L) 60 K‐aspartate, 65 KCl, 1 KH2PO4, 2 MgCl2, 3 EDTA, 3 ATP, 5 HEPES (pH 7.2 with KOH). The electrode resistance ranged from 1.0 to 2.5 MΩ when filled with pipette solution. Data acquisition was carried out using an Axopatch 200B amplifier and pCLAMP10.3 software (Molecular Devices, Sunnyvale, CA, USA). Currents were acquired at 20–50 kHz, and low pass‐filtered at 5 kHz using an analog‐to‐digital interface (Digidata 1440A acquisition system, Molecular Devices). Currents were evoked by 100 ms test pulses applied in 10 mV increments to potentials ranging from −140 mV to +50 mV from a holding potential of −60 mV. Current–voltage (I‐V) relationships were obtained by the current amplitudes at the end of 100 ms pulses against test potentials.
2.5. Catecholaminergic stimulation
To resemble the catecholaminergic effect (protein kinase A (PKA) activation) on KCNJ2 WT and KCNJ2 E118D, IK1 was also recorded 5 min after the presence of a PKA cocktail containing 100 μmol/L forskolin (Sigma‐Aldrich, Missouri, USA) and 10 μmol/L 3‐isobutyl‐1‐methylxanthine (Sigma‐Aldrich, Missouri, USA) in the Bath solution and 10 min after wash out of it as the same method reported by Vega et al.(Vega, Tester, Ackerman, & Makielski, 2009).
2.6. Statistical analysis
All data are expressed as mean ± SE, and statistical comparisons were tested using the unpaired Student's t test with p < 0.05 considered to be statistically significant.
3. RESULTS
3.1. Phenotypic manifestations
The proband (Ⅲ‐3) (Figure 1), a 40‐year‐old female, experienced cardiopulmonary arrest (CPA) while cycling and then was resuscitated. Her 12‐lead ECG recorded previously at medical examination showed sinus bradycardia (42 bpm) with prominent U‐waves (≥0.2 mV) in the anterior precordial leads (Figure 2a). Echocardiogram showed no structural heart diseases except for the presence of hypertrabeculation localized at left ventricular apex (Figure 2b) with preserved left ventricular systolic function (ejection fraction (EF): 73%). Exercise stress test induced frequent premature ventricular contractions (PVCs) and polymorphic VAs (Figure 2c). Notably, isoproterenol infusion test induced augmentation of U‐waves, followed by PVC (Figure 2d). Coronary angiography and subsequent acetylcholine provocation to coronary arteries revealed no significant stenosis in coronary arteries. Programmed right ventricular electrical stimulation did not induce VAs. Thus, the proband presented with typical phenotypic features of CPVT. An implantable cardioverter defibrillator (ICD) was implanted. However, it was extracted because of an ICD infection, then a subcutaneous ICD was implanted. Although medical therapies such as β‐blockers and flecainide were taken into consideration, those were not administered because of the sinus bradycardia phenotype. Under strict exercise restriction, a defibrillator shock has never been delivered. On family history, her elder sister (Ⅲ‐1) died suddenly while bouldering at the age of 35 (Figure 1). Her father (Ⅱ‐2) and paternal aunt (Ⅱ‐4), whose ECGs also showed prominent U‐waves at rest (Figures 3a,b), received permanent pacemaker (PPM) at the age of 69 and 70, respectively, due to SND (Figure 1). Her father (Ⅱ‐2) also had atrial tachycardia (AT) with atrioventricular block (AVB) (Figure 3c). Her three children (Ⅳ‐1, Ⅳ‐2, and Ⅳ‐3) had been asymptomatic. Her daughter (Ⅳ‐3)’s ECG showed normal sinus rhythm (72 bpm) with no prominent U‐waves but inverted T‐waves in V1–V3 leads (Figure 3d). Exercise stress test induced only one premature ventricular contraction. Her echocardiogram also showed hypertrabeculation localized at LVA (Figure 3e) with preserved systolic function (EF: 67%). Remaining family members in the pedigree have been asymptomatic.
Figure 2.
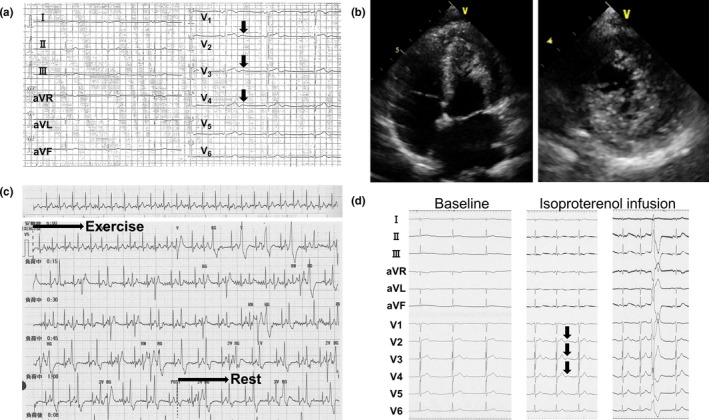
Clinical characteristics of the proband (Ⅲ‐3) (Figure 1). (a) 12‐lead ECG recorded at medical examination before cardiopulmonary arrest. Arrows indicate prominent U‐waves. (b) Four‐chamber view (left panel) and short‐axis view (right panel) during systole of transthoracic echocardiograms. Hypertrabeculation was recognized at left ventricular apex. (c) Exercise stress test using ergometer. D. Isoproterenol infusion test before (Baseline) and during isoproterenol infusion. Arrows indicate augmentation of U‐waves
Figure 3.
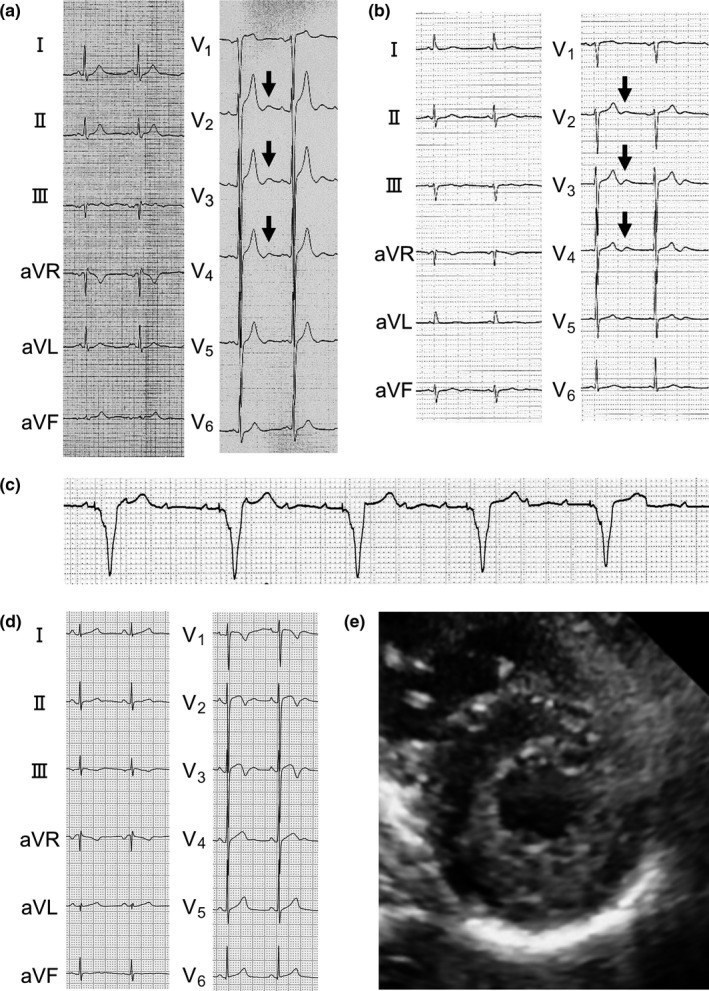
Clinical characteristics of the family (Figure 1). (a, b, and d) 12‐lead ECG of the proband's father (Ⅱ‐2) (a), paternal aunt (Ⅱ‐4) (b), and daughter (Ⅳ‐3) (d). Arrows indicate prominent U‐waves. (c) V1 lead of the proband's father (Ⅱ‐2) during ventricular pacing due to atrial tachycardia with atrioventricular block. (e) Short‐axis view during systole of transthoracic echocardiogram of the proband's daughter (Ⅳ‐3)
3.2. Identification of a rare KCNJ2 E118D variant and exon3 deletion of RYR2
On the first genetic screening by the targeted panel sequencing of the proband (Ⅲ‐3), we identified only a rare KCNJ2 (NM_000891.1) E118D variant (Figure 4a) (rs538725136, allele frequency in East Asian: 0.0004622 in Exome Aggregation Consortium Browser [http://exac.broadinstitute.org/] and that in Japanese: 0.0017 in Human Genetic Variation Database [http://www.hgvd.genome.med.kyoto-u.ac.jp/]) and failed to identify any other disease‐causing variants including RYR2.
Figure 4.
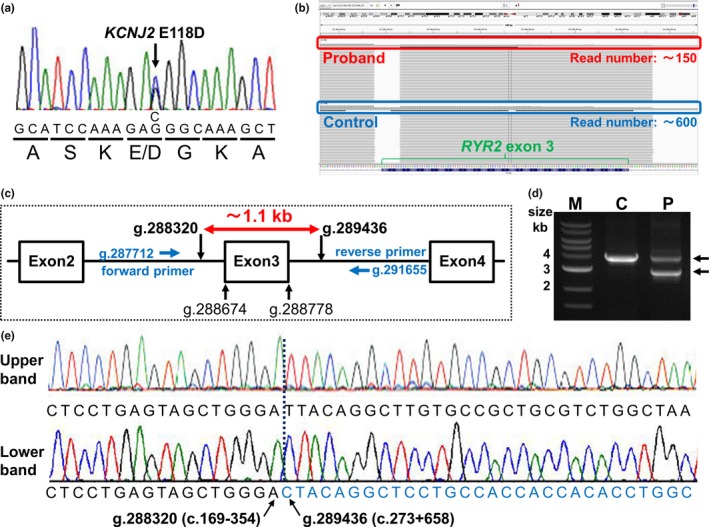
Identification of a KCNJ2 E118D variant and a novel exon3 deletion of RYR2. (a) A sequence electropherogram of KCNJ2 gene of the proband (Ⅲ‐3) (Figure 1). Nucleotide and amino acid substitutions were indicated below the electropherogram. (b) A result of targeted panel sequencing of the proband and control showing read number around exon3 of RYR2. Red and blue enclosing lines indicate read number of the proband and control, respectively. (c) Schematic representation of the position of exon3 of RYR2 (NCBI Reference Sequence NC_000001.10), deletion sites, and primers used for long‐range PCR (d). (d). Electrophoresis of long‐range PCR products. M, marker; C, control; P, proband. (e) Direct sequencings of the upper band (upper panel) and lower band (lower panel) from the proband (d).
Since the proband presented with typical CPVT phenotype, although KCNJ2 variants can be the cause of an atypical type of CPVT,(Tester et al., 2006) we next proceeded to conduct pair analysis to detect the presence of CNVs.(Ohno et al., 2014) Read numbers obtained by the targeted panel sequencing were compared between control and the proband, then we found that the read number around exon3 in RYR2 of the proband was apparently reduced compared to that of control (Figure 4b), suggested the presence of CNV, massive exon3 deletion, in the proband.
To confirm it, we conducted long‐range PCR using primers designed in intron 2 (forward primer) and intron 3 (reverse primer) of RYR2 as shown in Methods and Figure 4c. Electrophoresis of the PCR products from the control showed single band which size was consistent with an expected one (approximately 3.9 kb) whereas those from the proband showed two distinct bands (Figure 4d). The size of the upper band from the proband was approximately 3.9 kb identical to that from the control, and the size of the lower band was approximately 2.8 kb (Figure 4d), suggesting the presence of ~1.1 kb deletion of RYR2 including exon3. Direct sequencing of the upper band showed the sequence of wild‐type RYR2 including exon3 (Figure 4e). On the other hand, that of the lower band displayed the site of the deletion of RYR2 from intron2 to intron3 (Figure 4e). Thus, we finally identified a novel exon3 deletion of RYR2, NC_000001.10 (NM_001035.2):c.169‐353_273+657del (RYR2‐exon3 deletion) which was different from previous reports. (Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros‐Domingo et al., 2009; Ohno et al., 2014; Szentpali, Szili‐Torok, & Caliskan, 2013).
As shown in the pedigree (Figure 1), her father (Ⅱ‐2) and paternal aunt (Ⅱ‐4) were also found to carry both the RYR2‐exon3 deletion and KCNJ2 E118D. Her asymptomatic 11‐year‐old daughter (Ⅳ‐3) carried the RYR2‐exon3 deletion alone, but her sons (Ⅳ‐1 and Ⅳ‐2) carried neither the RYR2‐exon3 deletion nor the KCNJ2 E118D.
3.3. Functional analysis of the KCNJ2 E118D variant
Since KCNJ2 variants can be the cause of an atypical type of CPVT,(Tester et al., 2006) there remained a possibility that the KCNJ2 E118D variant might cause the CPVT phenotype of this family. Therefore, we performed functional analysis of the variant. We expressed KCNJ2 WT or KCNJ2 E118D in COS‐1 cells and recorded whole‐cell currents (IK1). KCNJ2 E118D exhibited typical IK1 indistinguishable from KCNJ2 WT (Figure 5a). Inward rectification property and IK1 density for KCNJ2 E118D were almost the same as those for KCNJ2 WT (Figure 5b). IK1 density at −140 mV for WT and E118D was −430 ± 79 pA/pF (n = 21) and −484 ± 70 pA/pF (n = 30), respectively (p = 0.17).
Figure 5.
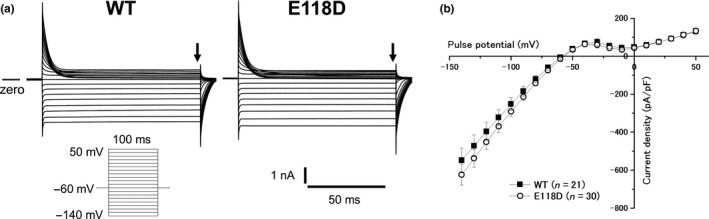
Electrophysiological properties for KCNJ2 WT and KCNJ2 E118D. (a) Representative currents obtained by a pulse protocol shown in the inset for KCNJ2 WT (left panel) and KCNJ2 E118D (right panel). (b) Current–voltage relationships for KCNJ2 WT (filled squares, n = 21) and KCNJ2 E118D (open circles, n = 30). Current amplitudes were evaluated at the end of the test pulses (arrows in A). WT, wild‐type
We next examined the effects of protein kinase A (PKA) activation on IK1 to resemble the catecholaminergic effect which occurs during exercise‐induced VAs. After recording IK1 at baseline condition, IK1 was also recorded in the presence of PKA cocktail (100 μmol/L forskolin +10 μmol/L 3‐isobutyl‐1‐methylxanthine) and after wash out of it, as reported by Vega et al.(Vega et al., 2009) As shown in Figure 6, PKA activation affected neither KCNJ2 WT nor KCNJ2 E118D. Contrary to the report by Vega et al.,(Vega et al., 2009) PKA activation did not reduce even IK1 produced by KCNJ2 WT.
Figure 6.
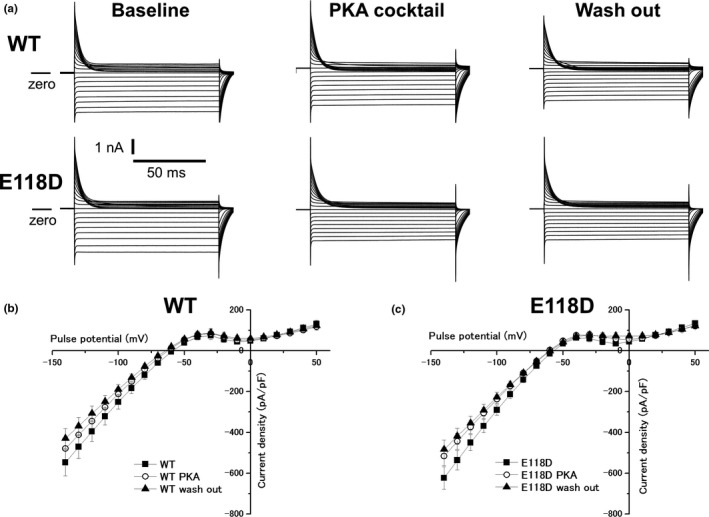
Effects of catecholaminergic stimulation on expressed currents (IK1) for KCNJ2 WT and KCNJ2 E118D. (a) Representative currents for KCNJ2 WT (upper panels) and KCNJ2 E118D (lower panels) obtained by the pulse protocol shown in the inset of Figure 5a. Currents were recorded at baseline (left panels), in the presence of a PKA cocktail (middle panels) and after wash out of it (right panels). (b) Current–voltage relationships for KCNJ2 WT obtained at baseline (filled squares, n = 21), in the presence of the PKA cocktail (open circles, n = 21) and after wash out of it (filled triangles, n = 21). (c) Current–voltage relationships for KCNJ2 E118D obtained at baseline (filled squares, n = 30), in the presence of the PKA cocktail (open circles, n = 30) and after wash out of it (filled triangles, n = 30). WT, wild‐type; PKA, protein kinase A
4. DISCUSSION
4.1. Identification of a rare KCNJ2 E118D variant and a novel exon3 deletion of RYR2
Our genetic screening by the initial targeted panel sequencing on the proband with CVPT identified only a rare KCNJ2 E118D variant. However, focusing on the read numbers of the targeted panel sequencing led us think of the presence of the CNV, then we could finally identify a novel massive deletion of RYR2 including exon3.
An MLPA method has been reported to be useful for identifying CNVs for many diseases including arrhythmogenic disorders. However, it is noteworthy that we identified CNV in RYR2 by focusing on the read numbers of the targeted panel sequencing and subsequent long‐range PCR without performing the MLPA method. (Barc et al., 2011; Cox et al., 2011; Sonoda et al., 2017, 2018) Therefore, our study underlies the diagnostic importance of examining the read numbers in targeted panel sequencing to rule out the presence of pathological CNVs, even when a conventional method identifies candidate variants that are not actually disease‐causing.
Although there have been several reports regarding massive deletion of RYR2 including exon3, direct sequencing of the PCR products (Figure 4e) from the proband revealed that the deletion site of our RYR2‐exon3 deletion was different from those of previous reports.(Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros‐Domingo et al., 2009; Ohno et al., 2014; Szentpali et al., 2013).
4.2. Pathophysiological roles of U‐waves in patients with CPVT
In patients with RYR2‐related CPVT, intracellular Ca2+ overload or oscillations in the heart are thought to be induced by catecholaminergic stimulation through an increased diastolic Ca2+ leakage from SR, which may lead to DAD or DAD‐induced triggered activity.(Jiang et al., 2004; Katra & Laurita, 2005; Katra et al., 2007; Paavola et al., 2007) Clinical examinations of CPVT patients reported that DADs recorded in monophasic action potential of ventricular myocardium could be augmented by catecholaminergic stimulation, and concomitant U‐wave changes on ECG were associated with the DADs.(Nakajima et al., 1997; Paavola et al., 2007) In the present case, catecholaminergic stimulation by isoproterenol infusion augmented U‐waves, followed by PVC. These findings appear to be consistent with these concepts. Intriguingly, Aizawa et al. reported that U‐waves in CPVT patients could also be affected by some specific conditions such as after ventricular pacing, after the exercise test and after sinus arrest.(Aizawa et al., 2006) Therefore, there may be a high probability of the relationship between U‐waves and arrhythmogenicity in CPVT patients; however, it needs to be further investigated.
4.3. Phenotypic manifestations of this family
The majority of RYR2 variants identified in typical CPVT patients are missense mutations. However, carriers of exon3 deletions of RYR2 have been reported to show a distinct phenotype of CPVT characterized by concomitant SND, AVB, atrial fibrillation, atrial standstill, dilated cardiomyopathy, and LVNC/hypertrabeculation.(Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros‐Domingo et al., 2009; Ohno et al., 2014; Szentpali et al., 2013).
In this family, the proband and her elder sister experienced cardiac events during exercise. The proband had sinus bradycardia, prominent U‐waves at rest, and hypertrabeculation localized at LVA. Her father and paternal aunt received PPM due to SND, and had prominent U‐waves at rest. Her father also had AT with AVB. These family members, except for her deceased sister, carried both the RYR2‐exon3 deletion and the KCNJ2 E118D, which obscured pathophysiological roles of each variant. On the other hand, ECG of her daughter (age 11), who carried only the RYR2‐exon3 deletion, showed no prominent U‐waves but inverted T‐waves. We speculate that normal juvenile pattern of T‐waves might obscure U‐waves.(Surawicz, 1998) She had been asymptomatic, and exercise stress test induced only one premature ventricular contraction. Intriguingly, she also had hypertrabeculation at LVA. These various phenotypic manifestations, except for prominent U‐waves at rest, of this family appeared to be consistent with those with exon3 deletion of RYR2 in previous reports.(Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros‐Domingo et al., 2009; Ohno et al., 2014; Szentpali et al., 2013).
4.4. Possible functional role of the RYR2‐exon3 deletion and the KCNJ2 E118D variant
The RYR2‐exon3 deletion may produce mutant RyR2 proteins without frameshift, thus may cause their functional abnormalities. Actually, functional analysis of exon3 deletion of RyR2 using heterologous expression in HEK293 cells or HL‐1 cells revealed that it reduced the endoplasmic reticulum or SR luminal Ca2+ threshold at which Ca2+ release terminates and increased the fractional Ca2+ release, and also enhanced the amplitude of store overload‐induced Ca2+ transients.(Tang, Tian, Wang, Fill, & Chen, 2012) Considering it together with clinical reports of patients with exon3 deletion of RYR2, various phenotypic manifestations in this family could be attributable to the RYR2‐exon3 deletion.(Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros‐Domingo et al., 2009; Ohno et al., 2014; Szentpali et al., 2013).
Regarding the KCNJ2 E118D variant, it caused no functional abnormalities of IK1 based on the patch‐clamp study, even in the presence of PKA activation. These findings suggested that prominent U‐waves at rest in this family might be associated with the RYR2‐exon3 deletion.
5. CONCLUSIONS
A novel RYR2‐exon3 deletion and a rare KCNJ2 E118D variant were identified in a family with CPVT. The RYR2‐exon3 deletion could be identified by focusing on the read numbers of the targeted panel sequencing without performing the MLPA method, which emphasizes the importance of focusing on the read numbers when performing genetic analysis using NGS. The RYR2‐exon3 deletion may produce mutant RyR2 proteins without frameshift, thus cause RyR2 dysfunction, but the KCNJ2 E118D variant did not cause IK1 dysfunction. Therefore, the novel RYR2‐exon3 deletion may be associated with phenotypic features of this family.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank to Ms. Miki Matsui and Ms. Takako Kobayashi for their technical expertise.
Dharmawan T, Nakajima T, Ohno S, et al. Identification of a novel exon3 deletion of RYR2 in a family with catecholaminergic polymorphic ventricular tachycardia. Ann Noninvasive Electrocardiol. 2019;24:e12623 10.1111/anec.12623
Funding information
This work was supported by a Grant‐in‐Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (23591030 and 17K09487 to T.N.) and Japan Agency for Medical Research and Development (JP18ek0109202 to S.O.).
REFERENCES
- Aizawa, Y. , Komura, S. , Okada, S. , Chinushi, M. , Aizawa, Y. , Morita, H. , & Ohe, T. (2006). Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT). International Heart Journal, 47, 381–389. 10.1536/ihj.47.381 [DOI] [PubMed] [Google Scholar]
- Barc, J. , Briec, F. , Schmitt, S. , Kyndt, F. , Le Cunff, M. , Baron, E. , … Schott, J. J. (2011). Screening for copy number variation in genes associated with the long QT syndrome: Clinical relevance. Journal of the American College of Cardiology, 57, 40–47. 10.1016/j.jacc.2010.08.621 [DOI] [PubMed] [Google Scholar]
- Bhuiyan, Z. A. , van den Berg, M. P. , van Tintelen, J. P. , Bink‐Boelkens, M. T. , Wiesfeld, A. C. , Alders, M. , … Wilde, A. A. (2007). Expanding spectrum of human RYR2‐related disease: New electrocardiographic, structural, and genetic features. Circulation, 116, 1569–1576. 10.1161/CIRCULATIONAHA.107.711606 [DOI] [PubMed] [Google Scholar]
- Cox, M. G. , van der Zwaag, P. A. , van der Werf, C. , van der Smagt, J. J. , Noorman, M. , Bhuiyan, Z. A. , … Hauer, R. N. (2011). Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Pathogenic desmosome mutations in index‐patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype‐phenotype follow‐up study. Circulation, 123, 2690–2700. 10.1161/CIRCULATIONAHA.110.988287 [DOI] [PubMed] [Google Scholar]
- Imai, M. , Nakajima, T. , Kaneko, Y. , Niwamae, N. , Irie, T. , Ota, M. , … Kurabayashi, M. (2014). A novel KCNQ1 splicing mutation in patients with forme fruste LQT1 aggravated by hypokalemia. Journal of Cardiology, 64, 121–126. 10.1016/j.jjcc.2013.11.014 [DOI] [PubMed] [Google Scholar]
- Jenni, R. , Oechslin, E. , Schneider, J. , Attenhofer Jost, C. , & Kaufmann, P. A. (2001). Echocardiographic and pathoanatomical characteristics of isolated left ventricular non‐compaction: A step towards classification as a distinct cardiomyopathy. Heart, 86, 666–671. 10.1136/heart.86.6.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, D. , Xiao, B. , Yang, D. , Wang, R. , Choi, P. , Zhang, L. , … Chen, S. R. (2004). RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store‐overload‐induced Ca2+ release (SOICR). Proceedings of the National Academy of Sciences, 101, 13062–13067. 10.1073/pnas.0402388101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katra, R. P. , & Laurita, K. R. (2005). Cellular mechanism of calcium‐mediated triggered activity in the heart. Circulation Research, 96, 535–542. 10.1161/01.RES.0000159387.00749.3c [DOI] [PubMed] [Google Scholar]
- Katra, R. P. , Oya, T. , Hoeker, G. S. , & Laurita, K. R. (2007). Ryanodine receptor dysfunction and triggered activity in the heart. American Journal of Physiology. Heart and Circulatory Physiology, 292, H2144–2151. 10.1152/ajpheart.00924.2006 [DOI] [PubMed] [Google Scholar]
- Kimura, H. , Zhou, J. , Kawamura, M. , Itoh, H. , Mizusawa, Y. , Ding, W. G. , … Horie, M. (2012). Phenotype variability in patients carrying KCNJ2 mutations. Circulation: Cardiovascular Genetics, 5, 344–353. 10.1161/CIRCGENETICS.111.962316 [DOI] [PubMed] [Google Scholar]
- Kushnir, A. , & Marks, A. R. (2010). The ryanodine receptor in cardiac physiology and disease. Advances in Pharmacology, 59, 1–30. 10.1016/S1054-3589(10)59001-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitinen, P. J. , Brown, K. M. , Piippo, K. , Swan, H. , Devaney, J. M. , Brahmbhatt, B. , … Kontula, K. (2001). Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation, 103, 485–490. 10.1161/01.CIR.103.4.485 [DOI] [PubMed] [Google Scholar]
- Leenhardt, A. , Lucet, V. , Denjoy, I. , Grau, F. , Ngoc, D. D. , & Coumel, P. (1995). Catecholaminergic polymorphic ventricular tachycardia in children. A 7‐year follow‐up of 21 patients. Circulation, 91, 1512–1519. 10.1161/01.CIR.91.5.1512 [DOI] [PubMed] [Google Scholar]
- Lieve, K. V. , van der Werf, C. , & Wilde, A. A. (2016). Catecholaminergic polymorphic ventricular tachycardia. Circulation Journal, 80, 1285–1291. 10.1253/circj.CJ-16-0326 [DOI] [PubMed] [Google Scholar]
- Marjamaa, A. , Laitinen‐Forsblom, P. , Lahtinen, A. M. , Viitasalo, M. , Toivonen, L. , Kontula, K. , & Swan, H. (2009). Search for cardiac calcium cycling gene mutations in familial ventricular arrhythmias resembling catecholaminergic polymorphic ventricular tachycardia. BMC Medical Genetics, 10, 12 10.1186/1471-2350-10-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros‐Domingo, A. , Bhuiyan, Z. A. , Tester, D. J. , Hofman, N. , Bikker, H. , van Tintelen, J. P. , … Ackerman, M. J. (2009). The RYR2‐encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise‐induced long QT syndrome: A comprehensive open reading frame mutational analysis. Journal of the American College of Cardiology, 54, 2065–2074. 10.1016/j.jacc.2009.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata, K. , Ohno, S. , Itoh, H. , & Horie, M. (2018). Bradycardia is a specific phenotype of catecholaminergic polymorphic ventricular tachycardia induced by RYR2 mutations. Internal Medicine, 57, 1813–1817. 10.2169/internalmedicine.9843-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima, T. , Kaneko, Y. , Taniguchi, Y. , Hayashi, K. , Takizawa, T. , Suzuki, T. , & Nagai, R. (1997). The mechanism of catecholaminergic polymorphic ventricular tachycardia may be triggered activity due to delayed afterdepolarization. European Heart Journal, 18, 530–531. 10.1093/oxfordjournals.eurheartj.a015281 [DOI] [PubMed] [Google Scholar]
- Nguyen, H. L. , Pieper, G. H. , & Wilders, R. (2013). Andersen‐Tawil syndrome: Clinical and molecular aspects. International Journal of Cardiology, 170, 1–16. 10.1016/j.ijcard.2013.10.010 [DOI] [PubMed] [Google Scholar]
- Ohno, S. , Omura, M. , Kawamura, M. , Kimura, H. , Itoh, H. , Makiyama, T. , … Horie, M. (2014). Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non‐compaction. Europace, 16, 1646–1654. 10.1093/europace/eut382 [DOI] [PubMed] [Google Scholar]
- Paavola, J. , Viitasalo, M. , Laitinen‐Forsblom, P. J. , Pasternack, M. , Swan, H. , Tikkanen, I. , … Laine, M. (2007). Mutant ryanodine receptors in catecholaminergic polymorphic ventricular tachycardia generate delayed afterdepolarizations due to increased propensity to Ca2+ waves. European Heart Journal, 28, 1135–1142. 10.1093/eurheartj/ehl543 [DOI] [PubMed] [Google Scholar]
- Plaster, N. M. , Tawil, R. , Tristani‐Firouzi, M. , Canun, S. , Bendahhou, S. , Tsunoda, A. , … Ptacek, L. J. (2001). Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell, 105, 511–519. 10.1016/S0092-8674(01)00342-7 [DOI] [PubMed] [Google Scholar]
- Postma, A. V. , Denjoy, I. , Kamblock, J. , Alders, M. , Lupoglazoff, J. M. , Vaksmann, G. , … Wilde, A. A. (2005). Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. Journal of Medical Genetics, 42, 863–870. 10.1136/jmg.2004.028993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori, S. G. , Napolitano, C. , Tiso, N. , Memmi, M. , Vignati, G. , Bloise, R. , … Danieli, G. A. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation, 103, 196–200. 10.1016/S1062-1458(01)00326-9 [DOI] [PubMed] [Google Scholar]
- Sonoda, K. , Ohno, S. , Otuki, S. , Kato, K. , Yagihara, N. , Watanabe, H. , … Horie, M. (2017). Quantitative analysis of PKP2 and neighbouring genes in a patient with arrhythmogenic right ventricular cardiomyopathy caused by heterozygous PKP2 deletion. Europace, 19, 644–650. 10.1093/europace/euw038 [DOI] [PubMed] [Google Scholar]
- Sonoda, K. , Ohno, S. , Ozawa, J. , Hayano, M. , Hattori, T. , Kobori, A. , … Horie, M. (2018). Copy number variations of SCN5A in Brugada syndrome. Heart Rhythm: the Official Journal of the Heart Rhythm SocietyThe Official Journal of the Heart Rhythm Society, 15, 1179–1188. 10.1016/j.hrthm.2018.03.033 [DOI] [PubMed] [Google Scholar]
- Surawicz, B. (1998). U wave: Facts, hypotheses, misconceptions, and misnomers. Journal of Cardiovascular Electrophysiology, 9, 1117–1128. 10.1111/j.1540-8167.1998.tb00890.x [DOI] [PubMed] [Google Scholar]
- Szentpali, Z. , Szili‐Torok, T. , & Caliskan, K. (2013). Primary electrical disorder or primary cardiomyopathy? A case with a unique association of noncompaction cardiomyopathy and cathecolaminergic polymorphic ventricular tachycardia caused by ryanodine receptor mutation. Circulation, 127, 1165–1166. 10.1161/CIRCULATIONAHA.112.144949 [DOI] [PubMed] [Google Scholar]
- Tang, Y. , Tian, X. , Wang, R. , Fill, M. , & Chen, S. R. (2012). Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circulation Research, 110, 968–977. 10.1161/CIRCRESAHA.111.256560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester, D. J. , Arya, P. , Will, M. , Haglund, C. M. , Farley, A. L. , Makielski, J. C. , & Ackerman, M. J. (2006). Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm: the Official Journal of the Heart Rhythm SocietyThe Official Journal of the Heart Rhythm Society, 3, 800–805. 10.1016/j.hrthm.2006.03.025 [DOI] [PubMed] [Google Scholar]
- Vega, A. L. , Tester, D. J. , Ackerman, M. J. , & Makielski, J. C. (2009). Protein kinase A‐dependent biophysical phenotype for V227F‐KCNJ2 mutation in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol, 2, 540–547. 10.1161/CIRCEP.109.872309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viitasalo, M. , Oikarinen, L. , Vaananen, H. , Kontula, K. , Toivonen, L. , & Swan, H. (2008). U‐waves and T‐wave peak to T‐wave end intervals in patients with catecholaminergic polymorphic ventricular tachycardia, effects of beta‐blockers. Heart Rhythm: the Official Journal of the Heart Rhythm SocietyThe Official Journal of the Heart Rhythm Society, 5, 1382–1388. 10.1016/j.hrthm.2008.06.011 [DOI] [PubMed] [Google Scholar]