

Abstract
Over the last decade, anaplastic lymphoma kinase (ALK), a receptor tyrosine kinase (RTK), has been identified as a fusion partner in a diverse variety of translocation events resulting in oncogenic signaling in many different cancer types. In tumors where the full‐length ALK RTK itself is mutated, such as neuroblastoma, the picture regarding the role of ALK as an oncogenic driver is less clear. Neuroblastoma is a complex and heterogeneous tumor that arises from the neural crest derived peripheral nervous system. Although high‐risk neuroblastoma is rare, it often relapses and becomes refractory to treatment. Thus, neuroblastoma accounts for 10–15% of all childhood cancer deaths. Since most cases are in children under the age of 2, understanding the role and regulation of ALK during neural crest development is an important goal in addressing neuroblastoma tumorigenesis. An impressive array of tyrosine kinase inhibitors (TKIs) that act to inhibit ALK have been FDA approved for use in ALK‐driven cancers. ALK TKIs bind differently within the ATP‐binding pocket of the ALK kinase domain and have been associated with different resistance mutations within ALK itself that arise in response to therapeutic use, particularly in ALK‐fusion positive non‐small cell lung cancer (NSCLC). This patient population has highlighted the importance of considering the relevant ALK TKI to be used for a given ALK mutant variant. In this review, we discuss ALK in neuroblastoma, as well as the use of ALK TKIs and other strategies to inhibit tumor growth. Current efforts combining novel approaches and increasing our understanding of the oncogenic role of ALK in neuroblastoma are aimed at improving the efficacy of ALK TKIs as precision medicine options in the clinic.
Keywords: Anaplastic lymphoma kinase, ALKAL, TKIs, neuroblastoma, non‐small cell lung cancer, ALK‐positive tumours, signal transduction
The ALK receptor tyrosine kinase can be activated in a wide range of human cancers by both chromosomal translocations leading to ALK‐fusion proteins and mutation in the context of full‐length ALK (Fig. 1). In addition to these two main mechanisms of ALK activation, ALK overexpression and activation in the absence of genetic aberration has also been described.
Figure 1.
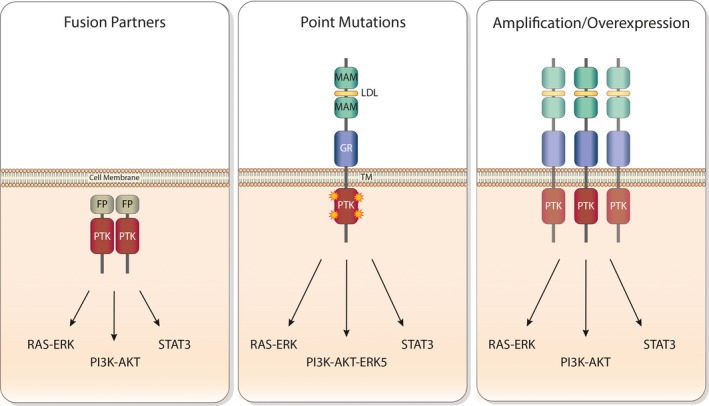
Oncogenic anaplastic lymphoma kinase (ALK) signaling. Schematic representation of different oncogenic forms of ALK (fusions, point mutations, amplification/overexpression) and downstream signaling pathways. FP, fusion protein; GR, glycine‐rich domain; LDL, low‐density lipoprotein class A domain; MAM, meprin, A‐5 protein and receptor protein tyrosine phosphatase Mu domain; PTK, protein tyrosine kinase domain; TM, transmembrane region.
ALK‐Fusion Proteins in Human Cancer – Identification of Chromosomal Translocations
Anaplastic lymphoma kinase was originally described as a fusion partner with nucleophosmin (NPM) in anaplastic large cell lymphoma (ALCL) 1. Since then, almost 30 different ALK‐fusion partners have been reported, identifying the ALK locus as a ‘hot spot’ for translocation events that occur in a wide range of cancers 2, 3. ALK‐fusion proteins share common features, including: (i) regulation of expression by the promotor of the fusion partner, (ii) modulation of subcellular localization by the fusion partner and (iii) ALK‐fusion dimerization/oligomerization by the fusion partner, leading to trans‐autophosphorylation of the ALK kinase domain and subsequent signaling to downstream targets 4, 5, 6, 7. Here, we briefly introduce ALK fusions in three of the more studied cancers: ALCL, inflammatory myofibroblastic tumors (IMTs) and non‐small cell lung cancer (NSCLC).
ALK Fusions in ALCL, IMT and NSCLC
Anaplastic large cell lymphoma
Anaplastic large cell lymphoma (ALCL) is a rare type of Non‐Hodgkin lymphoma involving T‐cell receptor rearrangement that commonly occurs in children and young adults 8. In ALCL, the predominant ALK translocation fusion partner is NPM‐ALK, which occurs in approximately 80% of ALK‐positive ALCL cases (Fig. 2) 9, 10. The molecular characterization of NPM‐ALK was first reported in ALCL in 1994, with a number of other ALK translocation fusions since reported in ALCL, including MSN‐ALK, ALO17‐ALK, TFG‐ALK, TPM3‐ALK, TPM4‐ALK, MYH9‐ALK, ATIC‐ALK, CLTC‐ALK and TRAF1‐ALK 3, 8.
Figure 2.
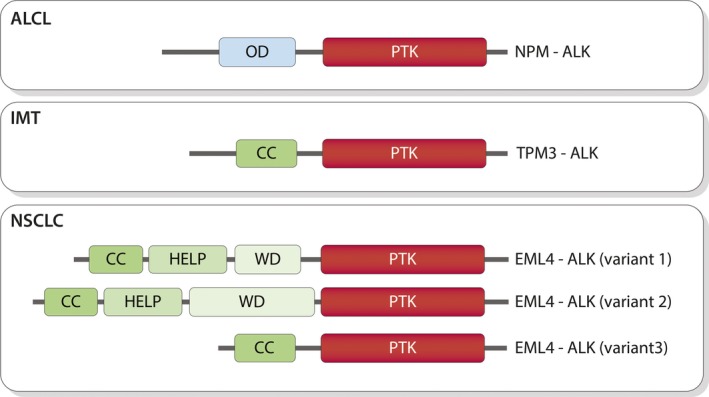
Schematic depicting the domain structure of the most common anaplastic lymphoma kinase (ALK) fusions found in anaplastic large cell lymphoma (ALCL), inflammatory myofibroblastic tumors (IMTs) and non‐small cell lung cancer (NSCLC). Fusion partners mediate dimerization of the ALK‐fusion proteins resulting in constitutive activation of the ALK tyrosine kinase. Domains are highlighted as: oligomerization domain (OD, in blue), ALK protein tyrosine kinase (PTK, in red), coiled coil (CC, in green), HELP (lime green), WD40 (light green).
Inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumors (IMTs) are rare mesenchymal neoplasms that frequently originate in the lung, abdomen and retroperitoneal region and mostly affect young adults 11. Almost 50% of IMT cases exhibit rearrangement of the ALK locus at 2p23, of which half are fusions with TPM3 that result in the TPM3‐ALK fusion protein (Fig. 2) 12, 13. ALK translocations in both ALCL and IMT are associated with better prognosis 14, 15, 16. Similar to ALCL, other ALK fusions, such as TPM4‐ALK, SEC31A‐ALK, PPFIBP1‐ALK, RANBP2‐ALK, CARS‐ALK, ATIC‐ALK, CLTC‐ALK, TFG‐ALK, EML4‐ALK, PRKAR1A‐ALK, LMNA‐ALK, FN1‐ALK and NUMA1‐ALK, are also found 3, 17, 18.
Non‐small cell lung cancer
Lung cancer is one of the leading causes of cancer death worldwide, which is classified into two subgroups: (i) small cell lung cancer (SCLC) and (ii) non‐small cell lung cancer (NSCLC) 19, 20. Almost 80% of lung carcinoma belongs to the NSCLC subgroup. The EML4‐ALK fusion protein accounts for around 2–9% of NSCLC adenocarcinoma cases, and ALK‐positive NSCLCs therefore represent the largest ALK‐positive patient group 2, 5, 21, 22. EML4‐ALK is the product of an inversion event at chromosome 2p, which results in the fusion of N‐terminal region containing coiled coil domain of the EML4 gene with the tyrosine kinase domain of the ALK gene 5, 21. At least 15 different EML4‐ALK variants have been described to date, with variants 1, 2 and 3a and 3b being most common (Fig. 2) 23, 24. Almost all EML4‐ALK variants contain exons 20–29 of ALK encoding the intracellular kinase domain; however, they contain different portions of EML4, which are thought to play a role in the stability or activity of the resulting fusion protein 7, 23, 24, 25. In addition to EML4‐ALK, other translocations reported in NSCLC are HIP1‐ALK, STRN‐ALK, PTPN3‐ALK, TFG‐ALK, KLC1‐ALK, KIF5B‐ALK and TPR‐ALK 3, 21, 26, 27, 28, 29, 30, 31. ALK‐targeted therapies are routinely employed clinically for ALK‐positive NSCLC; however, understanding the resistance mechanisms that arise in response to ALK inhibitor therapy is currently a major clinical challenge 2, 32, 33, 34, 35.
ALK Point Mutations in Human Cancer
Activation of ALK, whether by ALKAL ligands or by mutation, leads to downstream signaling via MEKK2/3‐MEK5‐ERK5, PI3K‐AKT‐mTOR, RAS‐MAPK and PLC‐γ pathways (Fig. 3) 7, 36, 37. The signaling initiated by ALK varies depending on the cell or tumor type as well as the method of ALK activation, whether by ligand, fusion partner, overexpression or activating mutation. A number of cancers have been associated with activating point mutations in ALK, including anaplastic thyroid tumors (ATC), NSCLC and neuroblastoma 2, 7, 38, 39, 40, 41, 42, 43. While the ALK point mutations ALK‐L1198F and ALK‐G1201E were described as gain‐of‐function activating point mutations in anaplastic thyroid tumor (ATC) 43, a recent report has shown that neither is constitutively active, thus questioning the role of ALK as an oncogenic driver in ATC 44. We focus in the following sections on ALK variants in neuroblastoma and patients, predominantly with ALK‐positive NSCLC, who have been treated with ALK TKIs.
Figure 3.
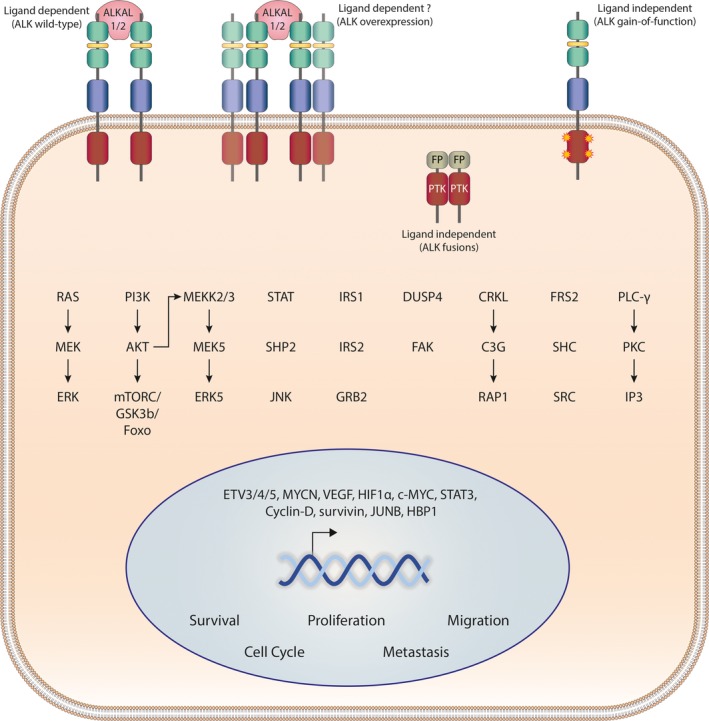
General overview of anaplastic lymphoma kinase (ALK) downstream signaling. ALK signaling can be activated in a ligand‐dependent (ALK wild‐type) or a ligand‐independent manner (ALK gain‐of‐function, ALK fusions, overexpression/amplification). ALK signals through multiple downstream pathways and stimulates the initiation of transcription to regulate specific cellular processes. The range of signaling pathways and cellular responses activated in response to ALK activation varies with cell type and ALK status (such as ALK fusion, overexpression or point mutation).
Neuroblastoma – A Brief Overview
Neuroblastoma is a childhood cancer that arises in the sympathetic nervous system. It accounts for 8–10% of all childhood cancer deaths and is the most commonly diagnosed cancer in infants under one year 45. Neuroblastoma is a complex and heterogeneous disease which affects very young children with a median age of 22 months at diagnosis 46, 47. Children can develop tumors at any point along the sympathetic chain; however, neuroblastoma most frequently originates in the area of the adrenal medulla, disseminating to tissues of the abdomen, chest, pelvis and neck region 45, 47, 48, 49. Neuroblastoma is classified in five clinical stages (stages 1–4 and 4S) according to the International Neuroblastoma Staging System (INSS) 50, 51, 52. In neuroblastoma, as in other pediatric cancers, the mutation load is low 53, 54, 55. In contrast, chromosomal aberrations are important for prognosis in neuroblastoma, with the most common genetic anomalies being deletions of parts of chromosome arms 1p and 11q, 17q gain, triploidy, as well as MYCN and ALK amplifications 47, 56, 57, 58, 59, 60. Amplification of MYCN on chromosome 2p24 is one of the main hallmarks of neuroblastoma, observed in 20–30% of all neuroblastoma cases and associated with poor survival 47, 51, 61. MYCN is involved in cell proliferation, apoptosis, survival and differentiation 62. Neuroblastoma models in which MYCN is overexpressed in the neural crest lead to neuroblastoma tumor development, that is accelerated by cooperation with other oncogenes and tumor suppressor genes, such as ALK, NF1, TP53, LIN28B and LMO1, driving increased penetrance and earlier onset of neuroblastoma 63, 64, 65, 66, 67, 68. Other factors, which also contribute to neuroblastoma tumorigenesis, are loss of heterozygosity (LOH) for chromosome 14 (14q), loss of NF1 and CDKN2A, amplification of DDX1 and MDM2, aberrant expression of neurotrophin receptors, ganglioside GD2, polycomb complex protein Bmi‐1, micro RNAs (miR‐10b, miR‐29a/b, miR‐335), as well as mutations in PHOX2B, ATRX, CHEK2 and BARD1 53, 69, 70, 71, 72, 73, 74, 75, 76, 77. In addition to protein coding genes, long noncoding RNAs, such as neuroblastoma associated transcript‐1 (NBAT‐1) and Cancer Susceptibility 15 (CASC15), regulate neuroblastoma tumorigenesis via cell proliferation and neuronal differentiation 78, 79.
ALK Mutations in Neuroblastoma
Initial reports of ALK gene amplification and ALK protein overexpression suggested a role of ALK in neuroblastoma 80, 81. This was firmly established with the identification of ALK point mutations in both familial and sporadic neuroblastoma 56, 57, 82, 83, 84. In addition, ALK activating deletions and translocations have been described 40, 85. The majority of the reported mutations are located within the ALK kinase domain and are present in 7–8% of all neuroblastoma cases 58, 86. While a large range of mutations are observed, the most frequently found ‘hotspot’ mutations are ALK‐F1174 (V, L, S, I, C), ALK‐F1245 (C, I, L, V) and ALK‐R1275 (L or Q) in the kinase domain, which account for around 85% of all ALK mutant cases (Table 1 and Fig. 4A). ALK‐F1174, ALK‐F1245 and ALK‐R1275 mutant variants are transforming when expressed in either nude mice or NIH3T3 cells 82, 83, 87. Furthermore, ALK drives the transcription of MYCN and ALK‐F1174L has been shown to cooperate with MYCN to enhance the tumorigenic activity in neuroblastoma mouse models 3, 64, 88, 89, 90. Subsequent analyses of the remaining 15% of ALK mutations found in neuroblastoma patients have highlighted differential activity, and ligand‐dependent/ligand‐independent characteristics of mutant variants, many of which remain to be characterized fully in the context of neuroblastoma development 86, 90, 91. Mutations at residues ALK‐G1128, ‐M1166, ‐I1170, ‐I1171, ‐R1192, ‐L1196 (gatekeeper), ‐L1240 and ‐Y1278 have also been shown to be activating neuroblastoma mutations (Fig. 4A). These mutations are in close proximity to important structures within the kinase domain and likely regulate the activation of ALK activity, such as the alpha‐C‐helix, and the activation loop 3, 58, 86. More recent analysis of relapsed neuroblastoma has highlighted an increased frequency of activating ALK point mutations in these patients 92, 93, 94, 95.
Table 1.
Frequency of ALK mutations in neuroblastoma
| Bresler et al. | De Brouwer et al. | |
|---|---|---|
| Number of neuroblastoma investigated | 1596 | 709 |
| Number of neuroblastoma with ALK mutations (%) | 126 (8) | 49 (6.9) |
| Number of ALK‐F1174L/V/S/I/C (% of ALK‐positive) | 38 (30) | 17 (34.7) |
| Number of ALK‐F1245C/I/L/V (% of ALK‐positive) | 15 (12) | 3 (6.1) |
| Number of ALK‐R1275L/Q (% of ALK‐positive) | 54 (43) | 24 (49) |
Figure 4.
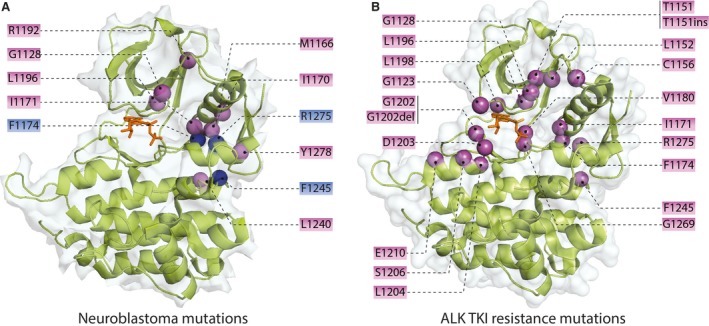
Anaplastic lymphoma kinase (ALK) kinase domain point mutations in neuroblastoma and resistance mutations from patients treated with ALK tyrosine kinase inhibitors (TKIs). (A) ALK kinase domain showing activating mutations reported in neuroblastoma patients (purple spheres). The three hotspot mutations (F1174, F1245 and R1275, blue spheres) are indicated. (B) ALK tyrosine kinase domain resistance mutations from patients treated with ALK TKIs are shown (purple spheres). ATP is shown in the ATP‐binding site of the kinase domain (orange).
Studies of ALK germline mutations in familial neuroblastoma have shown that neuroblastoma has an incomplete penetrance and the risk of developing the disease likely depends on other players, such as segmental chromosomal aberrations 57, 84, 96, 97, 98. As illustration, a recent study reported two siblings both carrying a germline ALK‐R1275Q mutation that exhibited very different neuroblastoma aggressiveness and chemotherapy response. Genetic analysis identified several differing segmental chromosomal aberrations including the amplification of MYCN between the siblings that potentially impacted on the progression of their disease 98. Indeed, studies in model systems such as mice or zebrafish suggest that activating ALK mutations alone do not drive neuroblastoma, rather ALK works together with other oncogenes to promote neuroblastoma tumor development 64, 89, 99, 100, 101, 102. A better understanding of the developmental processes that regulate the penetrance of ALK germline mutations should aid in unraveling of the underlying mechanisms of oncogenesis of familial neuroblastoma.
Targeting ALK
Since oncogenic ALK signaling is involved in several cancer forms, it stands to reason that targeting ALK and its downstream partners would be therapeutically beneficial in ALK‐positive cancer patients. ALK downstream signaling involves multiple known pathways, such as MEKK2/3‐MEK5‐ERK5, PI3K‐AKT‐mTOR, RAS‐MAPK and PLC‐γ (Fig. 3) 7. NVP‐TAE684 was one of the first ALK‐specific inhibitors identified to target the ATP‐binding site of ALK 103, and initial studies identified reduced cell proliferation in ALK‐positive ALCL, NSCLC and neuroblastoma cell lines on treatment with NVP‐TAE684 87, 104. While NVP‐TAE684 is not used therapeutically, a number of other ALK tyrosine kinase inhibitors (TKIs) have been developed and employed clinically in ALK‐positive patient populations 2, 105, 106.
Crizotinib
Crizotinib was the first ALK‐targeted TKI to enter the clinic (Fig. 5) 22. In 2011, the FDA approved crizotinib for the treatment of ALK‐fusion positive NSCLC patients based on the results from phase I/II clinical studies 2, 22, 107. In subsequent clinical studies, crizotinib was shown to be superior to conventional chemotherapy in advanced ALK‐fusion positive NSCLC 2, 108. The efficacy of crizotinib has been tested in other ALK‐fusion positive cancer forms, including pediatric and adult ALCL with good responses 109, 110, 111. However, responses in patients with ALK‐positive neuroblastoma and IMT were less encouraging 110. Response to crizotinib in ALK‐fusion positive NSCLC is transient due to the acquisition of secondary mutations in the kinase domain of the ALK fusions themselves (Fig. 4B) or by ALK copy number gain or bypass survival signaling via alternative oncogenes 2, 32, 34. Crizotinib is also less effective on brain metastases in ALK‐fusion positive NSCLC patients 112. Next‐generation ALK TKIs have been developed that address activity in the brain and secondary resistance mutations.
Figure 5.
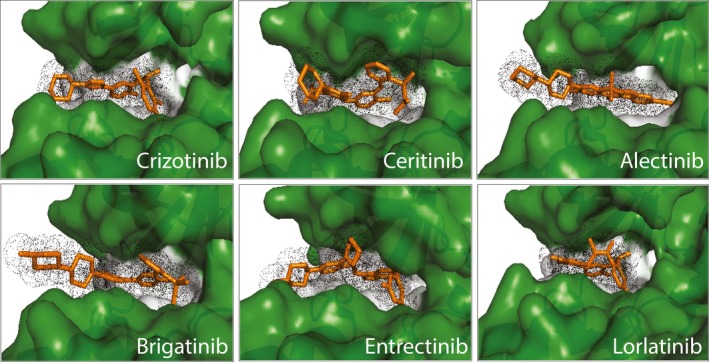
A selection of FDA approved anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors (TKIs) in the ATP‐binding site of the ALK kinase domain. The different ALK TKIs (orange) bind in the ATP‐binding pocket with varying contact sites resulting in unique profiles of inhibition of the ALK mutant variants observed in neuroblastoma patients as well as in ALK TKI resistant non‐small cell lung cancer (NSCLC) and inflammatory myofibroblastic tumor (IMT) patients.
Ceritinib
In 2014, the FDA approved the second‐generation ALK TKI ceritinib for crizotinib resistance ALK‐fusion positive NSCLC patients 32, 113, 114. Like crizotinib, ceritinib is an ATP competitive inhibitor which binds in the ALK ATP‐binding pocket (Fig. 5). Ceritinib is a derivative of NVP‐TAE684 and in addition to inhibiting ALK is effective against insulin‐like growth factor receptor‐1 (IGF‐1R), STKK22D and INSR 115, 116. Ceritinib is able to overcome both ALK‐crizotinib resistance mutations (G1269A, L1196M, I1171T/N and S1206C/Y) and ALK‐alectinib resistance mutations (I1171T/N/S and V1180L) (Fig. 4B) 116, 117. Ceritinib is also effective in the treatment of ALK‐rearranged ALCL 118. The median progression‐free survival (PFS) with ceritinib in ALK‐fusion positive NSCLC is 7–8 months, after which ALK secondary mutations arise and the response to ceritinib significantly decreases 119. While few reports exist in ALK‐positive neuroblastoma, one patient with a complete response to ceritinib has been described 120.
Alectinib
Alectinib is a potent ALK TKI which displays activity toward crizotinib resistance mutations including L1196M, F1174L, R1275Q and C1156Y (Fig. 5) 32, 121. A phase I/II trial in Japanese patients with ALK‐rearranged NSCLC led to the approval of alectinib in Japan and in 2015 the FDA granted breakthrough therapy designation for ALK‐fusion positive NSCLC patients who have progressed with crizotinib 122, 123. In a recent phase III clinical study of untreated advanced ALK‐fusion positive NSCLC, treatment with alectinib showed superior efficacy, with a 12‐month survival of 66.4%, and lower toxicity when compared with crizotinib 124. Further, alectinib demonstrated superior central nervous system (CNS) activity and delayed CNS progression in ALK‐fusion positive NSCLC relative to crizotinib 125. Alectinib is an effective inhibitor of the gatekeeper mutation ALK‐L1196M as well as the ALK‐R1275Q and ALK‐F1174L neuroblastoma hotspot mutations 121. Similar to other ALK TKIs, alectinib treatment leads to resistance with the ALK‐G1202R, ALK‐V1180L and ALK‐I1171T mutations reported as well as other ALK‐independent mechanisms 117, 126. Alectinib has been studied preclinically in neuroblastoma and has been employed in one heavily pretreated, refractory, metastatic ALK‐F1245C neuroblastoma case, where a partial clinical response was observed 127, 128.
Brigatinib
Brigatinib, FDA approved in 2017, is a potent inhibitor of ALK that is also effective against epidermal growth factor receptor (EGFR) and ROS Proto‐Oncogene 1, RTK (ROS1), and is effective against a range of ALK resistance mutations (Fig. 5) 129. In phase I/II trials in crizotinib resistance ALK‐positive NSCLC patients, brigatinib showed 72% overall response with a median PFS of 11–13 months 32. A recently published phase III trial showed a superior efficacy of brigatinib as compared with crizotinib in the treatment‐naïve ALK‐fusion positive NSCLC, with an estimated 12‐month event‐free survival of 67% 130. The predominant resistance mutation seen in response to alectinib therapy is ALK‐G1202R which is resistant to most ALK TKIs, with the exception of lorlatinib (see below). Sequential treatment with crizotinib and brigatinib has been reported to result in dual mutations such as ALK‐E1210K+ALK‐S1206C as well as ALK‐E1210K+ALK‐D1203N 2, 131. Brigatinib has been explored in preclinical neuroblastoma models, where it has been shown to inhibit ALK more effectively than crizotinib 132.
Entrectinib
Entrectinib, FDA approved as breakthrough therapy 2017, is a potent inhibitor of ALK, NTRK and ROS1 (Fig. 5), that is currently being evaluated in phase I/II trials for patients with ALK, ROS1, NTRK alterations 32, 133. A recent report identified entrectinib as effective in the reduction of neuroblastoma cell proliferation and tumor growth 134. Entrectinib has orphan drug designation for treating neuroblastoma patients as well as for NTRK, ALK, ROS1 alterations in NSCLC and metastatic colorectal cancer. Entrectinib has been studied in preclinical models, where it has been shown to have activity toward the ALK‐G1202R mutant 135.
Lorlatinib
Lorlatinib, FDA accelerated approval in 2018, is a novel, highly potent ALK/ROS1 inhibitor that can pass the blood–brain barrier (Fig. 5). Lorlatinib overcomes almost all known ALK resistance mutations observed with other ALK TKIs, including the ALK‐G1202 mutation 2, 136, 137. In both in vitro and in vivo systems, lorlatinib is more potent than other ALK TKIs 138. It has been shown that lorlatinib exhibits superior potency toward ALK in preclinical neuroblastoma tumor models 139, 140. Lorlatinib is currently being investigated in trails for ALK/ROS1‐positive NSCLC as well as in neuroblastoma, where it shows strong antitumor and CNS activity both in treatment‐naïve and previously ALK TKI treatment ALK‐positive NSCLC patients 32, 141. Due to its high efficacy, lorlatinib may serve as a useful partner for combinatorial treatments to overcome the emergence of resistance clones in ALK‐positive cancers.
ALK Kinase Domain Resistance Mutations in Response to ALK TKI Treatment
Based on in vitro drug screens, in vivo models and patient data, ALK TKI resistance mechanisms can be classified into two major groups 2, 34, 142. The first group is ALK dependent, which includes ALK secondary resistance mutations (Fig. 4B). The ALK gatekeeper mutations L1196M and C1156Y were the first reported resistance mutations in ALK‐fusion positive NSCLC 39. The other common resistance mutations seen in NSCLC are I1151Tins, F1174C/L/V, L1152P/R, G1202R, G1269A/S, D1203N, S1206C/Y 2, 34, 39, 116, 117 (Fig. 4B). In most cases, ALK secondary mutations can be overcome by second‐ and third‐generation ALK TKIs. However, consecutive treatment with ALK TKIs in patients can lead to dual mutational loads (such as E1210K/D1203N, C1156Y/L1198F and C1156Y/I1171N) which confer resistance to third‐generation ALK TKIs 2, 131. Much effort is being expended to match the individual ALK resistance mutations with the most effective inhibitor in ALK‐fusion positive NSCLC 2, 33, 35. Individual patient treatments can become complex involving therapeutic approaches that can include multiple ALK TKI treatments, illustrated by a report of therapeutic use of lorlatinib in a patient with crizotinib‐resistant ALK‐fusion positive NSCLC that led to the appearance of the ALK‐L1198F+C1156Y resistance mutation 143. In the same study, it was shown that this lorlatinib resistance mutation is sensitive to crizotinib treatment 143, highlighting the importance of understanding the dynamics of resistance mutations that arise in response to ALK TKIs. Interestingly, secondary resistance mutations have not yet been reported in neuroblastoma, where mutations in ALK are already present as primary mutations. The second group of resistance mutations is ALK independent and includes the activation of alternative oncogenes (such as EGFR, IGFR, MET, KIT) and lineage alterations described in NSCLC 2, 34, 142. In neuroblastoma, activation of alternative oncogenic drivers Axl and ErbB4 has been reported to lead to ALK TKI resistance in preclinical analyses 144, 145.
Targeting ALK in Neuroblastoma
The identification of ALK mutations in neuroblastoma, both at initial diagnosis and at increased frequency in relapsed neuroblastoma cases, has driven efforts to effectively employ ALK TKIs clinically. A phase I trial looking at the safety and activity of crizotinib included 11 neuroblastoma patients with identified ALK mutant variant status, including the three hotspot mutations (F1174, R1245 and R1275), as well as one patient with an ALK‐Y1278S mutation 110. Only one complete response was observed in a patient harboring a germline ALK‐R1275 mutation. Unfortunately, little is known about other somatic or companion mutations carried by these patients. A number of studies have investigated the effect of ALK TKIs in a neuroblastoma setting 86, 87, 90, 120, 132, 139, 140, 146, 147. Table 2 shows a selection of FDA approved ALK TKIs, brigatinib, ceritinib, lorlatinib and crizotinib 120, 132, 140, that vary in their efficacy to inhibit the different ALK mutant variants observed in neuroblastoma in preclinical models. Although not performed side‐by‐side, the accumulated results illustrate potential differences, important as the different ALK TKIs bind differentially within the ATP‐binding pocket of the ALK kinase domain (Fig. 5). Since ALK mutations found in both initial and in relapsed neuroblastoma cases are located mostly in the alpha‐C‐helix and the activation loop, some overlap exists with the reported NSCLC resistance mutations (Fig. 4) 7. Comparing inhibition of the hotspot mutations F1174, F1245 and R1275 in in vitro assays shows that ceritinib is generally twofold more effective than crizotinib, which inhibits the hotspot mutations in the range of 25–35 nM (Table 2). Brigatinib and lorlatinib abrogate the activity of ALK neuroblastoma hotspot mutations within a single digit nanomolar (nM) range (Table 2). The ALK‐I1171N mutant variant presents challenges for many ALK TKIs, and both ceritinib and brigatinib are less efficient at inhibiting this variant than lorlatinib (Table 2). This likely reflects sensitivity to steric effects of the ALK‐I1171N mutation on the binding surfaces in the ATP‐binding pocket. A recent molecular dynamics study shows dislocation of the ALK TKI alectinib triggered by the ALK‐I1171N mutation, which induces conformational changes at the inhibitor binding site that impact on the interaction between alectinib and the ALK‐I1171N mutant 148. The third‐generation ALK TKI lorlatinib shows strong activity toward all tested ALK neuroblastoma mutant variants, supporting the current clinical testing of lorlatinib in neuroblastoma 139, 140. Based on the efficacy in preclinical systems, both lorlatinib and brigatinib appear to be good options for the targeting of ALK in neuroblastoma.
Table 2.
IC50 values for inhibition of ALK Y1604 phosphorylation in the context of full‐length ALK expressed in PC12 cells by either brigatinib, lorlatinib, ceritinib or crizotinib
| ALK mutation | ALK TKI IC50s | ||||
|---|---|---|---|---|---|
| Brigatinib | Lorlatinib | Ceritinib | Crizotinib* | Crizo SD* | |
| Wildtype | 2.60 | 0.80 | 5.30 | 16.40 | 4.17 |
| G1128A | 2.00 | 1.20 | 19.80 | 33.53 | 10.15 |
| I1171N | 10.30 | 3.20 | 46.60 | 97.53 | 50.00 |
| I1171T | 7.50 | 6.80 | 17.40 | 193.00 | |
| F1174L | 1.50 | 0.70 | 17.00 | 23.16 | 5.8 |
| R1192P | 2.50 | 0.90 | 26.10 | 42.50 | 19.95 |
| F1245V | 6.60 | 0.90 | 24.70 | 32.02 | 6.78 |
| G1269A | 3.40 | 6.40 | 29.80 | 130.13 | 24.11 |
| R1275Q | 4.20 | 0.80 | 10.80 | 30.63 | 1.72 |
| Y1278S | 4.60 | 2.30 | 20.50 | 53.7 | 29.91 |
ALK, anaplastic lymphoma kinase; TKI, tyrosine kinase inhibitor.
The table is compiled from three independent articles, all of which have employed investigated crizotinib for comparison with other ALK TKIs 120, 132, 140. Crizotinib* indicates average of crizotinib treatment based on these studies and crizo* SD indicates standard deviation of these values. Results for ALK‐I1171T are taken from 120.
Combinatorial Treatments in Neuroblastoma
Given the complex pattern of ALK resistance mutations that arise in response to ALK TKI treatment, combinatorial targeting of downstream targets or other bypass pathway components could offer therapeutic benefit for ALK‐positive neuroblastoma patients and also hinder the development of resistance. Choosing the right target for poly‐therapy is complicated, considering the issue of toxicity and the fact that negative feedback signaling events may lead to the development of resistance 149, 150. Successful combinatorial treatments should not only show efficacy superior to mono‐treatment but also be tolerable at effective doses in patients. Several combinations of ALK TKI with chemotherapy agents, immunotherapy agents and downstream target agents have been evaluated (Table 3). These include the use of ERK5, mTOR, CDK4/6 and RET inhibitors together with ALK for improved inhibition of tumor growth in preclinical models 146, 147, 151, 152, 153. Recent proteomics‐based studies have also identified additional targets, such as IGF‐1R/INSR, identifying combined ALK and IGFR inhibition as effective in reducing the proliferation of ALK‐positive neuroblastoma cell lines 154, 155. Efforts are currently being concentrated on understanding how ALK TKIs can be employed therapeutically in combination so in the future patients are treated accordingly.
Table 3.
Different combinatorial targets in ALK‐positive cancers
| Molecular targets | Pre‐clinical experimental model | Combinatorial effects |
|---|---|---|
| ALK + MEK | EML4‐ALK (v1) positive NSCLC | Inhibition of NSCLC cell proliferation and tumor growth |
| ALK‐positive neuroblastoma | Increased AKT activity | |
| ALK + mTORC1/C2 | NPM‐ALK‐positive ALCL (mTORC1) | Inhibition of ALCL cell proliferation and tumor growth |
| ALK‐positive neuroblastoma (mTORC1/C2) | Inhibition of neuroblastoma cell proliferation and tumor growth | |
| ALK‐positive neuroblastoma (mTORC1) | Increased activation of AKT via Rictor | |
| ALK + ERK5 | ALK‐positive neuroblastoma | Inhibition of neuroblastoma cell proliferation and tumor growth |
| ALK + HSP90 | EML4‐ALK‐positive NSCLC | Inhibition of NSCLC cell proliferation and tumor growth |
| ALK + CDK4/6 | ALK‐positive neuroblastoma | Inhibition of neuroblastoma cell proliferation and tumor growth |
| ALK + IGF‐1R | EML4‐ALK‐positive NSCLC | Inhibition of NSCLC cell proliferation and tumor growth |
| ALK‐positive neuroblastoma | Inhibition of neuroblastoma cell proliferation |
ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; IGF‐1R, insulin‐like growth factor receptor‐1; NSCLC, non‐small cell lung cancer.
Concluding Remarks
Much current activity in the field of ALK inhibition in neuroblastoma centers around understanding how, when and if, the impressive arsenal of ALK TKIs can be usefully employed therapeutically in this patient population. This is an important challenge for the research field to tackle since current therapeutic regimes for high‐risk neuroblastoma come with significant side‐effects and morbidity. Those neuroblastoma cases that have shown responses to ALK TKI treatment would indicate that there is a window of opportunity to be better clarified. It is clear that tumor complexity in terms of heterogeneity and genetic background plays significant roles in neuroblastoma and that there is much to be learned. Improved understanding of the underlying biology of ALK in the neural crest during development and how that function is perturbed in neuroblastoma will be important. The combination of novel techniques now available, including single cell‐based approaches, offers unique opportunities to define heterogeneity, immune cell tumor infiltration and tumor microenvironments, ranging from tumor models in simple model systems to patient samples. These advanced analyses offer the power to address many challenging questions in the coming years, leading to improved treatments with precision medicine.
This work has been supported by grants from the Swedish Cancer Society (BH CAN18/718; RHP CAN18/729), the Swedish Childhood Cancer Foundation (BH 2015‐80 and 2014‐150; RHP 2015‐96; GU 2018‐0056), the Swedish Research Council (RHP 2015‐04466; BH 521‐2012‐2831), the Swedish Foundation for Strategic Research (RB13‐0204, http://www.nnbcr.se) and the Göran Gustafsson Foundation (RHP2016).
Conflicts of Interest
The authors declare that they have no competing interests.
Umapathy G, Mendoza‐Garcia P, Hallberg B, Palmer RH. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019; 127: 288–302.
Contributor Information
Bengt Hallberg, Email: bengt.hallberg@gu.se.
Ruth H. Palmer, Email: ruth.palmer@gu.se.
References
- 1. Morris S, Kirstein M, Valentine M, Dittmer K, Shapiro D, Saltman D, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non‐Hodgkin's lymphoma. Science 1994;263:1281–4. [DOI] [PubMed] [Google Scholar]
- 2. Lin JJ, Riely GJ, Shaw AT. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov 2017;7:137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013;13:685–700. [DOI] [PubMed] [Google Scholar]
- 4. Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non‐Hodgkin's lymphoma‐associated NPM‐anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol 1997;17:2312–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Soda M, Choi YL, Enomoto M, Shuji T, Yoshihiro Y, Shunpei I, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature 2007;448:561–6. [DOI] [PubMed] [Google Scholar]
- 6. Tort F, Pinyol M, Pulford K, Roncador G, Hernandez L, Nayach I, et al. Molecular characterization of a new ALK translocation involving moesin (MSN‐ALK) in anaplastic large cell lymphoma. Lab Invest 2001;81:419–26. [DOI] [PubMed] [Google Scholar]
- 7. Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol 2016;27(Suppl 3):iii4–15. [DOI] [PubMed] [Google Scholar]
- 8. Turner SD, Lamant L, Kenner L, Brugières L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br J Haematol 2016;173:560–72. [DOI] [PubMed] [Google Scholar]
- 9. Stein H, Foss HD, Dürkop H, Marafioti T, Delsol G, Pulford K, et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 2000;96:3681–95. [PubMed] [Google Scholar]
- 10. Amin HM, Lai R. Pathobiology of ALK+ anaplastic large‐cell lymphoma. Blood 2007;110:2259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meis JM, Enzinger FM. Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol 1991;15:1146–56. [DOI] [PubMed] [Google Scholar]
- 12. Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999;59:2776–80. [PubMed] [Google Scholar]
- 13. Lawrence B, Perez‐Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3‐ALK and TPM4‐ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011;117:5019–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chun YS, Wang L, Nascimento AG, Moir CR, Rodeberg DA. Pediatric inflammatory myofibroblastic tumor: anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer 2005;45:796–801. [DOI] [PubMed] [Google Scholar]
- 16. Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf‐Peeters C, et al. ALK+ lymphoma: clinico‐pathological findings and outcome. Blood 1999;93:2697–706. [PubMed] [Google Scholar]
- 17. Lovly CM, Gupta A, Lipson D, Otto G, Brennan T, Chung CT, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 2014;4:889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rao N, Iwenofu H, Tang B, Woyach J, Liebner DA. Inflammatory myofibroblastic tumor driven by novel NUMA1‐ALK fusion responds to ALK inhibition. J Natl Compr Canc Netw 2018;16:115–21. [DOI] [PubMed] [Google Scholar]
- 19. Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: diagnosis and management. Am Fam Physician 2007;75:56–63. [PubMed] [Google Scholar]
- 20. Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, et al. Cancer statistics, 2004. CA Cancer J Clin 2004;54:8–29. [DOI] [PubMed] [Google Scholar]
- 21. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007;131:1190–203. [DOI] [PubMed] [Google Scholar]
- 22. Kwak EL, Bang Y‐J, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N Engl J Med 2010;363:1693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sabir SR, Yeoh S, Jackson G, Bayliss R. EML4‐ALK variants: biological and molecular properties, and the implications for patients. Cancers (Basel) 2017;9:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woo CG, Seo S, Kim SW, Jang SJ, Park KS, Song JY, et al. Differential protein stability and clinical responses of EML4‐ALKfusion variants to various ALK inhibitors in advanced ALK‐rearranged non‐small cell lung cancer. Ann Oncol 2016;28:791–7. [DOI] [PubMed] [Google Scholar]
- 25. Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke‐Want H, Koker M, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res 2011;17:7394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM, Horlings HM, et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome‐centred RNA sequencing. J Pathol. 2013;230:270–6. [DOI] [PubMed] [Google Scholar]
- 27. Choi Y‐L, Lira ME, Hong M, Kim RN, Choi S‐J, Song J‐Y, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thorac Oncol 2014;9:563–6. [DOI] [PubMed] [Google Scholar]
- 28. Fang DD, Zhang B, Gu Q, Lira M, Xu Q, Sun H, et al. HIP1‐ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol 2014;9:285–94. [DOI] [PubMed] [Google Scholar]
- 29. Jung Y, Kim P, Jung Y, Keum J, Kim S‐N, Choi YS, et al. Discovery of ALK‐PTPN3 gene fusion from human non‐small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosom Cancer 2012;51:590–7. [DOI] [PubMed] [Google Scholar]
- 30. Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, et al. KIF5B‐ALK, a novel fusion oncokinase identified by an immunohistochemistry‐based diagnostic system for ALK‐positive lung cancer. Clin Cancer Res 2009;15:3143–9. [DOI] [PubMed] [Google Scholar]
- 31. Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R, et al. KLC1‐ALK: a novel fusion in lung cancer identified using a formalin‐fixed paraffin‐embedded tissue only. PLoS ONE 2012;7:e31323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dagogo‐Jack I, Shaw AT, Riely GJ. Optimizing treatment for patients with ALK positive lung cancer. Clin Pharmacol Ther 2017;101:625–33. [DOI] [PubMed] [Google Scholar]
- 33. Katayama R. Therapeutic strategies and mechanisms of drug resistance in anaplastic lymphoma kinase (ALK)‐rearranged lung cancer. Pharmacol Ther 2017;177:1–8. [DOI] [PubMed] [Google Scholar]
- 34. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non‐small cell lung cancer. Clin Cancer Res 2012;18:1472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR‐driven or ALK‐driven NSCLC: first‐generation or next‐generation TKI? Nat Rev Clin Oncol 2018;15:694–708. [DOI] [PubMed] [Google Scholar]
- 36. Guan J, Umapathy G, Yamazaki Y, Wolfstetter G, Mendoza P, Pfeifer K, et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. Elife 2015;4:e09811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reshetnyak AV, Murray PB, Shi X, Mo ES, Mohanty J, Tome F, et al. Augmentor alpha and beta (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: hierarchy and specificity of ligand‐receptor interactions. Proc Natl Acad Sci USA 2015;112:15862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cazes A, Louis‐Brennetot C, Mazot P, Dingli F, Lombard B, Boeva V, et al. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res 2013;73:195–204. [DOI] [PubMed] [Google Scholar]
- 39. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4‐ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734–9. [DOI] [PubMed] [Google Scholar]
- 40. Okubo J, Takita J, Chen Y, Oki K, Nishimura R, Kato M, et al. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene 2012;31:4667–76. [DOI] [PubMed] [Google Scholar]
- 41. Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, et al. The neuroblastoma‐associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK‐translocated cancers. Cancer Res 2010;70:10038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y‐W, Tu P‐H, Lin K‐T, Lin S‐C, Ko J‐Y, Jou Y‐S. Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer. Neoplasia 2011;13:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res 2011;71:4403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guan J, Wolfstetter G, Siaw J, Chand D, Hugosson F, Palmer RH, et al. Anaplastic lymphoma kinase L1198F and G1201E mutations identified in anaplastic thyroid cancer patients are not ligand‐independent. Oncotarget 2016;8:11566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010;362:2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kamijo T, Nakagawara A. Molecular and genetic bases of neuroblastoma. Int J Clin Oncol 2012;17:190–5. [DOI] [PubMed] [Google Scholar]
- 47. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 48. Tsubota S, Kadomatsu K. Origin and initiation mechanisms of neuroblastoma. Cell Tissue Res. 2018;372:211–221. [DOI] [PubMed] [Google Scholar]
- 49. Nakagawara A. Neural crest development and neuroblastoma: the genetic and biological link. Prog Brain Res 2004;146:233–42. [DOI] [PubMed] [Google Scholar]
- 50. Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–77. [DOI] [PubMed] [Google Scholar]
- 51. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007;369:2106–20. [DOI] [PubMed] [Google Scholar]
- 52. van Noesel MM, Versteeg R. Pediatric neuroblastomas: genetic and epigenetic ‘danse macabre’. Gene 2004;325:1–15. [DOI] [PubMed] [Google Scholar]
- 53. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high‐risk neuroblastoma. Nat Genet 2013;45:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 55. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan‐cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Caren H, Abel F, Kogner P, Martinsson T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J 2008;416:153–9. [DOI] [PubMed] [Google Scholar]
- 57. Janoueix‐Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008;455:967–70. [DOI] [PubMed] [Google Scholar]
- 58. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout E, et al. Meta‐analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 2010;16:4353–62. [DOI] [PubMed] [Google Scholar]
- 59. Michels E, Vandesompele J, De Preter K, Hoebeeck J, Vermeulen J, Schramm A, et al. ArrayCGH‐based classification of neuroblastoma into genomic subgroups. Genes Chromosom Cancer 2007;46:1098–108. [DOI] [PubMed] [Google Scholar]
- 60. Vandesompele J, Baudis M, De Preter K, Van Roy N, Ambros P, Bown N, et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J Clin Oncol 2005;23:2280–99. [DOI] [PubMed] [Google Scholar]
- 61. Schwab M, Ellison J, Busch M, Rosenau W, Varmus He, Bishop Jm. Enhanced expression of the human gene N‐myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc Natl Acad Sci USA 1984;81:4940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eilers M, Eisenman RN. Myc's broad reach. Genes Dev 2008;22:2755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 1997;16:2985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012;22:117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He S, Mansour MR, Zimmerman MW, Ki DH, Layden HM, Akahane K, et al. Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP‐related domain. Elife 2016;5:e14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chesler L, Goldenberg DD, Collins R, Grimmer M, Kim GE, Tihan T, et al. Chemotherapy‐induced apoptosis in a transgenic model of neuroblastoma proceeds through p53 induction. Neoplasia. 2008;10:1268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhu S, Zhang X, Weichert‐Leahey N, Dong Z, Zhang C, Lopez G, et al. LMO1 synergizes with MYCN to promote neuroblastoma initiation and metastasis. Cancer Cell 2017;32:310–23 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Molenaar JJ, Domingo‐Fernández R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let‐7 suppression. Nat Genet 2012;44:1199–206. [DOI] [PubMed] [Google Scholar]
- 69. Thompson PM, Seifried BA, Kyemba SK, Jensen SJ, Guo C, Maris JM, et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med Pediatr Oncol 2001;36:28–31. [DOI] [PubMed] [Google Scholar]
- 70. George RE, Kenyon RM, McGuckin AG, Malcolm AJ, Pearson AD, Lunec J. Investigation of co‐amplification of the candidate genes ornithine decarboxylase, ribonucleotide reductase, syndecan‐1 and a DEAD box gene, DDX1, with N‐myc in neuroblastoma. United Kingdom Children's Cancer Study Group. Oncogene 1996;12:1583–7. [PubMed] [Google Scholar]
- 71. Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 2004;75:727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012;307:1062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Inomistova MV, Svergun NM, Khranovska NM, Skachkova OV, Gorbach OI, Klymnyuk GI. Prognostic significance of MDM2 gene expression in childhood neuroblastoma. Exp Oncol 2015;37:111–5. [PubMed] [Google Scholar]
- 74. Chang HH, Chen CH, Chou CH, Liao YF, Huang MJ, Chen YH, et al. beta‐1,4‐Galactosyltransferase III enhances invasive phenotypes via beta1‐integrin and predicts poor prognosis in neuroblastoma. Clin Cancer Res 2013;19:1705–16. [DOI] [PubMed] [Google Scholar]
- 75. Hölzel M, Huang S, Koster J, Øra I, Lakeman A, Caron H, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 2010;142:218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. The I, Murthy AE, Hannigan GE, Jacoby LB, Menon AG, Gusella JF, et al. Neurofibromatosis type 1 gene mutations in neuroblastoma. Nat Genet 1993;3:62–6. [DOI] [PubMed] [Google Scholar]
- 77. Corvi R, Savelyeva L, Breit S, Wenzel A, Handgretinger R, Barak J, et al. Non‐syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene 1995;10:1081–6. [PubMed] [Google Scholar]
- 78. Pandey GK, Mitra S, Subhash S, Hertwig F, Kanduri M, Mishra K, et al. The risk‐associated long noncoding RNA NBAT‐1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014;26:722–37. [DOI] [PubMed] [Google Scholar]
- 79. Mondal T, Juvvuna PK, Kirkeby A, Mitra S, Kosalai ST, Traxler L, et al. Sense‐antisense lncRNA pair encoded by locus 6p22.3 determines neuroblastoma susceptibility via the USP36‐CHD7‐SOX9 regulatory axis. Cancer Cell 2018;33:417–34 e7. [DOI] [PubMed] [Google Scholar]
- 80. Osajima‐Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol 2005;167:213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Passoni L, Longo L, Collini P, Coluccia Aml, Bozzi F, Podda M, et al. Mutation‐independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Can Res 2009;69:7338–46. [DOI] [PubMed] [Google Scholar]
- 82. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008;455:971–4. [DOI] [PubMed] [Google Scholar]
- 83. George RE, Sanda T, Hanna M, Fröhling S, Luther W, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fransson S, Hansson M, Ruuth K, Djos A, Berbegall A, Javanmardi N, et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosom Cancer 2015;54:99–109. [DOI] [PubMed] [Google Scholar]
- 86. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014;26:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schonherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, et al. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP‐TAE684. Biochem J 2011;440:405–13. [DOI] [PubMed] [Google Scholar]
- 88. Schönherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012;31:5193–200. [DOI] [PubMed] [Google Scholar]
- 89. Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med. 2012;4:141ra91. [DOI] [PubMed] [Google Scholar]
- 90. Chand D, Yamazaki Y, Ruuth K, Schonherr C, Martinsson T, Kogner P, et al. Cell culture and Drosophila model systems define three classes of anaplastic lymphoma kinase mutations in neuroblastoma. Dis Model Mech 2013;6:373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schonherr C, Ruuth K, Eriksson T, Yamazaki Y, Ottmann C, Combaret V, et al. The neuroblastoma ALK(I1250T) mutation is a kinase‐dead RTK in vitro and in vivo. Transl Oncol 2011;4:258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Martinsson T, Eriksson T, Abrahamsson J, Caren H, Hansson M, Kogner P, et al. Appearance of the novel activating F1174S ALK mutation in neuroblastoma correlates with aggressive tumor progression and unresponsiveness to therapy. Cancer Res 2011;71:98–105. [DOI] [PubMed] [Google Scholar]
- 93. Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 2014;32:2727–34. [DOI] [PubMed] [Google Scholar]
- 94. Bellini A, Bernard V, Leroy Q, Rio Frio T, Pierron G, Combaret V, et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin Cancer Res 2015;21:4913–21. [DOI] [PubMed] [Google Scholar]
- 95. Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS‐MAPK pathway mutations. Nat Genet 2015;47:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol 2012;9:391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bourdeaut F, Ferrand S, Brugières L, Hilbert M, Ribeiro A, Lacroix L, et al. ALK germline mutations in patients with neuroblastoma: a rare and weakly penetrant syndrome. Eur J Hum Genet 2012;20:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kudo K, Ueno H, Sato T, Kubo K, Kanezaki R, Kobayashi A, et al. Two siblings with familial neuroblastoma with distinct clinical phenotypes harboring an ALK germline mutation. Genes Chromosom Cancer 2018;57:665–9. [DOI] [PubMed] [Google Scholar]
- 99. Zhu S, Lee J‐S, Guo F, Shin J, Perez‐Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012;21:362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fadeev A, Krauss J, Singh AP, Nüsslein‐Volhard C. Zebrafish Leucocyte tyrosine kinase controls iridophore establishment, proliferation and survival. Pigment Cell Melanoma Res 2016;29:284–96. [DOI] [PubMed] [Google Scholar]
- 101. Cazes A, Lopez‐Delisle L, Tsarovina K, Pierre‐Eugène C, De Preter K, Peuchmaur M, et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK‐mutated neuroblastoma. Oncotarget 2014;5:2688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ueda T, Nakata Y, Yamasaki N, Oda H, Sentani K, Kanai A, et al. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene 2016;35:4447–58. [DOI] [PubMed] [Google Scholar]
- 103. Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, et al. Identification of NVP‐TAE684, a potent, selective, and efficacious inhibitor of NPM‐ALK. Proc Natl Acad Sci USA 2007;104:270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008;68:3389–95. [DOI] [PubMed] [Google Scholar]
- 105. Rothenstein JM, Chooback N. ALK inhibitors, resistance development, clinical trials. Curr Oncol 2018;25(Suppl 1):S59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Millett RL, Elkon JM, Tabbara IA. Directed therapies in anaplastic lymphoma kinase‐rearranged non‐small cell lung cancer. Anticancer Res 2018;38:4969–75. [DOI] [PubMed] [Google Scholar]
- 107. US Food and Drug Administration . FDA Approves Crizotinib Capsules. US Food and Drug Administration; [online], (2016). https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm490391.htm. [Google Scholar]
- 108. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013;368:2385–94. [DOI] [PubMed] [Google Scholar]
- 109. Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase‐positive lymphoma patients. J Natl Cancer Inst 2014;106:djt378. [DOI] [PubMed] [Google Scholar]
- 110. Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large‐cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 2013;14:472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mosse YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children's Oncology Group Study. J Clin Oncol 2017;35:3215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maillet D, Martel‐Lafay I, Arpin D, Pérol M. Ineffectiveness of crizotinib on brain metastases in two cases of lung adenocarcinoma with EML4‐ALK rearrangement. J Thorac Oncol 2013;8:e30–1. [DOI] [PubMed] [Google Scholar]
- 113. US Food and Drug Administration . FDA broadens ceritinib indication to previously untreated ALK‐positive metastatic NSCLC [online] (2017). https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560873.html.
- 114. Soria J‐C, Tan DSW, Chiari R, Wu Y‐L, Paz‐Ares L, Wolf J, et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): a randomised, open‐label, phase 3 study. Lancet 2017;389:917–29. [DOI] [PubMed] [Google Scholar]
- 115. Katayama R, Shaw AT, Khan TM, Mino‐Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK‐rearranged lung Cancers. Sci Transl Med 2012;4:120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non‐small cell lung cancer. Cancer Discov 2014;4:662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, et al. Two novel ALK mutations mediate acquired resistance to the next‐generation ALK inhibitor alectinib. Clin Cancer Res 2014;20:5686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Richly H, Kim TM, Schuler M, Kim D‐W, Harrison SJ, Shaw AT, et al. Ceritinib in patients with advanced anaplastic lymphoma kinase‐rearranged anaplastic large‐cell lymphoma. Blood 2015;126:1257–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gainor JF, Tan DS, De Pas T, Solomon BJ, Ahmad A, Lazzari C, et al. Progression‐Free and Overall Survival in ALK‐Positive NSCLC Patients Treated with Sequential Crizotinib and Ceritinib. Clin Cancer Res. 2015;21:2745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Guan J, Fransson S, Siaw JT, Treis D, Van den Eynden J, Chand D, et al. Clinical response of the novel activating ALK‐I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb Mol Case Stud 2018;4:a002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90. [DOI] [PubMed] [Google Scholar]
- 122. US Food and Drug Administration . Alectinib approved for (ALK) positive metastatic non‐small cell lung cancer (NSCLC). [online] (2017). https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm584082.html.
- 123. Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, Seto T, et al. Alectinib versus crizotinib in patients with ALK‐positive non‐small‐cell lung cancer (J‐ALEX): an open‐label, randomised phase 3 trial. Lancet 2017;390:29–39. [DOI] [PubMed] [Google Scholar]
- 124. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK‐positive non‐small‐cell lung cancer. N Engl J Med 2017;377:829–38. [DOI] [PubMed] [Google Scholar]
- 125. Gadgeel S, Peters S, Mok T, Shaw AT, Kim DW, Ou SI, et al. Alectinib versus crizotinib in treatment‐naive anaplastic lymphoma kinase‐positive (ALK+) non‐small‐cell lung cancer: CNS efficacy results from the ALEX study. Ann Oncol 2018;29:2214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Isozaki H, Ichihara E, Takigawa N, Ohashi K, Ochi N, Yasugi M, et al. Non‐small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res 2016;76:1506–16. [DOI] [PubMed] [Google Scholar]
- 127. Lu J, Guan S, Zhao Y, Yu Y, Woodfield SE, Zhang H, et al. The second‐generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH‐MYCN transgenic neuroblastoma mouse model. Cancer Lett 2017;400:61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Heath JA, Campbell MA, Thomas A, Solomon B. Good clinical response to alectinib, a second generation ALK inhibitor, in refractory neuroblastoma. Pediatr Blood Cancer 2018;65:e27055. [DOI] [PubMed] [Google Scholar]
- 129. US Food and Drug Administration . Brigatinib. [online] (2017). https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.html.
- 130. Camidge DR, Kim HR, Ahn MJ, Yang JC, Han JY, Lee JS, et al. Brigatinib versus crizotinib in ALK‐positive non‐small‐cell lung cancer. N Engl J Med 2018;379:2027–39. [DOI] [PubMed] [Google Scholar]
- 131. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov 2016;6:1118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Siaw JT, Wan H, Pfeifer K, Rivera VM, Guan J, Palmer RH, et al. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK‐positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016;7:29011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. ClinicalTrials.gov . Basket study of entrectinib (RXDX‐101) for the treatment of patients with solid tumors harboring NTRK 1/2/3 (Trk A/B/C), ROS1, or ALK gene rearrangements (Fusions). Available from: https://ClinicalTrials.gov/show/NCT02568267.
- 134. Iyer R, Wehrmann L, Golden RL, Naraparaju K, Croucher JL, MacFarland SP, et al. Entrectinib is a potent inhibitor of Trk‐driven neuroblastomas in a xenograft mouse model. Cancer Lett 2016;372:179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. Entrectinib, a Pan‐TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther 2016;15:628–39. [DOI] [PubMed] [Google Scholar]
- 136. US Food and Drug Administration . FDA approves lorlatinib for second‐ or third‐line treatment of ALK‐positive metastatic NSCLC. [online](2018). https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm625027.htm.
- 137. Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel‐Vinay S, et al. Lorlatinib in non‐small‐cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open‐label, single‐arm first‐in‐man phase 1 trial. Lancet Oncol 2017;18:1590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF‐06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015;28:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF‐06463922 overcomes primary resistance to crizotinib in ALK‐driven neuroblastoma. Cancer Discov 2016;6:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF‐06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;9:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK‐positive non‐small‐cell lung cancer: results from a global phase 2 study. Lancet Oncol 2018;19:1654–67. [DOI] [PubMed] [Google Scholar]
- 142. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 2014;11:473–81. [DOI] [PubMed] [Google Scholar]
- 143. Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N Engl J Med. 2016;374:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Debruyne DN, Bhatnagar N, Sharma B, Luther W, Moore NF, Cheung N‐K, et al. ALK inhibitor resistance in ALK(F1174L)‐driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016;35:3681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, et al. Lorlatinib treatment elicits multiple on‐ and off‐target mechanisms of resistance in ALK‐driven cancer. Cancer Res 2018;78:6866–80. [DOI] [PubMed] [Google Scholar]
- 146. Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin Cancer Res 2017;23:2856–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Moore NF, Azarova AM, Bhatnagar N, Ross KN, Drake LE, Frumm S, et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK‐mutated neuroblastoma. Oncotarget 2014;5:8737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. He M, Li W, Zheng Q, Zhang H. A molecular dynamics investigation into the mechanisms of alectinib resistance of three ALK mutants. J Cell Biochem 2018;119:5332–42. [DOI] [PubMed] [Google Scholar]
- 149. Breuleux M, Klopfenstein M, Stephan C, Doughty Ca, Barys L, Maira S‐m, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther 2009;8:742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Umapathy G, Guan J, Gustafsson DE, Javanmardi N, Cervantes‐Madrid D, Djos A, et al. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)‐addicted neuroblastomas. Sci Signal 2017;10:eaam7550. [DOI] [PubMed] [Google Scholar]
- 151. Umapathy G, El Wakil A, Witek B, Chesler L, Danielson L, Deng X, et al. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal 2014;7:ra102. [DOI] [PubMed] [Google Scholar]
- 152. Lopez‐Delisle L, Pierre‐Eugène C, Louis‐Brennetot C, Surdez D, Raynal V, Baulande S, et al. Activated ALK signals through the ERK‐ETV5‐RET pathway to drive neuroblastoma oncogenesis. Oncogene 2018;37:1417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zage PE. Novel therapies for relapsed and refractory neuroblastoma. Children (Basel) 2018;5:E148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Emdal KB, Pedersen AK, Bekker‐Jensen DB, Lundby A, Claeys S, De Preter K, et al. Integrated proximal proteomics reveals IRS2 as a determinant of cell survival in ALK‐driven neuroblastoma. Sci Signal 2018;11:eaap9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Van den Eynden J, Umapathy G, Ashouri A, Cervantes‐Madrid D, Szydzik J, Ruuth K, et al. Phosphoproteome and gene expression profiling of ALK inhibition in neuroblastoma cell lines reveals conserved oncogenic pathways. Sci Signal 2018;11:eaar5680. [DOI] [PubMed] [Google Scholar]
- 156. Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, et al. RAS‐MAPK dependence underlies a rational polytherapy strategy in EML4‐ALK‐positive lung cancer. Nat Med 2015;21:1038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Redaelli S, Ceccon M, Antolini L, Rigolio R, Pirola A, Peronaci M, et al. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM‐ALK positive lymphoma. Oncotarget 2016;7:72886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non‐small cell lung cancer. Cancer Discov 2013;3:430–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Lovly CM, McDonald NT, Chen H, Ortiz‐Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co‐targeting IGF‐1R and ALK in ALK fusion‐positive lung cancer. Nat Med 2014;20:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]