

Summary
Metal‐sulfides are wide‐spread in marine benthic habitats. At deep‐sea hydrothermal vents, they occur as massive sulfide chimneys formed by mineral precipitation upon mixing of reduced vent fluids with cold oxygenated sea water. Although microorganisms inhabiting actively venting chimneys and utilizing compounds supplied by the venting fluids are well studied, only little is known about microorganisms inhabiting inactive chimneys. In this study, we combined 16S rRNA gene‐based community profiling of sulfide chimneys from the Manus Basin (SW Pacific) with radiometric dating, metagenome (n = 4) and metaproteome (n = 1) analyses. Our results shed light on potential lifestyles of yet poorly characterized bacterial clades colonizing inactive chimneys. These include sulfate‐reducing Nitrospirae and sulfide‐oxidizing Gammaproteobacteria dominating most of the inactive chimney communities. Our phylogenetic analysis attributed the gammaproteobacterial clades to the recently described Woeseiaceae family and the SSr‐clade found in marine sediments around the world. Metaproteomic data identified these Gammaproteobacteria as autotrophic sulfide‐oxidizers potentially facilitating metal‐sulfide dissolution via extracellular electron transfer. Considering the wide distribution of these gammaproteobacterial clades in marine environments such as hydrothermal vents and sediments, microbially accelerated neutrophilic mineral oxidation might be a globally relevant process in benthic element cycling and a considerable energy source for carbon fixation in marine benthic habitats.
Introduction
At deep‐sea hydrothermal vents, the oxidation of reduced compounds (e.g., hydrogen, methane, reduced sulfur, iron and manganese species) is the primary energy source for microbial life (Jannasch and Mottl, 1985; McCollom and Shock, 1997; Bach et al., 2006). These compounds are emitted by focused venting of hot (up to > 400 °C) hydrothermal fluid or by cooler, diffuse fluid flow (Tivey, 2007). Chemolithoautotrophic microorganisms are the only primary producers in the extensive ecosystems based on deep‐sea hydrothermal activity (e.g., Jannasch and Mottl, 1985). They are present in the form of microbial mats, planktonic cells or as symbionts of vent fauna and oxidize reduced mineral compounds dissolved in the hydrothermal fluids (reviewed in Nakagawa and Takai, 2008; Sievert and Vetriani, 2012).
In the majority of hydrothermal systems hosted in the volcanic basement, reduced sulfur species are the main source of energy for microbial growth (McCollom and Shock, 1997; Amend et al., 2011). However, a large fraction of the sulfur compounds delivered through hydrothermal venting precipitates with metal ions such as iron, copper or zinc upon contact with cold, oxygenated sea water. This results in the formation of massive sulfide deposits often occurring as hydrothermal chimneys or mounds (Haymon, 1983; Tivey, 2007). Recent studies have shown that these sulfide deposits remain abundantly colonized by chemolithoautotrophic microorganisms even after hydrothermal venting has ceased (Sylvan et al., 2012; Kato et al., 2015a; Barco et al., 2017). Massive sulfide deposits thus represent a secondary source of reduced compounds, enabling chemolithotrophic microorganisms to continue thriving even at inactive vent sites.
Bacteria and Archaea capable of oxidizing reduced compounds in mineral deposits are well known from industrial leaching and acid mine drainage studies (Colmer and Hinkle, 1947; Jensen and Webb, 1995; Boon et al., 1998; Bond et al., 2000; Edwards et al., 2000; Lopez‐Archilla et al., 2001; Gonzalez‐Toril et al., 2003). In marine sediments, bacterial oxidation of iron‐monosulfide (FeS) with nitrate as electron acceptor was shown by Schippers and Jørgensen (2002). Pyrite (FeS2), however, seems to be mainly oxidized by abiotic processes, whereas bacteria in marine sediments use dissolved Fe2+ and reduced sulfur species (Straub et al., 1996; Benz et al., 1998; Schippers and Jørgensen, 2002; Jørgensen and Nelson, 2004). At hydrothermal vents, the main research focus until now has been on microbes inhabiting active vent sites or vent fauna symbionts (reviewed in Sievert and Vetriani, 2012). Comparatively few studies have focused on microbial communities colonizing sulfide deposits not exposed to active venting. Wirsen et al. (1993) were able to show active CO2 fixation in microbial mats covering inactive hydrothermal chimneys on the East Pacific Rise and isolated Thiomicrospiraceae strains that grew autotrophically while oxidizing poly‐metal sulfides (Eberhard et al., 1995). Edwards et al. (2003) isolated several neutrophilic iron‐oxidizing Gamma‐ and Alphaproteobacteria from deep‐sea hydrothermal deposits on the Juan de Fuca Ridge. However, these few isolates were only partially characterized, and their genomes have not yet been sequenced. Later studies based on comparative 16S rRNA gene analysis documented a microbial community shift between active and inactive hydrothermal chimney structures (Suzuki et al., 2004; Kato et al., 2015a, 2010; Sylvan et al., 2012; Li et al., 2017). It was suggested that mineral‐sulfide oxidizing chemolithoautotrophs replace the bacteria oxidizing dissolved sulfur and other reduced compounds in hydrothermal fluids (Sylvan et al., 2012; Kato et al., 2015a). Supporting this hypothesis, lipid biomarker compositions of two chimneys from the Manus Basin, Papua New Guinea, indicated a shift in carbon fixation mechanisms from reverse tricarboxylic acid (rTCA) cycle‐dominated CO2 fixation on an actively venting chimney to a Calvin–Benson–Bassham (CBB) cycle‐like fingerprint on an inactive chimney, indicating a shift in the active chemolithoautotrophic microbial community (Reeves et al., 2014).
Although providing valuable insights, all the microbial community studies mentioned above were based on marker gene analysis (e.g., Sylvan et al., 2012; Kato et al., 2015a; Li et al., 2017). The taxonomic resolution provided in these studies was often poor, due to the high intrinsic endemicity of the analysed samples. The proposed in situ function of the found clades was usually inferred from the closest characterized relatives, which were often only distantly related to the detected phylotypes. Much of our current knowledge of the metabolism of deep‐sea iron‐sulfide oxidizing microbes stems from a few studies based on isolates (Eberhard et al., 1995; Edwards et al., 2003; Barco et al., 2017), which might not be representative for the in situ dominant microbial communities. The first metagenomic study of a similar habitat, massive sulfides buried below the sea floor in the Okinawa Trough, was only recently published by Kato et al. (2018).
To expand our understanding of the microbiome associated with inactive sulfide chimneys, we conducted metaproteogenomic analysis combined with chemical composition analysis, ore petrology and radiometric dating of multiple inactive hydrothermal chimneys. With this approach, we were able to characterize the affiliated microbial communities both taxonomically and functionally. In addition to previously published data from two inactive and six active chimneys (Meier et al., 2017), we analysed the microbial community composition of additional five inactive chimneys obtained from the PACManus and SuSu Knolls hydrothermal fields (Manus Basin, off Papua New Guinea). By analysing the metagenomes of four of these inactive chimneys, selected to represent a wide range of microbial community compositions and ages, we retrieved detailed insights into the metabolic potential of the hosted microorganisms, most of them belonging to yet uncultured phylogenetic groups. In addition, metaproteomic data obtained from the inactive chimney hosting the most diverse microbial community confirmed the majority of functional hypotheses inferred from the metagenomic data.
Results
Description of the inactive chimneys
The seven inactive massive sulfide chimneys analysed in this study span a range of textures and mineralogical as well as geochemical compositions (Fig. 1, Table 1, Supporting Information Table S1). Most samples are very porous and polymetallic in composition consisting of various proportions of chalcopyrite, sphalerite, barite and pyrite. Due to the weathering of the samples or due to interaction with lower temperature, hydrothermal fluids during waning stages of hydrothermal activity secondary Cu‐rich sulfides such as bornite, digenite and chalcocite are common. Some samples show elevated As‐ and Pb‐concentrations (up to 3.2 wt.% As; up to 2.0 wt.% Pb; Supporting Information Table S1), which is also reflected in the presence of tennantite and galena as well as other sulfosalts in thin sections. Sample SnC‐R2 differs from the rest of the samples in that it is massive and of almost monomineralic nature. This sample of a Cu‐rich chimney conduit is characterized by a thick, massive layer of chalcopyrite with only rare bornite, digenite and pyrite.
Figure 1.

Inactive chimneys collected during cruise SO216 to the Manus Basin. For each sample, the left photo shows the chimney piece at the sea floor before collection by ROV Quest, and the right photo shows the chimney piece after sampling on board. Pictures taken by the ROV Quest are copyright of MARUM, University of Bremen.
Table 1.
Hydrothermal chimney samples.
| Name | Sample | Location | Latitude/longitude | Venting status | Description | Performed analyses |
|---|---|---|---|---|---|---|
| NSu‐R1a | 12ROV01 | North Su (SuSu Knolls) | 03°47.946′S/152°06.043′E | Exposed to venting | Porous, fibrous baryte‐rich chimney next to white smoker, partially coated with yellow native sulfur, oxidized on the outside. Subsamples a and b taken for molecular analysis. | 16S rRNA genes |
| NSu‐R2a | 12ROV02 | North Su (SuSu Knolls) | 03°47.946′S/152°06.043′E | Exposed to venting | Porous, friable baryte‐rich chimney with an orifice/conduit partially coated with yellow native sulfur. | 16S rRNA genes |
| SnC‐R1a | 27ROV06 | Snowcap (PACMANUS) | 03°43.685′S/151°40.159′E | Exposed to venting | Fragments of porous polymetallic chimney conduits partially lined by pale brassy fine‐grained chalcopyrite; outer part with abundant sphalerite and tarnished marcasite. | 16S rRNA genes |
| RR‐R1a , b | 39ROV01 | Roman Ruins (PACMANUS) | 03°43.272′S/151°40.473′E | Focused venting | Top of active black smoker chimney measured at 338 °C with thin pipe‐like dense chalcopyrite conduits and thin coating of secondary Cu‐sulfides. | 16S rRNA genes |
| RR‐R2a , b | 53ROV03 | Roman Ruins (PACMANUS) | 03°43.252′S/151°40.499′E | Diffuse venting | Weakly shimmering venting chimney, pyrite, sphalerite, chalcopyrite, baryte, iron‐oxide crusts (Reeves et al., 2014). | 16S rRNA genes |
| StM‐R2 | 31ROV13 | Satanic Mills (PACMANUS) | 03°43.614′S/151°40.321′E | No visible venting | Polymetallic chimney (talus piece) dominated by porous sphalerite with minor chalcopyrite + bornite. | 16S rRNA genes, metagenome, dating |
| Sol6‐R1 | 53ROV02 | Solwara 6 (PACMANUS) | 03°43.686′S/151°40.788′E | No visible venting | Dark grey to black porous Cu‐rich chimney with abundant secondary chalcocite‐bornite and minor sphalerite and As‐rich tennantite. Zones outwards to marcasite and Fe‐Mn‐oxyhydroxide crust with minor occurrences of atacamite. | 16S rRNA genes, dating |
| SnC‐R2 | 27ROV08 | Snowcap (PACMANUS) | 03°43.686′S/151°40.160′E | No visible venting | Very dense Cu‐rich chimney (talus piece) with massive chalcopyrite conduit showing a purple‐bluish outer bornite rim. | 16S rRNA genes |
| Fw‐R1 | 29ROV16 | Fenway (PACMANUS) | 03°43.711′S/151°40.349′E | No visible venting | Porous polymetallic chimney with chalcopyrite associated with dark sphalerite and secondary Cu‐sulfides plus baryte; Fe‐Mn‐oxyhydroxide crust. | 16S rRNA genes, metagenome, dating |
| NSu‐R7 | 47ROV13 | North Su (SuSu Knolls) | 03°47.992′S/152°06.029′E | No visible venting | Porous Cu‐rich chimney with 0.5–1 cm open conduits lined by a thin layer of chalcopyrite followed by porous brassy chalcopyrite, rimmed by a thin layer (<0.5 cm) of pyrite/marcasite and a thin outer Fe‐Mn oxyhydroxide crust. | 16S rRNA genes, dating |
| Sol8‐R1a | 49ROV03 | Solwara 8 (PACMANUS) | 03°43.831′S/151°40.451′E | No visible venting | Porous, sphalerite‐rich chimney with thin Fe‐Mn oxyhydroxide crust. Abundant baryte and minor tennantite. | 16S rRNA genes metagenome, dating |
| StM‐R1a , b | 43ROV07 | Satanic Mills (PACMANUS) | 03°43.610′S/151°40.329′E | No visible venting | Weathered inactive chimney: porous chalcopyrite interior, sphalerite, baryte exterior (Reeves et al., 2014). Abundant bornite and minor tennantite in the interior. | 16S rRNA genes metagenome, metaproteome |
Dating of the inactive chimneys
Five of the seven collected inactive chimney samples were dated using the Ra226/Ba method (Table 2). The youngest, yet clearly inactive sample is StM‐R2 (0 ± 160 years) followed by Fw‐R1 (1400 ± 160 years), Sol8‐R1 (1800 ± 160 years) and Sol6‐R1 (2093 ± 267 years). The oldest dated chimney was NSu‐R7 (3183 ± 236 years). The barium concentration of StM‐R1 was too low to accurately determine its age. However, based on morphology and weathering (degree of oxidation and Mn‐oxide coating), it is likely the oldest chimney in the dataset.
Table 2.
Chimney dating based on radium isotopes of barium‐rich samples.
| Sample | Ba (wt.%) | 226Ra activity (Bq/kg) | N (Bq/kg/wt.% Ba) | Age (years) |
|---|---|---|---|---|
| StM‐R2 | 5.7 | 4228 ± 357 | 742 ± 357 | 0 |
| Fw‐R1 | 13.3 | 5253 ± 301 | 395 ± 301 | 1456 ± 262 |
| Sol8‐R1 | 7.4 | 2459 ± 183 | 332 ± 183 | 1854 ± 284 |
| Sol6‐R1 | 12.8 | 3836 ± 235 | 300 ± 235 | 2093 ± 267 |
| NSu‐R7 | 4.7 | 878 ± 25 | 187 ± 25 | 3183 ± 236 |
Microbial diversity
First, we analysed the microbial diversity in the outer 1–2 cm of all 13 hydrothermal chimney samples from different active and inactive vent sites across the PACManus and SuSu Knolls hydrothermal fields (Manus Basin) by 16S rRNA gene amplicon sequencing (Fig. 2). After clustering the sequences with SWARM v.2 (Mahe et al., 2015), we obtained 824 operational taxonomic units (OTUs) considered to represent different microbial species.
Figure 2.

Microbial diversity based on relative abundances of 16S rRNA gene amplicon sequences. The upper panel shows the hierarchical clustering of the samples with average linkage method based on a Bray–Curtis dissimilarity matrix. The central panel shows the relative abundances of 16S rRNA gene sequences according to their taxonomic classification. Black lines are delineating individual OTUs generated by SWARM. The lower panel shows the radiometric ages of the chimneys.
Hierarchical clustering of samples based on their microbial community composition revealed three major sample groups further referred to as ‘A’ (active), ‘I1’ (inactive 1) and ‘I2’ (inactive 2) (Fig. 2). Type‐A, comprising communities on chimneys visibly exposed to venting, was characterized by high relative abundances of Sulfurovum and Sulfurimonas (Epsilonbacteraeota) sequences (56% on average, up to 71%). Although Sulfurimonas sequences were also present in higher relative abundance in a single Type‐I2 sample (NSu‐R7, 18%), this Sulfurimonas OTU detected on the Type‐I2 sample did not surpass 0.1% relative abundance in the active chimney samples (Type‐A). The communities of inactive chimney structures without any visible exposure to fluid emission clustered into two completely different branches, Type‐I1 and Type‐I2. The two Type‐I1 samples showed high relative abundance of Nitrospirae sequences (83% and 38%) related to Thermodesulfovibrio. Although StM‐R2 was almost completely dominated by one Nitrospirae OTU (83% of the sequences), Sol6‐R1 also contained Thiogranum‐related Gammaproteobacteria sequences (10%), Leptospirillum (Nitrospirae, 8%), Candidatus Tenderia‐related Gammaproteobacteria (8%) and unclassified Hyphomicrobiaceae‐related Alphaproteobacteria sequences (7%). Type‐I2 communities were characterized by high relative abundances of sequences classified as Gammaproteobacteria, such as Thiogranum (8% on average, 24% max) and Thiogranum‐related sequences (21% on average, 44% max), Siboglinidae symbionts‐related (SSr, Dyksma et al., 2016) sequences (4% on average, 9% max) and Thiomicrospiraceae‐related sequences (2% on average, 9% max) (Fig. 2). The sample of a massive chalcopyrite conduit (SnC‐R2; Type‐I2) also showed high relative abundance of uncultured Rhodobacteraceae sequences (25%). Another prominent group of alphaproteobacterial sequences in Type‐I2 samples was classified as Emcibacter (7% on average, 15% max). Bacteroidetes sequences were relatively abundant in all sample types (Type‐A: 7% average, 23% max; Type‐I1: 2% in StM‐R2, 11% in Sol6‐R1; Type‐I2: 13% average, 27% max). Yet, the abundant Bacteroidetes OTUs on active and inactive chimneys were different.
The grouping of samples by their major and trace element composition (Supporting Information Table S1) did not reflect the clustering of microbial communities.
Also, the clustering of inactive chimney communities was not correlated with the determined age of the chimney samples. The communities dominated by Nitrospirae sequences were found in the youngest (StM‐R2) and the second oldest dated sulfide sample (Sol6‐R1).
16S rRNA gene reconstruction and phylogeny
We sequenced and analysed the metagenomes of the StM‐R2 (Type‐I1), Fw‐R1, Sol8‐R1 and StM‐R1 samples (Type‐I2). As a first analysis step, long 16S rRNA gene sequences (over 1200 bp) were reconstructed from raw metagenomic reads. Unassembled reads were mapped back to reconstructed sequences to determine their relative abundance in the datasets. In general, the metagenomic data confirmed the community composition determined by amplicon sequencing (Supporting Information Fig. S1). However, the relative abundance of gammaproteobacterial sequences was higher in the metagenomes than in the amplicon dataset.
We used the reconstructed long 16S rRNA gene sequences to calculate a phylogenetic tree for the Nitrospirae phylum (Supporting Information Fig. S2) to further resolve the phylogenetic affiliation of the Nitrospirae sequences dominating the Type‐I1 chimney samples. The two most highly covered 16S rRNA gene sequences in the StM‐R2 metagenome fell into a cluster of environmental sequences, which are classified as Thermodesulfovibrio in the current SILVA database. Yet, these sequences are closer related to Candidatus Magnetobacterium than to any cultured Thermodesulfovibrio strains. According to our phylogenetic analysis, these sequences represent a distinct family level clade within the Nitrospirae (Supporting Information Fig. S2). Closely related sequences originate from other studies of inactive hydrothermal sulfide deposits in the Pacific and Indian Oceans (Suzuki et al., 2004; Sylvan et al., 2012; Kato et al., 2015a).
Next, we calculated a phylogenetic tree of the Gammaproteobacteria to determine the precise phylogenetic affiliation of the many poorly classified gammaproteobacterial clades dominating most inactive chimneys (Supporting Information Fig. S3a,b). The tree showed that reconstructed 16S rRNA gene sequences retrieved from the metagenome affiliated with various clades of ‘basal’ Gammaproteobacteria (Williams et al., 2010). Many of these contained only environmental sequences from for example, coastal marine sediments (Lenk et al., 2011; Acosta‐Gonzalez et al., 2013; Dyksma et al., 2016), pyrite colonization experiments (Pjevac et al., 2014), iron hydroxide mats from Vailulu'u seamount (Sudek et al., 2009), as well as sequences from previous studies of active and inactive sulfide chimneys in the Atlantic and Pacific Oceans (Voordeckers et al., 2008; Sylvan et al., 2012, 2013; Kato et al., 2015a), mud volcanoes in the east Mediterranean (Pachiadaki et al., 2010), tube worm symbionts from Tsukumo Bay, an Arctic mud vulcano, and the Gulf of Guinea (Kubota et al., 2007; Losekann et al., 2008; Duperron et al., 2012). Clusters containing the majority of reconstructed gammaproteobacterial sequences appear to be novel family‐level clades (Yarza et al., 2014). The only cultured representatives of these clades are sulfur‐oxidizing bacteria of the genera Thiohalophilus, Thiogranum (currently Ectothiorhodospiraceae) and Sedimenticola, as well as of the hetero‐electrotrophic Candidatus Tenderia and the heterotrophic Woeseiaceae isolate Woeseia oceanii (Sorokin et al., 2002, 2007; Narasingarao and Haggblom, 2006; Flood et al., 2015; Mori et al., 2015; Du et al., 2016) (Supporting Information Fig. S3a,b). Throughout different tree calculations, the clusters remained stable and distinct from known gammaproteobacterial families. The clusters of Thiogranum sequences form a monophyletic family‐level branch (based on criteria by Yarza et al., 2014) with the Woeseiaceae, previously referred to as sediment JTB255‐clade (Du et al., 2016; Mußmann et al., 2017). Therefore, Thiogranum and related genera would be an extension to the Woeseiaceae family. Yet, although they were always found grouped together in the different tree calculations, their phylogenetic relationship to each other as well as to other Chromatiales families could not be reliably resolved, which is depicted as multifurcations in the phylogenetic tree (Supporting Information Fig. S3a,b). Two additional clades containing a significant proportion of gammaproteobacterial sequences are related to Siboglinidae symbionts and coastal sediment sequences (SSr clade, Dyksma et al., 2016) and have no cultured representatives.
Phylogenetic analysis of metagenomics bins
We performed a co‐assembly of the four metagenomes obtained from differently dated inactive chimneys. We binned the obtained contigs into a set of 64 bins of likely species‐level populations representing between 48% and 69% of the reads mapping to the initial assembly (Read mapping statistics in Supporting Information Table S2, bin statistics in Supporting Information Table S3).
The phylogenetic affiliation of the bins was estimated by placing them into the CheckM reference genome tree based on a concatenated alignment of 43 phylogenetically meaningful single‐copy marker genes (Parks et al., 2015). In addition, we calculated new phylogenomic trees for the Nitrospirae, Nitrospinae and Deltaproteobacteria (Fig. 3) as well as for the Gammaproteobacteria (Fig. 4).
Figure 3.
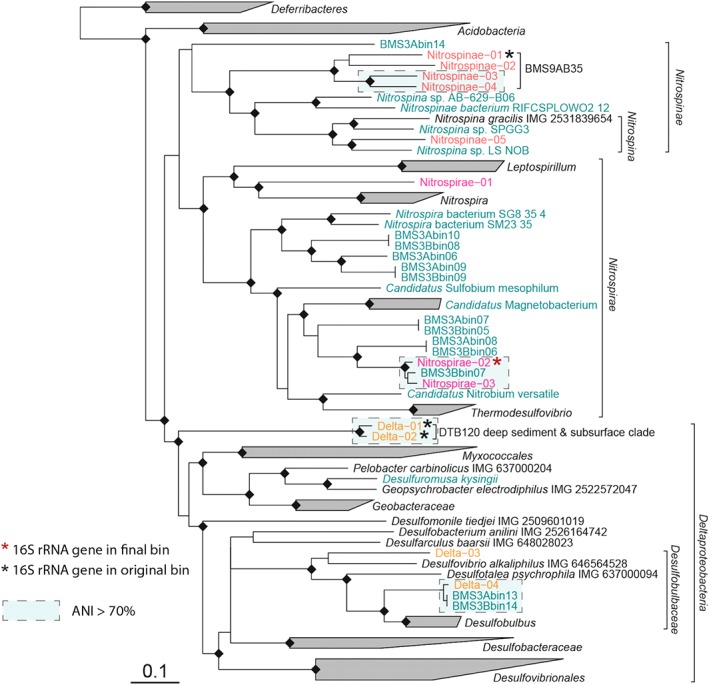
Phylogenetic tree of Nitrospinae, Nitrospirae and Deltaproteobacteria based on a concatenated alignment of translated conserved single‐copy genes (Parks et al., 2015). Only positions conserved in at least 25% of the sequences were considered. The tree was calculated using FastTree v 2.1.9 (Price et al., 2010) using weighted joints during initial tree calculation phase (‘‐bionj’), Le‐Gauscel substitution model and optimization of Gamma20 value (‘‐gamma’). Multifurcations were introduced for branchings with less than 50% support or shorter than 0.005. Black diamonds indicate branching support of 100%. Bins obtained in this study are marked in red, pink and orange. Additional reference genomes not present in the CheckM tree are marked in green (for accession numbers, see Supporting Information Table S5).
Figure 4.

Phylogenetic tree of Gammaproteobacteria bins. The tree was calculated based on a concatenated alignment of translated conserved single‐copy genes (Parks et al., 2015). Only positions conserved in at least 25% of the sequences were considered. The tree was calculated using FastTree v 2.1.9 (Price et al., 2010) using weighted joints during initial tree calculation phase (‘‐bionj’), Le‐Gauscel substitution model and optimization of Gamma20 value (‘‐gamma’). Multifurcations were introduced for branchings with less than 50% support or shorter than 0.005. Black diamonds indicate branching support of 100%. White circles indicate branching support 50% << 70%. Bins obtained in this study are marked in blue. Additional reference genomes not present in the CheckM tree are marked in green (for accession numbers, see Supporting Information Table S5).
The two closely related Nitrospirae bins Nitrospirae‐02 and Nitrospirae‐03 (average nucleotide identity, ANI = 89%) were placed on a long branch between Candidatus Magnetobacterium and Thermodesulfovibrio (Fig. 3), confirming the 16S rRNA gene inferred phylogeny (Supporting Information Fig. S2). This novel phylogenetic group of Nitrospirae also contained recently published bins from subsurface metal‐sulfide deposits in the Southern Mariana Trough (Kato et al., 2018). Based on 16S rRNA genes contained in the original bins from the bulk assembly, we could attribute the deep‐branching Deltaproteobacteria bins Delta‐01 and Delta‐02 to the DTB120 order‐level clade, consisting mainly of deep sediment and subsurface sequences. In the same fashion, four of the bins could be linked to the BMS9AB35 sequence cluster which is currently found within the Nitrospirae phylum in the SILVA database. However, based on our phylogenomic analysis, it clearly belongs to the Nitrospinae phylum (Fig. 3). The bins Delta‐03 and Delta‐04 were placed within the family Desulfobulbaceae. The Delta‐04 bin was very closely related to deltaproteobacterial bins retrieved from subsurface metal sulfide deposits (Kato et al., 2018).
The bins classified as Gammaproteobacteria were affiliated to various different clades yet poorly covered by sequenced genomes or isolates. According to our genome‐based phylogeny, bins Gamma‐01 and Gamma‐02 are representing Marinicella‐related bacteria and bins Gamma‐03 and Gamma‐04 belong to the Thiomicrorhabdus genus. The bins Gamma‐05 to Gamma‐11 constituted a novel family to order‐level clade with no sequenced relatives. Based on a 16S rRNA gene present in Gamma‐05, we attributed these bins to the uncultured SSr clade commonly found in coastal sediments.
Gamma‐14 together with Gamma‐13 and Gamma‐15 were attributed to the genus Thiogranum based on the 16S rRNA genes and ANI values (>70%) between the bins. Gamma‐14 is representing the most abundant Thiogranum OTU (Supporting Information Table S4). They provide first genomic information of this group. As in the 16S rRNA gene tree, the Thiogranum bins formed a monophyletic branch together with Woeseia oceanii, and the Gamma‐12 bin classified as Woeseiaceae/JTB255 based on a 16S rRNA gene present in the bin. This result confirmed Thiogranum being an extension of the Woeseiaceae family as suggested by 16S rRNA gene analysis (Fig. 4, Supporting Information Fig. S3a,b).
Finally, bins that were classified as Thiohalophilus‐related, based on a 16S rRNA gene present in the bin, formed a branch next to the Thiohalomonas denitrificans genome and a metagenomic bin retrieved from White Oak River estuary sediment (Baker et al., 2015).
Metabolic potential of the abundant bins
We compared the metabolic potential of the recovered bins (completeness, redundancy and coverage information in Supporting Information Table S3) with respect to their occurrence and abundance in the four metagenomes. The youngest chimney sample, the porous Zn‐rich chimney StM‐R2 (I1 type) was dominated by two Nitrospirae bins (Nitrospirae‐02 and Nitrospirae‐03) and a deltaproteobacterial bin (Delta‐04) (Fig. 5, Supporting Information Fig. S4). All three bins encode key genes for the reductive acetyl‐CoA pathway, sulfate reduction and potential extracellular electron transfer (EET) via multi‐heme cytochromes (Fig. 5, Supporting Information Fig. S4). Based on a brief phylogenetic analysis, multi‐heme cytochrome sequences from Nitrospirae and Deltaproteobacteria bins were mostly affiliated with proteins from genera containing metal‐reducing species such as Geobacter and Shewanella (Supporting Information Fig. S5). The most abundant bin as well as six other bins in StM‐R2 metagenome also encoded Cyc2‐homologues (Supporting Information Figs S4 and S6), suggested to be a candidate for an iron oxidase (Barco et al., 2015; Kato et al., 2015b). The Nitrospirae‐02 and Delta‐04 bins also contained nitrogenase‐encoding genes. Notably, apart from Nitrospirae‐02 and Delta‐04, there are four more bins in the StM‐R2 metagenome encoding the metabolic potential for nitrogen fixation (Fig. 5, Supporting Information Fig. S4). We have found two types of hydrogenases in the Nitrospirae‐02 and Nitrospirae‐03 bins. Based on HydDB search (Greening et al., 2016; Søndergaard et al., 2016), these are putatively classified as hydrogen‐evolving type 3b and supposedly oxygen‐protecting type 1f hydrogenases related to scavenging of hydrogen resulting from nitrogenase activity. The Delta‐04 bin encoded a respiratory oxygen‐sensitive hydrogenase of the 1c type. No genes indicative of aerobic respiration or nitrate/nitrite reduction could be found in these three bins. Other bins present at lower abundances in StM‐R2‐included gammaproteobacterial Thiogranum (Woeseiaceae) and Thiohalophilus‐related bins encoding the genomic potential for carbon fixation (CBB‐cycle), sulfur oxidation (SOX and rDsr genes) and denitrification.
Figure 5.
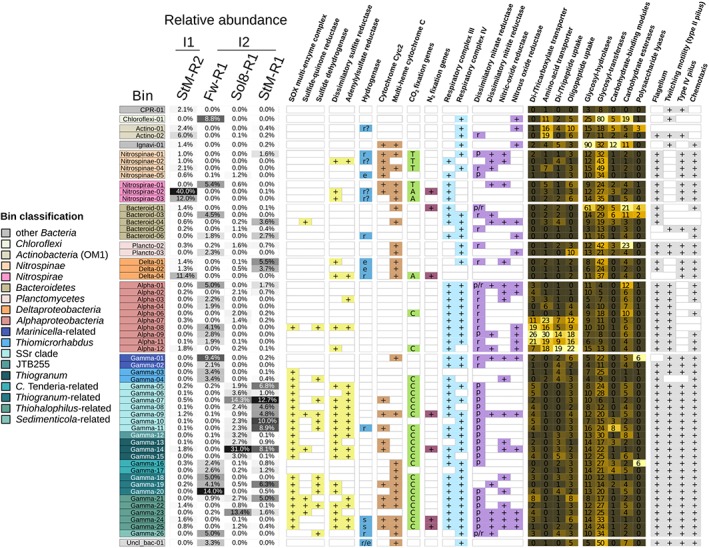
Metabolic potential of abundant microbial populations. Relative abundance of bins (>1%) in the metagenomes is shown in the first four columns of the figure. The presence or absence of a pathway/enzyme is based on presence of essential key enzymes/catalytic subunits. Hydrogenases were classified according to HydDB (Søndergaard et al., 2016) into ‘r’– respiratory/H2‐oxidizing, ‘r?’– unknown, probably H2‐oxidizing, reactive oxygen species‐protecting, ‘e’ – H2‐evolving and ‘s’ – H2‐sensing. For the carbon fixation pathways ‘A’ indicates acetyl‐CoA pathway, ‘R’ – Calvin–Benson–Bassham cycle, ‘T’–reverse TCA cycle. Nitrate reductases are denoted as ‘p’–periplasmic and ‘r’–respiratory/membrane bound. For genes expected to be found in most living organisms, numbers and a heatmap are used to indicate the amount of genes found in a given bin.
The most abundant bin in the porous, polymetallic Fw‐R1 chimney sample (I2‐type), Gamma‐20, represented an autotrophic sulfur‐oxidizing, nitric oxide‐ and oxygen‐respiring gammaproteobacterial population (Fig. 5, Supporting Information Fig. S4). Bins of other likely sulfur‐oxidizing and carbon‐fixing Gammaproteobacteria were also found in the Fw‐R1 metagenome at lower abundances (Gamma‐18 and Gamma‐19, Gamma‐04). The two most abundant bins (Gamma‐20, Gamma‐01) as well as further eight bins encode multi‐heme cytochromes potentially involved in EET. The gammaproteobacterial multi‐heme cytochromes largely clustered with the sequences from cathode‐oxidizing Candidatus Tenderia electrophaga and neutrophilic iron‐ and manganese‐oxidizing Leptothrix cholodnii (Adams and Ghiorse, 1987; Spring et al., 1996) (Supporting Information Fig. S5). Nitrospirae‐01 and Gamma‐19 bin also encoded homologues of Cyc2 cytochrome. The Cyc2‐homologous proteins found in Gamma‐19 and other gammaproteobacterial bins (Fig. 5) were also clustering with a Cyc2‐homologue from Candidatus Tenderia electrophaga (Supporting Information Fig. S6). Furthermore, the Fw‐R1 metagenome is characterized by the highest proportion of Alphaproteobacteria of all metagenomes (five bins representing 18% of the mapped reads). Based on the encoded 16S rRNA gene, the most abundant alphaproteobacterial bin (Alpha‐01) can be attributed to the most abundant alphaproteobacterial OTU in the amplicon analysis (Table 2). They classify as Emcibacter (Supporting Information Table S4), an alphaproteobacterium recently isolated from coastal sediments (Liu et al., 2015). The Alpha‐01 bin contains genes for an aerobic heterotrophic lifestyle and for complete denitrification. No bin in the Fw‐R1 contained genes annotated to encode nitrogen fixation proteins.
The 11 most abundant bins in the metagenome of the porous, Zn‐rich I2‐type chimney Sol8‐R1 belong to Woeseiaceae, Thiohalophilus‐related and SSr‐clade‐related Gammaproteobacteria. All of these bins contain genes encoding proteins for the oxidation of reduced sulfur compounds (SOX in all and rDsr in nine). In the majority of these bins, we also detected RuBisCO encoding genes, indicative of carbon fixation via the CBB cycle. The most abundant bin represents a Thiogranum population, also encoding the potential for nitrogen fixation, as well as aerobic respiration and denitrification. The two most abundant bins encode Cyc2 homologues. Cyc2‐homologues are also encoded in further three gammaproteobacterial bins and a Nitrospinae bin abundant in this metagenome (Supporting Information Figs S4 and S6). Multi‐heme cytochrome encoding genes were detected in three rather low abundant bins (Thiohalophilus‐related Gamma‐21 and Gamma‐25 and the Planctomyces bin Plancto‐02) in the Sol8‐R1b metagenome. Only two bins encode the metabolic potential for nitrogen fixation (Gamma‐14, Thiogranum and Gamma‐25, Thiohalophilus‐related).
The most abundant bins in the porous, polymetallic StM‐R1 chimney (I2 type), hosting the most diverse microbial community, belong to SSr‐clade‐, Woeseiaceae‐ and Thiohalophilus‐related Gammaproteobacteria. Similar to Sol8‐R1b, most of the 10 most abundant bins encode the potential for sulfur oxidation (SOX combined with rDSR) and carbon fixation (CBB cycle), as well as aerobic respiration. Two of the StM‐R1 bins, the Thiogranum bin Gamma‐14 and SSr clade bin Gamma‐09, encode a nitrogen fixation potential.
Protein expression in the porous, polymetallic StM‐R1 chimney
We analysed the proteome of the StM‐R1 chimney hosting the most diverse microbial community. In total, 947 proteins were identified. Of those, 528 were annotated with a function in the metagenome. Proteins of the most abundant bins recovered from the StM‐R1 metagenome were detected in the proteome with the exception of the Gamma‐23, Gamma‐17, Bacteroidetes and Deltaproteobacteria bins. However, proteins attributed to Deltaproteobacteria and Bacteroidetes based on BLAST hits and the classification of encoding contigs, but without an assignment to one of the final bins, were also found in the proteome. This indicates that most population genomes recovered from StM‐R1 represent active populations.
The two most prevalent proteins in the proteome were proteins of unknown function attributed to eukaryotes (Supporting Information Fig. S7) followed by two carboxysome shell proteins belonging to the bins Gamma‐18 (Woeseiaceae) and Gamma‐03 (Thiomicrospira) (Fig. 6, Supporting Information Fig. S7). The fifth most abundant protein was a multi‐heme cytochrome c not assigned to any bin (Fig. 6). The contig containing the encoding gene was classified as gammaproteobacterial. The closest hit in NCBI‐nr database (44% amino acid identity) was a tetraheme cytochrome c from Candidatus Tenderia electrophaga, a cathode oxidizing gammaproteobacterium enriched from coastal sediment (Eddie et al., 2016).
Figure 6.
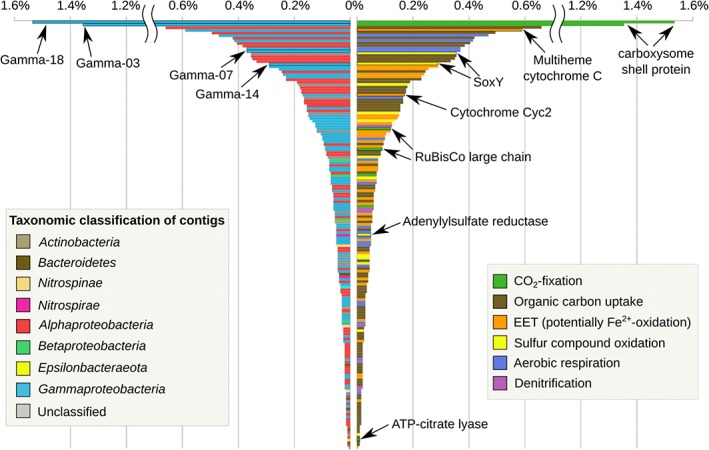
Relative abundance of proteins involved in carbon acquisition and energy generation in the metaproteome of the StM‐R1 chimney sample [displayed in Normalized Spectral Abundance Factors (NSAF)%]. The bars on the left are coloured by taxonomic classification of the encoding contig. Bars on the right are coloured by metabolic categories. For an overview of the overall 200 most expressed proteins, including unannotated proteins, see Supporting Information Fig. S5. All detected proteins and their annotations are listed in Supporting Information File S1.
Among the non‐hypothetical proteins related to a chemolithoautotrophic lifestyle, the vast majority was attributed to Gammaproteobacteria (Fig. 6). Three of four expressed RuBisCO proteins were attributed to Woeseiaceae and Thiomicrospiraceae (Gammaproteobacteria), and one was classified as Betaproteobacteria. Apart from the RuBisCO proteins, an ATP‐citrate lyase, the key enzyme in the reverse TCA carbon fixation pathway, assigned to Epsilonbacteraeota was detected (rank 893 of 947). Aerobic terminal oxidases were exclusively attributed to various Gammaproteobacteria, whereas nitrite reductases found in the proteome were attributed to various Gammaproteobacteria, the Alpha‐01 bin (Emcibacter) and Nitrospinae. SoxY proteins indicative of sulfur oxidation were attributed to Woeseiaceae and SSr‐clade Gammaproteobacteria. Proteins potentially involved in EET, like multi‐heme cytochrome c, Cyc2‐homologues, cytochrome c4 and Type IV pili were exclusively attributed to Gammaproteobacteria. Notably, the most highly expressed Cyc2‐homologue as well as two of three detected c4 cytochromes were attributed to bin Gamma‐19 (Woeseiaceae). The phylogenetic trees calculated for decaheme cytochromes and Cyc2‐homologous cytochromes place the expressed proteins, as well as the majority of decaheme and Cyc2‐like sequences detected in the metagenomes next to sequences from the iron‐oxidizing Leptothrix cholodnii and the cathode‐oxidizing Candidatus Tenderia electrophaga (Supporting Information Figs S5 and S6).
Furthermore, 68 of 75 proteins related to binding and import of small organic molecules, like amino acids, oligopeptides, sugars, etc., were attributed to Alphaproteobacteria, whereas all six detected SusC proteins involved in polysaccharide binding and import were exclusively attributed to Bacteroidetes.
Interestingly, we also detected 11 proteins attributed to Nitrospirae. Six of the encoding genes had the same coverage pattern as the Nitrospirae bins dominating the StM‐R2 metagenome: ~ 200–800× coverage in StM‐R2 and maximum 1× coverage in other metagenomes, including the StM‐R1 chimney from which this metaproteome was derived. Although the contigs carrying these genes were not binned, they likely belong to Nitrospirae‐02 or Nitrospirae‐03 genomes, based on this unique coverage pattern. The five annotated proteins among them are flagellin protein FlaA, nitrogen regulatory protein P‐II, adenylylsulfate reductase (beta subunit), ATP‐synthase (beta chain) and glyceraldehyde‐3‐phosphate dehydrogenase.
Discussion
Comparing the microbial community composition of seven inactive hydrothermal chimneys to six active chimneys published earlier (Reeves et al., 2014; Meier et al., 2017; Pjevac et al., 2018), all collected at various hydrothermal sites of the Eastern Manus Basin, we detected two clear‐cut shifts in the microbial community composition. The first shift was between communities of chimneys exposed to active venting and the communities on the inactive sulfides. The second shift was observed between two different community types found on inactive sulfides. The general community difference between active and inactive sulfides has been previously reported, for example, for the Southern Mariana Trough or East Pacific Rise (Kato et al., 2010; Sylvan et al., 2012). So far, it was mainly perceived as a shift from sulfur‐oxidizing Epsilonbacteraeota to Chromatiales‐related, putatively sulfur‐oxidizing Gammaproteobacteria. Also, microbial community differences driven by different host‐rocks or underlying mineralogy (sulfides vs. basalts) have been reported (Sylvan et al., 2013; Toner et al., 2013). However, the observed community difference within inactive chimneys of comparable mineralogical origin was rather unexpected. Neither radiometrically determined age of the samples nor the mineralogical composition could explain the clear clustering of microbial communities into two different types. Although the number of samples representing each community type was too low to infer correlations, it is possible that other, yet unconsidered environmental factors, such as organic matter content or porosity of the chimneys, have a strong effect on the community composition. Furthermore, heterogeneity of the chimneys on a small spatial scale might also contribute to discrepancies between samples.
Sulfate reducing Nitrospirae
Nitrospirae‐dominated inactive sulfides were so far rarely reported (Suzuki et al., 2004; Kato et al., 2015a). Kato et al. (2015a), for example, found large proportions of Nitrospirae on buried subsurface sulfides close to active venting sites in the Southern Mariana Trough, yet clearly not exposed to venting fluids. An earlier study of inactive sulfides from Iheya North (west Pacific) and Kairei (Indian Ocean) hydrothermal fields found the high abundance of Nitrospirae correlated with presence of barium sulfate (barite) (Suzuki et al., 2004). Later investigations of active calcium sulfate (anhydrite) rich chimneys from Juan de Fuca Ridge dominated by Epsilonbacteraeota revealed elevated abundances of a Thermodesulfovibrio‐related OTU (Olins et al., 2013). After 1 year of storage in ‘sulfidic effluent’, samples of the very same anhydrite chimneys were dominated by Thermodesulfovibrio‐like sequences and sulfate reduction rates were high (Frank et al., 2013). Although Frank et al. (2013) did not comment on the effect of storage, their work might represent an enrichment of these organisms by simulating the conditions within 1 year after the venting has ceased. The very young age of the StM‐R2 chimney, the low complexity of the microbial community and a high proportion of primary producers, in terms of both carbon and nitrogen fixation, suggest that the StM‐R2 sample represents the initial colonization stage of an inactive chimney after ceasing of venting and death of the fluid‐dependent Epsilonbacteraeota‐dominated community.
However, the young age of the chimney might not be the only determining factor for the community composition. High relative abundance of Nitrospirae‐related 16S rRNA gene OTUs was also detected in the second oldest chimney sample (Sol6‐R1). The Nitrospirae bins are not only lacking any aerobic terminal oxidases. The presence of the reductive Acetyl‐CoA pathway, indicative of either strictly anaerobic CO2‐fixation (reviewed in Hügler and Sievert, 2011) or a fermentative or sulfate‐reducing lifestyle (Rabus et al., 2013), in the three most abundant StM‐R2 bins is also striking. A microbial community largely composed of anaerobes might be dictated by the structure of the chimney. Sealed by a thick iron‐oxide crust anoxic spaces might exist within the chimney. A highly similar Nitrospirae bin was very recently recovered from massive sulfides buried below the sea floor surface by Kato et al. (2018). The StM‐R2 chimney however was collected from the sea floor surface itself.
In the Nitrospirae‐02 and Nitrospirae‐03 bins, we have only found genes for proteins catalysing sulfate reduction as an obvious electron sink. Confirming the sulfate reducing lifestyle, an adenylylsulfate reductase protein was detected in the StM‐R1 sample, although the coverage of the encoding bins in the StM‐R1 metagenome barely reaches one fold. The detection of proteins encoded by genes of the Nitrospirae bins from the young StM‐R2 chimney in the old StM‐R1 chimney indicates that these organisms continue to thrive in their niche also in more complex communities. Recently, new genomic sequences have been added to the branch of Nitrospirae in the phylogenetic tree, comprising Candidatus Magnetobacterium and Thermodesulfovibrio. These sequences originate from genomes of sulfate‐reducing microorganisms from rice paddy soils amended with gypsum (CaSO4·H2O) and from a denitrifying bioreactor (Arshad et al., 2017; Zecchin et al., 2018). Although Nitrospirae bins obtained in this study were placed in the same overall clade, we could not detect any Nitrospirae‐related denitrification genes. Phylogenetically, the Nitrospirae detected in I1‐type chimneys are also more closely related (same genus) to microorganisms found in inactive sulfides from the Pacific and Indian Oceans (Kato et al., 2018), where they were found associated with the barite layer (Suzuki et al., 2004). This correlation between a high relative abundance of Nitrospirae microorganisms and barite in inactive chimneys and their high abundances on active and stored anhydrite chimneys (Frank et al., 2013; Olins et al., 2013) together with our metagenomic and metaproteomic data characterizing them as sulfate reducers allow to speculate that they might use mineral sulfate as electron acceptor. However, microbial reduction of sulfate minerals has never been demonstrated for laboratory cultures. Another possible sulfate source might be the dissolved sulfate generally present in sea water or originating from mineral dissolution. However, these Nitrospirae organisms have never been found in environments of sea water sulfate reduction, such as anoxic sediments, mostly dominated by sulfate‐reducing Deltaproteobacteria. Barite, present in the sampled chimneys, on the one hand, is highly insoluble (Blount, 1977; Averyt and Paytan, 2003), and it is thus unlikely to be the source of increased dissolved sulfate concentrations. Anhydrite, on the other hand, dissolves fast after the venting stops, but the sampled inactive chimneys contain no large accumulation of anhydrite. However, we cannot exclude that it dissolved shortly before sampling. Therefore, a hypothetical dissolution of anhydrite may have been the source of dissolved sulfate enabling the Nitrospirae to thrive. Another electron sink might be encoded by the multi‐heme cytochrome c genes found in the Nitrospirae bins. Based on metagenomic data and thermodynamic modelling, Kato et al. (2018) suggested that sulfides inhabiting Nitrospirae might use these cytochromes for iron reduction.
We consider it likely that sulfate is the main electron acceptor, yet the potential electron donor for these organisms is not obvious from the encoded metabolic potential. The hydrogenases of Nitrospirae‐02 and Nitrospirae‐03 are of a yet poorly characterized type and are suggested to be involved in oxygen protection of the nitrogenase. Therefore, they are more likely to scavenge the hydrogen resulting from di‐nitrogen fixation (Robson and Postgate, 1980) than to oxidize environmental hydrogen gas.
Sulfide‐oxidizing Woeseiaceae and other Gammaproteobacteria
Unlike StM‐R2, the other inactive chimney metagenomes from our sample set were dominated by aerobic Gammaproteobacteria. The relative abundances of Gammaproteobacteria belonging to the Woeseiaceae family, and the SSr‐clade was higher in the metagenomic than in the amplicon data, probably due to primer bias against these clades. Inactive hydrothermal chimneys dominated by Gammaproteobacteria are known from several vent sites such as the Iheya North field, South Mariana Trough and Juan de Fuca Ridge (Suzuki et al., 2004; Kato et al., 2010, 2015a; Sylvan et al., 2012; Reeves et al., 2014; Li et al., 2017). Here, we provide first insights into their metabolic potential and activity as well as an improved phylogenetic placement of these diverse yet poorly described groups.
We also managed to obtain the first genomic information for the recently isolated genus Thiogranum (Mori et al., 2015). Contradicting the initial attribution of Thiogranum to Ectothiorhodospiraceae (Mori et al., 2015), the phylogenetic tree reconstructions of basal Gammaproteobacteria based on 16S rRNA genes and concatenated marker genes in this work affiliated it robustly as a sister branch to the Woeseiaceae/JTB255 clade (Du et al., 2016; Dyksma et al., 2016; Mußmann et al., 2017). Although the name giving isolate, Woeseia oceanii, is a heterotroph (Du et al., 2016), genomic bins attributed to Woeseiaceae in this and other studies exhibit a chemolithoautotrophic potential (Baker et al., 2015; Dyksma et al., 2016; Mußmann et al., 2017). Confirming this prediction, we detected proteins indicating that Woeseiaceae are the main carbon fixers in aerobic inactive chimney communities sampled in this study. We have also found indications of sulfide oxidizing activity and possibly EET. Based on the phylogeny of the expressed cyctochromes attributed to EET and the expression of aerobic terminal oxidases by the Gammaproteobacteria, it seems likely that the EET is an electron uptake. Thus these organisms could be actively facilitating the oxidation and dissolution of metal sulfides accelerating the release of their main energy source, sulfide. Furthermore, the most abundant Thiogranum bin also contained genes for nitrogen fixation, adding another key ecological function to the versatile functional repertoire of the group.
The second most abundant bin of chemolithoautotrophic Gammaproteobacteria affiliated with the SSr‐clade, which also has been shown to be a major contributor to CO2‐fixation in coastal sediments (Dyksma et al., 2016). According to our phylogenetic analysis, it is not closely related to any cultured Gammaproteobacteria and represents a family‐ to order‐level clade composed of environmental sequences. In the proteome of the polymetallic chimney StM‐R1, we found sulfur oxidation proteins attributed to the SSr clade indicating that they, together with the Woeseiaceae, are likely major sulfide oxidizers in this community.
Some of the abundant gammaproteobacterial sulfur‐oxidizer bins also possessed multi‐heme cytochrome c and Cyc2‐homologues encoding genes possibly indicating a capability to oxidize Fe2+ or actively facilitate metal‐sulfide dissolution. Apart from iron‐oxidizers isolated earlier from inactive sulfides in the East Pacific (Eberhard et al., 1995; Edwards et al., 2003), a Thiomicrospira strain capable of autotrophic growth with pyrrhotite as electron donor was recently isolated from surface sediment near Santa Catalina Island (Barco et al., 2017). Biotic acceleration of pyrite oxidation at neutral pH by freshwater Alpha‐ and Betaproteobacteria was also recently shown in borehole sediment (Washington, USA) enrichment cultures (Percak‐Dennett et al., 2017). Notably, the genomes obtained from these enrichments by Percak‐Dennett et al. (2017) encoded the same metabolic potential in terms of cytochromes, sulfur oxidation and carbon fixation pathways as the gammaproteobacterial bins obtained in this study. Supporting the hypothesis of active interaction with the mineral surface, we found a gammaproteobacterial multi‐heme cytochrome c to be the third most abundant annotated protein in the StM‐R1 metaproteome. The top hit in the NCBI‐nr database was a tetraheme cytochrome of Candidatus Tenderia electrophaga (Eddie et al., 2016) (44% amino acid identity). Recently, the respective gene was found to be transcribed during electrotrophic growth on a cathode (Eddie et al., 2017). Also, other gammaproteobacterial genes likely related to iron oxidation such as c4 cytochromes and type IV pili (Reguera et al., 2005; Barco et al., 2015; He et al., 2017) were detected in the proteome. The fact that Candidatus Tenderia electrophaga was enriched on a biocathode from estuary sediment indicates that active pyrite dissolution or the capability of EET in general might be a more wide‐spread trait of this group of Gammaproteobacteria also outside of ‘post‐hydrothermal’ environments.
Based on metabolic potential and detected proteins, the main heterotrophic organisms in inactive chimney communities belong to Bacteroidetes and Alphaproteobacteria. Alphaproteobacteria related to sediment species like Emcibacter nanhaiensis mainly specialize on utilization of small organic molecules like amino acids and oligopeptides as carbon and energy source, whereas Bacteroidetes utilize more complex polysaccharides like in other marine environments (Gomez‐Pereira et al., 2012; Teeling et al., 2012). Notably, the heterotrophic Proteobacteria and Bacteroidetes clades detected on inactive chimneys are different from the ones found on active chimneys or in diffuse venting environments (Stokke et al., 2015; Meier et al., 2016; Pjevac et al., 2018), likely due to relatively narrow substrate specialization.
Conclusions
With this study we expand the knowledge on marine microbial communities fuelled by the oxidation of sulfur minerals by revealing metabolic potential and function of yet poorly described microbial clades. Two major types of microbial communities seem to exist on inactive hydrothermal chimneys. One is dominated by anaerobic, autotrophic sulfate reducers similar to organisms found in subsurface sulfides from the Okinawa Trough. The other one is characterized by a dominance of aerobic sulfide‐oxidizing autotrophic Gammaproteobacteria. Phylogenetic analysis allowed us to link them to largely uncultured groups of microorganisms which are wide‐spread and abundant in marine sediments. The study underlines the importance of SSr‐clade and Woeseiaceae‐related Gammaproteobacteria as catalysts of sulfide mineral weathering and primary producers in inactive chimney communities. Considering the wide distribution of iron‐sulfide minerals on the ocean floor, these widespread bacteria might contribute significantly to turnover of iron and sulfur, as well as carbon fixation in deep‐sea habitats other than hydrothermal vent fields, such as marine sediments making microbial mineral oxidation a much more common process than previously assumed.
Experimental procedures
Site description and sample collection
Manus Basin is a fast‐spreading back‐arc basin with multiple basaltic‐ to felsic‐hosted hydrothermal fields located between the islands New Britain and New Ireland (Bismarck Sea, Papua New Guinea). All samples (Table 1) were collected during R/V Sonne expedition SO‐216 in June/July 2011 from the PACManus and SuSu Knolls hydrothermal vent fields that emit highly sulfidic fluids and feature poly‐metallic sulfide chimneys and mounds (Binns and Scott, 1993; Reeves et al., 2011; Thal et al., 2014; Yeats et al., 2014). Active and inactive hydrothermal chimney samples were collected by the ROV QUEST (Marum Bremen) with the ROV's hydraulic arm. Hydrothermal sulfide structures were subsampled and directly frozen at −20 °C until further use.
Ore petrology was performed on polished thin sections and shows that the chimneys were mainly composed of chalcopyrite (CuFeS2), pyrite/marcasite (FeS2), sphalerite (Zn(Fe)S) and bornite (Cu5FeS4) (Table 1) with a suite of other minor to rare minerals. The bulk geochemistry of the samples was determined on representative samples (several tens to hundreds of grams) commercially at ActLabs (Ontario, Canada) by a combination of methods including ICP‐OES, ICP‐MS and Instrumental Neutron Activation (Supporting Information Table S1). StM‐R1, RR‐R1 and RR‐R2 samples have been used for lipid biomarker analysis and 16S rRNA gene amplicon pyro‐sequencing in Reeves et al. (2014). Metagenomes and proteomes of the active chimneys RR‐R2 and RR‐R2 were analysed in Pjevac et al. (2018). Illumina sequences of 16S rRNA gene amplicons from NSu‐R1a, NSu‐R1b, NSu‐R2, RR‐R1, RR‐R2, SnC‐R1, Sol8‐R1 and StM‐R1 were published in Meier et al. (2017)
Dating of inactive sulfides
Individual chimney samples were dated using the 226Ra/Ba method (de Ronde et al., 2005; Ditchburn et al., 2012; Jamieson et al., 2013). Radium‐226 (half‐life of 1600 years) is preferentially incorporated into hydrothermal barite. The amount of radioactive decay over time, measured as 226Ra/Ba, can be compared with the same ratio from an actively forming chimney, and the age calculated using the equation:
where t is the age, in years, of the sample, N and N 0 are the 226Ra/Ba ratios of the sample and the initial, ‘zero‐age’ ratio respectively and λ is the half‐life of 226Ra (1600 years). This dating technique relies on the assumption that the 226Ra/Ba of actively forming barite does not change over time, and the ratio measured in newly formed barite is representative of the initial ratios in barite from older samples. Barium concentrations of representative bulk material were determined using instrumental neutron activation at ActLabs (Ontario, Canada). Radium‐226 activities of powdered samples were measured on an Ortec gamma spectrometer with a high‐purity germanium detector in a well‐configuration at the University of Ottawa. Due to potential peak interferences with 235U, radium‐226 activity was measured by recording the activity of 214Pb, a daughter product through the decay of the intermediate 222Rn isotope. Samples were sealed with an epoxy cap for 3 weeks before analysis, to trap the Rn and allow for accumulation of 214Pb and attainment of secular equilibrium between 214Pb and 226Ra.
DNA extraction
DNA was extracted from crumbled and homogenized chimney material (1–2 cm outer layer) using the Power Soil kit (MO BIO Laboratories, Carlsbad, CA, USA) with an additional 1 h Proteinase K digestion step at 37 °C and a 2 h incubation at 65 °C after the addition of SDS containing buffer S1 before applying the kit protocol. Metagenomic DNA from the StM‐R1 sample was extracted as described in Pjevac et al. (2018).
16S rRNA gene amplicon sequencing and analysis
The V3–V4 region of the 16S rRNA gene was amplified as described in Meier et al. (2016) and sequenced on an Illumina MiSeq sequencer at the Max Planck Genome Centre (Cologne, Germany). After trimming of 3′‐ends with quality below q10, paired‐end reads were merged using BBmerge v. 36.32 (http://jgi.doe.gov/data-and-tools/bbtools/) with a minimum overlap of 50 bp.
Merged amplicon reads were de‐multiplexed using Mothur v.1.34 (Schloss et al., 2009). Reads of the whole dataset were decomposed into ‘nodes’ by MED v2.0 (Eren et al., 2015) with four discriminant locations and minimum substantive abundance (count of the most abundant sequence in a node) of 3. Finally, OTUs were generated based on the MED nodes using SWARM (Mahe et al., 2015) with the ‘fastidious’ option, 20 as the number of sequences in a node for it to be considered ‘big’ and otherwise default parameters. Representative sequences of the OTUs generated by SWARM (centroids of the swarms) were aligned to the SILVA seed database and classified by last common ancestor (LCA) using the SINA on‐line aligner v.1.2.11 (Pruesse et al., 2012; Quast et al., 2013). Sequences of OTUs which could not be classified by the pipeline were added to the SILVA reference tree by maximum parsimony in ARB and manually classified based on their placement.
Statistical analysis was performed using the Vegan package (Oksanen et al., 2013) in R. Influence of environmental factors on microbial community composition was assessed with a non‐parametric permutational multivariate analysis of variance (perMANOVA) using the ‘adonis’ function on a Bray‐Curtis distance matrix with 9999 permutations as well as distance‐based redundancy analysis (dbRDA) performed with the “capscale” function using a Bray‐Curtis distance matrix.
Metagenome sequencing, assembly and binning
Genomic DNA was shotgun sequenced on an Illumina HiSeq sequencer at the Max Planck Genome Centre (Cologne, Germany). The paired‐end reads were error‐corrected using Bayes‐Hammer implemented in SPAdes (Bankevich et al., 2012), merged with BBmerge v.36.32 and normalized to a k‐mer depth of 42 with BBnorm v.36.32. Bulk co‐assembly of the metagenomes was performed with MEGAHIT v.1.1.1 (Li et al., 2015). Assembled contigs were binned based on a weighted combination of tetranucleotide frequencies (>95% similarity), GC‐content and coverage values, paired reads mappings, the presence of 138 conserved single‐copy genes (Campbell et al., 2013) and similarity of taxonomic classification using MetaWatt v.3.5.3 (Strous et al., 2012). Bins generated by MetaWatt were inspected and refined with Anvi'o v.2.1 (Eren et al., 2015).
Bins classified as Gammaproteobacteria, Deltaproteobacteria, Nitrospirae and Nitrospina were extracted from the bulk assembly and used for read mapping with a minimum identity of 97% using BBmap v.36.32. This led to less complex subsets of reads for subsequent re‐assembly. For each taxonomic group a separate re‐assembly with SPAdes v.3.10.1 (Bankevich et al., 2012) was performed followed by a new round of binning with MetaWatt and manual refinement in Anvi'O. This procedure improves assembly metrics due to reduced complexity of the datasets and lower data volume allowing for use of a more thorough assembler (SPAdes). As a result also the bin metrics such as contig length and purity of bins are improved. Completeness and quality of final assemblies was assessed by CheckM v.1.0.7 (Parks et al., 2015).
Average nucleotide identities to the next sequenced relative and between the assemblies were calculated using the JSpeciesWS web service (Richter et al., 2016). The reference genomes were selected based on the affiliation of 16S rRNA sequences reconstructed from the metagenome, majority of diamond blastx hits against a database of reference genomes (one genome per genus) during the MetaWatt contig classification step and tetranucleotide‐based relative search in JSpeciesWS.
Gene annotation
All assembled contigs were annotated using the myRAST annotation pipeline (Aziz et al., 2008), HMMER3 search (Eddy, 2011) against the PfamA database (Finn et al., 2014) and a diamond blastp search against NCBI‐nr. The annotation of genes of interest was manually inspected by comparing the results of the RAST annotation to HMMER3 searches against the Pfam‐A database and dismond blastp searches against the NCBI‐nr database. Multi‐heme cytochromes were identified by the Pfam motif PF09699. Cyc2‐homologues were identified based on an HMM‐motif derived from a MAFFT alignment (Katoh and Standley, 2013) of sequences identified as Cyc2‐like in Kato et al. (2015b). Related sequences were downloaded from the UniProt database (Magrane and UniProt Consortium, 2011) based on orthologous clusters identified in the eggNOG database by eggNOG mapper (Huerta‐Cepas et al., 2016, 2017). To identify carbohydrate active enzymes (CAZYmes), additional annotation by the dbCAN on‐line tool (Zhang et al., 2018) was performed.
Phylogenetic tree construction
Long 16S rRNA gene sequences reconstructed by PhyloFlash (Gruber‐Vodicka et al., 2017) (>1200 bp) and related high‐quality sequences from the SILVA SSU128 NR99 database (sequence quality >95) were aligned to a curated SILVA SSU128 NR99 database (Quast et al., 2013), where all sequences with a pintail value below 50 and alignment quality below 70 were removed. Only sequences with an alignment quality >95 were kept for tree calculations. Tree calculations were done with neighbour‐joining algorithm implemented in ARB (Ludwig et al., 2004), PhyML (Guindon et al., 2010) and FastTree v.2.1.9 (Price et al., 2010) using 30%, 40% and 50% positional conservation filters and the bacterial positional variability filter of SILVA. FastTree calculations were performed with weighted joints during the initial tree calculation phase (‘‐bionj’), generalized time‐reversible model and optimization of Gamma20 value (‘‐gamma’). Multifurcations were created with ARB for branches with less than 50% support and branches shorter than 0.005 changes per base. OTU‐representatives of poorly classified Gammaproteobacteria from 16S rRNA gene amplicon sequencing were added to the calculated trees based on maximum parsimony without changes of the overall tree topology.
A concatenated alignment of 43 phylogenetically informative single‐copy genes was generated with HMMER3 (Eddy, 2011) in CheckM v.1.0.7 (Parks et al., 2015). Concatenated alignments were imported into ARB, and maximum likelihood trees were calculated using PhyML and FastTree v.2.1.9 without filters as well as based on positions conserved in 25%, 30% and 35% of the sequences. The topology of the tree remained largely stable throughout different calculations.
Metaproteome analysis
Details of protein extraction, sample preparation and mass spectrometric (MS) analysis were recently described by Pjevac et al. (2018). Briefly, proteins were extracted from chimney material using a chloroform‐based method and proteins were separated by 1D PAGE in two technical replicates (two gel lanes of the same sample). Protein‐containing gel lanes were divided into 10 equal sized subsamples per replicate, destained and subjected to in‐gel digestion using trypsin. Digested protein was purified with μ‐C18 ZipTips (Millipore, USA). The resulting peptide mix was separated by Nano HPLC (Easy‐nLCII HPLC system, Thermo Fisher Scientific, Germany) and analysed by tandem mass spectrometry in an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Germany) as described in Pjevac et al. (2018). Obtained MS/MS spectra were analysed with the Sorcerer‐Sequest software (Sorcerer v.3.5, Sage‐N Research; Sequest version v.27, rev. 11, Thermo‐Finnigan, Thermo Fisher Scientific, Germany). A target‐decoy database containing the translated protein sequences from the metagenome, as well as common laboratory contaminants, was searched for protein identification (search parameters: peptide tolerance: 10 ppm; tolerance for fragment ions: 1 amu; band y‐ion series; variable modification: oxidation of methionine, 15.99 Da, max three modifications per peptide). Evaluation of MS/MS‐based peptide and protein identifications was performed with Scaffold version 4.0.4 (http://www.proteomesoftware.com/) with the following filter settings: Xcorr for doubly charged peptides 2.2, for triply charged peptides 3.3 and for quadruply charged peptides 3.8; Cn score 0.1 (Pjevac et al., 2018). Protein false discovery rates (FDRs) were between 0.3% and 0.5%, and peptide FDRs were between 0.0% and 0.1% throughout all experiments. Only proteins or protein groups with at least two unique peptides were considered identified.
To calculate normalized spectral abundance factors (NSAFs), total spectral count values for each protein or protein group were normalized to protein size (molecular weight in kDa) and to the sum of all spectral counts in the sample (Zybailov et al., 2006), thus giving the relative abundance of each protein/protein group in percentage of all proteins in the same sample.
Nucleotide sequence accession numbers
All sequencing data have been deposited at the European Nucleotide Archive under project accession number: PRJEB27164. The amplicon reads for the inactive chimneys are available under accession numbers ERR2639345–ERR2639351. The unassembled error‐corrected metagenomic reads are available under accession numbers ERR2639421–ERR2639424. The annotated contigs of the final bins are available under accession numbers ERZ665729–ERZ665792.
Proteome data accession numbers
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifier PXD010074 and https://doi.org/10.6019/PXD010074.
Supporting information
Appendix S1: Supplementary Information
Appendix S2: Supplementary Information
Acknowledgements
We would like to thank officers, crew, shipboard scientific party and the technical team of the ROV Quest 4000 m (MARUM) on R/V Sonne cruise SO216, for their invaluable assistance. The cruise SO216 with R/V Sonne was an integral part of the Cluster of Excellence of the MARUM ‘The Ocean in the Earth System, Research Area GB: Geosphere‐Biosphere Interactions’ funded by the German Research Foundation (DFG). We thank Richard Reinhardt, Bruno Huettel and the team of the Max Planck Genome Centre in Cologne for sequencing and Hanno Teeling for help with computational analyses. Furthermore, we thank Kathrin Büttner for excellent technical assistance in the laboratory and Bledina Dede for a helping hand in the genome assembly and annotation. This work was supported by the Max Planck Society.
Contributor Information
Dimitri V. Meier, Email: meier@microbial-ecology.net.
Anke Meyerdierks, Email: ameyerdi@mpi-bremen.de.
References
- Acosta‐Gonzalez, A. , Rossello‐Mora, R. , and Marques, S. (2013) Characterization of the anaerobic microbial community in oil‐polluted subtidal sediments: aromatic biodegradation potential after the Prestige oil spill. Environ Microbiol 15: 77–92. [DOI] [PubMed] [Google Scholar]
- Adams, L.F. , and Ghiorse, W.C. (1987) Characterization of extracellular Mn2+‐oxidizing activity and isolation of an Mn2+‐oxidizing protein from Leptothrix discophora SS‐1. J Bacteriol 169: 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amend, J.P. , McCollom, T.M. , Hentscher, M. , and Bach, W. (2011) Catabolic and anabolic energy for chemolithoautotrophs in deep‐sea hydrothermal systems hosted in different rock types. Geochim Cosmochim Acta 75: 5736–5748. [Google Scholar]
- Arshad, A. , Dalcin Martins, P. , Frank, J. , Jetten, M.S.M. , Op den Camp, H.J.M. , and Welte, C.U. (2017) Mimicking microbial interactions under nitrate‐reducing conditions in an anoxic bioreactor: enrichment of novel Nitrospirae bacteria distantly related to Thermodesulfovibrio . Environ Microbiol 19: 4965–4977. [DOI] [PubMed] [Google Scholar]
- Averyt, K.B. , and Paytan, A. (2003) Empirical partition coefficients for Sr and Ca in marine barite: implications for reconstructing seawater Sr and Ca concentrations. Geochem Geophys Geosyst 4 10.1029/2002GC000426 [DOI] [Google Scholar]
- Aziz, R.K. , Bartels, D. , Best, A.A. , DeJongh, M. , Disz, T. , Edwards, R.A. , et al (2008) The RAST server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach, W. , Edwards, K.J. , Hayes, J.M. , Huber, J.A. , Sievert, S.M. , and Sogin, M.L. (2006) Energy in the dark: fuel for life in the deep ocean and beyond. Eos, Trans Am Geophys Union 87: 73–78. [Google Scholar]
- Baker, B.J. , Lazar, C.S. , Teske, A.P. , and Dick, G.J. (2015) Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. , et al (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J Comput Biol 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco, R.A. , Emerson, D. , Sylvan, J.B. , Orcutt, B.N. , Jacobson Meyers, M.E. , Ramirez, G.A. , et al (2015) New insight into microbial iron oxidation as revealed by the proteomic profile of an obligate iron‐oxidizing chemolithoautotroph. Appl Environ Microbiol 81: 5927–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco, R.A. , Hoffman, C.L. , Ramirez, G.A. , Toner, B.M. , Edwards, K.J. , and Sylvan, J.B. (2017) In‐situ incubation of iron‐sulfur mineral reveals a diverse chemolithoautotrophic community and a new biogeochemical role for Thiomicrospira . Environ Microbiol 19: 1322–1337. [DOI] [PubMed] [Google Scholar]
- Benz, M. , Brune, A. , and Schink, B. (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemoheterotrophic nitrate‐reducing bacteria. Arch Microbiol 169: 159–165. [DOI] [PubMed] [Google Scholar]
- Binns, R.A. , and Scott, S.D. (1993) Actively forming polymetallic sulfide deposits associated with felsic volcanic‐rocks in the Eastern Manus back‐arc basin, Papua‐New‐Guinea. Econ Geol 88: 2226–2236. [Google Scholar]
- Blount, C. (1977) Barite solubilities and thermodynamic quantities up to 300 C and 1400 bars. Am Mineral 62: 942–957. [Google Scholar]
- Bond, P.L. , Druschel, G.K. , and Banfield, J.F. (2000) Comparison of acid mine drainage microbial communities in physically and geochemically distinct ecosystems. Appl Environ Microbiol 66: 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon, M. , Heijnen, J.J. , and Hansford, G.S. (1998) The mechanism and kinetics of bioleaching sulphide minerals. Miner Process Extr Metall Rev 19: 107–115. [Google Scholar]
- Campbell, J.H. , O'Donoghue, P. , Campbell, A.G. , Schwientek, P. , Sczyrba, A. , Woyke, T. , et al (2013) UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci U S A 110: 5540–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmer, A.R. , and Hinkle, M.E. (1947) The role of microorganisms in acid mine drainage: a preliminary report. Science 106: 253–256. [DOI] [PubMed] [Google Scholar]
- de Ronde, C.E.J. , Hannington, M.D. , Stoffers, P. , Wright, I.C. , Ditchburn, R.G. , Reyes, A.G. , et al (2005) Evolution of a submarine magmatic‐hydrothermal system: brothers volcano, southern Kermadec arc, New Zealand. Econ Geol 100: 1097–1133. [Google Scholar]
- Ditchburn, R.G. , De Ronde, C.E.J. , and Barry, B.J. (2012) Radiometric dating of volcanogenic massive sulfides and associated iron oxide crusts with an emphasis on 226Ra/Ba and 228Ra/226Ra in volcanic and hydrothermal processes at intraoceanic arcs. Econ Geol 107: 1635–1648. [Google Scholar]
- Du, Z.J. , Wang, Z.J. , Zhao, J.X. , and Chen, G.J. (2016) Woeseia oceani gen. nov., sp.nov., a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. nov. Int J Syst Evol Microbiol 66: 107–112. [DOI] [PubMed] [Google Scholar]
- Duperron, S. , Rodrigues, C.F. , Leger, N. , Szafranski, K. , Decker, C. , Olu, K. , and Gaudron, S.M. (2012) Diversity of symbioses between chemosynthetic bacteria and metazoans at the Guiness cold seep site (Gulf of Guinea, West Africa). Microbiology 1: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyksma, S. , Bischof, K. , Fuchs, B.M. , Hoffmann, K. , Meier, D. , Meyerdierks, A. , et al (2016) Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J 10: 1939–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard, C. , Wirsen, C.O. , and Jannasch, H.W. (1995) Oxidation of polymetal sulfides by chemolithoautotrophic bacteria from deep‐sea hydrothermal vents. Geomicrobiol J 13: 145–164. [Google Scholar]
- Eddie, B.J. , Wang, Z. , Hervey, W.J. , Leary, D.H. , Malanoski, A.P. , Tender, L.M. , et al (2017) Metatranscriptomics supports the mechanism for biocathode electroautotrophy by “Candidatus Tenderia electrophaga”. mSystems 2: e00002–e00017. Rabaey K (ed). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddie, B.J. , Wang, Z. , Malanoski, A.P. , Hall, R.J. , Oh, S.D. , Heiner, C. , et al (2016) “Candidatus Tenderia electrophaga”, an uncultivated electroautotroph from a biocathode enrichment. Int J Syst Evol Microbiol 66: 2178–2185. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. (2011) Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, K.J. , Bond, P.L. , Gihring, T.M. , and Banfield, J.F. (2000) An archaeal iron‐oxidizing extreme acidophile important in acid mine drainage. Science 287: 1796–1799. [DOI] [PubMed] [Google Scholar]
- Edwards, K.J. , Rogers, D.R. , Wirsen, C.O. , and McCollom, T.M. (2003) Isolation and characterization of novel psychrophilic, neutrophilic, Fe‐oxidizing, chemolithoautotrophicα‐ and γ‐Proteobacteria from the deep sea. Appl Environ Microbiol 69: 2906–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren, A.M. , Esen, O.C. , Quince, C. , Vineis, J.H. , Morrison, H.G. , Sogin, M.L. , and Delmont, T.O. (2015) Anvi'o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3: e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Bateman, A. , Clements, J. , Coggill, P. , Eberhardt, R.Y. , Eddy, S.R. , et al (2014) Pfam: the protein families database. Nucleic Acids Res 42: D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood, B.E. , Jones, D.S. , and Bailey, J.V. (2015) Sedimenticola thiotaurini sp.nov., a sulfur‐oxidizing bacterium isolated from salt marsh sediments, and emended descriptions of the genus Sedimenticola and Sedimenticola selenatireducens . Int J Syst Evol Microbiol 65: 2522–2530. [DOI] [PubMed] [Google Scholar]
- Frank, K.L. , Rogers, D.R. , Olins, H.C. , Vidoudez, C. , and Girguis, P.R. (2013) Characterizing the distribution and rates of microbial sulfate reduction at Middle Valley hydrothermal vents. ISME J 7: 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Pereira, P.R. , Schüler, M. , Fuchs, B.M. , Bennke, C. , Teeling, H. , Waldmann, J. , et al (2012) Genomic content of uncultured Bacteroidetes from contrasting oceanic provinces in the North Atlantic Ocean. Environ Microbiol 14: 52–66. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Toril, E. , Llobet‐Brossa, E. , Casamayor, E.O. , Amann, R. , and Amils, R. (2003) Microbial ecology of an extreme acidic environment, the Tinto River. Appl Environ Microbiol 69: 4853–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greening, C. , Biswas, A. , Carere, C.R. , Jackson, C.J. , Taylor, M.C. , Stott, M.B. , et al (2016) Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J 10: 761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber‐Vodicka, H.R. , Pruesse, E. , and Seah Brandon K (2017) phyloFlash. Online [DOI] [PMC free article] [PubMed]
- Guindon, S. , Dufayard, J.F. , Lefort, V. , Anisimova, M. , Hordijk, W. , and Gascuel, O. (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321. [DOI] [PubMed] [Google Scholar]
- Haymon, R.M. (1983) Growth history of hydrothermal black smoker chimneys. Nature 301: 695–698. [Google Scholar]
- He, S. , Barco, R.A. , Emerson, D. , and Roden, E.E. (2017) Comparative genomic analysis of neutrophilic iron(II) oxidizer genomes for candidate genes in extracellular electron transfer. Front Microbiol 8: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Forslund, K. , Coelho, L.P. , Szklarczyk, D. , Jensen, L.J. , von Mering, C. , and Bork, P. (2017) Fast genome‐wde functional annotation through orthology assignment by eggNOG‐mapper. Mol Biol Evol 34: 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Szklarczyk, D. , Forslund, K. , Cook, H. , Heller, D. , Walter, M.C. , et al (2016) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44: D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hügler, M. , and Sievert, S.M. (2011) Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann Rev Mar Sci 3: 261–289. [DOI] [PubMed] [Google Scholar]
- Jamieson, J.W. , Hannington, M.D. , Clague, D.A. , Kelley, D.S. , Delaney, J.R. , Holden, J.F. , et al (2013) Sulfide geochronology along the endeavour segment of the Juan de Fuca Ridge. Geochem Geophys Geosyst 14: 2084–2099. [Google Scholar]
- Jannasch, H.W. , and Mottl, M.J. (1985) Geomicrobiology of deep‐sea hydrothermal vents. Science 229: 717–725. [DOI] [PubMed] [Google Scholar]
- Jensen, A.B. , and Webb, C. (1995) Ferrous sulphate oxidation using Thiobacillus ferrooxidans: a review. Process Biochem 30: 225–236. [Google Scholar]
- Jørgensen, B.B. , and Nelson, D.C. (2004) Sulfide oxidation in marine sediments: geochemistry meets microbiology. Geol Soc Am Spec Paper 379: 63–81. [Google Scholar]
- Kato, S. , Ikehata, K. , Shibuya, T. , Urabe, T. , Ohkuma, M. , and Yamagishi, A. (2015a) Potential for biogeochemical cycling of sulfur, iron and carbon within massive sulfide deposits below the seafloor. Environ Microbiol 17: 1817–1835. [DOI] [PubMed] [Google Scholar]
- Kato, S. , Ohkuma, M. , Powell, D.H. , Krepski, S.T. , Oshima, K. , Hattori, M. , et al (2015b) Comparative genomic insights into ecophysiology of neutrophilic, microaerophilic iron oxidizing bacteria. Front Microbiol 6: 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, S. , Shibuya, T. , Takaki, Y. , Hirai, M. , Nunoura, T. , and Suzuki, K. (2018) Genome‐enabled metabolic reconstruction of dominant chemosynthetic colonizers in deep‐sea massive sulfide deposits. Environ Microbiol 20: 862–877. [DOI] [PubMed] [Google Scholar]
- Kato, S. , Takano, Y. , Kakegawa, T. , Oba, H. , Inoue, K. , Kobayashi, C. , et al (2010) Biogeography and biodiversity in sulfide structures of active and inactive vents at deep‐sea hydrothermal fields of the Southern Mariana Trough. Appl Environ Microbiol 76: 2968–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota, N. , Kanemori, M. , Sasayama, Y. , Aida, M. , and Fukumori, Y. (2007) Identification of endosymbionts in Oligobrachia mashikoi (Siboglinidae, Annelida). Microbes Environ 22: 136–144. [Google Scholar]
- Lenk, S. , Arnds, J. , Zerjatke, K. , Musat, N. , Amann, R. , and Mussmann, M. (2011) Novel groups of Gammaproteobacteria catalyse sulfur oxidation and carbon fixation in a coastal, intertidal sediment. Environ Microbiol 13: 758–774. [DOI] [PubMed] [Google Scholar]
- Li, D. , Liu, C. , Luo, R. , Sadakane, K. , and Lam, T. (2015) MEGAHIT: an ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31: 1674–1676. [DOI] [PubMed] [Google Scholar]
- Li, J. , Cui, J. , Yang, Q. , Cui, G. , Wei, B. , Wu, Z. , et al (2017) Oxidative weathering and microbial diversity of an inactive seafloor hydrothermal sulfide chimney. Front Microbiol 8: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Li, G. , Lai, Q. , Sun, F. , Du, Y. , and Shao, Z. (2015) Emcibacter nanhaiensis gen. nov. sp.nov., isolated from sediment of the South China Sea. Antonie Van Leeuwenhoek 107: 893–900. [DOI] [PubMed] [Google Scholar]
- Lopez‐Archilla, A.I. , Marin, I. , and Amils, R. (2001) Microbial community composition and ecology of an acidic aquatic environment: the Tinto River, Spain. Microb Ecol 41: 20–35. [DOI] [PubMed] [Google Scholar]
- Losekann, T. , Robador, A. , Niemann, H. , Knittel, K. , Boetius, A. , and Dubilier, N. (2008) Endosymbioses between bacteria and deep‐sea siboglinid tubeworms from an Arctic Cold Seep (Haakon Mosby Mud Volcano, Barents Sea). Environ Microbiol 10: 3237–3254. [DOI] [PubMed] [Google Scholar]
- Ludwig, W. , Strunk, O. , Westram, R. , Richter, L. , Meier, H. , Yadhukumar, B. A. et al (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane, M. , and Consortium, U . (2011) UniProt knowledgebase: a hub of integrated protein data. Database 2011: bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahe, F. , Rognes, T. , Quince, C. , de Vargas, C. , and Dunthorn, M. (2015) Swarm v2: highly‐scalable and high‐resolution amplicon clustering. PeerJ 3: e1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollom, T.M. , and Shock, E.L. (1997) Geochemical constraints on chemolithoautotrophic metabolism by microorganisms in seafloor hydrothermal systems. Geochim Cosmochim Acta 61: 4375–4391. [DOI] [PubMed] [Google Scholar]
- Meier, D.V. , Bach, W. , Girguis, P.R. , Gruber‐Vodicka, H.R. , Reeves, E.P. , Richter, M. , et al (2016) Heterotrophic Proteobacteria in the vicinity of diffuse hydrothermal venting. Environ Microbiol 18: 4348–4368. [DOI] [PubMed] [Google Scholar]
- Meier, D.V. , Pjevac, P. , Bach, W. , Hourdez, S. , Girguis, P.R. , Vidoudez, C. , et al (2017) Niche partitioning of diverse sulfur‐oxidizing bacteria at hydrothermal vents. ISME J 11: 1545–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, K. , Suzuki, K. , Yamaguchi, K. , Urabe, T. , and Hanada, S. (2015) Thiogranum longum gen. nov., sp.nov., an obligately chemolithoautotrophic, sulfur‐oxidizing bacterium of the family Ectothiorhodospiraceae isolated from a deep‐sea hydrothermal field, and an emended description of the genus Thiohalomonas . Int J Syst Evol Microbiol 65: 235–241. [DOI] [PubMed] [Google Scholar]
- Mußmann, M. , Pjevac, P. , Krüger, K. , and Dyksma, S. (2017) Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J 11: 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, S. , and Takai, K. (2008) Deep‐sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol Ecol 65: 1–14. [DOI] [PubMed] [Google Scholar]
- Narasingarao, P. , and Haggblom, M.M. (2006) Sedimenticola selenatireducens, gen. Nov., sp.nov., an anaerobic selenate‐respiring bacterium isolated from estuarine sediment. Syst Appl Microbiol 29: 382–388. [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Kindt, R. , Legendre, P. , Michin, P.R. , O'Hara, R.B. , et al.(2013) vegan: Community Ecology Package. R package version 2.0–10.
- Olins, H.C. , Rogers, D.R. , Frank, K.L. , Vidoudez, C. , and Girguis, P.R. (2013) Assessing the influence of physical, geochemical and biological factors on anaerobic microbial primary productivity within hydrothermal vent chimneys. Geobiology 11: 279–293. [DOI] [PubMed] [Google Scholar]
- Pachiadaki, M.G. , Lykousis, V. , Stefanou, E.G. , and Kormas, K.A. (2010) Prokaryotic community structure and diversity in the sediments of an active submarine mud volcano (Kazan mud volcano, East Mediterranean Sea). FEMS Microbiol Ecol 72: 429–444. [DOI] [PubMed] [Google Scholar]
- Parks, D.H. , Imelfort, M. , Skennerton, C.T. , Hugenholtz, P. , and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percak‐Dennett, E. , He, S. , Converse, B. , Konishi, H. , Xu, H. , Corcoran, A. , et al (2017) Microbial acceleration of aerobic pyrite oxidation at circumneutral pH. Geobiology 15: 690–703. [DOI] [PubMed] [Google Scholar]
- Pjevac, P. , Kamyshny, A., Jr. , Dyksma, S. , and Mussmann, M. (2014) Microbial consumption of zero‐valence sulfur in marine benthic habitats. Environ Microbiol 16: 3416–3430. [DOI] [PubMed] [Google Scholar]
- Pjevac, P. , Meier, D.V. , Markert, S. , Hentschker, C. , Schweder, T. , Becher, D. , et al (2018) Metaproteogenomic profiling of microbial communities colonizing actively venting hydrothermal chimneys. Front Microbiol 9: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. , and Arkin, A.P. (2010) FastTree 2 ‐ approximately maximum‐likelihood trees for large alignments. PLoS One 5: e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse, E. , Peplies, J. , and Glockner, F.O. (2012) SINA: accurate high‐throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28: 1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabus, R. , Hansen, T.A. , and Widdel, F. (2013)Dissimilatory sulfate‐ and sulfur‐reducing prokaryotes . In The Prokaryotes. Berlin, Heidelberg: Springer Berlin Heidelberg, pp.309–404. [Google Scholar]
- Reeves, E.P. , Prieto, X. , Hentscher, M. , Rosner, M. , Seewald, J.S. , Hinrichs, K.U. , and Bach, W. (2011) Phase separation, degassing and anomalous methane at the Menez Gwen hydrothermal field. Mineral Mag 75: 1702. [Google Scholar]
- Reeves, E.P. , Yoshinaga, M.Y. , Pjevac, P. , Goldenstein, N.I. , Peplies, J. , Meyerdierks, A. , et al (2014) Microbial lipids reveal carbon assimilation patterns on hydrothermal sulfide chimneys. Environ Microbiol 16: 3515–3532. [DOI] [PubMed] [Google Scholar]
- Reguera, G. , McCarthy, K.D. , Mehta, T. , Nicoll, J.S. , Tuominen, M.T. , and Lovley, D.R. (2005) Extracellular electron transfer via microbial nanowires. Nature 435: 1098–1101. [DOI] [PubMed] [Google Scholar]
- Richter, M. , Rossello‐Mora, R. , Glöckner, F. O. , and Peplies, J. (2016) J Species WS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32: 929–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson, R.L. , and Postgate, J.R. (1980) Oxygen and hydrogen in biological nitrogen fixation. Annu Rev Microbiol 34: 183–207. [DOI] [PubMed] [Google Scholar]
- Schippers, A. , and Jørgensen, B.B. (2002) Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments. Geochim Cosmochim Acta 66: 85–92. [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , et al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievert, S.M. , and Vetriani, C. (2012) Chemoautotrophy at deep‐sea vents: past, present, and future. Oceanography 25: 218–233. [Google Scholar]
- Søndergaard, D. , Pedersen, C.N.S. , and Greening, C. (2016) HydDB: a web tool for hydrogenase classification and analysis. Sci Rep 6: 34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin, D.Y. , Tourova, T.P. , Braker, G. , and Muyzer, G. (2007) Thiohalomonas denitrificans gen. Nov., sp.nov. and Thiohalomonas nitratireducens sp.nov., novel obligately chemolithoautotrophic, moderately halophilic, thiodenitrifying Gammaproteobacteria from hypersaline habitats. Int J Syst Evol Microbiol 57: 1582–1589. [DOI] [PubMed] [Google Scholar]
- Sorokin, D.Y. , Tourova, T.P. , Kolganova, T.V. , Sjollema, K.A. , and Kuenen, J.G. (2002) Thioalkalispira microaerophila gen. nov., sp.nov., a novel lithoautotrophic, sulfur‐oxidizing bacterium from a soda lake. Int J Syst Evol Microbiol 52: 2175–2182. [DOI] [PubMed] [Google Scholar]
- Spring, S. , Kämpfer, P. , Ludwig, W. , and Schleifer, K.H. (1996) Polyphasic characterization of the genus Leptothrix: new descriptions of Leptothrix mobilis sp.nov. and Leptothrix discophora sp.nov. nom. rev. and emended description of Leptothrix cholodnii emend. Syst Appl Microbiol 19: 634–643. [Google Scholar]
- Stokke, R. , Dahle, H. , Roalkvam, I. , Wissuwa, J. , Daae, F.L. , Tooming‐Klunderud, A. , et al (2015) Functional interactions among filamentous Epsilonproteobacteria and Bacteroidetes in a deep‐sea hydrothermal vent biofilm. Environ Microbiol 17: 4063–4077. [DOI] [PubMed] [Google Scholar]
- Strous, M. , Kraft, B. , Bisdorf, R. and Tegetmeyer, H. E. (2012) The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub, K.L. , Benz, M. , Schink, B. , and Widdel, F. (1996) Anaerobic, nitrate‐dependent microbial oxidation of ferrous iron. Appl Environ Microbiol 62: 1458–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudek, L. A. , Templeton, A. S. , Tebo, B. M. , and Staudigel, H. (2009) Microbial ecology of Fe (hydr)oxide mats and basaltic rock from Vailulu'u seamount, American Samoa. Geomicrobiol. J. 26: 581–596. [Google Scholar]
- Suzuki, Y. , Inagaki, F. , Takai, K. , Nealson, K.H. , and Horikoshi, K. (2004) Microbial diversity in inactive chimney structures from deep‐sea hydrothermal systems. Microb Ecol 47: 186–196. [DOI] [PubMed] [Google Scholar]
- Sylvan, J.B. , Sia, T.Y. , Haddad, A.G. , Briscoe, L.J. , Toner, B.M. , Girguis, P.R. , and Edwards, K.J. (2013) Low temperature geomicrobiology follows host rock composition along a geochemical gradient in Lau Basin. Front Microbiol 4: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvan, J.B. , Toner, B.M. , and Edwards, K.J. (2012) Life and death of deep‐sea vents: bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. MBio 3: e00279–e00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, H. , Fuchs, B.M. , Becher, D. , Klockow, C. , Gardebrecht, A. , Bennke, C.M. , et al (2012) Substrate‐controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611. [DOI] [PubMed] [Google Scholar]
- Thal, J. , Tivey, M. , Yoerger, D. , Jons, N. , and Bach, W. (2014) Geologic setting of PACManus hydrothermal area—high resolution mapping and in situ observations. Mar Geol 355: 98–114. [Google Scholar]
- Tivey, M.K. (2007) Generation of seafloor hydrothermal vent fluids and associated mineral deposits. Oceanography 20: 50–65. [Google Scholar]
- Toner, B.M. , Lesniewski, R.A. , Marlow, J.J. , Briscoe, L.J. , Santelli, C.M. , Bach, W. , et al (2013) Mineralogy drives bacterial biogeography of hydrothermally inactive seafloor sulfide deposits. Geomicrobiol.J. 30: 313–326. [Google Scholar]
- Vizcaíno, J.A. , Csordas, A. , Del‐Toro, N. , Dianes, J.A. , Griss, J. , Lavidas, I. , et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voordeckers, J.W. , Do, M.H. , Hugler, M. , Ko, V. , Sievert, S.M. , and Vetriani, C. (2008) Culture dependent and independent analyses of 16S rRNA and ATP citrate lyase genes: a comparison of microbial communities from different black smoker chimneys on the Mid‐Atlantic Ridge. Extremophiles 12: 627–640. [DOI] [PubMed] [Google Scholar]
- Williams, K.P. , Gillespie, J.J. , Sobral, B.W. , Nordberg, E.K. , Snyder, E.E. , Shallom, J.M. , and Dickerman, A.W. (2010) Phylogeny of Gammaproteobacteria . J Bacteriol 192: 2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza, P. , Yilmaz, P. , Pruesse, E. , Glockner, F.O. , Ludwig, W. , Schleifer, K.H. , et al (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12: 635–645. [DOI] [PubMed] [Google Scholar]
- Yeats, C.J. , Parr, J.M. , Binns, R.A. , Gemmell, J.B. , and Scott, S.D. (2014) The SuSu Knolls hydrothermal field, eastern Manus Basin, Papua New Guinea: an active submarine high‐sulfidation copper‐gold system. Econ Geol 109: 2207–2226. [Google Scholar]
- Zecchin, S. , Mueller, R.C. , Seifert, J. , Stingl, U. , Anantharaman, K. , von Bergen, M. , et al (2018) Rice paddy Nitrospirae carry and express genes related to sulfate respiration: proposal of the new genus “Candidatus Sulfobium.”. Appl. Environ. Microbiol 84: AEM.02224–AEM.02217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Yohe, T. , Huang, L. , Entwistle, S. , Wu, P. , Yang, Z. , et al (2018) dbCAN2: a meta server for automated carbohydrate‐active enzyme annotation. Nucleic Acids Res 46: W95–W101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov, B. , Mosley, A.L. , Sardiu, M.E. , Coleman, M.K. , Florens, L. , and Washburn, M.P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae . J Proteome Res 5: 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Appendix S2: Supplementary Information