

Abstract
Background
Naldemedine (S‐297995) is a peripherally acting μ‐opioid receptor antagonist developed as a once‐daily oral drug for opioid‐induced constipation (OIC) in adults with chronic noncancer or cancer pain. This study characterized the pharmacological effects of naldemedine in vitro and in vivo.
Methods
The binding affinity and antagonist activity of naldemedine against recombinant human μ‐, δ‐, and κ‐opioid receptors were assayed in vitro. Pharmacologic effects of naldemedine were investigated using animal models of morphine‐induced inhibition of small and large intestinal transit, castor oil‐induced diarrhea, antinociception, and morphine withdrawal.
Key Results
Naldemedine showed potent binding affinity and antagonist activities for recombinant human μ‐, δ‐, and κ‐opioid receptors. Naldemedine significantly reduced opioid‐induced inhibition of small intestinal transit (0.03‐10 mg kg−1; P < 0.05) and large intestinal transit (0.3‐1 μmol L−1; P < 0.05). Naldemedine (0.03‐1 mg kg−1) pretreatment significantly reversed the inhibition of castor oil‐induced diarrhea by subcutaneous morphine (P < 0.01). Naldemedine (1‐30 mg kg−1) pretreatment (1 or 2 hours) did not alter the analgesic effects of morphine in a model measuring the latency of a rat to flick its tail following thermal stimulation. However, a significant delayed reduction of the analgesic effect of morphine was seen with higher doses of naldemedine (10‐30 mg kg−1). Some centrally mediated and peripherally mediated withdrawal signs in morphine‐dependent rats were seen with naldemedine doses ≥3 and ≥0.3 mg kg−1, respectively.
Conclusions & Inferences
Naldemedine displayed potent binding affinity to, and antagonistic activity against, μ‐, δ‐, and κ‐opioid receptors. Naldemedine tempered OIC in vivo without compromising opioid analgesia.
Keywords: naldemedine, opioid receptor, opioid‐induced constipation, pharmacology
Naldemedine displayed potent binding affinity to, and antagonistic activity against, μ‐, δ‐, and κ‐opioid receptors. Naldemedine tempered OIC in vivo without compromising opioid analgesia.
Abbreviations
- BBB
blood‐brain barrier
- cm
centimeter
- CNS
central nervous system
- DADLE
[D‐Ala2, D‐Leu5]‐enkephalin
- DAMGO
[D‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin
- EC50
the concentration that produces half the maximal effect
- ED
effective dose
- Emax
percent of maximal stimulation
- GIMM
Gastrointestinal Motility Monitoring system
- GTPγS
guanosine 5'‐O‐[gamma‐thio]triphosphate
- IC50
a concentration producing 50% inhibition
- Kb
binding constant
- Kd
dissociation constant
- Ki
inhibition constant
- MPE
maximal possible effect
- OIC
opioid‐induced constipation
- PAMORA
peripherally acting μ‐opioid receptor antagonist
Key Points.
Naldemedine is a peripherally acting μ‐opioid receptor antagonist developed to treat opioid‐induced constipation (OIC). This report examines the pharmacologic effects of naldemedine in vitro and in vivo.
Naldemedine displayed potent binding affinity to, and antagonistic activity against, μ‐, δ‐, and κ‐opioid receptors. Naldemedine tempered OIC in several in vivo models at doses that did not compromise opioid analgesia.
Naldemedine is approved for the treatment of adults with OIC in Japan and adults with OIC and chronic noncancer pain in the United States.
1. INTRODUCTION
Opioid analgesics are important for the management of moderate‐to‐severe chronic pain. The prevalence of long‐term opioid use in the United States is estimated to be 40‐46 people per 1000 individuals.1 However, the clinical benefit of opioid analgesics is compromised by their side effects, which include nausea, bowel dysfunction including opioid‐induced constipation (OIC), and central nervous system events such as confusion, headache, and hallucination.2, 3, 4, 5
OIC is one of the most common and debilitating side effects of opioids and is characterized by a reduction in bowel movement frequency, development or worsening of straining while passing stool, sense of incomplete bowel evacuation, and hard‐stool formation after initiation of opioid therapy.6, 7, 8, 9, 10 Opioid analgesics act via the μ‐, δ‐, and κ‐opioid receptors distributed widely in the central and peripheral nervous system. Although the role of δ‐ and κ‐opioid receptors in causing gastrointestinal adverse events is less clear, μ‐opioid receptors are expressed throughout the gastrointestinal tract and, upon opioid binding, decrease neural activity in the enteric nervous system. This impairs motility and transit throughout the gastrointestinal tract, reduces the secretion of gut fluid, and increases fluid absorption, resulting in OIC.8
Laxatives, often used as first‐line treatment for OIC, are associated with limited efficacy and do not address the underlying mechanism of OIC.2, 7 Peripherally acting μ‐opioid receptor antagonists (PAMORAs) aim to reverse OIC by blocking opioid actions at peripheral μ‐opioid receptors in the gastrointestinal tract without adversely affecting analgesia.8 Currently, three PAMORAs are approved for OIC: naldemedine,11 naloxegol (oral),12, 13 and methylnaltrexone (oral or subcutaneous).14 Another PAMORA, alvimopan, is approved for postoperative ileus following partial or small bowel resection with primary anastomosis.15
Naldemedine (S‐297995) is a PAMORA indicated for the treatment of OIC, as a once‐daily oral drug, in adult patients with chronic noncancer pain in the United States and in patients with chronic noncancer pain and cancer in Japan. Naldemedine is an amide derivative of the opioid receptor antagonist naltrexone, but with structural modifications that limit its ability to cross the blood‐brain barrier (BBB). In fact, naldemedine showed high oral bioavailability but poor distribution throughout the CNS in pharmacokinetics studies in rats, (the bioavailability and the brain‐to‐plasma concentration ratio of naldemedine at a dose of 1 mg kg−1 were 29% and 0.03, respectively).16 The aims of the studies presented here were to determine the binding affinities and functional activities of naldemedine, to understand the pharmacologic effects of naldemedine in vitro and in vivo in animal models of OIC, and to determine the differences in the doses of naldemedine for treating OIC without impacting nociception or inducing morphine withdrawal.
2. MATERIALS AND METHODS
2.1. Experimental agents
Naldemedine tosylate (96.4% content, anhydrous basis) and methylnaltrexone were manufactured by Shionogi Research Laboratories (Osaka, Japan). Morphine hydrochloride (morphine; 100.7% content [subcutaneous‐administration studies] or 100.1% content [oral‐administration study]) for clinical use and oxycodone hydrochloride (oxycodone; [subcutaneous‐administration studies]) were manufactured by Shionogi & Co., Ltd. (Osaka, Japan). Saline, 5% Xylitol, and distilled water were obtained from Otsuka Pharmaceutical Factory, Inc (Tokushima, Japan). For in vitro studies, the radioligands [3H]‐[D‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin (DAMGO; for μ‐opioid receptors), [3H]‐[D‐Ala2, D‐Leu5]‐enkephalin (DADLE; for δ‐opioid receptors), [3H]‐U‐69,593 (for κ‐opioid receptors), and recombinant human μ‐, δ‐, and κ‐opioid receptors were purchased from PerkinElmer Life and Analytical Sciences, Inc (Kanagawa, Japan). Evans Blue dye, the vehicle used for Evans Blue dye (carboxymethyl cellulose sodium salt), castor oil, and the vehicle used for naldemedine (methylcellulose; 400 cP, 0.5% solution) were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
2.2. In vitro studies on specific binding affinities of naldemedine to opioid receptors and functional activities of naldemedine
The in vitro binding affinities and functional activities of naldemedine for recombinant human μ‐, δ‐, and κ‐opioid receptors were determined and compared with that of the reference compound, methylnaltrexone, as previously described,17 with some modifications.
Binding assays were used to determine the concentration of test substances that inhibited 50% of specific binding (IC50), and the inhibition constant (K i) value for each sample was calculated using the following equation: K i = IC50/(1 + L/K d), where L is the concentration of the radioactive ligand used and K d is the dissociation constant for the radioactive ligand.
A functional antagonist assay was used to determine the EC50 values of naldemedine and methylnaltrexone. Agonist activity as evaluated by a receptor assay was calculated as: Control (%) = {(c‐a)/(b‐a)} × 100; where a = average counts per minute of nonspecific binding (vehicle control), b = average counts per minute of control binding (agonist stimulation), and c = average counts per minute in the presence of naldemedine or methylnaltrexone. In the [35S]‐guanosine 5'‐O‐[gamma‐thio]triphosphate (GTPγS) binding assay, a compound was defined as an inverse agonist if the percent stimulation produced by naldemedine was less (<30%) than basal GTPγS binding levels.
In the functional antagonist assay, the K b value for the cellular assay was calculated as: K b = IC50/{(agonist/EC50) + 1}; where IC50 = the concentration of the antagonist producing 50% inhibition in the presence of agonist, and EC50 = the concentration that produces half the maximal effect of the agonist.
2.3. In vitro study of naldemedine specificity: enzyme inhibition and radioligand receptor binding assays
Naldemedine binding at a single concentration of 10 μmol L−1 was measured against receptors, channels, and transporters, and in functional enzyme assays, by Sekisui Medical Co., Ltd. (Tokyo, Japan). Radioligand binding and enzyme inhibition studies were conducted using cell lines (recombinant or endogenous target expression), animal tissue, or purified enzymes.
2.4. In vivo and ex vivo experiments: animals and procedures
All studies were conducted at Shionogi & Co., Ltd., except for the study involving the oxycodone‐induced small intestinal transit model, which was conducted at the Shiga Laboratory of Nissei Bilis Co., Ltd. (Osaka, Japan). The studies were conducted in accordance with standards of the Institutional Animal Care and Use Committee, Institutional Animal Ethics Committee, and Shionogi's Institutional Animal Care and Use Committee.
All Crlj: WI (Wistar) and Crl: CD (Sprague‐Dawley) male rats were obtained from Charles River Laboratories Japan, Inc (Kanagawa, Japan). Jcl: Wistar rats were supplied by CLEA Japan, Inc (Tokyo, Japan). Hartley guinea pigs were obtained from Japan SLC, Inc (Shizuoka, Japan). All animals were maintained on 12‐hour light/dark cycles and had free access to food and water. Rats were fasted for at least 20 hours for the small transit study and castor oil model, or 15‐24 hours for the antinociceptive model to render the stomach, small intestine, and colon empty prior to the experiments, but received tap water ad libitum. On each experimental day, naldemedine (oral) and opioids were administered at a volume of 2 mL kg−1 at the doses and routes described below.
2.5. Small intestinal transit
The antagonistic effect of naldemedine on constipation caused by morphine‐ or oxycodone‐induced inhibition of small intestinal transit was determined and compared with methylnaltrexone, as previously described,17 with some modifications. Briefly, 6‐week‐old Crlj: WI male rats were allocated into groups (10‐12 per group) based on body weight and administered naldemedine 0.001‐10 mg kg−1 or vehicle, followed by morphine (15 minutes later; 3 mg kg−1 subcutaneously or 20 mg kg−1 orally) or oxycodone (30 minutes later; 1 mg kg−1 subcutaneously). Evans Blue dye was administered intragastrically (0.5% in 2 mL) 45 minutes postdose. Rats were euthanized 15 minutes later by cervical dislocation, and the stomach and small intestine were quickly removed; the mesentery was completely separated to avoid circling. The distance traveled by the dye relative to the total length of the small intestine was measured. Small intestinal transition was calculated as: small intestine transition (%) = (moved distance of coloring matter [cm]/total length of small intestine [cm]) × 100. The effects of naldemedine, methylnaltrexone, or vehicle on morphine‐ or oxycodone‐induced inhibition of small intestinal transit were assumed to be the percent maximal possible effect (MPE). Percent MPEij for the jth individual in Group i (%MPEij) = ([Yij‐Ȳ 2 .]/[Ȳ 1‐Ȳ 2.]) × 100; where Yijis the small intestine transition for the jth individual in Group i, and Ȳ 1 is the average of the small intestine transition in the vehicle control group (Group 1) and Ȳ 2 is the average of the small intestine transition in the morphine or oxycodone control group (Group 2). The mean effective doses (ED50) were also calculated.
2.6. Large intestinal transit
The antagonistic effect of naldemedine on the velocity inhibition of the propulsion constipation caused by morphine was determined as described previously18, 19 with some modifications. Briefly, the large intestine was isolated from the guinea pig and placed in ice‐cold Krebs solution until use. Five centimeters of distal colon was isolated and pinned on either end in the organ bath of the Gastrointestinal Motility Monitoring system (GIMM; Catamount Research and Development; St. Albans, VT, USA), and continuously perfused with 37°C warmed oxygenated Krebs solution at 10 mL min−1 as detailed previously.18, 19 After >25 minutes of incubation, propulsion of a fecal pellet inserted from the oral end of the colon toward the anal end was monitored by a GIMM digital video camera. The velocity of the propulsion was measured on a 2‐cm section of the colon using GIMM software. To measure the effect of naldemedine on the basal rate of propulsion, 1 μmol L−1 naldemedine was added to the perfused Krebs solution for 10 minutes before fecal pellet insertion. To measure the effect of naldemedine (1, 0.3, 0.1, 0.03, and 0.01 μmol L−1) or vehicle control on morphine‐induced delayed fecal propulsion, naldemedine was exposed to Krebs solution for 10 minutes, and 3 μmol L−1 morphine (the morphine dose previously determined to significantly delay feces propulsion) was then added to the same Krebs solution and perfused for 15 minutes. The colon was washed with perfused Krebs solution for 10 minutes after each naldemedine dose, and then re‐used to test the next dose level. For each dose tested, propulsion was measured twice with 5‐minute intervals between runs, and the average velocity was calculated. If the fecal pellet did not emerge after 3 minutes, velocity was regarded as 0 mm s−1.
2.7. Castor oil‐induced diarrhea model
The antagonistic effect of naldemedine in a castor oil‐induced diarrhea model was determined and compared with methylnaltrexone, as previously described20 with some modifications. Briefly, 6‐week‐old Crl: CD male rats were allocated into nine groups (11 per group) based on body weight. Naldemedine (0.003‐1 mg kg−1) or vehicle was administered, followed by 2 mL of castor oil intragastrically 45 minutes later, and subcutaneous morphine 1 mg kg−1 or saline 15 minutes thereafter.
Evaluations were conducted 60 minutes after morphine or saline administration. Each rat was individually transferred to a transparent observation cage with the floor covered with filter paper to absorb moisture. Castor oil‐induced diarrhea was evaluated using a validated 3‐point scale21 for scoring symptoms: 0 = no diarrhea, 1 = mild diarrhea with loose bowel movements, and 2 = intense liquefied diarrhea. The ED50 was also calculated.
2.8. Antinociceptive model
The antianalgesic effect of naldemedine was determined with the tail‐flick test—using a Tail‐Flick Unit (model 7360; Ugo Basile, Italy)—and compared with methylnaltrexone, as previously described17 with some modifications. Briefly, 6‐week‐old Crlj: WI male rats were allocated into seven groups (10‐11 per group), based on the average latency of tail‐flick at pretest and body weight, before the administration of naldemedine (1‐30 mg kg−1) or vehicle, followed by subcutaneous morphine at 6 mg kg−1 or saline. To examine the influence of naldemedine on the analgesic effect of morphine, the rat tail‐flick test was conducted as previously described.22 Thermal stimulation was applied to the ventral surface of the tail, and the latency of the tail withdrawal reflex was measured at 1, 2, 4, 6, 8, and 24 hours after naldemedine administration, following the dosing regimen shown in Figure S1. A cutoff time of 20 seconds was set to prevent tissue damage. The antianalgesic effect was estimated as: normalized latency = postdose latency − predose latency.
2.9. Morphine withdrawal model
An osmotic pump (Model 2ML1, ALZET; Durect Corp., Cupertino, CA, USA) that injected subcutaneous morphine hydrochloride at a rate of 0.25 mg h−1 was implanted in the neck of anesthetized 6‐week‐old Jcl: Wistar male rats (8 per group) to induce morphine dependence. After 5 days, naldemedine 0.01‐7 mg kg−1 or vehicle was administered. Rats were observed for withdrawal signs immediately after dosing, and 1, 2, 4, 6, and 8 hours after dosing. Central withdrawal signs (jumping, wet‐dog shakes, and teeth chattering) and peripheral withdrawal signs (diarrhea and loss of body weight) were recorded. The number of times a rat jumped or had wet‐dog shakes were counted for 20 minutes during each observation period. Diarrhea and teeth chattering were scored as follows: 0 = normal, 1 = slight to moderate, and 2 = marked. Loss of body weight was defined as the difference in body weight from before dosing to 8 hours after dosing (measured at the end of a 20‐minutes observation period).
2.10. Statistical analyses
For in vitro specific binding and functional assays of naldemedine to opioid receptors, K d values of the radioligands for μ‐, δ‐, κ‐opioid receptors were determined by Scatchard plot analysis. In experiments with recombinant human κ‐receptors, K d was obtained from historical values on assay validity. In the human receptor studies, IC50 values were determined with nonlinear regression analysis of each curve using MathIQTM (ID Business Solutions Ltd., Guildford, UK), within the Eurofins Panlabs SMART system (Redmond, WA, USA). For the [35S]‐GTPγS binding functional assays, the EC50, EC95, and Emax (percent of maximal stimulation in the [35S]‐GTPγS binding) values were estimated from saturation analysis of agonist‐stimulated [35S]‐GTPγS binding using the XLfit program (Microsoft Corp., Redmond, WA, USA). When fitting was not performed for abnormal values, and IC50 and K b values were not able to be calculated, the data were excluded from analysis. Data were expressed as the mean ± standard error and/or standard deviation of duplicate measurements, in two or three independent experiments.
For in vitro binding studies of naldemedine to various receptors, channels, transporters, and enzymes, the acceptance criteria of assay values were (a) ≥80% inhibition ratio of the positive control substance, and (b) the difference between duplicate assay values of naldemedine and positive control substance (inhibition ratio [%]) to be within 10% of the mean value of duplicate assay values. If the above criteria were met, re‐assay was not performed. The mean inhibition ratios of naldemedine and positive control substances calculated from duplicate samples were expressed as percentages. Microsoft Excel 2003 (Microsoft Corp.) was used for data processing of in vitro data.
For all in vivo and ex vivo experiments, a two‐sided test was performed, assuming a significance level of 0.05. Statistical analysis was performed using the SAS system (SAS Institute Inc, Cary, NC, USA). The IC50 was estimated, unless indicated otherwise. For the small intestinal transit model, statistical analysis of differences between the vehicle control group and morphine or oxycodone control group was performed using Welch's t test. Statistical analysis of differences between the morphine or oxycodone control group and the groups simultaneously treated with morphine or oxycodone and naldemedine was performed using Dunnett's test.
For the large intestinal transit experiment, differences of fecal pellet propulsive velocity between the basal conditions and vehicle with morphine‐treated conditions were analyzed using a paired t test. Differences between each dose of naldemedine with morphine and morphine‐alone treated condition were analyzed using layout of randomized block design and Dunnett's test. Simple linear regression was used to estimate the IC50 of naldemedine.
For the castor oil‐induced diarrhea model, summary statistics for diarrhea symptom score in each group were calculated. The Wilcoxon rank‐sum test was conducted to determine the difference in diarrhea symptom score between the two groups. The Steel multiple comparison test was used to determine statistical significance for both this model and the morphine withdrawal model. For the antinociceptive model, if the cutoff time of 20 seconds was observed, then the postdose latency was censored. Data exceeding the cutoff time (>20 seconds) were considered as 20 seconds. Frequency of censored data was summarized for each group. Fisher's exact test was used to determine statistical significance, and the Dunn‐Šidák method was used for multiplicity adjustments. When more than one noncensored data point was observed in postdose trials, the log‐rank test was conducted.
3. RESULTS
3.1. In vitro binding and enzyme inhibition activities of naldemedine and methylnaltrexone
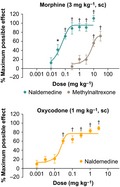
Naldemedine showed potent binding affinities and antagonist activities for recombinant human μ‐, δ‐, and κ‐opioid receptors (Table 1 and Figure 1), which were comparable between species (rats and humans; Table 1). By comparison, methylnaltrexone showed selective binding affinities and antagonist activities for the recombinant human μ‐opioid receptor (Table 1). The EC50 values of naldemedine were >10 μmol L−1 for all three receptors, irrespective of the species, indicating a lack of agonist activity. The inhibition ratio of naldemedine at 10 μmol L−1 for opioid (nonselective) receptor was 100%. However, the inhibition ratios of naldemedine at 10 μmol L−1 for other receptors, ion channels, transporters, and enzymes tested were <50%. Results from the positive control substances, which were measured simultaneously, showed inhibition ratios ≥80%, confirming the validity of the measurement systems.
Table 1.
Specific binding affinities and functional activities of naldemedine to human and rat recombinant opioid receptors and methylnaltrexone to human recombinant opioid receptors
| Drug/Source | Binding (nmol L−1) | Agonist activity (μmol L−1) | Antagonist activity (nmol L−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μ | δ | κ | μ | δ | κ | μ | δ | κ | ||||
| Naldemedine | ||||||||||||
| Human | IC50 | 1.15 ± 0.09a | 1.11 ± 0.21a | 1.50b | EC50 | >10 | >10 | >10 | IC50 | 25.57 ± 2.61a | 7.09 ± 0.74a | 16.1b |
| K i | 0.34 ± 0.03a | 0.43 ± 0.08a | 0.94b | K b | 0.50 ± 0.05a | 0.27 ± 0.03a | 0.44b | |||||
| Rat | IC50 | 3.21 ± 0.54a | 2.13 ± 0.14a | 5.43 ± 0.62a | EC50 | >10 | >10 | >10 | IC50 | 16.08 ± 6.39a | 10.64 ± 0.79a | 12.19 ± 0.57a |
| K i | 1.40 ± 0.24a | 0.96 ± 0.06a | 2.16 ± 0.24a | K b | 0.56 ± 0.02a | 0.22 ± 0.02a | 0.49 ± 0.02a | |||||
| Methylnaltrexone | ||||||||||||
| Human | IC50 | 18.26 ± 3.73a | 8835.89 ± 702.10a | 51.3 ± 2.30b | EC50 | >10 | >10 | >10 | IC50 | 2843.07 ± 308.94a | 28256.28 ± 6114.89a | >10000b |
| K i | 5.50 ± 1.11a | 3453.80 ± 305.23a | 32.1 ± 1.44b | K b | 55.86 ± 6.07a | 1082.71 ± 234.31a | >270.68b | |||||
Values shown are the mean ± standard error.
EC50, the concentration that produces half the maximal effect of the agonist; IC50, the concentration producing 50% inhibition.
K b = IC50/{(agonist/EC50) + 1}; where IC50 = the concentration of the antagonist producing 50% inhibition in the presence of agonist, and EC50 = as described above.
K i = IC50/(1 + [Ligand]/K d); where IC50 = the concentration of the antagonist producing 50% inhibition in the presence of agonist, [Ligand] = concentration of radioligand used in the assay, and K d = dissociation constant for the binding of radioligand to the receptor.
Mean of three independent experiments carried out in duplicate.
Mean of two independent experiments carried out in duplicate.
Figure 1.
(A) Naldemedine and methylnaltrexone binding affinities. μ‐, δ‐, and κ‐opioid receptor binding sites were labeled using [3H]‐DAMGO, [3H]‐DADLE, and [3H]‐U‐69, 593; (B) Agonistic activity of naldemedine and methylnaltrexone for human μ‐, δ‐, and κ‐opioid receptors; (C) Antagonistic activity of naldemedine and methylnaltrexone for human μ‐, δ‐, and κ‐opioid receptors. Functional assays were performed using the [35S]‐GTPγS binding assay. Each point represents the mean ± standard error of 3 (μ‐ and δ‐opioid receptors) or 2 (κ‐opioid receptor) independent experiments performed in duplicate
3.2. In vivo effects of naldemedine and methylnaltrexone on small intestinal transit in rats
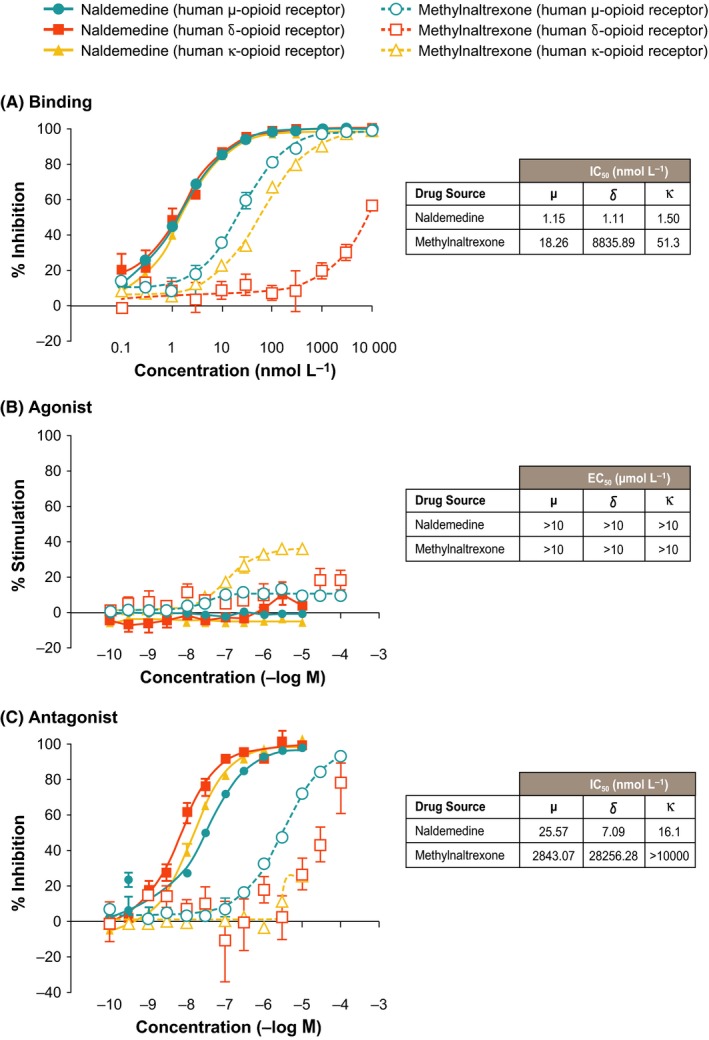
The in vivo effects of naldemedine were examined in a small intestinal transit model, where subcutaneous morphine (3 mg kg−1) or oxycodone (1 mg kg−1) was administered in rat models to induce OIC. The statistical analysis of the %MPE data showed that, compared with the vehicle control, subcutaneous morphine and oxycodone significantly inhibited small intestine transit (all P < 0.01). Naldemedine significantly repressed the opioid‐induced inhibition of small intestinal transit in rats by subcutaneous morphine (P < 0.05 or P < 0.01 for naldemedine 0.03‐10 mg kg−1), and oxycodone (P < 0.01 for naldemedine 0.03‐3 mg kg−1; Figure 2). In the subcutaneous morphine‐induced inhibition model, the ED50 values ± standard error of the mean were 0.03 ± 0.02 and 4.47 ± 2.44 mg kg−1 for naldemedine and methylnaltrexone, respectively; and 0.02 ± 0.015 mg kg−1 for naldemedine in the oxycodone‐induced inhibition model. Additionally, naldemedine significantly repressed the opioid‐induced inhibition of small intestinal transit in rats by oral morphine (dose: 20 mg kg−1) with an ED50 ± standard error of the mean of 0.23 ± 0.087 mg kg−1 (P < 0.01 for naldemedine 0.1, 0.3, 1, 3, and 10 mg kg−1; data not shown).
Figure 2.
The effect of naldemedine and methylnaltrexone on morphine‐induced inhibition of small intestine transit in rats and the effect of naldemedine on oxycodone‐induced inhibition of small intestine transit in rats. Each point represents the mean ± standard error for 10 rats in each group. *P < 0.05; † P < 0.01 compared with vehicle control
3.3. Ex vivo effects of naldemedine on guinea pig large intestinal transit
To directly visualize the effect of naldemedine on large intestinal motility, we conducted a GIMM study using guinea pig distal colons. Fecal pellets propelled smoothly from the oral to the anal side of the colons in the basal state without the presence of either test compound. Vehicle treatment with 3 μmol L−1 morphine was found to cause a significant decrease in the basal propulsive velocity (P < 0.01; Figure S2); therefore, 3 μmol L−1 morphine was selected as the morphine dose for the following experiments. Naldemedine (1 μmol L−1) alone had no effect on the basal propulsion rate (Figure S3).
The effects of naldemedine (0.01‐1 μmol L−1) or vehicle against 3 μmol L−1 morphine on fecal pellet propulsion were examined (Figure 3A). We first analyzed the statistical difference between basal propulsive velocity and the velocity under vehicle‐ and morphine‐treated condition and found a significant decrease in the latter group (P < 0.001). In addition, naldemedine antagonized morphine‐induced delayed propulsion in a dose‐dependent manner, with an IC50 value of 0.31 μmol L−1 (P < 0.05). Figure 3B shows representative results from a single colon. By exposing 1 μmol L−1 naldemedine with 3 μmol L−1 morphine, propulsive motility was not affected. However, when the same colon was exposed to vehicle with 3 μmol L−1 morphine, the propulsive motility was dramatically decreased, and the feces stopped at the oral side (Figure 3B; Video S1).
Figure 3.
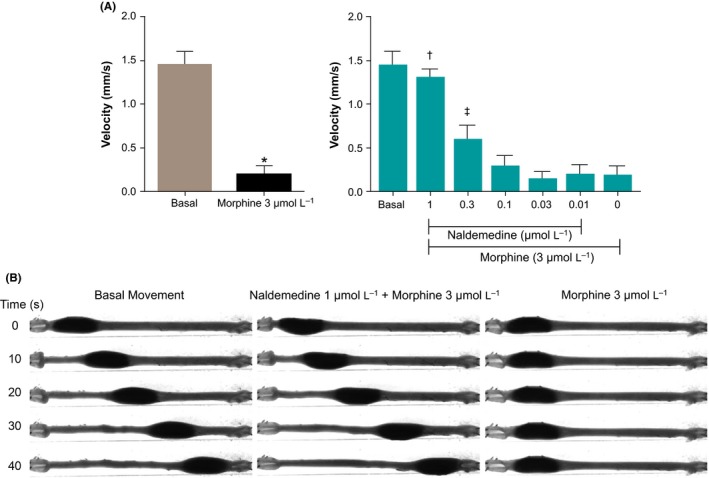
Effect of naldemedine on morphine‐delayed propulsion of guinea pig distal colon. After measuring the basal movement, 1 μmol L−1 naldemedine was added to the perfused Krebs solution for 10 min. 3 μmol L−1 morphine was then added to the same Krebs solution and perfused for 15 min and fecal pellet propulsion was measured. The same method was performed to measure the effect of naldemedine (0.3, 0.1, 0.03, 0.01 μmol L−1 or vehicle) against morphine (3 μmol L−1). (A; Left) Velocity of the basal‐ and vehicle with morphine‐treated conditions. (A: Right) Normalized velocity under several dose of naldemedine and morphine (3 μmol L−1). Velocity was normalized by the basal velocity of each colon. *P < 0.001 versus basal velocity; † P < 0.001, ‡ P < 0.05 versus naldemedine 0 μmol L−1 (vehicle) and morphine (3 μmol L−1) treated condition, respectively; n = 10. Each bar represents the mean ± standard error (B) Representative results of the fecal pellet propulsion from the same colon. Also viewable online as Video S1
3.4. Effect of naldemedine and methylnaltrexone on castor oil‐induced diarrhea
Subcutaneous morphine‐inhibited castor oil‐induced diarrhea (P < 0.01) in rats, and pretreatment with naldemedine 0.03‐1 mg kg−1 or methylnaltrexone 1‐10 mg kg−1 significantly reversed this effect (Figure 4). Naldemedine 0.1‐1 mg kg−1 resulted in a diarrhea symptom score of 2 for all rats (intense liquefied diarrhea). The ED50 values for naldemedine and methylnaltrexone were 0.01 and 0.585 mg kg−1, respectively.
Figure 4.
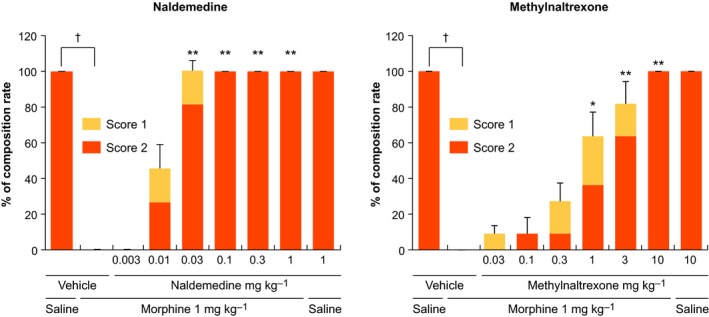
Effect of naldemedine and methylnaltrexone on morphine‐induced inhibition of castor oil‐induced diarrhea in rats. Each bar represents the mean ± standard error for 11 rats in each group. *P < 0.05, **P < 0.01 versus vehicle + morphine. † P < 0.01 for vehicle + saline versus vehicle + morphine. P‐values for each bar consider the summed scores of 1 and 2
3.5. Effect of naldemedine and methylnaltrexone on opioid analgesia
The influence of naldemedine on the analgesic effect of morphine was evaluated by measuring the latency of a rat to flick its tail following thermal stimulation. Subcutaneous morphine significantly prolonged the escape latency (P < 0.01 vs controls). Pretreatment (1 or 2 hours) with naldemedine (3‐30 mg kg−1) did not alter the analgesic effects of morphine (Table 2). Conversely, a significant but delayed inhibition of the analgesic effect of morphine was observed in rats treated with naldemedine at 10‐30 mg kg−1 for longer intervals, as summarized in Table 2. Naldemedine at a dose range of 3‐7 mg kg−1 did not affect the analgesic effect of morphine at any of the time points tested. These results suggest that naldemedine (at 7 mg kg−1) and methylnaltrexone (at 10 mg kg−1; Table S1) do not affect the analgesic effect of morphine in a rat tail‐flick test.
Table 2.
Effect of naldemedine on morphine‐induced analgesic effect measured by tail‐flick test in rats
| Group | n | Normalized latency (s) | |||||
|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 4 h | 6 h | 8 h | 24 h | ||
| Vehicle + saline | 11 | 1.82 (0.85‐2.90) | |||||
| Vehicle + morphine 6 mg kg−1 | 10‐11 | 15.20†† (14.77‐15.60) | 14.97†† (14.77‐15.57) | 15.10†† (14.73‐15.57) | 15.17†† (14.93‐15.40) | 15.50†† (15.17‐15.70) | 15.20†† (14.77‐15.30) |
| Naldemedine + morphine 6 mg kg−1 | |||||||
| 3 mg kg−1 | 11 | 15.27 (14.73‐15.67) | 15.50 (14.57‐15.57) | 15.43 (15.20‐15.67) | 15.40 (15.07‐15.70) | 15.03 (14.70‐15.47) | 15.00 (14.70‐15.50) |
| 5 mg kg−1 | 11 | 15.50 (15.00‐15.83) | 15.13 (14.67‐15.53) | 15.30 (14.83‐15.50) | 15.23 (14.93‐15.60) | 15.20 (14.83‐15.37) | 15.23 (14.70‐15.37) |
| 7 mg kg−1 | 11 | 15.13 (14.60‐15.77) | 15.13 (14.83‐15.83) | 15.37 (15.00‐15.60) | 15.00 (14.50‐15.63) | 15.33 (15.17‐15.53) | 15.07 (14.77‐15.23) |
| 10 mg kg−1 | 11 | 15.03 (14.80‐15.60) | 15.07 (14.30‐15.77) | 14.50 (9.17‐15.30) | 10.52* (5.87‐14.87) | 14.60 (11.65‐15.27) | 14.87 (14.23‐15.67) |
| 30 mg kg−1 | 11 | 15.30 (14.63‐15.67) | 14.17 (5.02‐15.23) | 3.65** (2.77‐4.87) | 4.82** (3.77‐15.63) | 4.50** (4.23‐5.47) | 14.57 (11.40‐15.10) |
The values are the median of normalized latency for tail withdrawal response in each group. In parentheses, quartiles of 25% and 75% are shown.
n indicates the number of rats/group/time point.
P < 0.01 compared to the vehicle‐saline group (log‐rank test).
P < 0.05, **P < 0.01 compared with the vehicle‐morphine group (log‐rank test followed by Dunn‐Sidak method for multiplicity adjustments).
3.6. Effect of naldemedine on opioid withdrawal
The influence of naldemedine on opioid withdrawal was evaluated by examining possible naldemedine‐precipitated withdrawal symptoms in morphine‐dependent rats. Treatment with naldemedine at oral doses of 0.01‐3 mg kg−1 did not result in jumping behavior in morphine‐dependent rats. However, naldemedine 1 mg kg−1 increased diarrhea scores (P < 0.05 at 2 hours postdose vs control); and naldemedine 3 mg kg−1 increased teeth chattering (P < 0.05 at 1, 4, and 8 hours postdose vs control) and diarrhea scores (P < 0.05 at 1 hour postdose vs control; Figure 5). Loss of body weight was noted at naldemedine doses ≥0.3 mg kg−1 (P < 0.05 for 0.3 mg kg−1; P < 0.01 for 1 and 3 mg kg−1 vs control; Figure 5). In another study evaluating possible naldemedine‐precipitated central withdrawal symptoms at higher doses of up to 7 mg kg−1, naldemedine did not result in jumping behavior in morphine‐dependent rats, although a slight increase in the number of wet‐dog shakes was observed at oral doses ≥5 mg kg−1 (P < 0.05 for the 5‐ and 7‐mg kg−1 doses at 4 hours postdose; P < 0.01 for the 5‐ and 7‐mg kg−1 doses at 6 hours postdose). The no‐observed‐effect levels of naldemedine for central and peripheral withdrawal signs are shown in Table 3.
Figure 5.
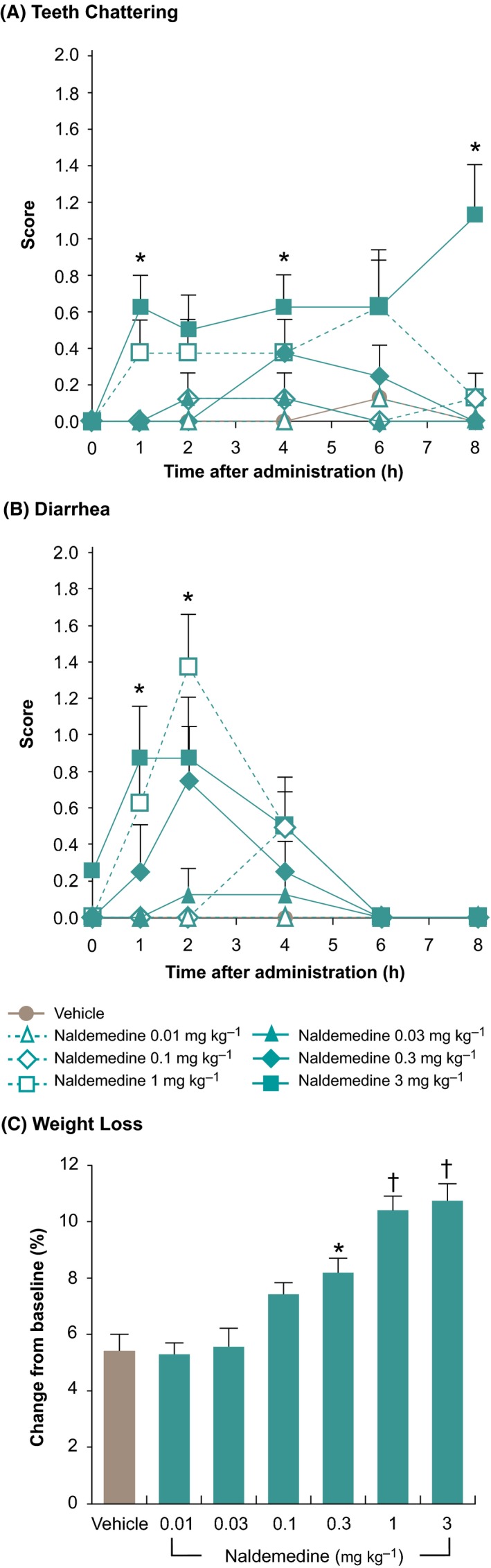
Naldemedine‐precipitated opioid withdrawal signs in morphine‐dependent rats (A) Teeth chattering, (B) Diarrhea, (C) Weight loss (change from baseline at 8 h postdose). Each point or bar represents the mean ± standard error. *P < 0.05, † P < 0.01 compared with the vehicle control group (Steel multiple comparison test)
Table 3.
No‐observed‐effect levels of naldemedine for central‐ and peripheral withdrawal signs, and loss of body weight in morphine‐dependent rats
| Withdrawal signs | No‐observed‐effect level of naldemedine (mg kg−1) |
|---|---|
| Central | |
| Jumping | ≥7 |
| Wet‐dog shakes | 3 |
| Teeth chattering | 1 |
| Peripheral | |
| Diarrhea | 0.3 |
| Loss of body weight | 0.1 |
4. DISCUSSION
The key findings from this study are that naldemedine elicits anticonstipation‐like effects induced by an opioid in three constipation models. Naldemedine also showed antianalgesic and withdrawal effects caused by morphine, but the dose range of these two effects is higher than that for anticonstipation. This difference might be related to the site of action (peripheral and central) to μ‐opioid receptors. These results indicate that naldemedine tempered OIC without causing inhibition of opioid analgesia and withdrawal.
The results presented here confirm the potent binding affinities and antagonist activity of naldemedine to μ‐, δ‐, and κ‐opioid receptors, as well as a lack of agonist activity. No major binding affinity or activity of naldemedine was observed with more than 60 nonopioid targets tested. These results are similar to those for other approved PAMORAs (naloxegol and methylnaltrexone bromide), showing selectivity and affinity for the three opioid receptors.23 However, both naloxegol23 and methylnaltrexone (Table 1) reportedly have a higher affinity for the μ‐opioid (K i: 7.42 and 5.50 nmol L−1, respectively) and κ‐opioid receptors (K i: 8.65 and 32.1 nmol L−1, respectively), in comparison to the δ‐opioid receptor (K i: 203.0 and 3453.80 nmol L−1, respectively), unlike naldemedine, which has a similarly high affinity for all three opioid receptors. Hence, we conclude that naldemedine possesses binding affinity with high antagonistic activity for all three opioid receptors.
Opioids for clinical use, including morphine, oxycodone, hydrocodone, and fentanyl, are μ‐selective agonists,24 suggesting that OIC and the anticonstipation effects of naldemedine are mainly due to effects on the μ receptors. Correspondingly, morphine‐induced gastrointestinal transit inhibition is blocked by μ‐opioid antagonists, but not by δ‐ and κ‐opioid antagonists.25 On the other hand, all three opioid receptors are localized to the enteric nervous system.26 In addition, μ/δ but not μ/κ co‐expression has been observed in rat interstitial cells located adjacent to myenteric plexus structures.27 Furthermore, in μ/δ heteromers, 1 protomer can act as an allosteric modulator of the other protomer.28 Although morphine‐induced gastrointestinal transit inhibition is blocked by a μ‐opioid antagonist, but not by a δ‐ or κ‐opioid antagonist,25 there is a possibility that κ‐ or δ‐opioid antagonists have an effect with μ‐opioid antagonist on gastrointestinal transit inhibition. The contribution of δ‐ or κ‐antagonism of naldemedine to the μ‐antagonistic activity of naldemedine is currently unclear. However, the attenuation of opioid‐induced nausea and vomiting through antagonist activity at the δ‐opioid receptor is supported by preclinical studies of the δ‐opioid receptor selective antagonist TAN‐452, which has shown potent antiemetic effects in morphine‐treated ferrets.17 Further studies are required to understand the contribution of each opioid receptor to OIC and naldemedine activity.
Castor oil is known to release ricinoleic acid followed by alterations in jejunal, ileal, and colonic ion transport and water flux29, 30, 31 leading to increases in fecal output or diarrhea. Morphine suppresses ion transport and water flux,32 and decreases castor oil‐induced fecal output or diarrhea, as a result of this suppression. Naldemedine significantly reversed the inhibitory effect of morphine on castor oil‐induced diarrhea; indicating that naldemedine might improve morphine‐induced stimulation of jejunal, ileal, and colonic ion transport and water flux.
Experiments were also conducted to directly visualize the effect of naldemedine on morphine‐induced delayed propulsion of a fecal pellet in the large intestine. This method has previously been used to examine the restorative effect of naloxone on DAMGO‐suppressed propulsive motility.18 Our results showed that naldemedine improved delayed large intestinal transit caused by morphine in a dose‐dependent manner. As morphine is suggested to act on small and large intestinal muscle contraction, thereby reducing peristaltic movement and consequently inducing constipation,33 naldemedine may have restored propulsion by antagonizing such contraction.
In the preclinical setting, it has been reported that OIC in the small intestine may be regulated by central and peripheral opioid receptors, whereas OIC in the large intestine may be regulated by the peripheral action of opioid receptors.25, 34 Therefore, our methods for the small and large intestinal measurement were suitable to investigate the characteristics of each intestinal region. Furthermore, the small intestine (but not the large intestine) gains tolerance to opioids through chronic exposure. This difference is reflected in the observation that both small and large intestines contribute to the early stages of OIC, whereas chronic OIC is driven mainly by the large intestine.25, 34 Because naldemedine improved both small and large intestinal transits, and showed efficacy in patients with chronic OIC,35 we consider naldemedine as a potential antagonist that improves both acute and chronic stages of OIC.
The antinociceptive model confirmed that the analgesic effect of opioids was maintained even after administration of high doses of naldemedine (up to 7 mg kg−1). However, naldemedine at 10 and 30 mg kg−1 showed a delayed antianalgesic effect at 6 and 4‐8 hours postdose, respectively. This delayed antianalgesic effect is consistent with time‐course experiments of opioid receptor occupancy of 10 and 30 mg kg−1 of naldemedine in the rat cerebral cortex using [3H]‐diprenorphine (data not shown). However, unlike naloxone—a μ‐opioid receptor antagonist with the capability to cross the BBB—the naldemedine dose range for this delayed antianalgesic effect is notably higher than that for anticonstipation.36 Correspondingly, for naldemedine, the highest dose of drug without an observed antianalgesic effect was 233‐fold higher than the ED50 of naldemedine in the small intestinal transit study. By comparison, for methylnaltrexone—another PAMORA used to treat OIC—the highest dose of drug without an observed antianalgesic effect was only 2.24‐fold higher than the ED50 of methylnaltrexone in the small intestinal transit study. These results suggest lower BBB penetration by naldemedine compared with methylnaltrexone.
In a study describing the discovery of naldemedine, naldemedine showed high oral bioavailability but poor distribution throughout the CNS. Moreover, the maximum plasma concentration of naldemedine is enough to antagonize the peripheral μ‐opioid receptor based on a K b value of 0.5 nmol L−1 (antagonist activity). There was no enterohepatic recycling in bile duct‐cannulated tandem rats using [carbonyl14C]‐naldemedine, although hepatic portal vein/bile duct levels were not measured (data not shown). These data support a hypothesis that the predominant effects of naldemedine to enteric nerve are associated with systemic circulation, rather than the CNS and direct effect from intestinal lumen.
Central opioid withdrawal symptoms were not observed with up to 1 mg kg−1 of naldemedine. This result demonstrates that the no‐observed‐effect levels of naldemedine, which underlie its antianalgesic effects (3 mg kg−1), and centrally mediated withdrawal symptoms (1 mg kg−1), are at least 100 times and 30 times, respectively, as high as the naldemedine ED50 for anticonstipation effects (0.03 mg kg−1) under current experimental conditions. These wide margins reinforce naldemedine as a peripherally acting compound.
In conclusion, data from in vitro and in vivo studies indicate that naldemedine has a potent binding affinity and antagonistic activity to the μ‐, δ‐, and κ‐opioid receptors that is different from that of methylnaltrexone. The concentration of naldemedine necessary to inhibit the constipating effect of opioids is much lower than the concentration of naldemedine that interferes with opioid analgesia. The large difference between these concentrations seems to be even wider than the difference in concentrations seen with methylnaltrexone. These data support the clinical results of naldemedine as a treatment for patients with OIC, with minimal concern for interference of the action of opioids in the CNS.37
DISCLOSURES
All authors are employees of Shionogi & Co., Ltd., the manufacturer of naldemedine.
AUTHOR CONTRIBUTIONS
All authors reviewed the manuscript critically. TK designed the research study and wrote the manuscript; KK performed the research and analyzed data for all studies except large intestinal transit; TA performed the research for analgesia and withdrawal; HO performed the research for large intestinal transit; NH performed the research for small intestinal transit and castor oil study; HC performed the research for in vitro experiments and small intestinal transit; AN designed the research study for large intestinal transit; YM designed the research for all studies; TK designed the research for all studies; and MH designed the research for all studies.
Supporting information
ACKNOWLEDGMENTS
All authors had complete access to the data that supports this publication. We thank Dr. T. Suzuki (Hoshi University School of Pharmacy and Pharmaceutical Sciences, Tokyo, Japan) for helpful discussions.
Medical writing and editorial support was provided by Shilpa Aggarwal, PhD, of Oxford PharmaGenesis, Inc, and this support was funded by Shionogi & Co., Ltd.
Kanemasa T, Koike K, Arai T, et al. Pharmacologic effects of naldemedine, a peripherally acting μ‐opioid receptor antagonist, in in vitro and in vivo models of opioid‐induced constipation. Neurogastroenterol Motil. 2019;31:e13563 10.1111/nmo.13563
Presented in part at the American College of Gastroenterology Annual Meeting, October 16–21, 2015, Honolulu, HI, USA.
Funding information
This work was supported by Shionogi & Co., Ltd.
REFERENCES
- 1. Boudreau D, Von Korff M, Rutter CM, et al. Trends in long‐term opioid therapy for chronic non‐cancer pain. Pharmacoepidemiol Drug Saf. 2009;18:1166‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poulsen JL, Brock C, Olesen AE, Nilsson M, Drewes AM. Evolving paradigms in the treatment of opioid‐induced bowel dysfunction. Therap Adv Gastroenterol. 2015;8:360‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morlion B, Clemens KE, Dunlop W. Quality of life and healthcare resource in patients receiving opioids for chronic pain: a review of the place of oxycodone/naloxone. Clin Drug Investig. 2015;35:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lazzari M, Greco MT, Marcassa C, Finocchi S, Caldarulo C, Corli O. Efficacy and tolerability of oral oxycodone and oxycodone/naloxone combination in opioid‐naïve cancer patients: a propensity analysis. Drug Des Devel Ther. 2015;9:5863‐5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Camileri M. Opioid‐induced constipation: challenges and therapeutic opportunities. Am J Gastroenterol. 2011;106:835‐842; quiz 843. [DOI] [PubMed] [Google Scholar]
- 6. Bruner HC, Atayee RS, Edmonds KP, Buckholz GT. Clinical utility of naloxegol in the treatment of opioid‐induced constipation. J Pain Res. 2015;8:289‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bell TJ, Panchal SJ, Miaskowski C, Bolge SC, Milanova T, Williamson R. The prevalence, severity, and impact of opioid‐induced bowel dysfunction: results of a US and European Patient Survey (PROBE 1). Pain Med. 2009;10:35‐42. [DOI] [PubMed] [Google Scholar]
- 8. Camilleri M, Drossman DA, Becker G, Webster LR, Davies AN, Mawe GM. Emerging treatments in neurogastroenterology: a multidisciplinary working group consensus statement on opioid‐induced constipation. Neurogastroenterol Motil. 2014;26:1386‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kalso E, Edwards JE, Moore RA, McQuay HJ. Opioids in chronic non‐cancer pain: systematic review of efficacy and safety. Pain. 2004;112:372‐380. [DOI] [PubMed] [Google Scholar]
- 10. Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non‐malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res Ther. 2005;7:R1046‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Symproic (naldemedine) [package insert]. Florham Park, NJ: Shionogi, Inc; 2018. [Google Scholar]
- 12. Movantik (naloxegol) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2018. [Google Scholar]
- 13. Chey WD, Webster L, Sostek M, Lappalainen J, Barker PN, Tack J. Naloxegol for opioid‐induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387‐2396. [DOI] [PubMed] [Google Scholar]
- 14. Relistor (methylnaltrexone bromide) [package insert]. Raleigh, NC: Salix Pharmaceuticals, Inc.; 2017. [Google Scholar]
- 15. Entereg (alvimopan) [package insert]. Kenilworth, NJ: Merck Sharp & Dohme Corp.; 2015. [Google Scholar]
- 16. Inagaki M, Kume M, Tamura Y, et al. Discovery of naldemedine: a potent and orally available opioid receptor antagonist for treatment of opioid‐induced adverse effects. Bioorg Med Chem Lett. 2019; 29:73–77. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki T, Sawada T, Kawai K, Ishihara Y. Pharmacological profile of TAN‐452, a novel peripherally acting opioid receptor antagonist for the treatment of opioid‐induced bowel syndromes. Life Sci. 2018;215:246‐252. [DOI] [PubMed] [Google Scholar]
- 18. Wood MJ, Hyman NH, Mawe GM. The effects of daikenchuto (DKT) on propulsive motility in the colon. J Surg Res. 2010;164:84‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoffman JM, Brooks EM. Mawe GM. Gastrointestinal motility monitor (GIMM). J Vis Exp. 2010; 1:e2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chittrakarn S, Sawangjaroen K, Prasettho S, Janchawee B, Keawpradub N. Inhibitory effects of kratom leaf extract (Mitragyna speciosa Korth.) on the rat gastrointestinal tract. J Ethnopharmacol. 2008;116:173‐178. [DOI] [PubMed] [Google Scholar]
- 21. Piercey MF, Ruwart MJ. Naloxone inhibits the anti‐diarrhoeal activity of loperamide. Br J Pharmacol. 1979;66:373‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prado WA. Antinociceptive potency of intrathecal morphine in the rat tail flick test: a comparative study using acute lumbar catheter in rats with or without a chronic atlanto‐occipital catheter. J Neurosci Methods. 2003;129:33‐39. [DOI] [PubMed] [Google Scholar]
- 23. Naloxegol NDA 204‐760. Briefing Document for the Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC). http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndAnalgesicDrugProductsAdvisoryCommittee/UCM400209.pdf. Accessed December 7, 2016.
- 24. Peckham EM, Traynor JR. Comparison of the antinociceptive response to morphine and morphine‐like compounds in male and female Sprague‐Dawley rats. J Pharmacol Exp Ther. 2006;316:1195‐1201. [DOI] [PubMed] [Google Scholar]
- 25. Matsumoto K, Umemoto H, Mori T, et al. Differences in the morphine‐induced inhibition of small and large intestinal transit: Involvement of central and peripheral μ‐opioid receptors in mice. Eur J Pharmacol. 2016;771:220‐228. [DOI] [PubMed] [Google Scholar]
- 26. Sternini C, Patierno S, Selmer IS, Kirchgessner A. The opioid system in the gastrointestinal tract. Neurogastroenterol Motil. 2004;16(Suppl 2):3‐16. [DOI] [PubMed] [Google Scholar]
- 27. Gray AC, Coupar IM, White PJ. Comparison of opioid receptor distributions in the rat ileum. Life Sci. 2006;78:1610‐1616. [DOI] [PubMed] [Google Scholar]
- 28. Gomes I, Ijzerman AP, Ye K, Maillet EL, Devi LA. G protein‐coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol Pharmacol. 2011;79:1044‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ammon HV, Phillips SF. Inhibition of ileal water absorption by intraluminal fatty acids. Influence of chain length, hydroxylation, and conjugation of fatty acids. J Clin Invest. 1974;53:205‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ammon HV, Thomas PJ, Phillips SF. Effects of oleic and ricinoleic acids on net jejunal water and electrolyte movement. Perfusion studies in man. J Clin Invest. 1974;53:374‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bright‐Asare P, Binder HJ. Stimulation of colonic secretion of water and electrolytes by hydroxy fatty acids. Gastroenterology. 1973;64:81‐88. [PubMed] [Google Scholar]
- 32. Wood JD, Galligan JJ. Function of opioids in the enteric nervous system. Neurogastroenterol Motil. 2004;16(Suppl 2):17‐28. [DOI] [PubMed] [Google Scholar]
- 33. Ono H, Nakamura A, Matsumoto K, Horie S, Sakaguchi G, Kanemasa T. Circular muscle contraction in the mice rectum plays a key role in morphine‐induced constipation. Neurogastroenterol Motil. 2014;26:1396‐1407. [DOI] [PubMed] [Google Scholar]
- 34. Maguma HT, Dewey WL, Akbarali HI. Differences in the characteristics of tolerance to mu‐opioid receptor agonists in the colon from wild type and β‐arrestin2 knockout mice. Eur J Pharmacol. 2012;685:133‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hale M, Wild J, Reddy J, Yamada T, Arjona Ferreira JC. Efficacy and safety of naldemedine for the treatment of opioid‐induced constipation in subjects with chronic non‐cancer pain receiving opioid therapy: results from two phase 3 clinical trials. Presented at Digestive Disease Week, San Diego, CA, USA, May 21—24; 2016.
- 36. Center for Drug Evaluation and Research. Pharmacology/Toxicology NDA Review and Evaluation. Application number: 204,760. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204760Orig1s000PharmR.pdf. Accessed July 25, 2018.
- 37. Hale M, Wild J, Reddy J, Yamada T, Arjona Ferreira JC. Naldemedine versus placebo for opioid‐induced constipation (COMPOSE‐1 and COMPOSE‐2): two multicentre, phase 3, double‐blind, randomised, parallel‐group trials. Lancet Gastroenterol Hepatol. 2017;2:555‐564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials